

SUMMARY
G-protein-gated inwardly rectifying K+ (GIRK) channels are essential effectors of inhibitory neurotransmission in the brain. GIRK channels have been implicated in diseases with abnormal neuronal excitability, including epilepsy and addiction. GIRK channels are tetramers composed of either the same subunit (e.g., homotetramers) or different subunits (e.g., heterotetramers). Compounds that specifically target subsets of GIRK channels in vivo are lacking. Previous studies have shown that alcohol directly activates GIRK channels through a hydrophobic pocket located in the cytoplasmic domain of the channel. Here, we report the identification and functional characterization of a GIRK1-selective activator, termed GiGA1, that targets the alcohol pocket. GiGA1 activates GIRK1/GIRK2 both in vitro and in vivo and, in turn, mitigates the effects of a convulsant in an acute epilepsy mouse model. These results shed light on the structure-based development of subunit-specific GIRK modulators that could provide potential treatments for brain disorders.
In Brief
Zhao et al. identify GiGA1 through a virtual screen of ~750,000 chemical compounds using a high-resolution model of the alcohol pocket in GIRK channels. GiGA1 selectively activates GIRK1-containing channels and is G protein independent, like alcohol. GiGA1 activates endogenous GIRK channels in the brain and suppresses induced seizures.
Graphical Abstract
INTRODUCTION
As members of the large inwardly rectifying potassium (K+) channel family, activation of G-protein-gated inwardly rectifying K+ (GIRK) channels hyperpolarizes cells by conducting a small outward K+ current near the resting membrane potential (RMP). This outward current underlies the metabotropic-dependent, slow inhibitory postsynaptic currents found in most brain regions (Lüscher and Slesinger, 2010; Luján and Aguado, 2015). Upon neurotransmitter-dependent activation of G-protein-coupled receptors (GPCRs), Gαi/o-βγ heterotrimeric G proteins dissociate, allowing Gβγ subunits to directly activate GIRK channels (Aryal et al., 2009; Dascal, 1997; Reuveny et al., 1994; Yamada et al., 1998). Four mammalian GIRK channel subunits assemble as heterotetrameric channels (e.g., GIRK1/GIRK2, GIRK1/GIRK4, and GIRK2/GIRK3) and, in the case of GIRK2 and GIRK4, form homotetramers. GIRK1, GIRK2, and GIRK3 comprise the major GIRK subunits in the brain, whereas GIRK1/GIRK4 are expressed in the heart (Lesage et al., 1995; Wickman et al., 2000; Hibino et al., 2010; Lüscher and Slesinger, 2010). In the brain, different GIRK subunit combinations show distinct localization and expression patterns (Kobayashi et al., 1995; Karschin et al., 1996). GIRK1/GIRK2 heterotetramers are the predominant form of GIRK channels in the brain (Liao et al., 1996), except in the ventral tegmental area (VTA) dopamine neurons, which only express GIRK2 and GIRK3 subunits, and substantia nigra dopamine neurons, which only express GIRK2a/c subunits (Inanobe et al., 1999; Cruz et al., 2004). Recent high-throughput screening (HTS) studies have identified some GIRK modulators (Kaufmann et al., 2013), but the limited number of GIRK subtype-specific compounds has hindered our understanding of the physiological role of GIRK channels in specific brain regions.
GIRK channels play an essential role in maintaining the RMP (Lüscher et al., 1997) and have been implicated in the pathophysiology of several diseases. For example, excessive GIRK channel activity can considerably reduce neuronal activity, such as described in Down syndrome, which contains a duplication of the chromosome containing KCNJ6 (GIRK2) (Reeves et al., 1995; Harashima et al., 2006; Best et al., 2007). The involvement of GIRK channels in neuronal death in Parkinson’s disease was first characterized in the weaver GIRK2 mutant mouse (Liao et al., 1996; Surmeier et al., 1996; Slesinger et al., 1997). Moreover, several studies have identified mutations in GIRK2 that cause loss of ion selectivity. Aberrant Na+ flux through mutant GIRK channels trigger cell death, leading to physical abnormalities, developmental delay, and intellectual disabilities in severe hyperkinetic movement disorder patients, such as in Keppen-Lubinsky syndrome (Masotti et al., 2015; Horvath et al., 2018). In addition to gain-of-function-related diseases, knockout (KO) experiments in mice establish the importance of GIRK channels in preventing abnormal neuronal excitability. For instance, GIRK2 KO mice show susceptibility to γ-aminobutyric acid type A (GABAA) receptor antagonist-induced seizures (Signorini et al., 1997). To date, none of the commercially available anticonvulsive drugs specifically target the GIRK channel (Goldenberg, 2010).
In addition to G-protein-mediated activation, the GIRK channel can be directly activated by alcohols, such as ethanol and propanol (PrOH), through a G-protein-independent pathway (Kobayashi et al., 1999; Lewohl et al., 1999). An alcohol binding pocket was initially observed in an atomic structure of the Kir2.1 cytoplasmic domain (Pegan et al., 2006). Subsequently, the homologous pocket was identified in GIRK2 and characterized by mutagenesis and alcohol tagging studies, underscoring the significance of this pocket in the activation of GIRK channels (Aryal et al., 2009; Bodhinathan and Slesinger, 2013). These findings presented us with an opportunity to rationally design small molecules that target this alcohol binding pocket and modulate the activity of GIRK channels.
Here we describe the discovery and characterization of a GIRK1-specific channel modulator, which we refer to as G-protein-independent GIRK channel activator 1 (GiGA1). GiGA1 activates the GIRK1/GIRK2 channel in both heterologous expression systems and native neurons. Unlike previously characterized GIRK1 activators (Kaufmann et al., 2013), GiGA1 possesses alcohol-like kinetics of fast on and off rates. Moreover, our results suggest GiGA1 has a distinct mechanism of activation involving amino acids located within and near the alcohol pocket. By activating GIRK1/GIRK2 heterotetramers, GiGA1 can suppress the excitability of hippocampal CA1 pyramidal neurons. Moreover, in a pentylenetetrazol (PTZ)-induced model of epilepsy, we show in vivo efficacy for GiGA1 as an anticonvulsant.
RESULTS
Identification of GIRK Modulators from Homology Modeling and Structure-Based Virtual Screening against an Alcohol Pocket in GIRK2
To search for potential modulators of GIRK channels (Aryal et al., 2009; Bodhinathan and Slesinger, 2013), we first constructed a structural model of an alcohol pocket in GIRK2 for virtual screening. The alcohol pocket in GIRK channels is composed of three cytoplasmic domains (N terminus, βL-βM loop, βD-βE loop) at the interface of two adjoining subunits (Aryal et al., 2009). The alcohol pocket is partially visible in the full-length GIRK2 structures (PDB: 3SYA, 3SYQ, and 4KFM), but the region of the N terminus that forms one side of the alcohol binding pocket (residues 50–55) is disordered (PDB: 3SYA, 3SYQ, and 4KFM) (Figure 1Ai). To visualize a more complete GIRK2 alcohol pocket, we built a homology model of the GIRK2 cytoplasmic domain using MODELLER (Šali and Blundell, 1993) from the Kir2.1-methyl-pentane-diol (MPD) crystal structure (PDB: 2GIX) (Pegan et al., 2006) as a modeling template (Figure 1Aii). The cytoplasmic domain of Kir2.1 shares a sequence identity of 61% and a nearly identical structure with GIRK2 (root-mean-square deviation [RMSD] of 0.58 Å), with the residues corresponding to GIRK2 residues 50–55 resolved. To identify small molecules that are similar in size to MPD and would fit the allosteric site, we virtually screened the fragment-like subset of the ZINC15 (Sterling and Irwin, 2015) small-molecule library against the model of the GIRK2 alcohol pocket, with constraints focusing on including a hydrophobic interaction constraint near leucines at position 257 (Leu257), an essential position for alcohol action (Aryal et al., 2009), as well as hydrogen-bond constraints to backbone of Lys52 and Pro256 (Figure 1B). Compounds with a favorable docking score were examined and prioritized for experimental testing to assess their functional effects on GIRK channels transiently expressed in HEK293T cells using whole-cell patch-clamp recordings.
Figure 1. Discovery of GIRK Modulators through Structure-Based Virtual Screening of the GIRK2 Alcohol Pocket.
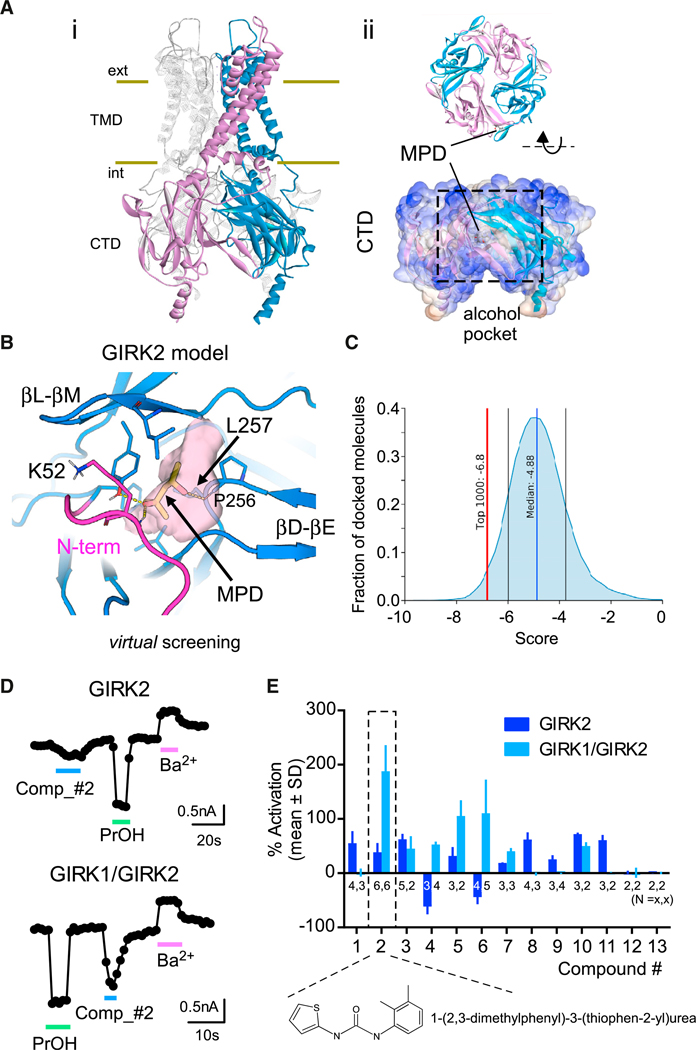
(A) (i) Ribbon and surface views of homotetrameric GIRK2 (Whorton and MacKinnon, 2013). (ii) The alcohol pocket is located in the cytoplasmic domain (CTD) of the GIRK2 homology model.
(B) Close view of the hydrophobic alcohol pocket (pink surface) between two subunits in the GIRK2 (blue cartoon) model (this study). The modeled N terminus is in magenta. MPD is indicated by yellow sticks.
(C) Graph shows the normal distribution of docking scores of the screened NCATS compounds (56,834 molecules). The red line indicates the cutoff for the top 1,000 compounds.
(D) Whole-cell patch-clamp recordings show the current response to PrOH (100 mM), compound 2 (100 μM), and Ba2+ (1 mM) in cells transfected with GIRK2c cDNA (top) or GIRK1/GIRK2c tandem dimer cDNA (bottom). Currents were measured at −120 mV.
(E) Bar graph shows the mean response, normalized to the Ba2+-sensitive basal current (% activation), of compounds identified through virtual screening (see Table S1). Negative numbers indicate inhibition of GIRK channels. Compound 2 produced the largest activation of GIRK1/GIRK2 channels and was used for subsequent experiments (dashed box). n is indicated on the graph. Error bars represent SD on the graph.
Nine compounds were selected and purchased based on commercial availability from the ZINC15 library, and two were found to have moderate inhibitory effects on GIRK2 homotetrameric channels at high concentrations (500 μM) (data not shown). We then expanded our search by virtually screening a subset of the National Center for Advancing Translational Sciences (NCATS) small-molecule library using the model of the GIRK2 alcohol binding pocket. Of the 1,000 top compounds, we selected and tested 13 compounds based on their docking pose, using selection criteria similar to that described earlier (Figure 1C). Nine of the 13 compounds activated GIRK2 channels (Figures 1D and 1E; Table S1). We also tested the effect on GIRK1/GIRK2 heterotetramers and discovered that 1-(2,3-di-methylphenyl)-3-(thiophen-2-yl)urea (i.e., compound 2) activated GIRK1/GIRK2 channels by ~200% (Figures 1D and 1E), making this a promising candidate for modulation of GIRK1/ GIRK2 channel in vivo.
To develop an initial structure-activity relationship (SAR) model and improve the potency and specificity of compound 2, we tested 4 analogs, varying the position or number of methyl groups on the phenyl ring (Figure 2A). The analogs were identified by performing a similarity search with the NCATS in-house application and applying a similarity threshold of Tanimoto similarity ≥ 0.85 (Tanimoto, 1969). One of the analogs, compound 2.3, which we refer to as GiGA1, exhibited high specificity for activating GIRK1/GIRK2 channels (Figures 2A and 2B). The reversal potential measured for GiGA1-induced GIRK1/GIRK2 currents was close to the Nernst potential for K+ and comparable to basal and PrOH-induced currents (Figure 2B, arrow). Moreover, the inward rectification property of GIRK1/GIRK2 was maintained in the presence of GiGA1 (Figure 2B). To explore possible mechanisms for GiGA1’s selectivity for activating GIRK1/GIRK2 channels, we performed an induced-fit docking (IFD) of GiGA1 and its analogs to the GIRK1/GIRK2 alcohol pocket (see STAR Methods). The predicted binding mode of GiGA1 placed the o-methylphenyl moiety of GiGA1 close to GIRK1L233 and GIRK1F46, thereby forming a π-π interaction (Figure 2C). This tight fitting of GiGA1 in the GIRK1/GIRK2 alcohol pocket may explain the weaker binding of GiGA1 analogs, compounds 2, 2.1, 2.2, and 2.4, because they all have an extra methyl group that would potentially clash with GIRK1 F46 and L233, rendering them less optimal in binding and activating GIRK1/GIRK2 channels.
Figure 2. Characterization of a GIRK1-Specific Activator, GiGA1.
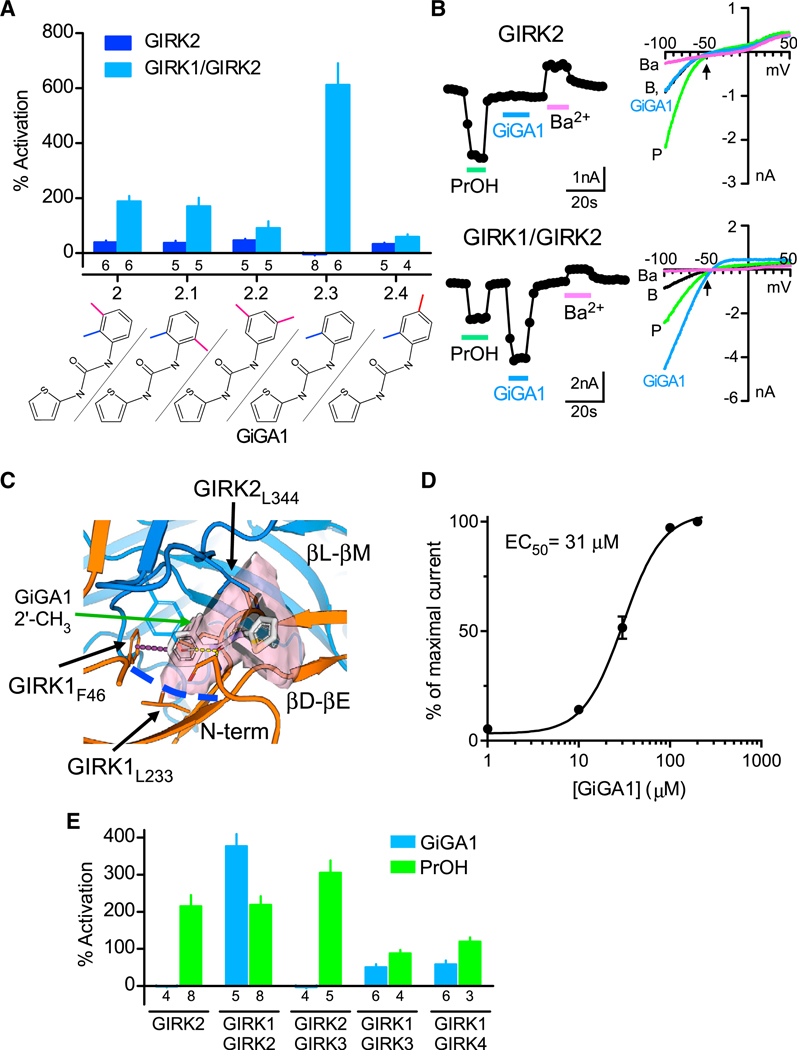
(A) Bar graph shows the mean percentage of activation responses for GIRK2 and GIRK1/GIRK2 channels to compound 2 and its analogs, varying the position or number of methyl groups on the phenyl ring (100 μM). The chemical structure for each compound is shown on the bottom. Currents are normalized to the Ba2+-sensitive basal current. Compound 2.3 showed specificity for GIRK1/GIRK2 (called GiGA1). n is indicated on the graph.
(B) Traces show the current response to PrOH (100 mM), GiGA1 (100 μM), and Ba2+ (1 mM) for GIRK2 (top trace) and GIRK1/GIRK2 channels (bottom trace). Current-voltage plots for GIRK2 channels (top) and GIRK1/GIRK2 channels (bottom) recorded in the absence (black) and presence of PrOH (green), GiGA1 (blue), and Ba2+ (magenta). The arrow indicates EK = −50 mV, with 20 mM Kout.
(C) Predicted binding mode of GiGA1 (white sticks) to a model of the GIRK1/GIRK2 (orange and blue cartoons, respectively) alcohol pocket from IFD. The binding pocket is depicted as a pink surface; the upper boundary of the pocket is limited by GIRK2L344, and the lower boundary is marked by GIRK1L233 (blue dashed line). o-methylphenyl moiety of GiGA1 is predicted to form π-π interactions with GIRK1F46 (purple dashed line). Note the position in the pocket of 2′ CH3 on the phenyl ring of GiGA1.
(D) Dose-response fit with the Hill equation is shown for activation of GIRK1/GIRK2 channels by GiGA1 (n = 6 cells). EC50 = 31 μM and Hill coefficient = 2.0.
(E) Bar graph shows the mean change in percentage of activation for subtypes of GIRK channels to 25 μM GiGA1 (blue) and 100 mM PrOH (green). Note the specificity for GIRK1/GIRK2 subunits. n is indicated on the graph.
Error bars represent SEM on the graphs.
Although GiGA1 was selected based on the binding mechanism of alcohol, it has significantly higher potency compared with PrOH (>100 mM), with an EC50 (i.e., 50% activation concentration) of 31 μM (Figure 2D). Unlike alcohol activation, which does not saturate (Bodhinathan and Slesinger, 2014), the GiGA1 activaiton of GIRK1/GIRK2 channels does saturate at ~100 μM (Figure 2D). Because GIRK subunits also assemble into other forms of GIRK channels in the brain and the heart, we next tested the effects of GiGA1 on GIRK2/GIRK3, GIRK1/GIRK3, and GIRK1/GIRK4 channels expressed in HEK293T cells. GiGA1 activated other GIRK1-containing GIRK channels, but not to the same extent as GIRK1/GIRK2 channels (Figure 2E). The specificity of PrOH was also tested on different GIRK channels and, consistent with previous studies (Kobayashi et al., 1999; Lewohl et al., 1999), PrOH was more potent in activating GIRK2-containing GIRK channels than other subtypes (Figure 2E).
Mechanism of GiGA1 Action on GIRK1-Containing GIRK Channels
GIRK channels are activated via a G-protein-dependent pathway in native cells (Dascal, 1997), and the alcohol pocket is located near the Gβγ binding domain of GIRK2 (PDB: 4KFM) (Bodhinathan and Slesinger, 2014) (Figure 3A). To investigate whether the effect of GiGA1 requires receptor-dependent G proteins, we co-expressed the pertussis toxin (PTX) S1 subunit with the γ-aminobutyric acid type B (GABAB) heterodimer receptor and GIRK1/GIRK2 channels. The PTX S1 subunit ribosylates Gαi/o G proteins and uncouples them from receptors (Reisine, 1990) (Figure 3A). Whereas baclofen-dependent activation was absent from cells expressing PTX S1, GiGA1 continued to activate GIRK1/GIRK2, similarly to PrOH, suggesting that G protein activation is not involved (Figures 3B and 3C).
Figure 3. GiGA1 Activates GIRK1/GIRK2 Channels through an Alcohol Pocket in a G-Protein-Independent Manner.
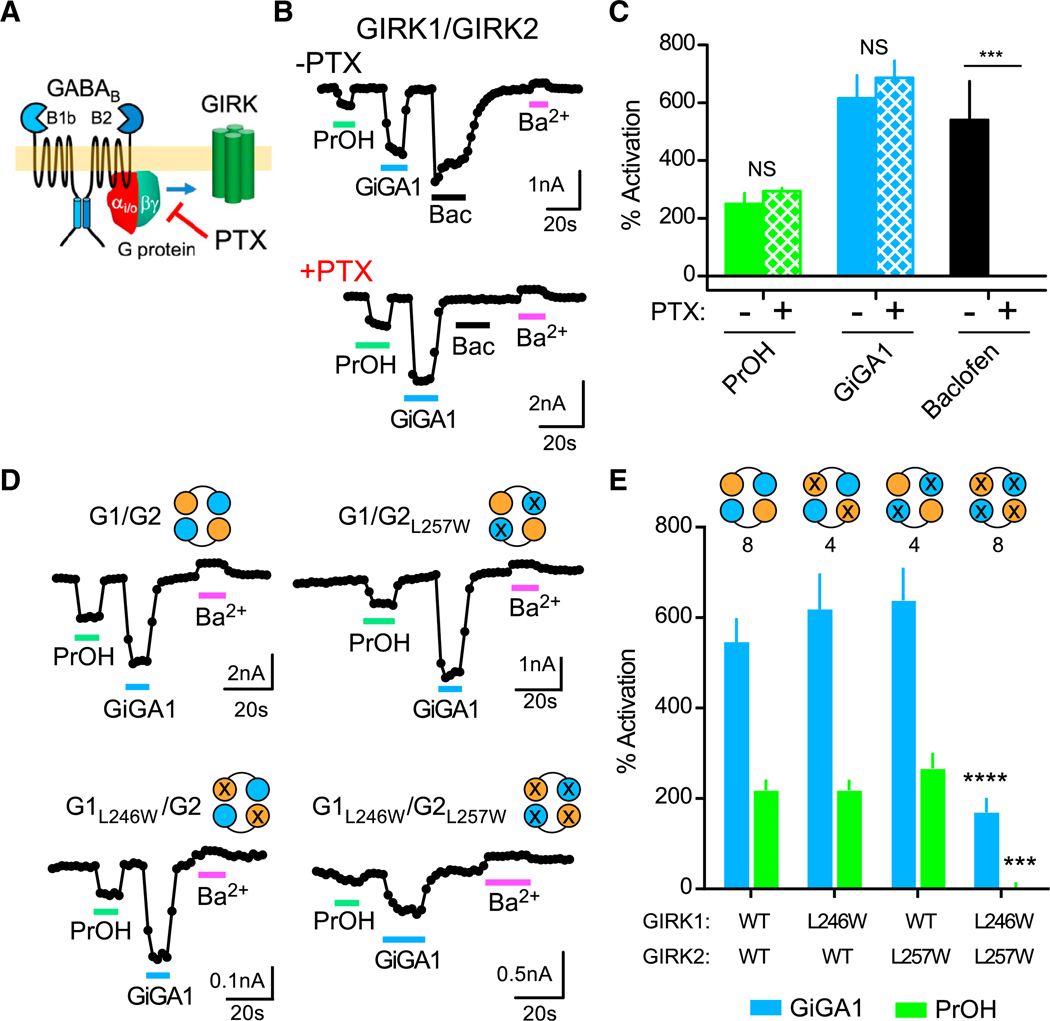
(A) Cartoon shows activation of the GIRK channel via the G-protein-dependent pathway, which is blocked by PTX.
(B) Traces show the current response to PrOH (100 mM), GiGA1 (100 μM), baclofen (100 μM), and Ba2+ (1 mM) in HEK cells transfected with cDNA for GABAB1b/B2 receptors and GIRK1/GIRK2c tandem dimer with or without the PTX S1 subunit.
(C) Bar graph shows the mean change in percentage of activation for GIRK1/GIRK2 channels with PrOH (green), GiGA1 (blue), or baclofen (black) in the absence or presence of the PTX S1 subunit. ***p = 0.0006; F(2, 5) = 25.52 (baclofen × PTX); mixed-effects model (restricted maximum likelihood [REML]) with Bonferroni post hoc (n = 4 (−PTX), 5 (+PTX)).
(D) Traces for the WT GIRK1/GIRK2 channel and the indicated alcohol pocket mutation (GIRK1L246W/GIRK2L257W) (Aryal et al., 2009) in response to PrOH (100 mM), GiGA1 (100 μM), and Ba2+ (1 mM). The schematic indicates the subunit that has a mutation; the orange circle is GIRK1, and the cyan circle is GIRK2.
(E) Bar graph shows the mean percentage of activation response to GiGA1 and PrOH. ***p = 0.0004, ****p < 0.0001 versus GIRK1/GIRK2 WT; two-way repeated-measures ANOVA with Dunnett’s multiple comparison test (F(3, 20) = 5.298; ***p = 0.0075 activator × mutation). n is indicated on the graph.
Error bars represent SEM on the graphs.
To determine whether the site of action for GiGA1 involves the alcohol pocket of GIRK1/GIRK2, we examined the effects of a tryptophan substitution at Leu257, which was shown previously to directly attenuate alcohol activation (Figure S1) (Aryal et al., 2009). We sequentially mutated one or both Leu257 in GIRK2 and the homologous position 246 in GIRK1. Although neither activation of GiGA1 nor activation of PrOH was altered by a single mutation, the GIRK1L246W/GIRK2L257W double mutation significantly reduced both PrOH- and GiGA1-activated currents (Figures 3D and 3E), suggesting involvement of all four alcohol pockets.
A GIRK1-selective activator, ML297, was identified through HTS, and its mechanism has been characterized (Kaufmann et al., 2013; Wydeven et al., 2014). Two unique amino acids, Phe137 and Asp173, located within the gating region of GIRK1 are necessary and sufficient to determine the specificity and action of ML297 (Wydeven et al., 2014) (Figure 4A). Because GiGA1-induced channel activity is also specific to GIRK1, we tested whether GiGA1 shares a similar mechanism of action with ML297. We examined the effect of GIRK1F137S (FS) and GIRK1D173N (DN) mutations, and as shown previously (Wydeven et al., 2014), each mutation significantly reduced the activation by ML297. By contrast, neither mutation in GIRK1 abolished the GiGA1-induced current (Figures 4B–4E). In addition to a different site of action, the kinetics of the GiGA1- and ML297-induced currents are different. GiGA1 has a rapid deactivation rate, comparable to that of alcohol modulation of GIRK channels, whereas the off rate of ML297 is significantly slower (Figure S2A). We also tested the effect of Ba2+ to inhibit GiGA1 activation of GIRK1/GIRK2 by co-application of GiGA1 and Ba2+ (Figure S2B). Ba2+ blocked GIRK1/GIRK2 currents to the same extent and did not affect the reversal of GiGA1 activation.
Figure 4. GiGA1 Activates GIRK1/GIRK2 Channels through a Different Mechanism from ML297.
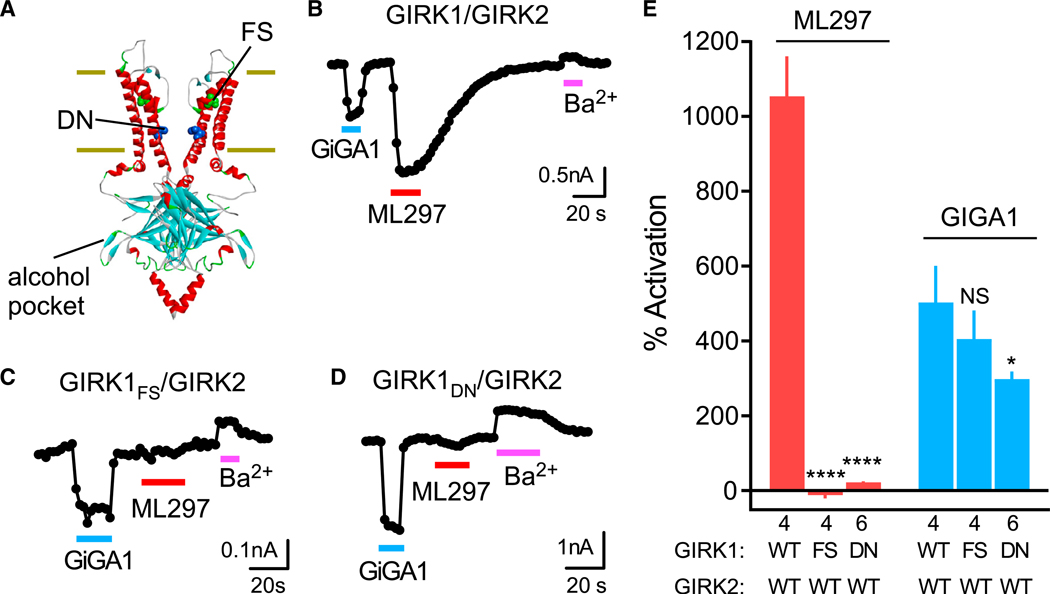
(A) Structural model of GIRK2 (PDB: 4KFM) shows the alcohol pocket and location of homologous amino acids in GIRK1 implicated previously in ML297 activation (FS (green), GIRK1F137 to ~GIRK2S148; DN (blue), GIRK1D173 to ~GIRK2N184).
(B–D) Traces show the current response of the WT GIRK1/GIRK2 (B), GIRK1FS/GIRK2 (C), and GIRK1DN/GIRK2 (D) channels to GiGA1 (100 μM), ML297 (10 μM), and Ba2+ (1 mM). Both mutants abolished the ML297 response, but not the activation with GiGA1.
(E) Bar graph shows the mean percentage of activation with GiGA1 and ML297. ****p < 0.0001, *p = 0.0374 versus GIRK1/GIRK2 WT; two-way repeated-measures ANOVA with Dunnett’s multiple comparison test (F(2, 11) = 106.3; p < 0.0001; activator × mutation). n is indicated on the graph. Error bars represent SEM on the graph.
We therefore focused on the alcohol pockets of GIRK1 and GIRK2 subunits (see sequence alignment, Figure S1) to further investigate the preferential activation of GIRK1/GIRK2 heterotetramers by GiGA1 (Figure 2). To identify key amino acids in the pocket that determine specificity, we first evaluated the effect of a series of mutations in the GIRK1 subunit. The alcohol pocket is composed of the N terminus and βD-βE loop from one subunit and βL-βM loop from the adjacent subunit and is therefore located at the interface of two adjoining subunits; thus, there are two different pairs of alcohol pockets in GIRK1/GIRK2 heterotetramers. The features of GIRK1 that support GiGA1-selective activation of GIRK1/GIRK2 channels could be distributed in all four pockets on different regions of GIRK1 and GIRK2 (Figure 5A). By comparing protein sequences of GIRK1 and GIRK2 subunit within the alcohol pocket, we selected amino acids that are different between GIRK1 and GIRK2, and could potentially affect binding of GiGA1 (Figure S1). To test this, we examined the effect of individually replacing five amino acids of GIRK1 that face the pocket with the corresponding amino acid in GIRK2. To quantify effects of mutations on GiGA1 activation, we calculated the GiGA1-induced current as the percentage of the ML297-induced response (GiGA1/ML297), which is not expected to change with the mutation. F46Y, C230A, L233I, E250D, and F338Y mutations in GIRK1 did not alter the activation by GiGA1 (% normalized activation), in contrast to the GIRK1L246W/GIRK2L257W double mutation in the alcohol pocket (Figure S3).
Figure 5. Identification of Amino Acids in GIRK1 that Contribute to GIRK1 Specificity of GiGA1.
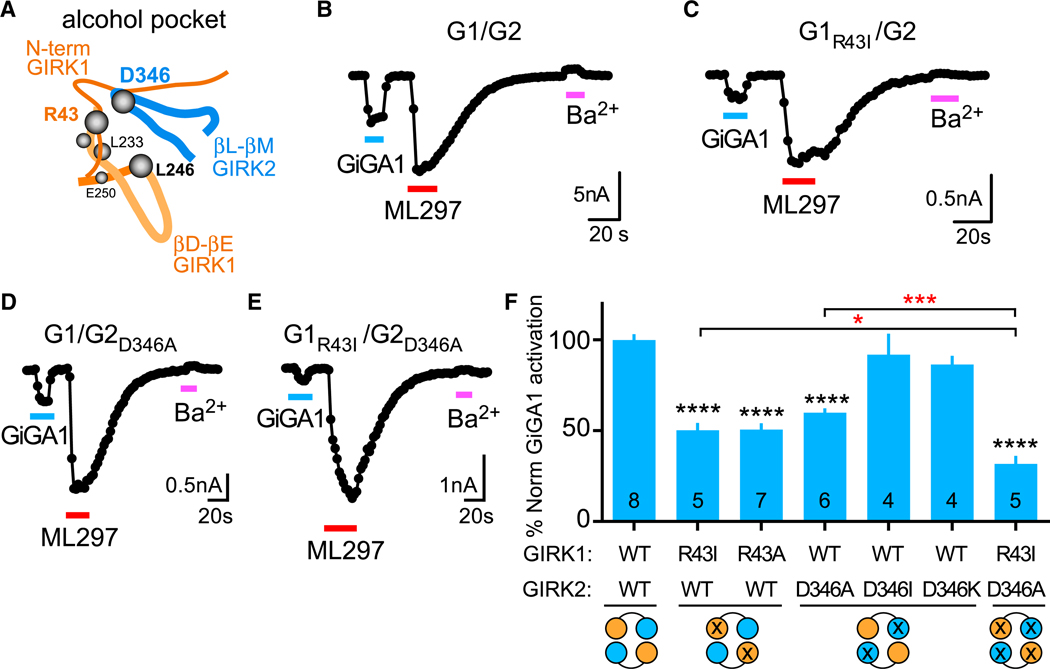
(A) Schematic depicts the alcohol pocket in one of the GIRK1/GIRK2 subunit interfaces, composed of the N-terminal domain and βD-βE loop from the GIRK1 subunit and βL-βM loop from the GIRK2 subunit.
(B–E) Whole-cell patch-clamp recordings show the current response of WT G1/G2 (B), G1R43I/G2 (C), G1/G2D346A (D) ,and G1R43I/G2D346A (E) channels to GiGA1 (100 μM), ML297 (10 μM), and Ba2+ (1 mM).
(F) Bar graph shows the mean percentage of GiGA1 activation normalized to ML297-induced currents and WT (see STAR Methods). ****p < 0.0001 versus GIRK1/GIRK2 WT, *p = 0.0385 GIRK1R43I, ***p = 0.0006 GIRK2D346A versus GIRK1R43I/GIRK2D346A; one-way ANOVA with Bonferroni’s multiple comparison test (F(6, 32) = 30.96; p < 0.0001). n is indicated on the graph. Error bars represent SEM on the graph.
To identify other amino acids near the alcohol pocket, we generated a structural model of the GIRK1/GIRK2 alcohol pocket (see STAR Methods) (Figure 2C). One unique pair of amino acids was ideally positioned to potentially alter the alcohol pocket: Arg43 in GIRK1 and Asp346 in GIRK2. Arg43 in GIRK1 is localized outside of the alcohol pocket but could stabilize the pocket by forming a salt bridge with Asp346 in GIRK2. Mutation GIRK1R43I significantly decreased the % normalized GiGA1-activated current, compared with WT channels (Figures 5B, 5C, and 5F). An alanine substitution produced a similar decrease in the % normalized GiGA1-activated current. We next examined an Ala or Ile substitution at GIRK2D346. Interestingly, GIRK2D346A, but not GIRK2D346I, reduced % normalized GiGA1 activation (Figures 5D and 5F). To probe whether a salt bridge was essential, we examined the effect of charge reversal at Asp346. GIRK2D346K was indistinguishable from that of WT (Figure 5F). Thus, the side-chain length, and not the charge, appeared to be important for conferring specificity of GiGA1 activation of GIRK1/GIRK2 channels. Lastly, we examined the effect of mutating all four pockets in the heterotetramers. The double-mutant GIRK1R43I/GIRK2D346A exhibited a statistically smaller % normalized GiGA1-activated current, compared with each of the single mutations (GIRK1R43I or GIRK2D346A) (Figures 5E and 5F). To validate that ML297 currents were not altered by the mutations, we analyzed current density for each of the mutants; only one of six mutants (D346I) significantly reduced ML297 current (Figure S6).
To gain insights into the effect of the GIRK1R43I/GIRK2D346A mutations on GiGA1 activation of the GIRK1/GIRK2 heterotetramer, we used Gaussian accelerated molecular dynamics (GaMD) simulations (see STAR Methods) that can assess the secondary structure elements of the residues lining the alcohol pocket. We observed that both GIRK1R43I and GIRK2D346A single mutations and the GIRK1R43I/GIRK2D346A double mutation have little impact on the secondary structure of the residue in the alcohol pocket, GIRK2L342 (Figure S4). However, these mutations perturbed each other’s secondary structure (Figure S4), as well as that of GIRK2L344 (Figure S5), a residue implicated in Gβγ activation (Figures 1B and 2C). Thus, the combined effect of GIRK1R43I/GIRK2D346A mutations could alter the structure of the βL-βM loop, which forms one side of the alcohol pocket, or access to the pocket, and could be important for modulating GiGA1 activation of GIRK1/GIRK2 heterotetramers.
Role of GiGA1 on Ex Vivo Hippocampal Neurons
To investigate whether GiGA1 could activate natively expressed GIRK channels, we examined the effect of GiGA1 on GIRK currents in hippocampal CA1 pyramidal neurons, which express mostly GIRK1/GIRK2 heterotetramers (Liao et al., 1996; Leaney, 2003). In acutely prepared hippocampal slices, we found that a saturating concentration of baclofen (300 μM) induced an outward current of ~120 pA and was selectively reversed with the GABAB receptor (GABABR) antagonist CGP55845 (5 μM). We then bath applied GiGA1 (100 μM), which also activated an outward current of ~120 pA and was sensitive to inhibition with Ba2+ (Figures 6A and 6B). In contrast, there was no change in the voltage-gated outward K+ currents measured at 0 mV (Figure 6B). GABABR-activated GIRK currents have been shown previously to hyperpolarize resting potential and reduce neuronal excitability (Yamada et al., 1998). In current-clamp recordings, GiGA1 significantly hyperpolarized the RMP following CGP55845 inhibition, to the same level as baclofen (Figure 6C). In addition, GiGA1 significantly suppressed the number of spikes induced by current injection in pyramidal neurons (Figures 6D and 6E).
Figure 6. GiGA1 Induces GIRK Currents in Hippocampal CA1 Pyramidal Neurons.
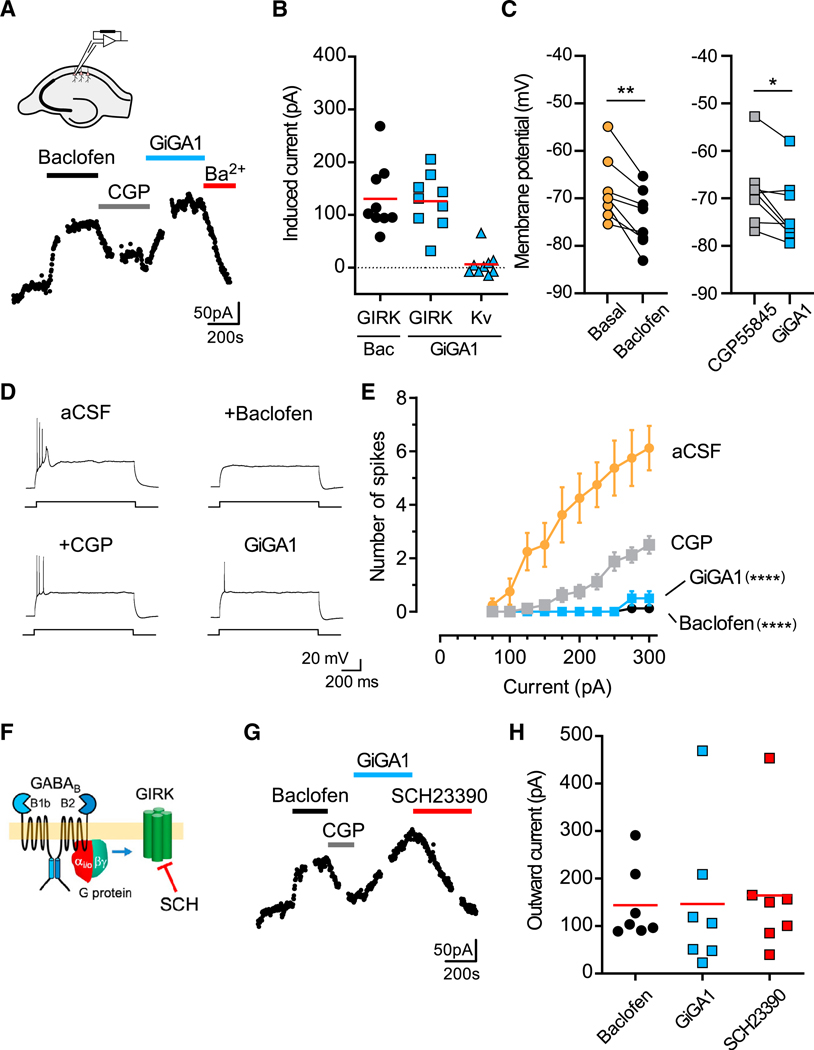
(A) Whole-cell patch-clamp recording from a hippocampal CA1 pyramidal neuron shows baclofen-induced (300 μM) and GiGA1-induced (100 μM) outward currents and inhibition by CGP55845 (5 μM) and Ba2+ (1 mM), respectively. VH = −40 mV. Schematic shows the hippocampal slice and placement of the recording pipette.
(B) Scatterplot shows baclofen- and GiGA1-induced GIRK currents measured at −40 mV, as well as outward voltage-gated K (Kv) currents measured at 0 mV. n = 9 cells. The ed bar indicates the mean.
(C) Plot shows the individual RMP (I = 0) in the absence (basal) and presence of baclofen (left graph) and the presence of CGP55845 followed by GiGA1 (right graph). **p = 0.0028 baclofen versus basal, *p = 0.0182 GiGA1 versus CGP55845; paired t test. n = 8 cells.
(D) Voltage traces show induced action potentials (+275 pA) in the presence of aCSF alone or with baclofen, CGP55845, or GiGA1.
(E) Input-output plots show the firing frequency increases as a function of current injections for artificial cerebrospinal fluid (aCSF) (basal) or CGP55845 (5 μM) that are significantly suppressed by either baclofen (300 μM) or GiGA1 (100 μM), respectively. ****p < 0.0001 for basal versus baclofen and CGP55845 versus GiGA1 (n = 8); two-way repeated-measures ANOVA with Tukey’s multiple comparisons post hoc test for interaction between drug and current injection. Error bars represent SEM on the graph.
(F) Cartoon shows the inhibition of the GIRK channel by SCH23390 (Kuzhikandathil and Oxford, 2002; Montalbano et al., 2015).
(G) Whole-cell patch-clamp recording from the hippocampal CA1 pyramidal neuron shows baclofen-induced (300 μM) and GiGA1-induced (100 μM) outward currents and the response to CGP55845 (5 μM) and a GIRK-selective inhibitor (SCH23390; 30 μM), respectively.
(H) Scatterplot shows baclofen- and GiGA1-induced GIRK currents, as well as SCH23390-inhibited GIRK currents measured at −40 mV (n = 7 cells). The red bar indicates the mean (there is no significant difference).
Ba2+ is known to inhibit several types of K+ channels (Piasta et al., 2011). We therefore explored whether the GiGA1-activated current could be selectively reversed with a GIRK-selective inhibitor. Kuzhikandathil and Oxford, 2002 reported that the D1 receptor antagonist SCH23390 inhibits GIRK channels (Figure 6F). Similar to Ba2+, bath application of SCH23390 (30 μM) inhibited GiGA1-induced outward current (Figures 6G and 6H). Importantly, there was good agreement between the amplitude of the GiGA1-induced current and that of the SCH23390-inhibited current (Figure 6H). Altogether, these results demonstrate that GiGA1 may control the excitability of a CA1 pyramidal neuron by activating GIRK1/GIRK2 channels.
Potential Protective Role of GiGA1 in Epilepsy
Because activation of GIRK channels in the brain is expected to reduce membrane excitability, and GIRK2 KO mice show spontaneous convulsions (Signorini et al., 1997), we asked whether GiGA1 possesses antiseizure properties in an in vivo epilepsy model. We induced acute epilepsy in mice by intraperitoneal (i.p.) injection of PTZ. PTZ is thought to generate tonic-clonic seizure by antagonizing GABAA receptors (Shimada and Yamagata, 2018). We first determined that 60 mg/kg PTZ induced distinct stages of seizure intensity without being lethal. We also investigated the pharmacokinetic (PK) properties of GiGA1 in vivo. GiGA1 displayed a higher concentration in the brain sample than the plasma sample throughout and reached a maximum brain concentration at 15 min after injection, which decreased significantly after 1 h (Figures S7A–S7C). To test the potential antiseizure property, we first injected GiGA1 (20, 40, or 60 mg/ kg), vehicle (Veh), or the positive control valproic acid (VPA) (200 mg/kg) and then waited 15–30 min before injecting PTZ to induce seizures (Figure 7A). Seizure scores were analyzed for 30 min based on the modified Racine score (Shimada and Yamagata, 2018), and the time in a score of 3 and above was used as the time in the tonic seizure. Two doses of GiGA1 (40 and 60 mg/kg) significantly lowered the seizure score and reduced the time in tonic seizure by ~80% (Figures 7B and 7C). We also observed some dose-dependent sedative effects of GiGA1 (Figure S7D).
Figure 7. Anticonvulsive Effect of GiGA1 in the PTZ-Induced Epilepsy Model.
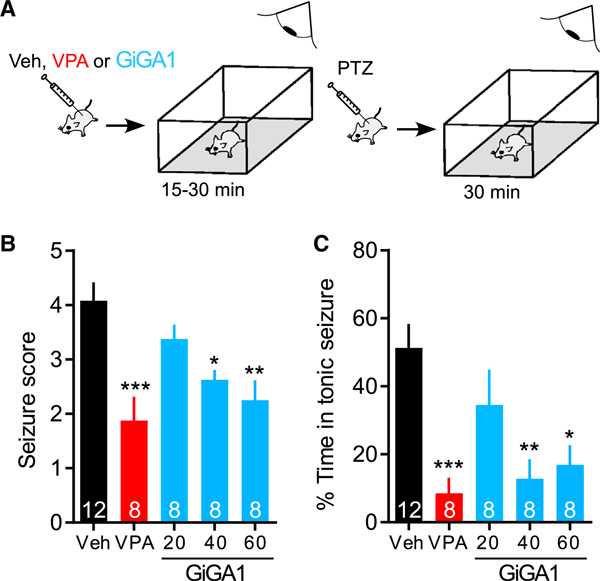
(A) Schematic shows the time course of the experiment. WT C57BL/6J mice first received an i.p. injection of Veh, valproic acid (VPA, 200 mg/kg), or GiGA1 (20, 40, and 60 mg/kg), and then locomotor activity was monitored for 15–30 min. Mice then received an i.p. injection of PTZ (60 mg/kg) to induce acute seizures and were monitored for another 30 min.
(B) Bar graph shows mean seizure scores based on the modified Racine score (Shimada and Yamagata, 2018). *p = 0.0278, **p = 0.0026, ***p = 0.0002 versus Veh; one-way ANOVA with Bonferroni’s multiple comparison test (F(4, 39) = 7.709; p = 0.0001).
(C) Bar graph shows the mean percentage of time with seizure scores ≥ 3. *p = 0.0101, **p = 0.0029, ***p = 0.0008 versus Veh; one-way ANOVA with Bonferroni’s multiple comparison test (F(4, 39) = 7.067; p = 0.0002).
n is indicated on the graphs. Error bars represent SEM on the graphs.
DISCUSSION
In the present study, we used an integrated approach of identifying a GIRK1-specific activator through computational modeling and subsequent biochemical and physiological studies. We first developed a structural model of the putative alcohol binding site in GIRK2, allowing us to investigate the mode of interaction between GIRK2 and alcohol, as well as conduct a virtual screen of numerous compounds in a short amount of time. The atomic resolution model of an alcohol-bound GIRK2 channel provided us essential insights into the alcohol pocket, suggesting a key hydrophobic effect near Leu257, as well as additional hydrogen bonds with the backbone amides of Lys52 and Pro256 (Aryal et al., 2009; Pegan et al., 2006). These proposed interactions likely contributed to the accuracy of our initial virtual screening. Although the virtual screening was based on the alcohol pocket in GIRK2, we identified compounds that modulate GIRK channels other than GIRK2 homotetramers. This outcome may seem surprising at first, but the alcohol pocket is composed of structural elements (part of the N terminus and βD-βE and βL-βM loops) that are highly conserved among the GIRK channels. For example, conserved Pro256 and Leu257 in GIRK2 (P245 and L246 in GIRK1) are predicted to interact with the recently discovered modulators. Notably, through chemical modification of the initial hit (compound 2), we were able to improve the specificity for GIRK1.
Our results revealed that GiGA1 has a distinct site of action on GIRK1/GIRK2 channels. Under physiological conditions, Gβγ subunits dissociate from heterotrimeric Gαi/oβγ proteins upon GPCR activation and directly activate GIRK channels (Huang et al., 1995; Inanobe et al., 1995; Reuveny et al., 1994). The critical region for Gβγ binding was mapped to residues localized near the alcohol pocket (Bodhinathan and Slesinger, 2014; Finley et al., 2004; Ivanina et al., 2003; Whorton and MacKinnon, 2013). We tested the involvement of Gβγ in GiGA1-mediated GIRK channel activation by co-expressing the PTX catalytic subunit. Like alcohol, GiGA1 activated GIRK channels in a G-protein-independent manner and was not affected by PTX expression. Another GIRK1-specific modulator, ML297, was identified through a thallium-based flux screening assay (Kaufmann et al., 2013). Similar to GiGA1, this compound activates GIRK channels in a G-protein-independent manner, with a preference for GIRK1-containing channels (Kaufmann et al., 2013). Molecular studies identified two amino acids in the channel pore and membrane-spanning region of GIRK1 essential for its efficacy and selectivity (Wydeven et al., 2014). F137 is also implicated in the slow voltage-dependent gating property of the GIRK1 subunit (Kofuji et al., 1996). However, none of these sites appeared to be important for GiGA1-mediated activation. Furthermore, GiGA1 exhibits fast activation and deactivation properties that are similar to those of alcohol.
Does GiGA1 activate GIRK channels via the alcohol pocket? Mutation of a leucine shown previously to be important for alcohol activation (Aryal et al., 2009) significantly attenuated GiGA1-dependent activation, as well as decreasing PrOH-dependent activation when all four leucines were mutated to Trp in both GIRK1 and GIRK2 subunits. Although these leucines are important for alcohol and GiGA1 activation, these mutations did not reveal the mechanism of specificity of GiGA1 activation of GIRK1/GIRK2 channels over GIRK2. Mutagenesis of other amino acids in the pocket that differed between GIRK1 and GIRK2 also did not produce changes in GiGA1 activation. We hypothesized that GiGA1 requires additional structural features, besides GIRK1L246/GIRK2L257 in the pocket, to enable subunit specificity. We identified Arg43 in the N terminus of GIRK1 and Asp346 in the C-terminal βL-βM loop of GIRK2 that contribute to the specificity of GiGA1 activation of GIRK1/GIRK2 channels. Although these two residues carry opposite charges, and could therefore form a salt bridge, this interaction did not appear to be essential, because substituting Asp346 with the positively charged Lys did not alter GiGA1-dependent activation. The molecular dynamics simulations revealed that mutagenesis of either of these residues affects the secondary structure of each residue. In addition, mutation of these two residues appears to propagate to a residue within the pocket, GIRK2L344. These findings suggest that the mutations allosterically alter the local secondary structure of the βL-βM loop, interfering with the proper binding of GiGA1 to the alcohol pocket, although we cannot rule out an additional site of action for GiGA1.
Altogether, these observations support the conclusion that GiGA1 is a GIRK1-specific modulator that may activate GIRK1/ GIRK2 channels via the alcohol binding pocket. However, the precise binding mechanism of GiGA1 to GIRK1/GIRK2 channels should be explored by additional structural studies. One notable advantage over alcohol is the higher potency and saturating response of GiGA1, which is difficult to show with alcohols (Bodhinathan and Slesinger, 2014). The fast deactivation kinetics could be beneficial when an acute adjustment in channel activity is required; a slow off rate might result in undesirable excess activation of GIRK channels. GiGA1 showed good aqueous solubility (34 µg/mL) at pH 7.4. In in vivo PK studies, GiGA1 also produced a maximum brain-to-plasma ratio of 1.91 ± 0.86 when administered systemically (i.p.) to mice, which is an improvement over ML297 (0.2) (Kaufmann et al., 2013). Thus, GiGA1 could be evaluated for functional effects in vivo.
Previous studies implicated GIRK channels in controlling excitability and preventing seizures. GIRK2 KO mice showed spontaneous convulsions, as well as increased vulnerability to GABAA receptor antagonist-induced seizures (Signorini et al., 1997). Similarly, GIRK2−/−/GIRK3−/− KO mice developed spontaneous and lethal seizures (Torrecilla et al., 2002). Furthermore, downregulation of GIRK channel function and expression occurs following neuronal injury during prolonged seizures (Baculis et al., 2017). Seizure induction and increased severity in these studies may be explained by the overall reduction of GIRK channel function, leading to hyperexcitability in many regions in the brain. Conversely, augmentation of GIRK channel activity in an epilepsy model may exert a protective effect, especially if focused on GIRK1/GIRK2 channels, the predominant form in the brain (Liao et al., 1996). In the current study, we show that GiGA1 preferentially activates GIRK1/GIRK2 channels, as well as those channels natively expressed in hippocampal pyramidal neurons (Koyrakh et al., 2005; Luján and Aguado, 2015). Studies of temporal lobe epilepsy have reported the involvement of the hippocampus and suggest that the hippocampus plays an essential role in epilepsy occurring and spreading (McIntyre and Racine, 1986; Diehl et al., 2004; Cascino, 2005). Accordingly, systemic administration of GiGA1 significantly reduced seizure severity and frequency of tonic seizures. These effects were comparable to the antiseizure effects of VPA, a commonly used antiepileptic drug (Pellock, 1994). Notably, increased self-grooming was seen in VPA-treated mice, but not in GiGA1-treated mice, which may reflect the actions of VPA on glutamatergic transmission in the prefrontal cortex (PFC) (Mehta et al., 2011). However, GiGA1 did show a dose-dependent sedative effect, although this outcome was not unexpected because GIRK-channel-deficient mice demonstrate a higher level of locomotion compared with WT (Morgan et al., 2003; Labouèbe et al., 2007; Arora et al., 2010) and baclofen, which partly targets the GABABR-GIRK signaling pathway and produces sedation (Gray et al., 1987; Ong and Kerr, 2005; Li et al., 2013). Lastly, alcohol is a sedative (Hendler et al., 2013). Future optimization of GiGA1 may mitigate some of these side effects in vivo.
In addition to an antiepileptic effect of GiGA1, selective activation of GIRK1/GIRK2 channels could be useful for treating other brain disorders, including alcohol use disorder (AUD) and intractable pain. Alcohol affects multiple signaling pathways in the brain (Abrahao et al., 2017). Systemic administration of baclofen significantly reduced alcohol intake in a binge-drinking mouse model (Moore et al., 2007). In alcohol-addicted rats, the acute treatment of baclofen blocked the increase of alcohol intake after alcohol deprivation (Colombo et al., 2003, 2006). In humans, several randomized controlled trials have demonstrated beneficial effects of baclofen in AUD (Addolorato et al., 2002, 2007; Müller et al., 2015; Morley et al., 2018). Furthermore, conditioning mechanisms, especially hippocampus-related ones, have been shown to play major roles in alcohol addiction (Kutlu and Gould, 2016). Because the major type of GIRK channels in the hippocampus is GIRK1/GIRK2, application of GiGA1 might prevent neuronal plasticity that is associated with alcoholdependent contextual learning (Valyear et al., 2017). GiGA1 or a variant could be effective at treating AUD.
GIRK channels are also implicated in drug-induced analgesia. Many of these drugs activate GIRK channels either directly or through a G-protein-dependent pathway, including ethanol, baclofen, and opioids (Ikeda et al., 2000; Blednov et al., 2003). Lack of acute ethanol-induced analgesia was first demonstrated in weaver mice, which carry a mutation in the GIRK2 subunit (Patil et al., 1995). A subsequent study has shown that deletion of GIRK2 in mice reduced the antinociceptive effects of some analgesic drugs (Mitrovic et al., 2003). In GIRK2−/−/GIRK3−/− KO mice, morphine analgesia potency decreased, but efficacy was preserved (Cruz et al., 2008). In humans, single-nucleotide polymorphisms (SNPs) in GIRK2 were associated with decreased opioid sensitivity (Nishizawa et al., 2014), and subjects with these SNPs required an increased dose of opioid analgesics after abdominal surgery (Nishizawa et al., 2009). Because GiGA1 directly activates the GIRK1/GIRK2 channel in a G-protein-independent manner, it may offer non-opioid therapy for producing analgesia. Furthermore, because GiGA1 does not activate GIRK2/GIRK3 channels, the predominant form in the VTA (Cruz et al., 2004), it is less likely to show reinforcing effects. In conclusion, pharmacologically targeting subsets of GIRK channels in the brain may afford new opportunities for developing advanced therapeutics for treating various human neurological disorders.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Paul Slesinger (paul.slesinger@mssm.edu).
Materials Availability
Plasmids generated in this study are available from the lead contact upon request. Compounds disclosed in this study are freely available upon request to NCATS and completion of regular NIH procedures in the form of a material transfer agreement (MTA). NCATS repository holds limited amounts of compounds. Additional amounts can be acquired from commercial sources. This study did not generate new unique reagents.
Data and Code Availability
The published article includes all data generated or analyzed during this study. This study did not generate any unique code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cultures of HEK293 cells
Human Embryonic Kidney 293T (HEK293T) cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% (v/v) Fetal Bovine Serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin and 1X Glutamax (ThermoFisher) in a humidified 37°C incubator with 5% CO2. Cells were plated onto poly-D-lysine (100 μg/ml; SigmaAldrich) coated 12-mm glass coverslips in 24-well plates and transiently transfected with cDNA using Lipofectamine 2000 (ThermoFisher).
Mice
All animal experiments were performed in accordance with institutional guidelines and approved protocol at Icahn School of Medicine at Mount Sinai’s Institutional Animal Care and Use Committee. For brain slice recordings, both male and female C57BL/6 mice (8–12 weeks; Jackson labs) were used. For in vivo seizure study and the locomotor activity analysis, male C57BL/6 mice (3–4 months old, approximately 30 g; Jackson labs) were used. In vivo mouse pharmacokinetic (PK) studies were performed by Pharmaron Inc, using male CD1 mice (~6–8 weeks of age and a weight of about 20–26 g). All animals were housed 3–5 per cage and maintained on a 12-hour light/dark cycle, in a humidity- and temperature-controlled room with water and food available ad libitum. Littermates of the same sex were randomly assigned to experimental groups.
METHOD DETAILS
Homology Modeling
In all crystal structures of mouse GIRK2 (PDB: 3SYA (Whorton and MacKinnon, 2011), 3SYQ (Whorton and MacKinnon, 2011), 4KFM (Whorton and MacKinnon, 2013)) thus far, the N terminus of GIRK2 (residues 50–55), which forms a part of the alcohol-binding pocket in the cytoplasmic domain, is disordered. Therefore, we constructed a homology model of GIRK2 based on the crystal structure of the homotetrameric mouse Kir2.1 (sequence identity of the cytoplasmic domain: 61%; PDB: 2GIX (Pegan et al., 2006)), which has a structured N terminus and a bound alcohol, methyl-pentane-diol (MPD), in each interface between the subunits. As this construct had the transmembrane helices removed, the N-terminal region of subunit 1 (residues 41–64) was modeled to attach to the core of subunit 2 (residues 189–370). To correct this rearrangement for the subsequent homology modeling, the N-terminal region of the subunits in the template structure were reassigned to their corresponding subunits. Initially, 50 models of homodimeric GIRK2 cytoplasmic domain (residues 52–74, 205–376), in absence of MPD, were generated using MODELLER (Šali and Blundell, 1993) v9.18 with the default settings. One model of the GIRK2 pocket with the least steric clashes with the crystallographic pose of MPD in the known structure (i.e., 2GIX) was used for virtual screening. Subsequently, models of heterodimeric GIRK1 (residues 40–63, 194–365)/GIRK2 cytoplasmic domain (with two alternative arrangements: GIRK1/2 and GIRK2/1) were generated and used for identifying important sites for GiGA1 binding.
Virtual Screening
All docking calculations were performed with the Schrödinger suites (Schrödinger, 2017a, 2017b). The model of homodimeric GIRK2 cytoplasmic domain was prepared by Glide Grid with the following settings: (i) aromatic CH hydrogens and halogen atoms were treated as hydrogen-bond donor and acceptor, respectively; (ii) hydrophobic constraint to Leu257 side-chain was used; (iii) hydrogen-bond constraints to Lys52 amide NH and Pro256 carbonyl O were used; and (iv) van der Waals radius was softened (scaled to 0.8) for atoms with partial charge exceeding 0.25e. Docking calculations were performed with Glide (Schrödinger, 2017a) using the SP accuracy mode. The OPLS3 force field (Harder et al., 2016) was used to parameterize both the ligands and receptor.
We virtually screened two small molecule libraries: the “Fragment-Like Tranche” from ZINC15 (Sterling and Irwin, 2015) (~714,000 “clean, in-stock” compounds, downloaded in 2016), and a subset of a small-molecule library (56,834 molecules) provided by the National Center for Advancing Translational Sciences (NCATS). The molecules were prepared and tautomerized at pH 7.2 by LigPrep (Schrödinger, 2017b). The 1,000 top-scoring compounds from each virtual screen were subjected to visual inspection, to discard likely false positive predictions related to the limitation of the docking programs, such as docking poses with high internal energies or unbound polar groups, as well as molecules with strained conformations (Schlessinger et al., 2011). Nine compounds from the ZINC15 library were initially purchased and tested. Subsequently, 13 compounds from the NCATS library were synthesized and tested.
Molecular Dynamics Simulation
Induced-fit docking (IFD) (Schrödinger, 2017a) was used to predict the binding mode of GiGA1 and its analogs in the alcohol binding site of GIRK1/GIRK2. We used Gaussian accelerated molecular dynamics (GaMD) simulations to predict the structural and functional effect of mutations in GIRK1/GIRK2 and its binding to GiGA1. To enhance the sampling of protein dynamics, we used Gaussian accelerated Molecular Dynamics (GaMD) simulation method (Miao, Feher and McCammon, 2015), available in Amber 18 (Case et al., 2005), to examine the cytosolic domain of the GIRK1/GIRK2 heterotetramer models generated by MODELLER (Šali and Blundell, 1993) in the forms of wild-type, R43I and D346A single-mutations, and R43I/D346A double-mutation. FF14SB force field (Case et al., 2005) and TIP3P water model (Jorgensen et al., 1983) were used to parameterize the simulation system. Bond to hydrogen atom was restrained to enable 2-fs time interval (Ryckaert et al., 1977) and a 10-Å cutoff for PME (Darden et al., 1993) and non-bond interactions were used. Two independent trajectories of the protein system were conducted with minimization and equilibration for 30 ns before the 300 ns NVT production run with PMEMD.CUDA (Lee et al., 2018). Backbone dihedral angle data of trajectories was extracted using CPPTRAJ (Roe and Cheatham, 2013) and was plotted as Ramachandran plot with an in-house script (https://github.com/schlessinger-lab/RAMAplot) to examine the distribution of secondary structure of a residue throughout the GAMD trajectories. An updated definition of amino acid secondary structures (Porter and Rose, 2011) is used to describe the distribution of backbone dihedral angles in an area between αR-helix and β sheet, the hydrogen-bonded inversed γ-turn centered at Phi(φ),Psi(Ψ)= −85o, 78o: NH(i + 1)…O=C(i - 1).
Molecular Biology and Cell Culture
cDNAs for rat GIRK1 (P63251), mouse GIRK2 (P48542), mouse GIRK3 (P48543), and rat GIRK4 (P48548) were used. Heteromeric channels were studied using either cDNAs for tandem dimers connected by a short linker (GIRK1_GIRK2c, GIRK1_GIRK3) or co-expression of individual subunits (GIRK1 + GIRK4, GIRK2c + GIRK3). All heterotetrameric channels in the results are referred to as GIRK1/GIRK2, GIRK1/GIRK3, and GIRK1/GIRK4. Mutations were introduced into GIRK2c and GIRK1/GIRK2c tandem cDNAs using site-directed mutagenesis (QuikChange II XL, Agilent Technology) and confirmed by DNA sequencing. Human Embryonic Kidney 293T (HEK293T) cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% (v/v) Fetal Bovine Serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin and 1X Glutamax (ThermoFisher) in a humidified 37°C incubator with 5% CO2. Cells were plated onto poly-D-lysine (100 μg/ml; SigmaAldrich) coated 12-mm glass coverslips in 24-well plates and transiently transfected with cDNA using Lipofectamine 2000 (ThermoFisher). HEK293T cells were transfected with GIRK channel cDNAs: GIRK2 (0.5 μg), GIRK1/GIRK2 dimer (0.5 μg), or a series of GIRK1/GIRK2 mutants (0.5 μg) and eYFP cDNA (0.02 μg; to identify transfected cells). For testing G-protein-dependent effect, GABAB1b (0.25 μg), GABAB2 (0.25 μg) and PTX S1 cDNA (0.25 μg) were also included.
HEK293T Electrophysiology
For HEK293T cell recordings, whole-cell patch-clamp recordings were made as described previously (Bodhinathan and Slesinger, 2013). Borosilicate glass electrodes (Warner Instruments) of 3–6 MΩ were filled with an intracellular solution containing 130 mM KCl, 20 mM NaCl, 5 mM EGTA, 5.46 mM MgCl2, 2.56 mM K2ATP, 0.3 mM Li2GTP and 10 mM HEPES (pH 7.4, ~300 mOsm). The extracellular ‘20K’ solution contained 20 mM KCl, 140 mM NaCl, 0.5 mM CaCl2, 2 mM MgCl2, and 10 mM HEPES (pH 7.4, ~318 mOsm) (Aryal et al., 2009). Currents were elicited at 0.5 Hz with a voltage step to −120 mV from a holding potential of −40 mV, followed by a ramp voltage protocol (−120 mV to +50 mV, EK = −50 mV with 20 mM Kout). K+ currents were measured at −120 mV with either extracellular solution or drug solution. Drugs were diluted into extracellular solution with a final concentration of 100 μM Baclofen, 1–200 μM GiGA1, 100 mM 1-propanol (PrOH), 10 μM ML297 or 1 mM BaCl2. The percent activation was calculated by measuring the amplitude of the basal (Ibasal) and drug-induced current (Idrug) using the equation I%= (Idrug – Ibasal)/ Ibasal. Ibasal was defined as the Ba2+-sensitive current. Dose-response curve was fit with the Hill equation, as described previously (Glaaser and Slesinger, 2017). In some plots, the GiGA1-dependent activation of mutants was calculated as the ratio of GiGA1 to ML297 [(GiGA1-induced)/(ML297-induced)], and then normalized as a percentage of the WT ratio (% Normalized GiGA1 activation). The voltage-dependent activation was measured in response to a voltage-step from −40 mV to –120 mV.
Ex Vivo Slice Electrophysiology
Mice were sacrificed with isoflurane euthanasia and perfused with ice-cold recovery artificial cerebrospinal fluid (aCSF) containing the following: NMDG 92 mM, KCl 2.5 mM, NaHCO3 30 mM, NaH2PO4 1.25 mM, HEPES 20 mM, Glucose 25 mM, sodium ascorbate 5 mM, Thiourea 2 mM, sodium pyruvate 3 mM, CaCl2 0.5 mM (pH 7.3), and MgSO4 10 mM, bubbled with 95% O2/5% CO2 (Ting et al., 2018). All aspects of animal care and experimentation were approved by the Institutional Animal Care and Use Committee (IACUC) at the Icahn School of Medicine at Mount Sinai. Coronal slices containing hippocampus (300 μm) were prepared in recovery aCSF as well. Next, brain slices were equilibrated for 15 min at 33°C, and 1h at room temperature (RT, ~22°C) in recovery aCSF, and then transferred to a recording chamber equipped with constant perfusion of aCSF (2ml/min) contained the following: NaCl 119 mM, D-glucose 11 mM, NaHCO3 26.2 mM, KCl 2.5 mM, MgCl2 1.3 mM, NaH2PO4 1 mM, CaCl2 2.5 mM (pH 7.3) (Cruz et al., 2004). We added 100 μM GiGA1, 300 μM Baclofen (Sigma-Aldrich), 5 μM CGP55845 (Sigma-Aldrich) or 30 μM SCH23390 (Sigma-Aldrich) directly to the aCSF (Kuzhikandathil and Oxford, 2002; Montalbano et al., 2015; Rifkin et al., 2018). Neurons were visualized by infrared illumination on an Olympus fluorescent scope (BX51WI) and whole-cell patch-clamp recordings were made from neurons in the CA1 region of hippocampus. Pyramidal neurons were identified by morphology and location. For measuring drug-induced currents, whole-cell voltage-clamp recordings were used, and currents were measured at −40 mV. Whole-cell current-clamp recordings were used to measure the resting membrane potential and firing patterns, induced injecting current steps, in response to drug application.
In Vivo Seizure Model
Mice were pre-injected i.p. with either GiGA1 (20, 40, 60 mg/kg), Valproic acid (VPA; 200 mg/kg), or vehicle (10% DMSO, 40% PEG300, and 5% Solutol). After 15–30 min, 60 mg/kg PTZ was injected i.p. to induce acute seizure and mice behaviors were recorded for 30 min with a video camera. This dose induced generalized seizures but was sublethal (Itoh and Watanabe, 2009). Tonic-clonic seizures in PTZ-injected mice occurred within the first 10 min. Convulsions were classified and scored according to modified Racine score (Racine, 1972; Itoh et al., 2004; Shimada and Yamagata, 2018) as follows: 0, normal; 1, immobilization, sniffing; 2, head nodding, facial and forelimb clonus (short myoclonic jerk); 3, continuous myoclonic jerk, tail rigidity; 4, generalized limbic seizures with kangaroo posture or violent convulsion; 5, continuous generalized seizures (tonic or clonic–tonic convulsions); 6, death. The mean seizure score and percent of time in tonic seizure were compared between the VPA (+ control for anticonvulsant), GiGA1, and the vehicle group (n = 8 mice/group).
Locomotor Activity Analysis
The locomotion data was obtained for 15 min following i.p. injection of either vehicle or GiGA1 (20, 40, 60 mg/kg). In brief, same groups of mice from in vivo seizure study were placed in the center of an activity field arena (30 × 30 cm) equipped with a camera above for 5 min to habituate. The initial video was transformed in an images-sequence corresponding to a reduced frame rate (2 fps). The distance traveled 5 min after drug administration as analyzed using Spot tracker function from FIJI.
In vivo Pharmacokinetics
20 mg/kg of GiGA1 was administered intraperitoneally (i.p.) with a single dose (2 mg/mL) prepared in 10% DMSO, 40% PEG300, 5% solutol and 45% water on the day of dosing or directly before dosing. Each cohort had three mice, and plasma was collected at 5 min, 15 min, 30 min, 1 h, 2 h, 4 h, 8 h, 12 h, and 24h post-dose. Approximately 0.025 mL of blood was collected via the dorsal metatarsal vein at each time point. Blood samples were then transferred into plastic microcentrifuge tubes containing heparin—Na as an anti-coagulant. Samples were then centrifuged at 4000 g for 5 min at 4°C to obtain plasma. Plasma samples were then stored in poly-propylene tubes, quickly frozen, and kept at −75°C until analyzed by LC/MS/MS. The mouse was fully exsanguinated before brain collection. The brains were collected at specific time points, quick-frozen in an ice box and kept at −75 ± 15°C. The tissue samples were weighed and homogenized with water by tissue weight (g) to water volume (mL) ratio 1:3 before analysis. The actual concentration is the detected value multiplied by the dilution factor. The following pharmacokinetic parameters were measured: T1/2, Cmax, Tmax, AUClast, and AUCinf and calculated brain/plasma ratio. Animals were also monitored during the in-life phase by once-daily cage-side observations.
QUANTIFICATION AND STATISTICAL ANALYSIS
For single or multiple comparisons, statistical differences were assessed using Student’s Paired t test, one-way analysis of variance (ANOVA) or Mixed-effects model (REML) followed by Bonferroni post hoc test, two-way repeated-measured ANOVA with Dunnett’s or Tukey’s post hoc tests. All values are reported as mean ± SEM or S.D., as indicate in legend. Statistical tests and details along with sample size are described in the figure legends/figures. All statistics were performed using Prism 8 (GraphPad).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Baclofen | Sigma-Aldrich | Catalog #: B5399 |
| ML297 | Sigma-Aldrich | Catalog #: SML0836 |
| SCH23390 | Sigma-Aldrich | Catalog #: D054 |
| CGP55845 | Sigma-Aldrich | Catalog #: SML0594 |
| 1-Propanol | Sigma-Aldrich | Catalog #: 402893 |
| Barium Chloride | Sigma-Aldrich | Catalog #: 342920 |
| Valproic acid | Sigma-Aldrich | Catalog #: P4543 |
| Pentylenetetrazol | Sigma-Aldrich | Catalog #: P6500 |
| Critical Commercial Assays | ||
| QuikChange II XL | Agilent Technology | |
| Experimental Models: Cell Lines | ||
| HEK293T cells | ATCC | Catalog #: CRL-3216 |
| Experimental Models: Organisms/Strains | ||
| C57BL6/J mice (male and female) | Jackson Labs | Wild-type |
| CD1 mice (male) used for in vivo pharmacokinetic (PK) studies | Pharmaron labs | https://www.pharmaron.com/ |
| Oligonucleotides | ||
| DNA primers for mutagenesis | This study | N/A |
| Recombinant DNA | ||
| rat GIRK1_mouse GIRK2c dimer cDNA | Horvath et al., 2018 | N/A |
| mouse GIRK2c cDNA | Horvath et al., 2018 | N/A |
| rat GIRK1_mouse GIRK3 dimer cDNA | This study | N/A |
| mouse GIRK3 cDNA | Cruz et al., 2004 | N/A |
| rat GIRK4 cDNA | Aryal et al., 2009 | N/A |
| human GABAB1b cDNA | Horvath et al., 2018 | N/A |
| human GABAB2 cDNA | Horvath et al., 2018 | N/A |
| PTX S1 cDNA | This study | N/A |
| Software and Algorithms | ||
| Prism 7.0 | Graphpad | RRID:SCR_002798 |
| Schrödinger (2017a, 2017b), LigPrep, Glide | Schrödinger | https://www.schrodinger.com/; RRID: SCR_014879 |
| MODELLER v9.18 | (Šali and Blundell, 1993) | salilab.org/modeller; RRID: SCR_008395 |
| MOE 2014.0901, 2016 | Chemical Computing Group | https://www.chemcomp.com/; RRID: SCR_014882 |
| OEDocking 3.2.0.2, FRED, OMEGA | OpenEye | https://www.eyesopen.com/; RRID: SCR_014880 |
| KNIME | https://www.knime.com/; RRID: SCR_006164 | |
| JChem Cartridge | ChemAxon | https://chemaxon.com; RRID: SCR_004111 |
| StarDrop | Optibrium | https://www.optibrium.com/stardrop/; RRID: SCR_014902 |
| FIJI | ImageJ | http://fiji.sc; RRID: SCR_002285 |
| PyMOL | https://pymol.org/2/; RRID: SCR_000305 | |
Highlights.
Virtual screening with a GIRK alcohol pocket reveals a GIRK1-selective activator
GiGA1 activates the GIRK channel directly without G proteins
Natively expressed GIRK channels in the hippocampus are activated by GiGA1
Systemic GiGA1 application reduces seizures in a convulsant animal model
ACKNOWLEDGMENTS
This work has been supported in part by National Institutes of Health (NIH) grants from the National Institute on Alcohol Abuse and Alcoholism, USA (R01AA018734) to P.A. Slesinger and from the National Institute of General Medical Sciences, USA (R01 GM108911) to A. Schlessinger. and P.M.- U.Ung. This work was supported in part through the computational resources and staff expertise provided by the Department of Scientific Computing at the Icahn School of Medicine at Mount Sinai and by the Intramural Research Program of the National Center for Advancing Translational Sciences (NCATS), NIH. The graphical abstract was created with BioRender.com.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107770.
DECLARATION OF INTERESTS
Schlessinger is a co-founder of AIchemy.
REFERENCES
- Abrahao KP, Salinas AG, and Lovinger DM (2017). Alcohol and the Brain: Neuronal Molecular Targets, Synapses, and Circuits. Neuron 96, 1223–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Addolorato G, Caputo F, Capristo E, Domenicali M, Bernardi M, Janiri L, Agabio R, Colombo G, Gessa GL, and Gasbarrini G (2002). Baclofen efficacy in reducing alcohol craving and intake: a preliminary double-blind randomized controlled study. Alcohol Alcohol. 37, 504–508. [DOI] [PubMed] [Google Scholar]
- Addolorato G, Leggio L, Ferrulli A, Cardone S, Vonghia L, Mirijello A, Abenavoli L, D’Angelo C, Caputo F, Zambon A, et al. (2007). Effectiveness and safety of baclofen for maintenance of alcohol abstinence in alcoholdependent patients with liver cirrhosis: randomised, double-blind controlled study. Lancet 370, 1915–1922. [DOI] [PubMed] [Google Scholar]
- Arora D, Haluk DM, Kourrich S, Pravetoni M, Fernandez-Alacid L, Nicolau JC, Luján R, and Wickman K (2010). Altered neurotransmission in the mesolimbic reward system of Girk mice. J. Neurochem 114, 1487–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aryal P, Dvir H, Choe S, and Slesinger PA (2009). A discrete alcohol pocket involved in GIRK channel activation. Nat. Neurosci 12, 988–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baculis BC, Weiss AC, Pang W, Jeong HG, Lee JH, Liu DC, Tsai NP, and Chung HJ (2017). Prolonged seizure activity causes caspase dependent cleavage and dysfunction of G-protein activated inwardly rectifying potassium channels. Sci. Rep 7, 12313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best TK, Siarey RJ, and Galdzicki Z (2007). Ts65Dn, a mouse model of Down syndrome, exhibits increased GABAB-induced potassium current. J. Neurophysiol 97, 892–900. [DOI] [PubMed] [Google Scholar]
- Blednov YA, Stoffel M, Alva H, and Harris RA (2003). A pervasive mechanism for analgesia: activation of GIRK2 channels. Proc. Natl. Acad. Sci. USA 100, 277–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodhinathan K, and Slesinger PA (2013). Molecular mechanism underlying ethanol activation of G-protein-gated inwardly rectifying potassium channels. Proc. Natl. Acad. Sci. USA 110, 18309–18314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodhinathan K, and Slesinger PA (2014). Alcohol modulation of G-protein-gated inwardly rectifying potassium channels: From binding to therapeutics. Front. Physiol 5, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascino GD (2005). Temporal lobe epilepsy: more than hippocampal pathology. Epilepsy Curr. 5, 187–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case DA, Cheatham TE 3rd, Darden T, Gohlke H, Luo R, Merz KM Jr., Onufriev A, Simmerling C, Wang B, and Woods RJ (2005). The Amber biomolecular simulation programs. J. Comput. Chem 26, 1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo G, Serra S, Brunetti G, Vacca G, Carai MA, and Gessa GL (2003). Suppression by baclofen of alcohol deprivation effect in Sardinian alcohol-preferring (sP) rats. Drug Alcohol Depend. 70, 105–108. [DOI] [PubMed] [Google Scholar]
- Colombo G, Serra S, Vacca G, Carai MAM, and Gessa GL (2006). Baclofen-induced suppression of alcohol deprivation effect in Sardinian alcohol-preferring (sP) rats exposed to different alcohol concentrations. Eur. J. Pharmacol 550, 123–126. [DOI] [PubMed] [Google Scholar]
- Cruz HG, Ivanova T, Lunn ML, Stoffel M, Slesinger PA, and Lüscher C (2004). Bi-directional effects of GABA(B) receptor agonists on the mesolimbic dopamine system. Nat. Neurosci 7, 153–159. [DOI] [PubMed] [Google Scholar]
- Cruz HG, Berton F, Sollini M, Blanchet C, Pravetoni M, Wickman K, and Lüscher C (2008). Absence and rescue of morphine withdrawal in GIRK/Kir3 knock-out mice. J. Neurosci 28, 4069–4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darden T, York D, and Pedersen L (1993). Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J. Chem. Phys 98, 10089. [Google Scholar]
- Dascal N (1997). Signalling via the G protein-activated K+ channels. Cell. Signal 9, 551–573. [DOI] [PubMed] [Google Scholar]
- Diehl B, Najm I, LaPresto E, Prayson R, Ruggieri P, Mohamed A, Ying Z, Lieber M, Babb T, Bingaman W, and Lüders HO (2004). Temporal lobe volumes in patients with hippocampal sclerosis with or without cortical dysplasia. Neurology 62, 1729–1735. [DOI] [PubMed] [Google Scholar]
- Finley M, Arrabit C, Fowler C, Suen KF, and Slesinger PA (2004). βL-βM loop in the C-terminal domain of G protein-activated inwardly rectifying K(+) channels is important for Gβγ subunit activation. J. Physiol 555, 643–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaaser Ian, and Slesinger Paul A. (2017). Dual activation of neuronal G protein-gated inwardly rectifying potassium (GIRK) channels by cholesterol and alcohol. Scientific Reports 7, 4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldenberg MM (2010). Overview of drugs used for epilepsy and seizures: etiology, diagnosis, and treatment. P&T 35, 392–415. [PMC free article] [PubMed] [Google Scholar]
- Gray JA, Goodwin GM, Heal DJ, and Green AR (1987). Hypothermia induced by baclofen, a possible index of GABAB receptor function in mice, is enhanced by antidepressant drugs and ECS. Br. J. Pharmacol 92, 863–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harashima C, Jacobowitz DM, Witta J, Borke RC, Best TK, Siarey RJ, and Galdzicki Z (2006). Abnormal expression of the G-protein-activated inwardly rectifying potassium channel 2 (GIRK2) in hippocampus, frontal cortex, and substantia nigra of Ts65Dn mouse: a model of Down syndrome. J. Comp. Neurol 494, 815–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder E, Damm W, Maple J, Wu C, Reboul M, Xiang JY, Wang L, Lupyan D, Dahlgren MK, Knight JL, et al. (2016). OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput 12, 281–296. [DOI] [PubMed] [Google Scholar]
- Hendler RA, Ramchandani VA, Gilman J, and Hommer DW (2013). Stimulant and sedative effects of alcohol. Curr. Top. Behav. Neurosci 13, 489–509. [DOI] [PubMed] [Google Scholar]
- Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, and Kurachi Y (2010). Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol. Rev 90, 291–366. [DOI] [PubMed] [Google Scholar]
- Horvath GA, Zhao Y, Tarailo-Graovac M, Boelman C, Gill H, Shyr C, Lee J, Blydt-Hansen I, Drögemöller BI, Moreland J, et al. (2018). Gain-of-function KCNJ6 Mutation in a Severe Hyperkinetic Movement Disorder Phenotype. Neuroscience 384, 152–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CL, Slesinger PA, Casey PJ, Jan YN, and Jan LY (1995). Evidence that direct binding of Gβγ to the GIRK1 G protein-gated inwardly rectifying K+ channel is important for channel activation. Neuron 15, 1133–1143. [DOI] [PubMed] [Google Scholar]
- Ikeda K, Kobayashi T, Kumanishi T, Niki H, and Yano R (2000). Involvement of G-protein-activated inwardly rectifying K (GIRK) channels in opioid-induced analgesia. Neurosci. Res 38, 113–116. [DOI] [PubMed] [Google Scholar]
- Inanobe A, Morishige KI, Takahashi N, Ito H, Yamada M, Takumi T, Nishina H, Takahashi K, Kanaho Y, Katada T, and Kurachi Y (1995). Gβγ directly binds to the carboxyl terminus of the G protein-gated muscarinic K+ channel, GIRK1. Biochem. Biophys. Res. Commun 212, 1022–1028. [DOI] [PubMed] [Google Scholar]
- Inanobe A, Yoshimoto Y, Horio Y, Morishige KI, Hibino H, Matsumoto S, Tokunaga Y, Maeda T, Hata Y, Takai Y, and Kurachi Y (1999). Characterization of G-protein-gated K+ channels composed of Kir3.2 subunits in dopaminergic neurons of the substantia nigra. J. Neurosci 19, 1006–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K, and Watanabe M (2009). Paradoxical facilitation of pentylenetetrazole-induced convulsion susceptibility in mice lacking neuronal nitric oxide synthase. Neuroscience 159, 735–743. [DOI] [PubMed] [Google Scholar]
- Itoh K, Watanabe M, Yoshikawa K, Kanaho Y, Berliner LJ, and Fujii H (2004). Magnetic resonance and biochemical studies during pentylenetetrazole-kindling development: the relationship between nitric oxide, neuronal nitric oxide synthase and seizures. Neuroscience 129, 757–766. [DOI] [PubMed] [Google Scholar]
- Ivanina T, Rishal I, Varon D, llner C, Frohnwieser-Steinecke B, Schreibmayer W, Dessauer CW, and Dascal N (2003). Mapping the Gβγ-binding sites in GIRK1 and GIRK2 subunits of the G protein-activated K+ channel. J. Biol. Chem 278, 29174–29183. [DOI] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, and Madura JD (1983). Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 79, 926–935. [Google Scholar]
- Karschin C, Dissmann E, Stühmer W, and Karschin A (1996). IRK(1–3) and GIRK(1–4) inwardly rectifying K+ channel mRNAs are differentially expressed in the adult rat brain. J. Neurosci 16, 3559–3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann K, Romaine I, Days E, Pascual C, Malik A, Yang L, Zou B, Du Y, Sliwoski G, Morrison RD, et al. (2013). ML297 (VU0456810), the first potent and selective activator of the GIRK potassium channel, displays antiepileptic properties in mice. ACS Chem. Neurosci 4, 1278–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Ikeda K, Ichikawa T, Abe S, Togashi S, and Kumanishi T (1995). Molecular cloning of a mouse G-protein-activated K+ channel (mGIRK1) and distinct distributions of three GIRK (GIRK1, 2 and 3) mRNAs in mouse brain. Biochem. Biophys. Res. Commun 208, 1166–1173. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Ikeda K, Kojima H, Niki H, Yano R, Yoshioka T, and Kumanishi T (1999). Ethanol opens G-protein-activated inwardly rectifying K+ channels. Nat. Neurosci 2, 1091–1097. [DOI] [PubMed] [Google Scholar]
- Kofuji P, Doupnik CA, Davidson N, and Lester HA (1996). A unique P-region residue is required for slow voltage-dependent gating of a G protein-activated inward rectifier K+ channel expressed in Xenopus oocytes. J. Physiol 490, 633–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyrakh L, Luján R, Colón J, Karschin C, Kurachi Y, Karschin A, and Wickman K (2005). Molecular and cellular diversity of neuronal G-protein-gated potassium channels. J. Neurosci 25, 11468–11478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutlu MG, and Gould TJ (2016). Effects of drugs of abuse on hippocampal plasticity and hippocampus-dependent learning and memory: contributions to development and maintenance of addiction. Learn. Mem 23, 515–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzhikandathil EV, and Oxford GS (2002). Classic D1 dopamine receptor antagonist R-(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride (SCH23390) directly inhibits G protein-coupled inwardly rectifying potassium channels. Mol. Pharmacol 62, 119–126. [DOI] [PubMed] [Google Scholar]
- Labouèbe G, Lomazzi M, Cruz HG, Creton C, Luján R, Li M, Yanagawa Y, Obata K, Watanabe M, Wickman K, et al. (2007). RGS2 modulates coupling between GABAB receptors and GIRK channels in dopamine neurons of the ventral tegmental area. Nat. Neurosci 10, 1559–1568. [DOI] [PubMed] [Google Scholar]
- Leaney JL (2003). Contribution of Kir3.1, Kir3.2A and Kir3.2C subunits to native G protein-gated inwardly rectifying potassium currents in cultured hippocampal neurons. Eur. J. Neurosci 18, 2110–2118. [DOI] [PubMed] [Google Scholar]
- Lee TS, Cerutti DS, Mermelstein D, Lin C, LeGrand S, Giese TJ, Roitberg A, Case DA, Walker RC, and York DM (2018). GPU-Accelerated Molecular Dynamics and Free Energy Methods in Amber18: Performance Enhancements and New Features. J. Chem. Inf. Model 58, 2043–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage F, Guillemare E, Fink M, Duprat F, Heurteaux C, Fosset M, Romey G, Barhanin J, and Lazdunski M (1995). Molecular properties of neuronal G-protein-activated inwardly rectifying K+ channels. J. Biol. Chem 270, 28660–28667. [DOI] [PubMed] [Google Scholar]
- Lewohl JM, Wilson WR, Mayfield RD, Brozowski SJ, Morrisett RA, and Harris RA (1999). G-protein-coupled inwardly rectifying potassium channels are targets of alcohol action. Nat. Neurosci 2, 1084–1090. [DOI] [PubMed] [Google Scholar]
- Li X, Risbrough VB, Cates-Gatto C, Kaczanowska K, Finn MG, Roberts AJ, and Markou A (2013). Comparison of the effects of the GABAB receptor positive modulator BHF177 and the GABAB receptor agonist baclofen on anxiety-like behavior, learning, and memory in mice. Neuropharmacology 70, 156–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao YJ, Jan YN, and Jan LY (1996). Heteromultimerization of G-protein-gated inwardly rectifying K+ channel proteins GIRK1 and GIRK2 and their altered expression in weaver brain. J. Neurosci 16, 7137–7150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luján R, and Aguado C (2015). Localization and Targeting of GIRK Channels in Mammalian Central Neurons. Int. Rev. Neurobiol 123, 161–200. [DOI] [PubMed] [Google Scholar]
- Lüscher C, and Slesinger PA (2010). Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat. Rev. Neurosci 11, 301–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, Jan LY, Stoffel M, Malenka RC, and Nicoll RA (1997). G protein-coupled inwardly rectifying K+ channels (GIRKs) mediate postsynaptic but not presynaptic transmitter actions in hippocampal neurons. Neuron 19, 687–695. [DOI] [PubMed] [Google Scholar]
- Masotti A, Uva P, Davis-Keppen L, Basel-Vanagaite L, Cohen L, Pisaneschi E, Celluzzi A, Bencivenga P, Fang M, Tian M, et al. (2015). Keppen-Lubinsky syndrome is caused by mutations in the inwardly rectifying K+ channel encoded by KCNJ6. Am. J. Hum. Genet 96, 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre DC, and Racine RJ (1986). Kindling mechanisms: current progress on an experimental epilepsy model. Prog. Neurobiol 27, 1–12. [DOI] [PubMed] [Google Scholar]
- Mehta MV, Gandal MJ, and Siegel SJ (2011). mGluR5-antagonist mediated reversal of elevated stereotyped, repetitive behaviors in the VPA model of autism. PLoS ONE 6, e26077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y, Feher VA, and McCammon JA (2015). Gaussian Accelerated Molecular Dynamics: Unconstrained Enhanced Sampling and Free Energy Calculation. J. Chem. Theory Comput 11, 3584–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrovic I, Margeta-Mitrovic M, Bader S, Stoffel M, Jan LY, and Basbaum AI (2003). Contribution of GIRK2-mediated postsynaptic signaling to opiate and α 2-adrenergic analgesia and analgesic sex differences. Proc. Natl. Acad. Sci. USA 100, 271–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montalbano A, Corradetti R, and Mlinar B (2015). Pharmacological characterization of 5-HT1A autoreceptor-coupled GIRK channels in rat dorsal raphe 5-HT neurons. PLoS ONE 10, e0140369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore EM, Serio KM, Goldfarb KJ, Stepanovska S, Linsenbardt DN, and Boehm SL, 2nd. (2007). GABAergic modulation of binge-like ethanol intake in C57BL/6J mice. Pharmacol. Biochem. Behav 88, 105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan AD, Carroll ME, Loth AK, Stoffel M, and Wickman K (2003). Decreased cocaine self-administration in Kir3 potassium channel subunit knockout mice. Neuropsychopharmacology 28, 932–938. [DOI] [PubMed] [Google Scholar]
- Morley KC, Baillie A, Fraser I, Furneaux-Bate A, Dore G, Roberts M, Abdalla A, Phung N, and Haber PS (2018). Baclofen in the treatment of alcohol dependence with or without liver disease: multisite, randomised, double-blind, placebo-controlled trial. Br. J. Psychiatry 212, 362–369. [DOI] [PubMed] [Google Scholar]
- Müller CA, Geisel O, Pelz P, Higl V, Krüger J, Stickel A, Beck A, Wernecke KD, Hellweg R, and Heinz A (2015). High-dose baclofen for the treatment of alcohol dependence (BACLAD study): a randomized, placebo-controlled trial. Eur. Neuropsychopharmacol 25, 1167–1177. [DOI] [PubMed] [Google Scholar]
- Nishizawa D, Nagashima M, Katoh R, Satoh Y, Tagami M, Kasai S, Ogai Y, Han W, Hasegawa J, Shimoyama N, Sora I, Hayashida M, and Ikeda K (2009). Association between KCNJ6 (GIRK2) gene polymorphisms and postoperative analgesic requirements after major abdominal surgery. PLoS ONE 4, e7060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizawa D, Fukuda K, Kasai S, Ogai Y, Hasegawa J, Sato N, Yamada H, Tanioka F, Sugimura H, Hayashida M, and Ikeda K (2014). Association between KCNJ6 (GIRK2) gene polymorphism rs2835859 and postoperative analgesia, pain sensitivity, and nicotine dependence. J. Pharmacol. Sci 126, 253–263. [DOI] [PubMed] [Google Scholar]
- Ong J, and Kerr DIB (2005). Clinical potential of GABAB receptor modulators. CNS Drug Rev. 11, 317–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil N, Cox DR, Bhat D, Faham M, Myers RM, and Peterson AS (1995). A potassium channel mutation in weaver mice implicates membrane excitability in granule cell differentiation. Nat. Genet 11, 126–129. [DOI] [PubMed] [Google Scholar]
- Pegan S, Arrabit C, Slesinger PA, and Choe S (2006). Andersen’s syndrome mutation effects on the structure and assembly of the cytoplasmic domains of Kir2.1. Biochemistry 45, 8599–8606. [DOI] [PubMed] [Google Scholar]
- Pellock JM (1994). Standard approach to antiepileptic drug treatment in the United States. Epilepsia 35 (Suppl 4), S11–S18. [DOI] [PubMed] [Google Scholar]
- Piasta KN, Theobald DL, and Miller C (2011). Potassium-selective block of barium permeation through single KcsA channels. J. Gen. Physiol 138, 421–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter LL, and Rose GD (2011). Redrawing the Ramachandran plot after inclusion of hydrogen-bonding constraints. Proc. Natl. Acad. Sci. USA 108, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine RJ (1972). Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr. Clin. Neurophysiol 32, 281–294. [DOI] [PubMed] [Google Scholar]
- Reeves RH, Irving NG, Moran TH, Wohn A, Kitt C, Sisodia SS, Schmidt C, Bronson RT, and Davisson MT (1995). A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat. Genet 11, 177–184. [DOI] [PubMed] [Google Scholar]
- Reisine T (1990). Pertussis toxin in the analysis of receptor mechanisms. Biochem. Pharmacol 39, 1499–1504. [DOI] [PubMed] [Google Scholar]
- Reuveny E, Slesinger PA, Inglese J, Morales JM, lñiguez-Lluhi JA, Lefkowitz RJ, Bourne HR, Jan YN, and Jan LY (1994). Activation of the cloned muscarinic potassium channel by G protein βγ subunits. Nature 370, 143–146. [DOI] [PubMed] [Google Scholar]
- Rifkin RA, Huyghe D, Li X, Parakala M, Aisenberg E, Moss SJ, and Slesinger PA (2018). GIRK currents in VTA dopamine neurons control the sensitivity of mice to cocaine-induced locomotor sensitization. Proc. Natl. Acad. Sci. USA 115, E9479–E9488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe DR, and Cheatham TE 3rd. (2013). PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput 9, 3084–3095. [DOI] [PubMed] [Google Scholar]
- Ryckaert JP, Ciccotti G, and Berendsen HJC (1977). Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys 23, 327–341. [Google Scholar]
- Šali A, and Blundell TL (1993). Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol 234, 779–815. [DOI] [PubMed] [Google Scholar]
- Schlessinger A, Geier E, Fan H, Irwin JJ, Shoichet BK, Giacomini KM, and Sali A (2011). Structure-based discovery of prescription drugs that interact with the norepinephrine transporter, NET. Proc. Natl. Acad. Sci. USA 108, 15810–15815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger. (2017a). Glide, version 2 (Schrödinger; ). [Google Scholar]
- Schrödinger. (2017b). LigPrep, version 2 (Schrödinger; ). [Google Scholar]
- Shimada T, and Yamagata K (2018). Pentylenetetrazole-induced kindling mouse model. J. Vis. Exp 136, 56573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signorini S, Liao YJ, Duncan SA, Jan LY, and Stoffel M (1997). Normal cerebellar development but susceptibility to seizures in mice lacking G protein-coupled, inwardly rectifying K+ channel GIRK2. Proc. Natl. Acad. Sci. USA 94, 923–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slesinger PA, Stoffel M, Jan YN, and Jan LY (1997). Defective γ-amino-butyric acid type B receptor-activated inwardly rectifying K+ currents in cerebellar granule cells isolated from weaver and Girk2 null mutant mice. Proc. Natl. Acad. Sci. USA 94, 12210–12217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterling T, and Irwin JJ (2015). ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model 55, 2324–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Mermelstein PG, and Goldowitz D (1996). The weaver mutation of GIRK2 results in a loss of inwardly rectifying K+ current in cerebellar granule cells. Proc. Natl. Acad. Sci. USA 93, 11191–11195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimoto TT (1969). Classification by computers: a generalized pearsonyule contingency technique and its application to automatic classification. Ann. N Y Acad. Sci 161, 420–423. [Google Scholar]
- Ting JT, Lee BR, Chong P, Soler-Llavina G, Cobbs C, Koch C, Zeng H, and Lein E (2018). Preparation of Acute Brain Slices Using an Optimized N-Methyl-D-glucamine Protective Recovery Method. J. Vis. Exp 132, 53825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrecilla M, Marker CL, Cintora SC, Stoffel M, Williams JT, and Wickman K (2002). G-protein-gated potassium channels containing Kir3.2 and Kir3.3 subunits mediate the acute inhibitory effects of opioids on locus ceruleus neurons. J. Neurosci 22, 4328–4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valyear MD, Villaruel FR, and Chaudhri N (2017). Alcohol-seeking and relapse: A focus on incentive salience and contextual conditioning. Behav. Processes 141, 26–32. [DOI] [PubMed] [Google Scholar]
- Whorton MR, and MacKinnon R (2011). Crystal structure of the mammalian GIRK2 K+ channel and gating regulation by G proteins, PIP2, and sodium. Cell 147, 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whorton MR, and MacKinnon R (2013). X-ray structure of the mammalian GIRK2-βγ G-protein complex. Nature 498, 190–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickman K, Karschin C, Karschin A, Picciotto MR, and Clapham DE (2000). Brain localization and behavioral impact of the G-protein-gated K+ channel subunit GIRK4. J. Neurosci 20, 5608–5615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wydeven N, Marron Fernandez de Velasco E, Du Y, Benneyworth MA, Hearing MC, Fischer RA, Thomas MJ, Weaver CD, and Wickman K (2014). Mechanisms underlying the activation of G-protein-gated inwardly rectifying K+ (GIRK) channels by the novel anxiolytic drug, ML297. Proc. Natl. Acad. Sci. USA 111, 10755–10760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Inanobe A, and Kurachi Y (1998). G protein regulation of potassium ion channels. Pharmacol. Rev 50, 723–760. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all data generated or analyzed during this study. This study did not generate any unique code.