

Abstract
Background.
Immunotherapies targeting cellular immunity are currently approved for treatment of melanoma, renal cell carcinoma, and prostate cancer. Studies on the immunogenicity and immune responsiveness of pediatric tumors are limited, therefore, it remains unclear to what extent T-cell-based immunotherapy holds promise for pediatric solid tumors.
Procedure.
A new rhabdomyosarcoma cell line (M3–9-M) was derived from an embryonal rhabdomyosarcoma (ERMS) occurring in a C57BL/6 mouse transgenic for hepatocyte growth factor and heterozygous for mutated p53. Primary tumors and metastases derived from M3–9-M were studied for similarities to human ERMS, and for immunogenicity and immune responsiveness.
Results.
Primary and metastatic tumors develop after orthotopic injection of M3–9-M into immunocompetent C57BL/6 mice, which mirror human ERMS with regard to histology, gene expression, and metastatic behavior. Whole cell vaccination using irradiated M3–9-M cells or M3–9-M-pulsed dendritic cells (DC)-induced tumor-specific T-cell responses that prevent tumor growth following low-dose tumor injection, and slow tumor growth following higher doses. Administration of anti-CD25 moAbs to deplete CD4+CD25+FOXP3+ regulatory T cells prior to tumor vaccination enhanced the potency of the ERMS tumor vaccine. Adoptive immunotherapy with M3–9-M primed T cells plus DC-based vaccination resulted in complete eradication of day 10 M3–9-M derived tumors.
Conclusions.
M3–9-M derived murine ERMS is immunogenic and immunoresponsive; regulatory T cells contribute to immune evasion by murine rhabdomyosarcoma. Adoptive immunotherapy with DC vaccination can eradicate low tumor burdens. Future work will seek to identify the tumor-associated antigens that mediate protective and therapeutic immunity in this model.
Keywords: adoptive immunotherapy, embryonal rhabdomyosarcoma, immunotherapy, regulatory T cells, rhabdomyosarcoma, tumor antigen, tumor vaccine
INTRODUCTION
Rhabdomyosarcoma (RMS) is the most common soft tissue sarcoma in children and accounts for approximately 5% of childhood cancer. Over the last several decades, progressive improvements in multimodal therapy have improved overall survival rates to 60–70%, but many challenges remain. Numerous cytotoxic agents are active against rhabdomyosarcoma [1,2], but recent attempts at dose-escalation and incorporation of new cytotoxic agents into frontline regimens have not substantially changed outcomes [3]. Most children who present with metastatic disease or who relapse following primary therapy will eventually die from progressive disease [4,5], and short-term and long-term toxicities of current standard regimens are substantial [6]. Therefore, progress against rhabdomyosarcoma rests upon the development of novel targeted therapies with greater efficacy and less toxicity.
Proof-of-principle studies demonstrate that many cancers are immunogenic, as evidenced by their ability to induce measurable immune responses toward tumor-associated antigens. Further, some tumors are susceptible to immune-based targeting and can thus be considered immune responsive. What remains unclear is the extent to which immunogenicity and immune responsiveness of cancer varies based upon histology, host factors, stage and treatment regimen, and, ultimately, how likely it is that immune therapies will be effective across a wide range of tumor types. Immunogenicity and immune responsiveness have been most clearly demonstrated for malignant melanoma where tumor reactive T cells can frequently be grown from resected tumors [7,8] and where immunomodulators such as interleukin-2 [9] and anti-CTLA4 [10] induce reproducible response rates as single agents. Chronic myelogenous leukemia, in chronic phase, is also highly immune responsive based upon its susceptibility to donor leukocyte infusions following bone marrow transplant [11]. But it remains unclear to what extent malignant melanoma and chronic myelogenous leukemia are exceptional among cancers with regard to immunogenicity and/or immune responsiveness versus whether they represent one end of a spectrum on which most tumors lie. Recent data demonstrating improved survival rates in prostate cancer following dendritic cell (DC) vaccination [12], responses to donor leukocyte infusions sometimes seen in other leukemias and solid tumors [13], improved overall survival in patients receiving MTP-PE for non-metastatic osteosarcoma [14] and tumor regressions using genetically modified T cells in neuroblastoma [15], suggest that multiple tumor types are immunogeneic and/or immunoresponsive, at least to some degree.
Few studies have directly explored the immunogenicity and immune responsiveness of pediatric solid tumors and only one report has demonstrated a response of murine rhabdomyosarcoma to immune therapy [16]. The goals of this study were to describe a new mouse model for studying ERMS and to determine whether immunotherapy prevents or modulates growth of embryonal rhabdomyosarcoma (ERMS) in this model.
METHODS
Mice and Tumor Lines
Female C57BL/6 mice were obtained from the Animal Production Program, National Cancer Institute (NCI, Frederick, MD). Rag1−/− mice were purchased from The Jackson Laboratory (Bar Harbor, ME). C57BL/6 HGFTg+ mice were generated as previously described [17]. Mice were housed in a strict, pathogen-free environment at the NIH animal facility. All studies were approved by the NCI Animal Care and Use Committee. Experiments were initiated when mice were 6–10 weeks of age. The M3–9-M line was developed by harvesting a tumor occurring in an HGFTg+p53+/− mouse and implanting whole tumor fragments into the leg of a C57BL/6 recipient, then harvesting metastatic tumors from intra-abdominal lymph nodes. M3–9-M was transduced with cDNA encoding firefly luciferase (pMSCV-puro-luciferase, a gift from Dr. Sam Hwang, Medical College of Wisconsin) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Where noted, B16 melanoma (a gift from Nick Restifo, Surgery Branch, NCI, Bethesda, MD) and RMS 76–9 [16] (a gift from Brenda Weigel, University of Minnesota), or YAC-1 cells were used in Cr51 release assays and vaccine experiments. M3–9-M cells were cultured in RPMI 1640 with 10% heat-inactivated fetal calf serum, HEPES buffer, 1% non-essential amino acids, 1% sodium pyruvate, 1% penicillin/streptomycin, 1% l-glutamine, all Invitrogen; and 50 μ-M 2-mercaptoethanol (Sigm–Aldrich, St. Louis, MO). For orthotopic tumor inoculation, cells were harvested by trypsin digestion, a single cell suspension was prepared, mice were shaved on the left hindlimb area and tumor cells were injected into the gastrocnemius muscle in 0.1 ml PBS (Invitrogen) using a sterile 27.5 G needle.
Histology and Immunostaining
M3–9-M tumor was harvested from tumor-bearing mice and tissues were fixed in 10% neutral buffered formalin at 4°C overnight, dehydrated, and embedded in paraffin. Tissue blocks were sectioned at 5 μm and stained with hematoxylin and eosin (H&E). The ARK kit (Dako North America, Carpinteria, CA) was used for immunohistochemistry. In brief, tissue sections were cleared in xylene and rehydrated in serial concentrations of ethanol. After peroxidase blocking for 5 min, sections were incubated with biotinylated primary anti-mouse actin antibody (1:100, Dako North America Inc, Carpinteria) for 15 min. Immunoperoxidase staining was performed according to the manufacturer’s protocol. For immunofluorescent staining, tissues were immediately frozen in Tissue Tek O.C.T. compound (Sakura Finetek USA Inc, Torrence, CA) and sectioned at 5 μm, fixed in acetone, blocked for 30 min using 5% goat serum, then stained with rabbit polyclonal anti-desmin (IgG, Ab 8592; Abcam Inc, Cambridge, MA) as the primary antibody (1:40, overnight at 4°C), then anti-rabbit IgG Alexa Fluor 488 conjugate (Invitrogen; 1:400, 45 min at room temperature), then washed and DAPI was added (1:500 dilution for approximately 30 sec). Background controls were stained with rabbit polyclonal IgG (Ab27478; Abcam Inc) as the primary antibody for an isotype control. Sections were viewed using a Zeiss AxioObserver Z1 microscope using a 20× differential interference contrast objective and appropriate filter sets. Images were captured with a Zeiss AxioCam MRm monochrome digital camera (Carl Zeiss Microimaging GmbH, Jena, Germany) and analyzed using AxioVision version 4.6 software (Carl Zeiss, Microimaging GmbH).
Gene Expression Analysis
Total RNAs were extracted from M3–9-M tumors. Gene expression analysis was performed using Affymetrix Mouse 430.2 arrays (Affymetrix Inc, Santa Clara, CA) according to Affymetrix protocol. The .CEL files were exported from Affymetrix GCOS software and normalized using RMA. Metagene projection analysis was performed as previously described [18]. Both mouse datasets and published human tumor datasets were used in this analysis [19,20]. During the data processing, a median value was selected in the case of multiple probes representing the same gene. The datasets from different platforms were matched and merged based on gene symbols. The genes were ranked according to their expression levels and the value replaced by 10,000 × (rank(gene) − 1)/(number of genes − 1). Thresholds were set at 20 and 100,000 U, and gene filtering excluded genes with <5-fold and 500 U of maximum difference. Gene set enrichment analysis (GSEA; http://www.broad.mit.edu/gsea/) was completed using a weighted enrichment statistic. Mouse RMS cell line and tumor sample data were combined and genes were ranked using log 2 intensity. A gene set of human RMS up-regulated genes is built by collecting RMS genes from published articles [21,22].
Bioluminescence Imaging
Where indicated, bioluminescence imaging was performed weekly to monitor tumor progression. Mice received 100 μl of 30 mg/ml of D-luciferin (Caliper Life Sciences, Hopkinton, MA), and then were anesthetized and the area of the tumor shaved, then transferred to the imaging chamber. A cooled charge coupled device camera apparatus (Xenogen IVIS In Vivo Imaging, Xenogen Corporation, Alameda, CA) was used to detect photon emission from tumor-bearing mice with an acquisition time of 60 sec. The emitted photons were displayed as a pseudocolor graphic, superimposed over a conventional photographic image. Display and analyses of the images were performed using Living Image software (Xenogen Corporation, Alameda, CA) and Igor Image analysis software (Wave Metrics Inc, Portland, OR).
Vaccination, Cell Depletion, and Cytotoxicity Assays
M3–9-M cells were grown to confluence and a single cell suspension was prepared. Cells were irradiated to 15,000 cGy, then washed and resuspended at 25 × 106 cell/ml in PBS. Vaccinated mice received 5 × 106 irradiated M3–9-M cells in PBS IP on the days indicated. Treg depletion was accomplished using affinity-purified, sterile, rat anti-mouse CD25 (moAb PC61, Biologic Resources Branch, NCI), 0.5 mg/dose IP 3 days prior to vaccination as previously described [23]. Where indicated, some groups of mice were depleted of CD8 and/or CD4 T cells using 250 μg of purified anti-CD8 (moAb 2.43, National Cell Culture Center, Minneapolis, MN) and/or 250 μ of anti-CD4 (moAb GK1.5, National Cell Culture Center), respectively, administered IP at the time points indicated.
Following anti-CD4 and/or anti-CD8 peripheral blood was analyzed for CD8 and/or CD4 T cells which confirmed >95% subset depletion.
Dendritic cell vaccinations were administered as previously described [24]. Briefly, bone marrow was flushed from femurs and tibiae of C57BL/6 mice, filtered, and erythrocytes lysed. Cells expressing the lineage-specific antigens CD5, CD45R, CD11b, Gr-1, and Ter-119 were depleted by magnetic-activated cell sorting (MACS, Lineage Depletion Kit, Miltenyi Biotec Inc, Auburn, CA), followed by positive selection for Sca-1 (Sca-1 Microbeads, Miltenyi Biotec, Auburn, CA). Sca-1+/lin− cells were cultured for 7 days in media supplemented with GM-CSF (1,000 μ/iml) and IL-4 (1,000 μ/ml), FLT3 ligand (25 ng/ml), SCF (100 ng/ml), and IL-3 (20 ng/ml; all from PeproTech Inc, Rocky Hill, NJ). Cultures were maintained at 37°C, 5% CO2 in tilted flasks. Apoptotic M3–9-M cells were generated by irradiation with 15,000 cGy. Following irradiation, M3–9-M cells were washed, counted, and incubated in a 1:1 ratio with DCs. DCs were activated 20 to 22 hr after co-culture with apoptotic cells using 20 mg/ml anti-CD40 moAb (I-C10, R&D Systems, Minneapolis, MN).
Chromium release cytotoxicity assays were performed using standard techniques. Briefly, spleen single cell suspensions from control or immunized mice were enriched for T cells using immunomagnetic selection (Pan T cell selection kit, Miltenyi Biotec Inc) to generate effector cells. M3–9-M cells, B16, RMS 76–9, and YAC-1 cells were labeled with Cr51 (PerkinElmer, Waltham, MA). Effector cells were plated, and where indicated purified anti-mouse H-2Kb/H-2Db antibody (clone 28-8-6; BioLegend Inc, San Diego, CA) was used to block MHC class 1 or purified mouse IgG2a, k isotype control (clone MG2a-53, BioLegend Inc, San Diego, CA) was used as an isotype control. Labeled targets were added to the effector cells at the effector:target ratios indicated. After 4 hr, supernatant was removed and placed in Luma plates (PerkinElmer), then read using a MicroBeta scintillation counter (PerkinElmer). Percent-specific lysis was calculated using: 100 × (experimental – spontaneous release)/ (maximal – spontaneous release).
Adoptive Immunotherapy
To generate M3–9-M primed cells for use in adoptive immunotherapy, C57BL/6 mice received irradiated tumor vaccines on days −14 and −7, then 1 × 104 tumor cell inoculation on day 0. Control mice and primed mice were sacrificed between days 80 and 130 post-tumor challenge, single cell suspensions were generated from spleens and lymph nodes, and T cells were positively selected using immunomagnetic labeling [CD8a (Ly-2) and CD4 (L3T4) microbeads, Miltenyi Biotec Inc]. Cells were washed and resuspended in RPMI at 5.5 × 106 T cells in 0.2 ml RPMI (1:1 CD4:CD8 ratio), then injected IV.
Statistical Analysis
Statistical tests were performed using GraphPad Prism, Version 4.0a for Macintosh (GraphPad Software Inc, La Jolla, CA). Significant differences for comparisons of two groups were determined by the Mann-Whitney test and survival curves were analyzed using a Wilcoxon log-rank test. P-values <0.05 were considered significant.
RESULTS
M3–9-M Provides a Representative Model of Pediatric Embryonal RMS As Evidenced by Histology, Gene Expression, and Aggressive Growth In Vivo
To assess whether M3–9-M growth in vivo mirrors human embryonal RMS, primary tumor fragments, and lung metastases derived from orthotopic tumors were sectioned and analyzed by histology. As shown in Figure 1A,B, H&E staining of primary tumors and lung metastases derived from M3–9-M emulate human rhabdomyosarcoma, immunohistochemistry shows muscle-specific actin expression in the malignant cells, and the tumors stain strongly for desmin. Gene expression data generated by microarray analysis of orthotopic primary tumors was investigated using a metagene model, and trained using gene expression data of different human tumors. As shown in the Figure 1C, M3–9-M derived samples (both cell lines grown ex vivo and tumor samples harvested in vivo) cluster with human RMS (ARMS and ERMS). Figure 1D demonstrates that genes up-regulated in human RMS by GSEA are also significantly enriched in the tested mouse samples. Tumor titration experiments monitoring growth of M3–9-M derived tumors in C57BL/6 mice demonstrate that M3–9-M is an aggressive tumor, with latency to tumor development being dose dependent. Interestingly, 80% of mice succumbed to tumor even following inoculation of only 100 cells/mouse indicating a very high frequency of tumor initiating cells (Fig. 1E).
Fig. 1.
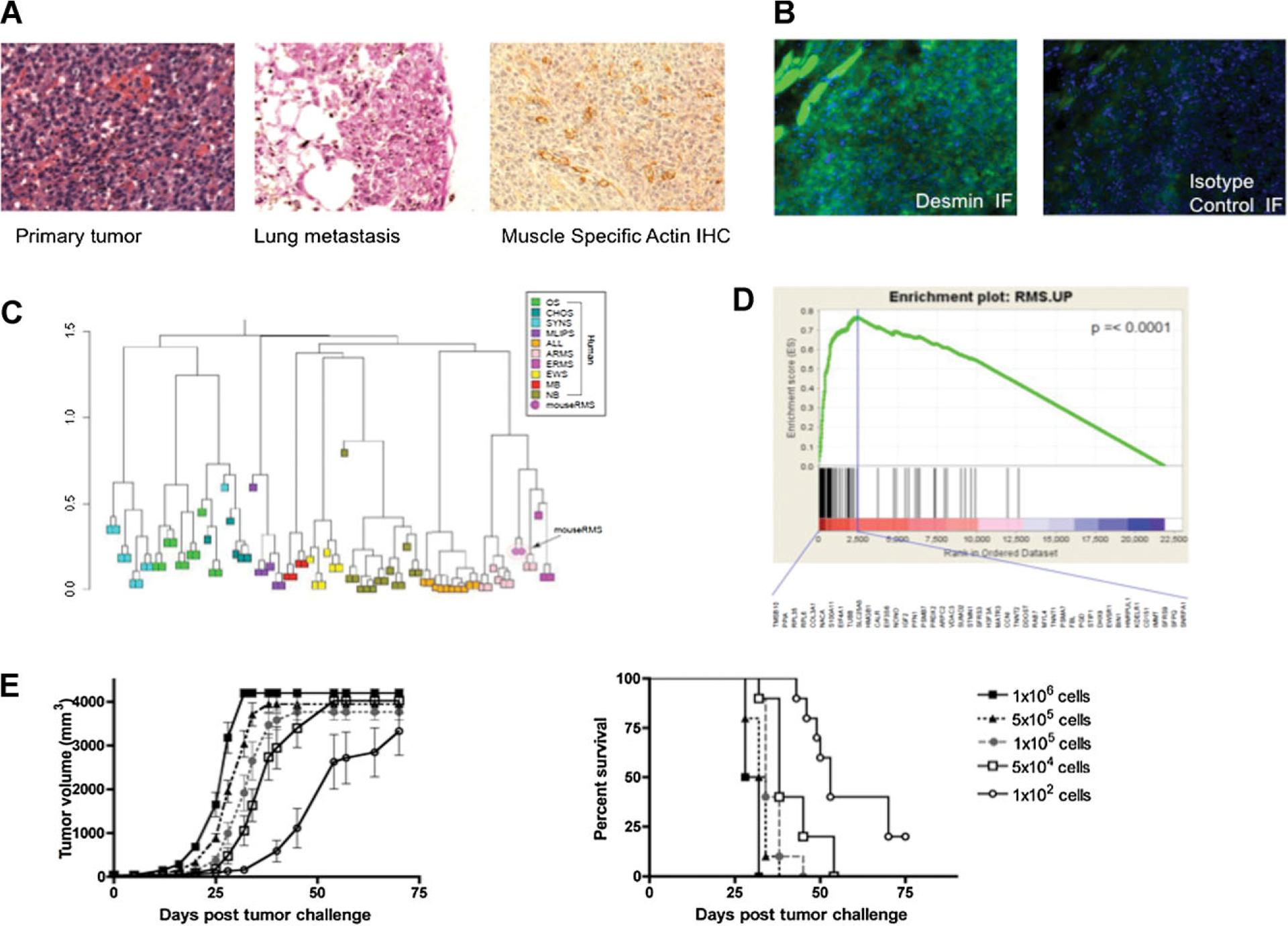
M3–9-M tumors accurately model pediatric rhabdomyosarcoma. A: H&E staining of primary and metastatic M3–9-M tumors and actin immunohistochemistry of primary M3–9-M tumors. B: Left panel: Desmin immunofluorescence and DAPI staining. Right panel: Isotype control immuofluorescence and DAPI staining (20× magnification). C: Hierarchical clustering after metagene projection. M3–9-M tumors (magenta circle) cluster with human embryonal RMS and alveolar RMS. D: Gene set enrichment analysis (GSEA) demonstrates that genes up-regulated in human RMS samples are significantly enriched in the M3–9-M tumors (P < 0.0001). The green curve represents the running sum of enrichment score for ranked genes. The long vertical line specifies the maximum ES score, and the genes listed under the plot are those in the leading edge subset. E: Dose-response tumor growth (left) and survival (right) curves of M3–9-M tumors following injection of the designated cell doses into the left gastrocnemius (n = 10/group).
T-Cell Responses Generated in Response to an Irradiated Whole-Cell Tumor Vaccine Prevent or Slow Growth of M3–9-M Tumors in a Tumor Dose-Dependent Manner
To determine whether vaccine-induced immune responses play any role in controlling growth of M3–9-M-derived tumors, we evaluated whether whole-cell vaccines could protect against primary tumor growth. 5 × 106 irradiated M3–9-M cells were administered IP on days −14 and −7, and mice were challenged with 5 × 105 viable M3–9-M cells on day 0. As shown in Figure 2A, mice receiving the irradiated tumor vaccine had significantly slower tumor growth (P = 0.005) and improved survival (P = 0.003) compared to mice receiving sham vaccine. This effect was completely abrogated by CD4 and CD8 depletion (P < 0.0001), and partially abrogated by depletion of CD8 cells alone (P = 0.001), or depletion of CD4 T cells alone (P = 0.018). Furthermore, vaccinated mice depleted of both CD8 and CD4 T cells showed diminished survival compared to control mice (P = 0.001), demonstrating that immune surveillance provides a modest deterrent to growth of M3–9-M.
Fig. 2.
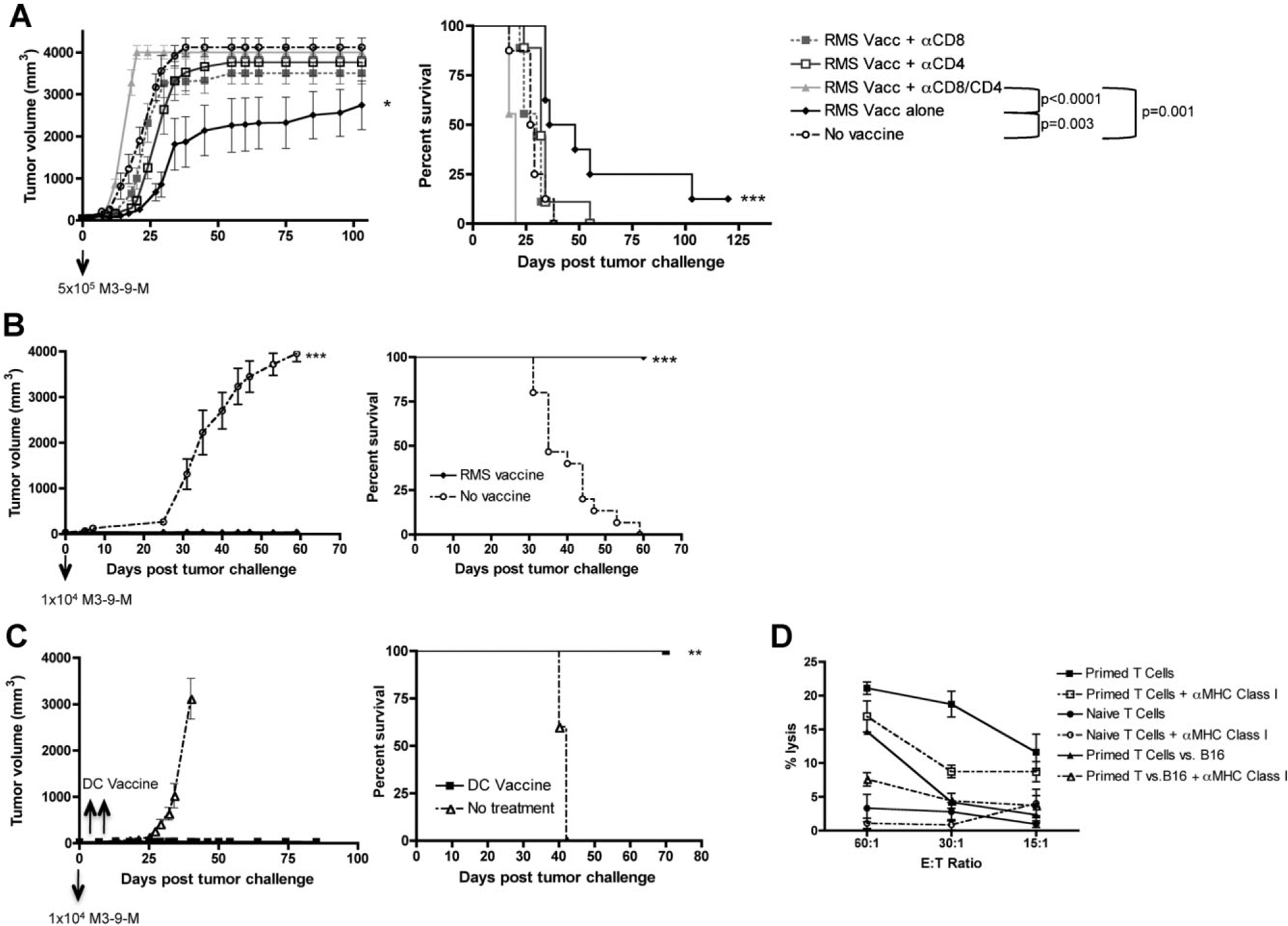
M3–9-M based whole tumor vaccines can prevent growth of M3–9-M-derived tumors and eradicate incipient M3–9-M tumors. A: Vaccinations comprising irradiated M3–9-M cells were administered on days −14 and −7, then mice were challenged with 5 × 105 viable M3–9-M tumor cells on day 0. Where indicated mice received anti-CD8 and/or anti-CD4 moAbs 3× weekly for 3 weeks beginning on day −4. Vaccinated mice showed significantly slower tumor growth (P = 0.005) and improved survival (P = 0.003) compared to controls. Vaccine mediated improvement in survival was abrogated by depletion of CD4 (P = 0.018) and/or CD8 (P = 0.001) T cells. Animals depleted of both CD4 and CD8 cells died more rapidly than vaccinated mice (P < 0.0001) and more rapidly than unvaccinated control animals (P = 0.001). Results are pooled from two independent experiments (n = 8–9/group). B: Animals were treated as in (A), except the dose of viable M3–9-M cell challenge was 1 × 104 cells. Vaccinated mice showed no tumor growth and 100% survival compared to progressive tumors (P < 0.001) and 0% survival (P < 0.0001) in all control mice. C: Mice received 1 × 104 viable M3–9-M cells on day 0, then vaccinations comprising IP injection of 1 × 106 bone marrow-derived dendritic cells co-cultured with irradiated M3–9-M cells on days 2 and 4 following tumor challenge. Mice receiving DC vaccination after tumor challenge show no tumor growth compared to controls (P = 0.0005), and improved survival compared to controls (P = 0.003). D: T cells from vaccinated mice (primed with M3–9-M-pulsed DCs then boosted with irradiated M3–9-M) show specific lysis of M3–9-M cells in a 4 hr Cr51 release assay, which is higher than T cells from naive mice. M3–9-M primed T cells did not kill B16 melanoma, and killing was inhibited by antibodies that block MHC class I.
Importantly, the tumor burden and/or the tumor latency period plays a critical role in determining the effectiveness of the prophylactic whole-cell vaccine in this model. When tumor challenge doses were lowered from 5 × 105 cells to 1 × 104 cells, the irradiated whole-cell vaccine completely prevented primary tumor growth of M3–9-M tumors in 100% of mice (P < 0.0001), resulting in 100% survival compared to 0% survival in mice given a sham vaccine (P < 0.0001; Fig. 2B). Consistent with the anti-tumor effect being associated with immune memory, five of the surviving mice treated in Figure 2B were rechallenged with 1 × 104 viable M3–9-M cells on day 67 following the first tumor challenge. All rechallenged mice survived without tumor while all untreated control mice were euthanized due to progressive tumors (Supplemental Appendix 1).
Preventative tumor vaccination is useful to demonstrate the presence of immune responses, but clinical application of immunotherapy requires that antitumor effects of vaccination be translated to a treatment model. We did not observe substantial differences in survival when irradiated tumor vaccines were administered after tumor challenge. Therefore, we sought to augment the efficacy of vaccination through the use of DCs pulsed with apoptotic tumor cells, an approach which has been potent in other models [24]. DC vaccines were given on days 2 and 4 post-tumor challenge of 1 × 104 cells into the left gastrocnemius muscle of C57BL/6 mice. None of the vaccinated mice developed tumors while all untreated control mice required euthanasia due to progressive tumor growth (Fig. 2C). The rate of tumor growth (P = 0.008) was significantly less in mice receiving DC vaccines compared to untreated control mice. Additionally, all mice receiving the DC vaccines survived compared to control mice (P = 0.003), which eventually had to be sacrificed due to maximum tumor burden (Fig. 2C). Furthermore, splenic T cells from mice vaccinated as in Figure 2C showed higher MHC class I restricted lysis of M3–9-M compared to T cells from non-vaccinated mice (Fig. 2D). Less killing of B16 melanoma was observed suggesting that the antigens involving in this immune-mediated protection could be RMS specific.
In summary, M3–9-M is immunogenic as evidenced by the activity of whole-cell-based vaccination in preventing primary tumor growth and in eradicating incipient tumors when used in the context of a DC vaccine. The immune effects are T cell mediated as evidenced by the demonstration of MHC class I restricted tumor lysis, abrogation of the effects with T cell depletion and long-term protection from tumor rechallenge consistent with immune memory.
Depletion of T-Regulatory Cells Enhances Anti-Tumor Effects of M3–9-M Whole-Cell Vaccines
FoxP3 expressing regulatory T cells have been implicated in many models of tumor escape and in clinical studies from tumor-bearing patients [25–27]. We sought to determine whether immune surveillance and/or the effectiveness of whole-cell vaccination in murine rhabdomyosarcoma could be augmented by depletion of Tregs. Cohorts of mice received anti-CD25 or sham injection on day −17, irradiated whole-cell vaccine or sham vaccines on days −14 and −7, and 1 × 105 M3–9-M tumor cell challenge on day 0. As shown in Figure 3A, administration of anti-CD25 prior to vaccination significantly enhanced the effectiveness of the tumor vaccine as evidenced by diminished tumor growth (P = 0.005) and improved overall survival (P = 0.013). Furthermore, even in the absence of tumor vaccination, depletion of Tregs significantly improved survival (33.4 vs. 44.5 days median survival) compared to controls (P-value of survival curves = 0.001; Fig. 3A). Bioluminescence imaging of M3–9-M tumor shown in Figure 3B provides visual evidence that anti-CD25 alone diminishes tumor growth.
Fig. 3.
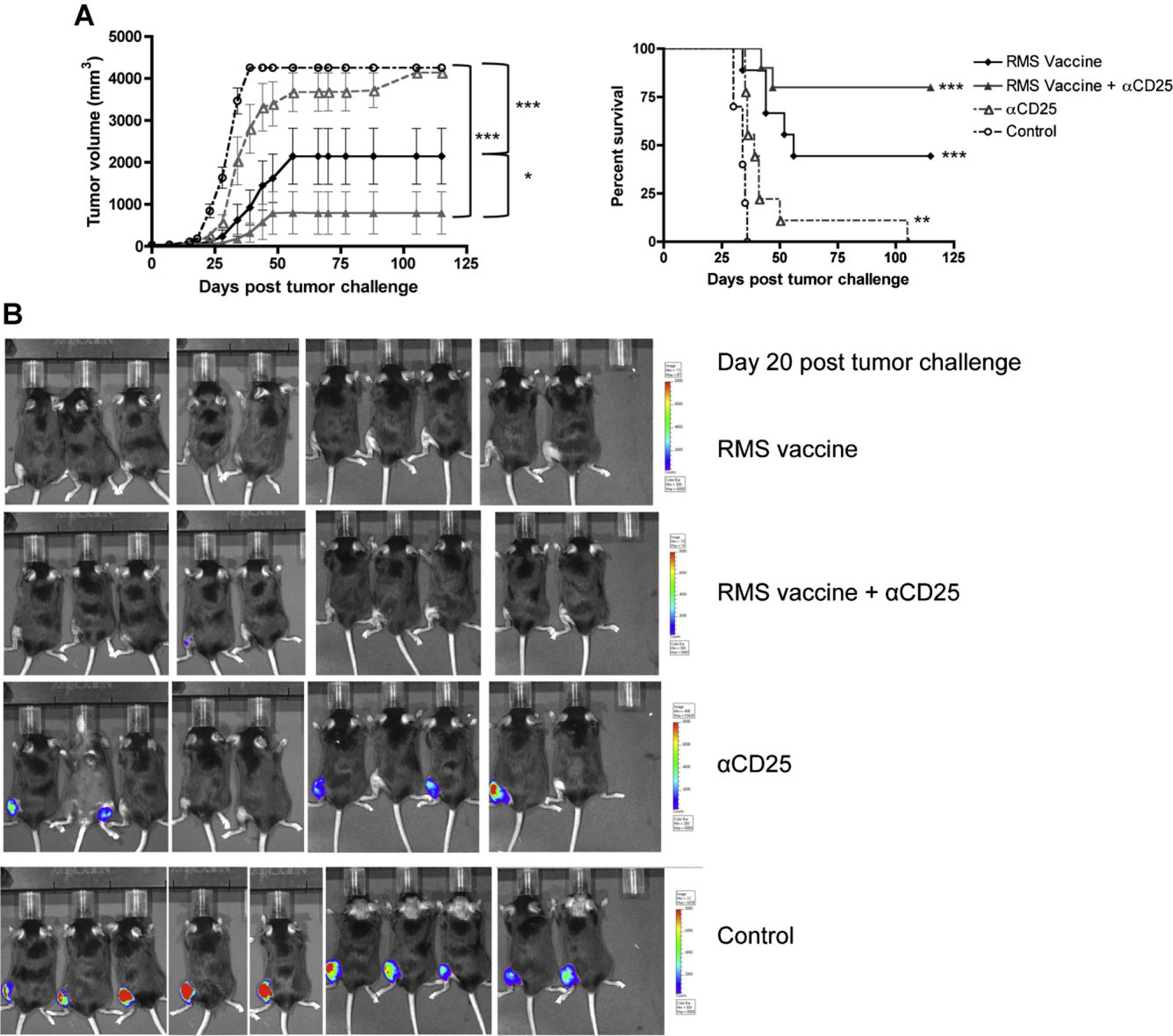
Depletion of T-regulatory cells enhances anti-tumor effects of the irradiated M3–9-M tumor vaccine. A: Cohorts of mice (5–10) received anti-CD25 on day −17 and/or irradiated M3–9-M vaccines on days −14 and −7, then were challenged with 1 × cells on day 0. Mice receiving vaccination plus anti-CD25 showed higher survival rates (75% vs. 50%) and slower tumor growth (P = 0.01) compared to mice receiving vaccination alone. Mice receiving anti-CD25 without vaccination also had improved survival compared to controls (P = 0.001). Results are pooled from two separate experiments (n = 9–10/group). B: Bioluminescence imaging of M3–9-M tumors in C57BL/6 mice on day 20 post-tumor challenge illustrates an absence of tumor at day 20 in both vaccinated groups and diminished tumor at day 20 in unvaccinated mice treated with anti-CD25. Bioluminescence was read as p/sec/cm2/sr with the minimum = 4.0 × 105 p/sec/cm2/sr and the maximum = 2.0 × 107 p/sec/cm2/sr.
Adoptive Transfer of M3–9-M Primed T Cells ± Dendritic Cell-Based Tumor Vaccination Diminishes Tumor Growth in a Treatment Model
To investigate whether adoptive therapy with M3–9-M primed T cells can impact growth of existent tumor, we adoptively transferred T cells from M3–9-M vaccinated and tumor challenged mice (treated as described in Fig. 2B) to T-cell replete and lymphopenic Rag1−/− mice on day 1 after inoculation with M3–9-M tumor. C57BL/6 mice receiving primed T cells showed almost no tumor growth compared to mice receiving either naive T cells (P = 0.002) or no T cells (P = 0.001; Fig. 4A). As further evidence for inherent immune surveillance impacting growth of rhabdomyosarcoma, we also observed that in the absence of adoptive transfer, the rate of tumor growth in immunocompetent C57BL/6 mice is significantly diminished compared to immunocompromised Rag1−/− mice (P = 0.025; Fig. 4A). As a result, primed T cells were more effective in controlling tumor growth in immunocompetent C57BL/6 mice as compared to Rag1−/− mice (P = 0.0011), similar to recently published results using other tumor models [28] (Fig. 4A).
Fig. 4.

Adoptive transfer with M3–9-M primed T cells plus dendritic cell vaccination eradicates 10 day M3–9-M tumors. A: T cells (2.25 × 106 CD4 and 2.25 × 106 CD8 cells) from mice vaccinated as in 2B (primed T cells, harvested on day 83 post-vaccine) versus control mice (naive T cells) were administered IV to Rag1−/− mice or C56BL/6 (F.B6) mice 1 day following administration of 1 × 104 viable M3–9-M cells. C57BL/6 mice receiving primed T cells show significantly diminished tumor growth and improved survival compared to mice receiving no T cells or naive T cells. C57BL/6 mice receiving no T cells survived significantly longer than Rag1−/− mice receiving no T cells, and C57BL/6 mice receiving primed T cells also had significantly diminished tumor growth and improved survival compared to Rag1−/− mice receiving primed T cells. B: Adoptive transfer of primed T cells (as in Fig. 2B) plus DC vaccine (as in Fig. 2C) on day 10 post-tumor challenge results in complete control of M3–9-M tumors. Tumor growth curves from mice treated on day 10 post-tumor challenge with either unpulsed DCs or M3–9-M-pulsed DCs and/or primed T cells or naïve T cells. Mice receiving M3–9-M-pulsed DC vaccination plus primed T cells showed complete control of tumors growth (P = 0.0006 compared to mice receiving unpulsed DCs vaccine or naive T cells). Mice receiving the M3–9-M-pulsed DCs only showed a significant delay in tumor growth (P = 0.026), and mice receiving only primed T cells showed a trend toward slower tumor growth, but it was not significant compared to controls. Mice receiving both M3–9-M-pulsed DCs plus primed T cells had 100% tumor-free survival compared to control mice (P = 0.007).
To investigate whether adoptive therapy with M3–9-M primed T cells in combination with a DC-based vaccine can impact growth of existent tumor at a late time point (10 days post-tumor challenge), we gave either pulsed or unpulsed DCs and/or adoptively transferred T cells from M3–9-M vaccinated and tumor challenged mice (treated as described in Fig. 2B) to T-cell replete mice on day 10 after inoculation with 1 × 104 M3–9-M tumor cells (Fig. 4B). C57BL/6 mice receiving primed T cells in combination with a pulsed DC vaccine showed no tumor growth compared to mice receiving unpulsed DCs and naive T cells (P = 0.0006) and compared to control mice (P = 0.0006; Fig. 4B). Mice receiving only a pulsed DC vaccine showed a significant delay in tumor growth compared to mice receiving unpulsed DCs (P = 0.026; Fig. 4B), but the effect was not as potent as when the vaccine was combined with adoptive immunotherapy. While the adoptive transfer of primed T cells appeared to slow tumor growth, the adoptive transfer alone was not enough to delay tumor growth significantly. Thus, combining adoptive immunotherapy with DC vaccine leads to significant control of murine ERMS, and this effect is more potent than with either modality alone.
DISCUSSION
Little is known regarding the potential for immune-based therapy to mediate anti-tumor effects in pediatric tumors, but emerging clinical studies suggest that immunotherapy may be effective in these diseases. The addition of MTP-PE to multiagent chemotherapy in osteosarcoma improved overall survival [14], tumor vaccines administered to patients with neuroblastoma and fibrosarcoma have mediated tumor shrinkage [29,30], and recent work using genetically engineered T cells demonstrated antitumor effects in neuroblastoma [15]. Still, T-cell tumor antigens have not been identified for pediatric tumors and there is little understanding of the degree to which endogenous immune responses mediate antitumor effects. Several studies have demonstrated a relationship between lymphocyte numbers and outcome in a variety of cancers, but it remains unclear if this is causal or simply correlative [31–33].
Preclinical models provide an opportunity to enhance our understanding of tumor:immune interactions and to optimize immune-based therapies of cancer [34]. The B16 melanoma model has been used successfully to identify human tumor antigens in melanoma and to optimize cell-based therapies for this disease [35]. With regard to pediatric tumors, a genetically engineered whole-cell vaccine was demonstrated to slow growth of neuroblastoma in the A/J neuroblastoma model [36], and syngeneic adoptive T-cell transfer was demonstrated to diminish metastases in K7M2 osteosarcoma [37]. These studies provide proof-of-principle that pediatric solid tumors can be immunogenic and immune responsive, but neither the A/J nor the K7M2 model is on the C57BL/6 background, rendering further immune modeling, and antigen identification challenging. Recently, genetically engineered models have become available that accurately mimic ERMS [17,38] and alveolar rhabdomyosarcoma [39], but neither has previously been available on the C57BL/6 background.
We sought to develop a murine model of human RMS in C57BL/6 mice as a first step toward detailed studies of rhabdomyosarcoma immunobiology. The M3–9-M tumor cell line is derived from a RMS that developed in an HGFTg+p53+/− mouse that was fully backcrossed to C57BL/6. It generates aggressive tumors in immunocompetent hosts, which on pathologic review confirm their morphologic similarity to ERMS. Moreover, their expression of muscle-specific actin, desmin, and genes enriched in human rhabdomyosarcoma are consistent with M3–9-M derived tumors providing a reasonable model of the human disease. Interesting, the M3–9-M derived tumor did not show overexpression of EGFR or Fibrillin-2, which were recently reported to be expressed in human ERMS [40], therefore some distinctions between the murine and human tumors exist. Even 100 cells reproducibly generated tumors, illustrating a very high frequency of tumor initiating cells. Using a whole-cell vaccine, we provide evidence that M3–9-M murine RMS is immunogenic. Similar protection was observed when an irradiated non-luciferase transduced M3–9-M line was used for vaccination (Supplemental Appendix 2), ruling out the possibility that these immune responses reflected potential immunogenicity of the luciferase gene. This is notable, since B16 melanoma, a prototype of an immunoresponsive tumor, is not immunogenic when used as an irradiated whole tumor vaccine. While vaccine mediated immune protection was overcome with higher tumor doses, we present clear evidence that T-cell-mediated immunity could prevent growth of M3–9-M, and could eradicate incipient tumors following low tumor doses. This does not appear to be unique to M3–9-M, as we observed similar protection using 76–9, another RMS cell line on C57BL/6 (Supplemental Appendix 3). Further, Weigel et al. [16] demonstrated that CpG oligodeoxynucleotides, stimulators of innate immunity, in combination with cyclophosphamide slowed growth of large palpable 76–9 derived tumors, indirectly implicating tumor-associated antigens in antitumor effects. Together, the data from our RMS studies as well as other studies in neuroblastoma [41] and osteosarcoma [37] are consistent with an inherent immunogenicity associated with pediatric solid tumors. Optimal clinical extrapolation of this insight would come from the molecular identification of the antigens involved and we are actively engaged in these studies.
This work provides other significant insights. First, the data demonstrate the existence of naturally acquired low-level anti-tumor responses toward M3–9-M, albeit weak. This implies that T-cell depletion in rhabdomyosarcoma-bearing hosts can adversely effect tumor control, a point relevant to current standard therapies for this disease, which reproducibly induce significant T-cell depletion [42]. Furthermore, the existence of low-level anti-tumor responses implies that immune responses sculpt RMS during early stages of tumor growth for immune evasive characteristics [43]. Second, we provide the first evidence that regulatory T cells contribute to immune escape in murine RMS, since Treg depletion enhances the effectiveness of tumor vaccine and diminishes tumor growth in non-vaccinated hosts (Fig. 3A,B). This is consistent with several recent reports demonstrating that Treg depletion enhances the effectiveness of tumor vaccines [28,36,44–46]. Consistent with previous reports, anti-CD25-based Treg depletion did not result in any clinical evidence autoimmunity or toxicity. Mice receiving anti-CD25 plus whole-cell vaccination lived past 250 days with no signs of weight loss or hair loss; necropsy was performed after 250 days and no evidence of tumor or autoimmunity was observed. The lack of toxicity with CD25-based approaches to Treg depletion likely reflects the incomplete depletion induced with this approach [28]. Nonetheless, the depletion was sufficient to enhance immune responses to rhabdomyosarcoma. We conclude that clinical strategies seeking to augment immune responses to RMS should consider incorporating approaches to deplete CD4+CD25+ regulatory T cells. Third, we demonstrate that adoptive cell therapy can prevent the outgrowth tumors in hosts with minimal tumor burdens, a result potentially relevant to patients with minimal residual disease. Interestingly, while some groups have concluded that lymphopenia is an optimal environment for adoptive cell therapy, in previously published studies by our group we observed that the loss of immune surveillance that occurs as a result of profound lymphopenia offset the benefits of changes in homeostatic milieu [28].
One of the challenges of targeting pediatric tumors with immune-based therapies is the fact that dominant tumor antigens have not yet been identified. Furthermore, the rarity of these diseases means that approaches targeting one HLA allele will be applicable for only a small numbers of patients. For these reasons, whole-cell vaccines have been proposed for this population. The optimal approach for whole-cell vaccination remains unknown, and the challenges of developing personalized whole-cell vaccines are substantial, but it is important to note that a personalized whole-cell vaccine was recently FDA approved for prostate cancer. Thus, individualized therapies that administer whole-cell vaccines therapies can potentially be applied to large populations.
In conclusion, we present a new orthotopic model of ERMS on a C57BL/6 background. Using the model to study the tumor:-immune interface, we demonstrate that RMS is an immunogenic tumor, for which whole-cell vaccination can protect against tumor growth and immunotherapy can eradicate low tumor burdens. We further demonstrate that immunocompetent hosts mediate weak immune surveillance against ERMS and that immune escape is mediated, in part, by CD4+CD25+ Tregs. These studies lay the foundation for future work aimed at molecular identification of RMS-associated tumor antigens, optimization of cell-based therapies, and further analysis of mediators of ERMS immune escape.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGMENT
The authors would like to thank Dr. Brenda Weigel for providing the 76-9 rhabdomyosarcoma cell line and Dr. Nicholas Restifo for providing B16 melanoma. We would also like to thank Dr. Christian Capitini and Dr. Terry Fry for helpful discussion and careful review of the manuscript.
Footnotes
Additional Supporting Information may be found in the online version of this article.
Conflict of interest: nothing to declare.
REFERENCES
- 1.Pappo AS, Lyden E, Breneman J, et al. Up-front window trial of topotecan in previously untreated children and adolescents with metastatic rhabdomyosarcoma: An intergroup rhabdomyosarcoma study. J Clin Oncol 2001;19:213–219. [DOI] [PubMed] [Google Scholar]
- 2.Cosetti M, Wexler LH, Calleja E, et al. Irinotecan for pediatric solid tumors: The Memorial Sloan-Kettering experience. J Pediatr Hematol Oncol 2002;24:101–105. [DOI] [PubMed] [Google Scholar]
- 3.Spunt SL, Smith LM, Ruymann FB, et al. Cyclophosphamide dose intensification during induction therapy for intermediate-risk pediatric rhabdomyosarcoma is feasible but does not improve outcome: A report from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group. Clin Cancer Res 2004;10:6072–6079. [DOI] [PubMed] [Google Scholar]
- 4.Breneman JC, Lyden E, Pappo AS, et al. Prognostic factors and clinical outcomes in children and adolescents with metastatic rhabdomyosarcoma—A report from the Intergroup Rhabdomyosarcoma Study IV. J Clin Oncol 2003;21:78–84. [DOI] [PubMed] [Google Scholar]
- 5.Pappo AS, Anderson JR, Crist WM, et al. Survival after relapse in children and adolescents with rhabdomyosarcoma: A report from the Intergroup Rhabdomyosarcoma Study Group. J Clin Oncol 1999;17:3487–3493. [DOI] [PubMed] [Google Scholar]
- 6.Oeffinger KC, Mertens AC, Sklar CA, et al. Chronic health conditions in adult survivors of childhood cancer. N Engl J Med 2006;355:1572–1582. [DOI] [PubMed] [Google Scholar]
- 7.Rosenberg SA, Yannelli JR, Yang JC, et al. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J Natl Cancer Inst 1994;86:1159–1166. [DOI] [PubMed] [Google Scholar]
- 8.Yannelli JR, Hyatt C, McConnell S, et al. Growth of tumor-infiltrating lymphocytes from human solid cancers: Summary of a 5-year experience. Int J Cancer 1996;65:413–421. [DOI] [PubMed] [Google Scholar]
- 9.Dillman RO, Oldham RK, Tauer KW, et al. Continuous interleukin-2 and lymphokine-activated killer cells for advanced cancer: A National Biotherapy Study Group trial. J Clin Oncol 1991;9:1233–1240. [DOI] [PubMed] [Google Scholar]
- 10.Beck KE, Blansfield JA, Tran KQ, et al. Enterocolitis in patients with cancer after antibody blockade of cytotoxic T-lymphocyte-associated antigen 4. J Clin Oncol 2006;24:2283–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Porter DL, Antin JH. Infusion of donor peripheral blood mononuclear cells to treat relapse after transplantation for chronic myelogenous leukemia. Hematol Oncol Clin North Am 1998;12:123–150. [DOI] [PubMed] [Google Scholar]
- 12.Finke LH, Wentworth K, Blumenstein B, et al. Lessons from randomized phase III studies with active cancer immunotherapies—Outcomes from the 2006 meeting of the Cancer Vaccine Consortium (CVC). Vaccine 2007;25: B97–B109. [DOI] [PubMed] [Google Scholar]
- 13.Loren AW, Porter DL. Donor leukocyte infusions after unrelated donor hematopoietic stem cell transplantation. Curr Opin Oncol 2006;18:107–114. [DOI] [PubMed] [Google Scholar]
- 14.Meyers PA, Schwartz CL, Krailo MD, et al. Osteosarcoma: The addition of muramyl tripeptide to chemotherapy improves overall survival—A report from the Children’s Oncology Group. J Clin Oncol 2008;26:633–638. [DOI] [PubMed] [Google Scholar]
- 15.Pule MA, Savoldo B, Myers GD, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. Nat Med 2008;14:1264–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weigel BJ, Rodeberg DA, Krieg AM, et al. CpG oligodeoxynucleotides potentiate the antitumor effects of chemotherapy or tumor resection in an orthotopic murine model of rhabdomyosarcoma. Clin Cancer Res 2003; 9:3105–3114. [PubMed] [Google Scholar]
- 17.Sharp R, Recio JA, Jhappan C, et al. Synergism between INK4a/ARF inactivation and aberrant HGF/SF signaling in rhabdomyosarcomagenesis. Nat Med 2002;8:1276–1280. [DOI] [PubMed] [Google Scholar]
- 18.Tamayo P, Scanfeld D, Ebert BL, et al. Metagene projection for cross-platform, cross-species characterization of global transcriptional states. Proc Natl Acad Sci USA 2007;104:5959–5964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Henderson SR, Guiliano D, Presneau N, et al. A molecular map of mesenchymal tumors. Genome Biol 2005;6:R76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neale G, Su X, Morton CL, et al. Molecular characterization of the pediatric preclinical testing panel. Clin Cancer Res 2008;14:4572–4583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen QR, Vansant G, Oades K, et al. Diagnosis of the small round blue cell tumors using multiplex polymerase chain reaction. J Mol Diagn 2007; 9:80–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Romualdi C, De Pitta C, Tombolan L, et al. Defining the gene expression signature of rhabdomyosarcoma by meta-analysis. BMC Genomics 2006; 7:287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson BD, Jing W, Orentas RJ. CD25+ regulatory T cell inhibition enhances vaccine-induced immunity to neuroblastoma. J Immunother 2007;30:203–214. [DOI] [PubMed] [Google Scholar]
- 24.Fry TJ, Shand JL, Milliron M, et al. Antigen loading of DCs with irradiated apoptotic tumor cells induces improved anti-tumor immunity compared to other approaches. Cancer Immunol Immunother 2009;58: 1257–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thistlethwaite FC, Elkord E, Griffiths RW, et al. Adoptive transfer of T(reg) depleted autologous T cells in advanced renal cell carcinoma. Cancer Immunol Immunother 2008;57:623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sportes C, Hakim FT, Memon SA, et al. Administration of rhIL-7 in humans increases in vivo TCR repertoire diversity by preferential expansion of naive T cell subsets. J Exp Med 2008;205:1701–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lacelle MG, Jensen SM, Fox BA. Partial CD4 depletion reduces regulatory T cells induced by multiple vaccinations and restores therapeutic efficacy. Clin Cancer Res 2009;15:6881–6890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cui Y, Zhang H, Meadors J, et al. Harnessing the physiology of lymphopenia to support adoptive immunotherapy in lymphoreplete hosts. Blood 2009;114:3831–3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rousseau RF, Haight AE, Hirschmann-Jax C, et al. Local and systemic effects of an allogeneic tumor cell vaccine combining transgenic human lymphotactin with interleukin-2 in patients with advanced or refractory neuroblastoma. Blood 2003;101: 1718–1726. [DOI] [PubMed] [Google Scholar]
- 30.Geiger JD, Hutchinson RJ, Hohenkirk LF, et al. Vaccination of pediatric solid tumor patients with tumor lysate-pulsed dendritic cells can expand specific T cells and mediate tumor regression. Cancer Res 2001;61:8513–8519. [PubMed] [Google Scholar]
- 31.De Angulo G, Hernandez M, Morales-Arias J, et al. Early lymphocyte recovery as a prognostic indicator for high-risk Ewing sarcoma. J Pediatr Hematol Oncol 2007;29:48–52. [DOI] [PubMed] [Google Scholar]
- 32.DuBois SG, Elterman K, Grier HE. Early lymphocyte recovery in Ewing sarcoma. J Pediatr Hematol Oncol 2007;29:351–352. [DOI] [PubMed] [Google Scholar]
- 33.Siddiqui M, Ristow K, Markovic SN, et al. Absolute lymphocyte count predicts overall survival in follicular lymphomas. Br J Haematol 2006;134: 596–601. [DOI] [PubMed] [Google Scholar]
- 34.Finkelstein SE, Heimann DM, Klebanoff CA, et al. Bedside to bench and back again: How animal models are guiding the development of new immunotherapies for cancer. J Leukoc Biol 2004;76:333–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Overwijk WW, Tsung A, Irvine KR, et al. gp100/pmel 17 is a murine tumor rejection antigen: Induction of “self”-reactive, tumoricidal T cells using high-affinity, altered peptide ligand. J Exp Med 1998;188:277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Orentas RJ, Kohler ME, Johnson BD. Suppression of anti-cancer immunity by regulatory T cells: Back to the future. Semin Cancer Biol 2006; 16:137–149. [DOI] [PubMed] [Google Scholar]
- 37.Merchant MS, Melchionda F, Sinha M, et al. Immune reconstitution prevents metastatic recurrence of murine osteosarcoma. Cancer Immunol Immunother 2007;56:1037–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu Y, Khan J, Khanna C, et al. Expression profiling identifies the cytoskeletal organizer ezrin and the developmental homeoprotein Six-1 as key metastatic regulators. Nat Med 2004;10:175–181. [DOI] [PubMed] [Google Scholar]
- 39.Nishijo K, Chen QR, Zhang L, et al. Credentialing a preclinical mouse model of alveolar rhabdomyosarcoma. Cancer Res 2009;69:2902–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grass B, Wachtel M, Behnke S, et al. Immunohistochemical detection of EGFR, fibrillin-2, P-cadherin and AP2beta as biomarkers for rhabdomyosarcoma diagnostics. Histopathology 2009;54:873–879. [DOI] [PubMed] [Google Scholar]
- 41.Johnson BD, Yan X, Schauer DW, et al. Dual expression of CD80 and CD86 produces a tumor vaccine superior to single expression of either molecule. Cell Immunol 2003;222:15–26. [DOI] [PubMed] [Google Scholar]
- 42.Mackall CL, Fleisher TA, Brown MR, et al. Lymphocyte depletion during treatment with intensive chemotherapy for cancer. Blood 1994;84:2221–2228. [PubMed] [Google Scholar]
- 43.Swann JB, Smyth MJ. Immune surveillance of tumors. J Clin Invest 2007; 117:1137–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Prasad SJ, Farrand KJ, Matthews SA, et al. Dendritic cells loaded with stressed tumor cells elicit long-lasting protective tumor immunity in mice depleted of CD4+CD25+ regulatory T cells. J Immunol 2005; 174:90–98. [DOI] [PubMed] [Google Scholar]
- 45.Goforth R, Salem AK, Zhu X, et al. Immune stimulatory antigen loaded particles combined with depletion of regulatory T-cells induce potent tumor specific immunity in a mouse model of melanoma. Cancer Immunol Immunother 2009;58:517–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Delluc S, Hachem P, Rusakiewicz S, et al. Dramatic efficacy improvement of a DC-based vaccine against AML by CD25 T cell depletion allowing the induction of a long-lasting T cell response. Cancer Immunol Immunother 2009;58:1669–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.