

Abstract
JQ1 is a small-molecule inhibitor of the bromodomain and extra terminal (BET) protein family that potently inhibits the bromodomain testis-specific protein (BRDT), which is essential for spermatogenesis. JQ1 treatment produces a reversible contraceptive effect by targeting the activity of BRDT in mouse male germ cells, validating BRDT as a male contraceptive target. Although JQ1 possesses favourable physical properties, it exhibits a short half-life. Because the details of xenobiotic metabolism play important roles in the optimization of drug candidates and in determining the role of metabolism in drug efficacy, we investigated the metabolism of JQ1 in human and mouse liver microsomes. We present the first comprehensive view of JQ1 metabolism in liver microsomes, distinguishing nine JQ1 metabolites, including three monohydroxylated, one de-tert-butylated, two dihydroxylated, one monohydroxylated/dehydrogenated, one monohydroxylated-de-tert-butylated and one dihydroxylated/dehydrogenated variant of JQ1. The dominant metabolite (M1) in both human and mouse liver microsomes is monohydroxylated on the fused three-ring core. Using recombinant cytochrome P450 (CYP) enzymes, chemical inhibitors and the liver S9 fraction of Cyp3a-null mice, we identify enzymes that contribute to the formation of these metabolites. Cytochrome P450 family 3 subfamily A member 4 (CYP3A4) is the main contributor to the production of JQ1 metabolites in vitro, and the CYP3A4/5 inhibitor ketoconazole strongly inhibits JQ1 metabolism in both human and mouse liver microsomes. Our findings suggest that JQ1 half-life and efficacy might be improved in vivo by co-administration of a selective CYP inhibitor, thereby impacting the use of JQ1 as a probe for BRDT activity in spermatogenesis and as a probe or therapeutic in other systems.
Keywords: JQ1, BRDT inhibitors, metabolite identification, liver microsomes, cytochrome P450s
Nine JQ1 metabolites were identified and characterized in liver microsomes, and the enzyme CYP3A4 mediates the formation of most JQ1 metabolites.
Introduction
(+)-JQ1, (tert-butyl (S)-2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo [4,3-a][1,4]diazepin-6-yl)acetate), is a potent and competitive inhibitor for the bromodomain and extra terminal (BET) protein family [1], epigenetic readers that regulate gene expression and play an important role in cell proliferation, pro-inflammatory events and immune responses [2]. In mouse and human, BET family consists of ubiquitously expressed BRD2, BRD3, and BRD4 and the testis-restricted BRDT [3, 4]. As a first-in-class potent inhibitor, JQ1 has become a tool for many preclinical studies on BET inhibition [1], and in collaboration with Drs. Jun Qi and James Bradner, our group reported that JQ1 potently inhibits the testis-specific member BRDT, which is essential for spermatogenesis [5]. JQ1 inhibits bromodomain activity and produces a reversible contraceptive effect by targeting male germ cell in mammals [5]. JQ1 was the first identified bromodomain inhibitor and has been used as a chemical probe in numerous in vivo biological studies. JQ1 can also induce apoptosis of diverse human cancer cells (e.g. liver fibrosis [6], lung adenocarcinoma I [7] and myeloid leukaemia [1, 8, 9]) and suppress HIV [10] by inhibiting BRD4 activity. However, JQ1 half-life in CD1 mice is only about 1 h, so its effects are short-lasting [1, 5, 11, 12], which limits the ability to translate preclinical findings into clinical benefit.
Drug metabolism generates metabolites that can be pharmacologically active, inactive or toxic [13]. Studying drug metabolism is essential in the validation of optimized compounds as both efficacy and safety depend on drug metabolism [14, 15]. Metabolite identification can be a critical step in identifying the ‘soft spot’, a chemical moiety that facilitates rapid metabolism of a given compound, which can be modified by medicinal chemists to increase the half-life of a candidate and during lead optimization phase in drug discovery [16]. For example, 3-(4-fluoropiperidin-3-yl)-2-phenyl-1H-indole is a h5-HT2A receptor antagonist that has a short half-life (1.4 h) in rats, and its major metabolite is formed by oxidation of the 6-position of the indole ring in vitro and in vivo. Replacing the hydrogen at 6-position of the indole with fluorine to give 6-fluoro-3-(4-fluoropiperidin-3-yl)-2-phenyl-1H-indole improved potency and resulted in an 8-fold increase in half-life (12 h) in rats [17]. Identifying the major enzymes that metabolize a drug can help predict likely drug–drug interactions that should be evaluated in the process of drug development [18]. Nevertheless, few studies of JQ1 metabolism have been performed to date. Here, we employ LC-MS-based metabolomic approaches to profile the in vivo fate of JQ1 in human and mouse liver microsomes. Mouse liver microsomes are included in this work because mice are commonly used animal models in the preclinical studies that use JQ1.
Metabolomics has proven to be a powerful and effective tool to investigate drug metabolism, bioactivation [19–23] and toxicity [24–27]. We used LC-MS-based metabolomic approaches to determine, for the first time, that nine metabolites are generated from JQ1 in human and mouse liver microsomes. Monohydroxylated-JQ1 (M1) is the dominant metabolite in both types of liver microsomes. Our studies show that CYP3A4 is the major enzyme contributing to the formation of JQ1 metabolites in human and mouse liver microsomes, provide information to predict possible drug–JQ1 interactions and suggest that the use of CYP3A4 inhibitors (but not Cyp3a-null mice) may have application in mapping the effects of JQ1 on spermatogenesis.
Materials and methods
Materials
JQ1, [tert-butyl (S)-2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetate], ketoconazole (KCZ), NADPH (β-nicotinamide adenine dinucleotide 2′-phosphate reduced tetrasodium salt hydrate) and formic acid were obtained from Sigma-Aldrich (St. Louis, MO). Nootkatone was purchased form Cayman Chemical Company (Ann Arbor, MI). Human and mouse liver microsomes and recombinant human cytochrome P450s (EasyCYP Bactosomes) were purchased from XenoTech (Lenexa, KS). All solvents for liquid chromatography and mass spectrometry were of the highest grade commercially available.
Metabolism of JQ1 in human and mouse liver microsomes and recombinant cytochrome P450s
Incubations were conducted in 1× phosphate-buffered saline (1 × PBS, pH 7.4) containing 20 μM JQ1 and 0.2 mg human or mouse liver microsomes or 2 pmol of each cDNA-expressed P450 enzyme (control, CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 and 3A4) in a final volume of 190 μl. After a 5 min pre-incubation at 37 °C, 10 μl of 20 mM NADPH was added (final concentration 1.0 mM), and incubation was continued for 30 min with gentle shaking. Incubations without NADPH were used as controls. Co-incubations of JQ1 (20 μM) and KCZ (CYP3A4 inhibitor, 2.0 μM) or nootkatone (CYP2C19 inhibitor, 10 μM) in human and mouse liver microsomes were performed to determine the role of CYP3A4 and CYP2C19 in the formation of JQ1 metabolites. Reactions were terminated by adding 200 μl of ice-cold methanol and vortexing for 30 s and then centrifuged at rcf 15 000 for 15 min. Each supernatant was transferred to an autosampler vial, and 3.0 μl was injected onto UHPLC-Q Exactive MS system for analysis. Incubations were performed in duplicate for cDNA-expressed enzymes and in triplicate for liver microsome experiments.
Metabolism of JQ1 in S9 fraction from Cyp3a-null and wild-type mice
Liver tissue (150 mg) (Cyp3a-null or WT) was homogenized in 600 ml of 1× PBS at 0 °C. The homogenate was centrifuged at 9000 × g for 20 min at 4 °C, and the resulting supernatant was designated the enzyme fraction (S9 fraction). JQ1 (25 μM) or MDZ (30 μM) was incubated in the 1× PBS in the presence of 60 ml of S9 fraction freshly prepared, a final volume of 190 ml. After 5 min of pre-incubation at 37 °C, the reaction was initiated by adding 10 ml of 20 mM NADPH (final concentration 1.0 mM) and continued for 30 min for JQ1 and 15 min for MDZ with gentle shaking. Reactions were stopped by adding 200 ml of ice-cold methanol and vortexing for 30 s. The reaction mixtures were centrifuged at 15 000 × g for 20 min. Each supernatant was transferred to an autosampler vial for metabolite analysis. Incubations were performed in triplicate.
UHPLC-Q Exactive Orbitrap MS analyses
JQ1 and its metabolites were resolved, identified and quantified (relatively) using UHPLC coupled with Q Exactive Orbitrap MS (Thermo Fisher Scientific, San Jose, CA) equipped with 100 mm × 2.1 mm column (BEH C-18, Agilent Technologies, Santa Clara, CA). The column temperature was maintained at 40 °C. The flow rate was 0.3 ml/min with a gradient ranging from 2 to 95% aqueous acetonitrile containing 0.1% formic acid in a 15 min run. Q Exactive MS was operated in positive mode with electrospray ionization. Ultrapure nitrogen was applied as the sheath (45 arbitrary unit), auxiliary (10 arbitrary unit), sweep (1.0 arbitrary unit) and collision gas. The capillary gas temperature was set at 275 °C and the capillary voltage was set at 3.7 kV. MS data were acquired from 80 to 1200 Da in profile mode; reference ions at m/z 371.1012 and 445.1200 in the positive mode were used as lock masses during acquisition. MS/MS of JQ1 metabolites was performed in targeted mode with an isolation width of 2 m/z with ramp collision energy being set at 15, 20 and 35 eV.
Data analysis
Mass chromatograms and mass spectra were acquired and processed using the Xcalibur software (Thermo Fisher Scientific, San Jose, CA) in profile formats from m/z 50 to 750. The acquired data were processed by Compound Discoverer 3.0 software (Thermo Fisher Scientific, San Jose, CA) to generate a multivariate data matrix. Data matrices were exported into SIMCA14 (Umetrics, Kinnelon, NJ) for multivariate data analysis [28]. Orthogonal projection to latent structure discriminant analysis (OPLS-DA) was conducted on Pareto-scaled data [29]. For chemometric analysis, matrix data were processed from m/z 100 to 750. Statistical analysis was conducted using Student’s independent t-test. Experimental data are presented as mean ± SEM.
Results
Profiling JQ1 metabolism in human liver microsomes using metabolomic approaches
The results of metabolomic analysis on the ions produced by UHPLC-QTOFMS of control and JQ1 group are shown in Figure 1. Metabolomic analysis revealed two clusters (Figure 1A) corresponding to the control and JQ1 group in the score plots, which indicates that the chemical components are different between control and JQ1 groups. The human liver microsome data S-plot (Figure 1B) generated from OPLS-DA displays ions that contribute to group separation. Many of the top-ranking ions were identified as JQ1 and its metabolites, as marked in the S-plots (Figure 1B). All nine distinguishable JQ1 metabolites were present in both human and mouse liver microsomes (Figure 1C and Table 1), including three monohydroxylated JQ1 (M1–M3), two acids (M4 and M8), two dihydroxylated JQ1 (M5 and M6), one monohydroxylated/dehydrogenated JQ1 (M7) and one dihydroxylated/dehydrogenated JQ1 (M9). The detailed information associated with JQ1 and its metabolites are summarized in Table 1. The relative abundance of ions for these metabolites in human and mouse liver microsomes is presented in Figure 1C. The putative structures of JQ1 metabolites were identified based on the exact mass, MS/MS fragments, and predicted formula, as the standards of these metabolites are not available.
Figure 1.
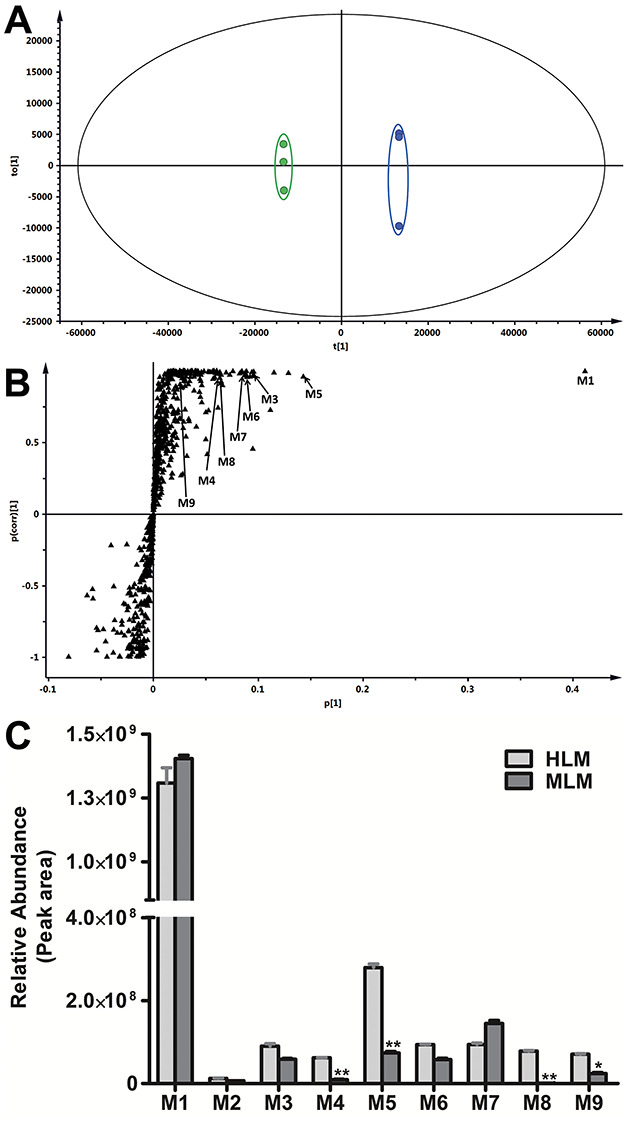
Metabolomic analysis of control and JQ1 groups in human liver microsomes and relative abundance of metabolites of JQ1. The incubation conditions of JQ1 were detailed in experimental procedures. All the samples were analysed using UHPLC-Q Exactive MS. (A) Separation of control and JQ1 group in OPLS-DA score plot. The t[1] and to[1] values represent the score of each sample in principal components 1 and 2, respectively. (B) Loading S-plot generated by OPLS-DA analysis. The x-axis is a measure of the relative abundance of ions, and the y-axis is a measure of the correlation of each ion to the model. The top-ranking ions are labelled. The number of ions (metabolite identification) shown was accordant with Table 1. (C) Relative abundance of metabolites of JQ1in human and mouse liver microsomes. Incubation conditions were detailed in Methods. The relative quantification was conducted based on the peak area. The data are expressed as mean ± SEM (n = 3). *P < 0.05, **P < 0.01.
Table 1.
Summary of metabolites of JQ1 in HLM and MLM.
| RT (min) | Observed m/z [M + H]+ | Calculated m/z [M + H]+ | Mass error (ppm) | Predicted molecular formula | Identification | Metabolite ID | Source |
|---|---|---|---|---|---|---|---|
| 12.02 | 457.1460 | 457.1459 | 0.22 | C23H26ClN4O2S | JQ1 | JQ1 | HLM, MLM |
| 10.16 | 473.1410 | 473.1409 | 0.21 | C23H26ClN4O3S | O + JQ1 | M1 | HLM, MLM |
| 10.37 | 473.1408 | 473.1409 | −0.21 | C23H26ClN4O3S | O + JQ1 | M2 | HLM, MLM |
| 11.38 | 473.1411 | 473.1409 | 0.43 | C23H26ClN4O3S | O + JQ1 | M3 | HLM, MLM |
| 9.32 | 401.0833 | 401.0834 | −0.25 | C19H18ClN4O2S | JQ1 [–C(CH3)3] | M4 | HLM, MLM |
| 8.28 | 489.1359 | 489.1358 | 0.20 | C23H26ClN4O4S | 2O + JQ1 | M5 | HLM, MLM |
| 9.74 | 489.1357 | 489.1358 | −0.20 | C23H26ClN4O4S | 2O + JQ1 | M6 | HLM, MLM |
| 11.07 | 471.1255 | 471.1252 | 0.64 | C23H24ClN4O3S | O + JQ1 (–2H) | M7 | HLM, MLM |
| 7.48 | 417.0781 | 417.0783 | −0.48 | C19H18ClN4O3S | O + JQ1 [–C(CH3)3] | M8 | HLM, MLM |
| 10.32 | 487.1202 | 487.1201 | 0.20 | C23H24ClN4O4S | 2O + JQ1 (–2H) | M9 | HLM, MLM |
JQ1; –C(CH3)3, de-tert-butylation; O+, monohydroxylation; 2O+, dihydroxylation; –2H, dehydrogenation; HLM/MLM, human/mouse liver microsomes.
Identification of monohydroxylated-JQ1 metabolites (M1–M3)
Three monohydroxylated JQ1 metabolites (M1–M3) whose formation is NADPH-dependent (Figure 3B) were identified and characterized in human and mouse liver microsomes. M1 is the dominant metabolite in both human and mouse liver microsomes. Metabolite M1 eluted at 10.16 min (Figure 2A) and was detected as a protonated molecule at m/z 473.1410, 16 Da higher than parent JQ1 (Figure 2C and D). The MS/MS of M1 produces major fragmental ions at m/z 455 (loss of H2O), 417 (loss of tert-butyl), 399 (loss of tert-butyl and H2O), 357 and 290. Comparison of the fragmental ions at m/z 357 and 290 with the fragmental ions of JQ1 (Figure 2C) indicates that the oxidation occurs on the fused three-ring moiety (boxed in Figure 2D). The M1 fragmental ions are interpreted in the inlaid structural diagram (Figure 2D). Metabolite M2 eluted at 10.37 min (Figure 2A) and was detected as a protonated molecule at m/z 284.1272 (Figure 2E). MS/MS of M2 produced major fragmental ions at m/z 455, 417, 399, 357 and 290, matching the pattern of M1 and indicating that the oxidation occurs on the same fused three-ring moiety as in M1 (Figure 2E), though the different elution positions of M1 and M2 mean different oxidation positions within that moiety. The M2 fragmental ions are interpreted in the inlaid structural diagram (Figure 2E). Metabolite M3 eluted at 10.38 min (Figure 2A) and was detected as a protonated molecule at m/z 473.1410 (Figure 2F). MS/MS of M3 produces major fragmental ions at m/z 455, 417, 399, 357 and 274 (Figure 2F). The fragmental ions at m/z 357 and 274 compared with the fragmental ions of JQ1 indicate that the oxidation in this metabolite occurs on the 4-chlorophenyl moiety (Figure 2C), as interpreted in the inlaid structural diagram (Figure 2F). The exact oxidation sites of metabolites were not defined because standards are not available for comparison by LC-MS.
Figure 3.
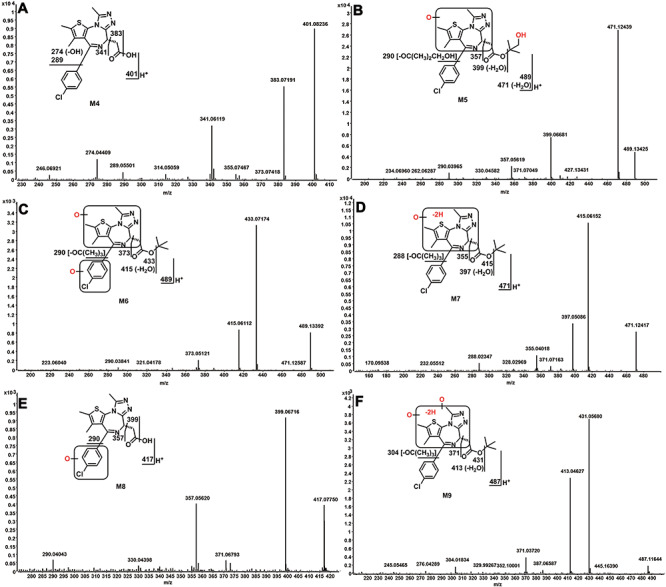
Identifying JQ1 metabolites (M4–M9). The incubation conditions of JQ1 were as described in Figure 1C. All the samples were analysed using UHPLC-Q Exactive MS. The metabolite elucidation conditions are as described in Figure 3 legend. (A) MS/MS of M4. (B) MS/MS of M5. (C) MS/MS of M6. (D) MS/MS of M7. (E) MS/MS of M8. (F) MS/MS of M9.
Figure 2.
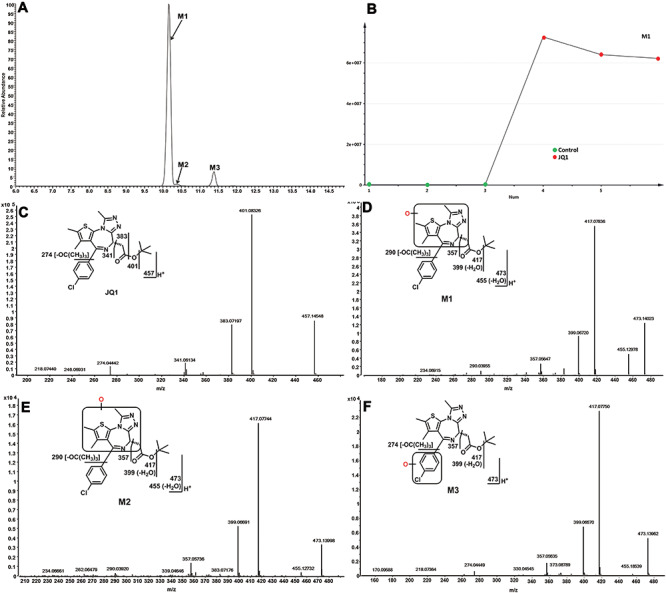
Identification of monohydroxylated-JQ1 metabolites (M1, M2 and M3). The incubation conditions of JQ1 were as described in Figure 1C. All the samples were analysed using UHPLC-Q Exactive MS. Structural elucidations were performed based on accurate mass (mass errors less than 5 ppm) and MS/MS fragmentation. MS/MS was performed with collision energy ramping from 10 to 35 V. The major fragmental ions are interpreted in the insets. (A) Extracted ion chromatograms of JQ1 metabolites M1, M2 and M3. (B) Representative trend plot of JQ1 metabolite M1. (C) MS/MS of JQ1. (D) MS/MS of M1. (E). MS/MS of M2. (F). MS/MS of M3.
Identification of JQ1 metabolites (M4–M9)
JQ1 metabolites (M4–M9) were detected in both human and mouse liver microsomes. The formation of M5–M9 is completely NADPH-dependent. However, low amounts of M4 were detected without addition of NADPH to human and mouse liver microsomes (1.7 and 3.5%, respectively, of the M4 levels seen with addition of NADPH; data not shown). The formation M4 in the control group is not likely due to slow ester hydrolysis in the original JQ1 stock, because did not observe the M4 in the JQ1 stock solution. The small amount of M4 formation may be mediated by esterases in liver microsomes. M4 eluted at 9.32 min, and was detected as a protonated molecule at m/z 401.0833 (Table 1). MS/MS of M4 produced major fragmental ions at m/z 383, 341, 289, and 274 (Figure 3A), which are interpreted in the inlaid structural diagram. These data identify M4 to be de-tert-butylated JQ1. Two doubly hydroxylated-JQ1 metabolites (M5 and M6) were identified in both human and mouse liver microsomes, though much more M5 is made by human liver microsomes than by mouse liver microsomes (Figure 1C). M5 eluted at 8.28 min, and was detected as a protonated molecule at m/z 489.1359 (Table 1), 32 Da higher than that of JQ1. MS/MS of M5 produced major fragmental ions at m/z 489, 471, 399, 357 and 290 (Figure 3B). The fragmental ions at m/z 399 and 290 (Figure 3B) indicate that one oxidation occurred on the tert-butyl group. The ions at m/z 357 and 290 suggested that the second oxidation occurred on the fused three-ring moiety. Fragmental ions from M5 are interpreted in the inlaid structural diagram in Figure 3B. M6 eluted at 9.74 min and was detected as a protonated molecule at m/z 489.1359 (Table 1). MS/MS of M6 produced major fragmental ions at m/z 489, 433, 415, 373 and 290 (Figure 3C). The fragmental ion at m/z 433 indicates that no oxidation occurred on the tert-butyl group. The ions at m/z 373 and 290 suggested that one oxidation occurred on the 4-chlorophenyl group and the other one occurred on the fused three-ring moiety. Fragmental ions from M6 are interpreted in the inlaid structural diagram in Figure 3C.
M7 eluted at 11.07 min and was detected as a protonated molecule at m/z 471.1255 (Table 1). MS/MS of M7 produced major fragmental ions at m/z 415, 397, 355 and 288 (Figure 3D). Compared to the fragments of monohydroxylated JQ1 (M1), the ions at m/z 355 and 288 indicate that monohydroxylation and dehydrogenation occurred on the fused three-ring moiety of JQ1; fragmental ions are interpreted in the inlaid structural diagram (Figure 3D). M8 eluted at 7.48 min and was detected as a protonated molecule at m/z 417.0782 (Table 1). MS/MS of M8 produced major fragmental ions at m/z 399, 357 and 290 (Figure 3E). Compared to the fragments of de-tert-butyl JQ1 (M4), the ions at m/z 357 and 290 indicated that hydroxylation occurred on the 4-chlorophenyl moiety. Fragmental ions from M8 are interpreted in the inlaid structural diagram in Figure 4E. M9 was observed at a retention time of 10.32 min, having a protonated molecular ion at m/z 487.1202 (Table 1). The MS/MS of M9 produced major fragmental ions at m/z 431, 413, 371 and 304 (Figure 3F). Compared to the fragments of dihydroxylated JQ1 (M5), ions at m/z 371 and 304 indicate that dihydroxylation and dehydrogenation all occurred on the fused three-ring moiety of JQ1. Fragmental ions from M9 are interpreted in the inlaid structural diagram in Figure 3F.
Figure 4.
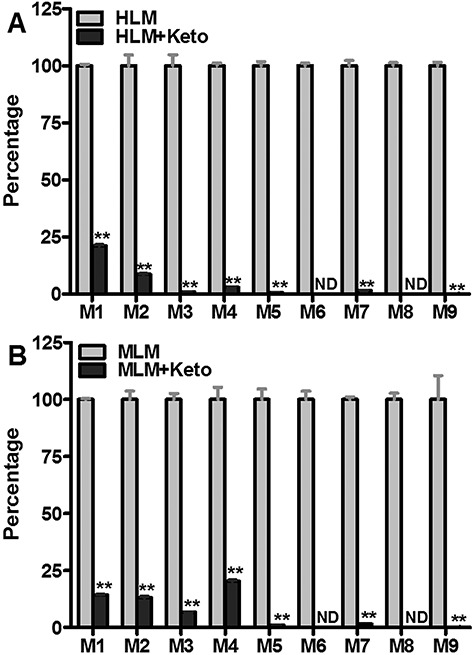
Roles of CYP3A4 in the formation of JQ1 metabolites in human and mouse liver microsomes. KCZ (2.0 μM, CYP3A4 inhibitor) was used in the inhibitory tests in human and mouse liver microsomes. The incubation conditions of JQ1 in human or mouse liver microsomes were detailed in experimental procedures. All samples were analysed by UHPLC-Q Exactive MS. (A) Effects of KCZ (2.0 μM) on the formations of M1–M9 in human liver microsomes. The relative abundance of JQ1 metabolites from the incubation with human liver microsomes without KCZ was set as 100%. (B) Effects of KCZ (2.0 μM) on the formations of the formations of M1–M9 in mouse liver microsomes. The relative abundance of JQ1 metabolite from the incubation in mouse liver microsomes in the absence of KCZ was set as 100%. All data are expressed as mean ± SEM (n = 3). *P < 0.05, **P < 0.01. HLM, human liver microsomes; MLM, mouse liver microsomes; KCZ, ketoconazole.
Role of cytochrome P450s in JQ1 metabolism
The roles of individual cytochrome P450s in the metabolism of JQ1 were determined by incubating JQ1 with nine human cDNA-expressed P450s (control, CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 and CYP3A4), by chemical inhibitory experiments in human and mouse liver microsomes and by incubation with liver S9 fractions of wild-type and Cyp3a-null mice. Incubating JQ1 with human cDNA-expressed P450s revealed that, of these enzymes, only CYP3A4 is able to generate metabolites M3–M6, M8 and M9 (Table 2) and both CYP3A4 and CYP2C19 can form M7. Six of the tested enzymes can form monohydroxylated metabolite M1, with strong activity demonstrated by CYP3A4, CYP2C8 and CYP2C19. None of the nine human cDNA-expressed P450s tested can generate the monohydroxylated metabolite M2.
Table 2.
Role of CYP450s in the formation of metabolites of JQ1.
| M1 | M2 | M3 | M4 | M5 | M6 | M7 | M8 | M9 | |
|---|---|---|---|---|---|---|---|---|---|
| Control | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| CYP1A2 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| CYP2A6 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| CYP2B6 | 0.6 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| CYP2C8 | 14.9 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| CYP2C9 | 8.8 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 24.6 | 0.0 | 0.0 |
| CYP2C19 | 27.9 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 100 | 0.0 | 0.0 |
| CYP2D6 | 2.3 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| CYP2E1 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| CYP3A4 | 100.0 | 0.0 | 100 | 100 | 100 | 100 | 88.2 | 100 | 100 |
cDNA-expressed CYP450s (control, CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 and 3A4) were used to determine the role of individual CYP450 in JQ1 metabolism. All samples were analysed by UHPLC-Q Exactive MS. The total peak area of the metabolite from all the CYP isozymes was set as 100%. All data are expressed as mean (n = 2).
The dependence of metabolite formation on CYP3A4 was verified by co-incubation of JQ1 with human or mouse liver microsomes and with ketoconazole (KCZ), a potent CYP3A4 inhibitor. In human liver microsomes, the formation of metabolites M1–M9 was suppressed by 79, 91, 99, 97, 99, 100, 98, 100 and 99%, respectively, by KCZ at 2.0 μM (Figure 4A). In mouse liver microsomes, the formation of M1–M9 was suppressed by 85, 87, 93, 80, 99, 100, 98, 100 and 99%, respectively, by 2.0 μM KCZ (Figure 4B). Incubation of cDNA-expressed P450 CYP2C19 with JQ1 shows that this enzyme is capable of forming both M1 and M7 (Table 2); the CYP2C19 inhibitor nootkatone (10 μM) has no effect on M7 formation in human liver microsomes (not shown) but suppresses M1 formation by 9%. Quercetin (30 μM), a CYP2C8 inhibitor, inhibited M1 formation in human liver microsomes by 78% (data not shown).
Because CYP3A4 is by far the major enzyme shown to contribute to JQ1 phase I metabolism (Table 2) and the CYP3A4 inhibitor ketoconazole greatly diminishes the generation of all detectable metabolites in human and mouse liver microsomes, we anticipated that Cyp3a-null mice might show greatly decreased metabolism of JQ1. Before undertaking long and expensive in vivo pharmacokinetic analyses, we tested the relative abilities of S9 fractions from wild-type and Cyp3a-null mice to generate the detected JQ1 metabolites. Most JQ1 metabolites are produced at lower levels by Cyp3a-null S9 fraction than by wild type (Figure 5), but the Cyp3a-null S9 fraction makes 2.67-fold more M4 than wild type and generates major metabolite M1 at much higher levels (65% of wild type) than would be expected from the ketoconazole inhibition data (Figure 4).
Figure 5.
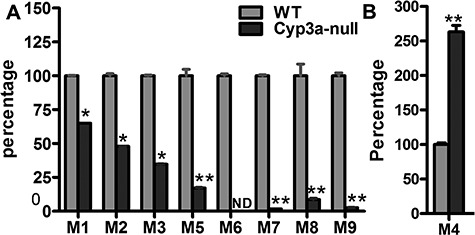
The formation of JQ1 metabolites in liver S9 fractions of WT and Cyp3a-null mice. S9 fractions were freshly prepared from WT and Cyp3a-null mouse liver. The incubation conditions of JQ1 in S9 fraction were detailed in experimental procedures. All samples were analysed by UHPLC-Q Exactive MS. (A) Role of CYP3A in the formation of M1–M3 and M5–M9. (B) Role of CYP3A in the formation of M4. Incubations with S9 fraction from WT and Cyp3a-null mice were performed in triplicates. The relative abundances of M1–M9 were set as 100% in S9 from WT mice, respectively. All data are expressed as mean (n = 3). N.D., not detected. *P < 0.05, **P < 0.01 vs WT.
Discussion
Nine JQ1 metabolites were distinguished in human and mouse liver microsomes, and the relative abundances of the ions of these metabolites in human and mouse liver microsomes are presented in Figure 1C. Our data indicated that the major monohydroxylated-JQ1 metabolite (M1) was generated by oxidizing one site in the fused three-ring moiety of JQ1 (Figure 2D), but we were not able to determine if hydroxylation occurred at one of the three methyl groups or the methane C6 position. M5 and M7, the second most abundant metabolites in human and mouse liver microsomes, respectively (Figure 1C), also show monohydroxylation of the JQ1 fused three-ring moiety (Figures 2D, 3B and D). We conclude that hydroxylation of the fused three-ring moiety comprises the primary metabolic pathway for JQ1 and the three methyl groups of this moiety are probable sites for oxidation. The precise identification of the major metabolic soft spot will be sought by synthesizing JQ1 variants modified at these positions; this work is ongoing. Once the exact site is known, medicinal chemists may be able to optimize the half-life of JQ1 by blocking the ‘soft spot’. The synthesis would also enable the activity of the major metabolite to be evaluated. In our study, we found that human liver microsomes generated more debutylated- (M4), dihydroxylated- (M5) and monohydroxylated-debutylated JQ1 (M8) compared to mouse liver microsomes, which indicated species differences exist for these JQ1 metabolites. These species difference may help us understand the pharmacokinetics of JQ1 in vivo.
Using recombinant CYP450 isoenzymes, we showed that CYP3A4 is the primary enzyme contributing to the formation of metabolites M3–M6, M8, and M9. Multiple P450s were involved in the formation of metabolites M1 and M7, mainly CYP2Cs and CYP3A4. Metabolism in the presence of CYP3A4 inhibitor ketoconazole confirmed that the formation of M1–M9 is largely mediated by CYP3A4 in human liver microsomes (Figure 4A). Since M2 was not produced by the recombinant CYP3A4 used in this study (Table 2), other CYP450 enzymes that are inhibited by ketoconazole (such as CYP3A5 [30]) are likely involved in M2 formation. Our findings with recombinant P450 enzymes (Table 2) show that M1 can be made by six of the tested enzymes, and the role for enzymes other than CYP3A4 in generating M1 is confirmed by ketoconazole blocking only 79% of M1 production in human liver microsomes. We demonstrated that CYP2C19 contributes to M1 formation in human liver microsomes as nootkatone, a CYP2C19 inhibitor, suppresses M1 formation by 9%. Quercetin, a CYP2C8 inhibitor, inhibited M1 formation as effectively (78%) as the selective CYP3A4 inhibitor ketoconazole (79%). A previous report showed that quercetin can inhibit CYP3A4 with a 50% inhibition concentration of 1.97 μM [31], so the role of CYP2C8 in M1 formation is not clear yet. Although recombinant CYP2C19 generated the metabolite M7 (Table 2), CYP2C19 inhibitor has no effect on the formation of M7 in human liver microsomes (data not shown). These results suggested that CYP2C19 is able to generate M7 but has a limited role in JQ1 metabolism, perhaps because of low affinity for JQ1. Because the formation of M7 was suppressed over 98% by ketoconazole in human liver microsomes, CYP3A4 is the primary enzyme responsible for M7 formation. These results indicated that CYP3A4 has a great potential to mediate drug–JQ1 interaction and further affect the pharmacokinetics of JQ1. CYP3A4 inducers might significantly decrease JQ1 efficacy by increasing its clearance. Conversely, CYP3A4 inhibitors might increase its efficacy (e.g. contraceptive activity) by blocking JQ1 metabolism. In vivo studies are needed to evaluate the effect of altering CYP3A4 activity on JQ1 pharmacokinetics and efficacy.
A previous study has shown that ketoconazole inhibits mouse Cyp3a [32], so we evaluated the effect of ketoconazole on the formation of JQ1 metabolites in mouse liver microsomes, which could suggest how ketoconazole might alter CYP3A4 activity in mice and thus affect JQ1 pharmacokinetics and efficacy in mice. Ketoconazole displayed similar inhibitory patterns in human and mouse liver microsomes for metabolites M1–M3 and M5–M9 (Figure 4B). However, only 80% of M4 formation was suppressed in mouse liver microsomes compared to 93% inhibition in human liver microsomes, suggesting that different enzymes may contribute to M4 formation in mouse liver microsomes than in human liver microsomes. Our data from the liver S9 fraction of Cyp3a-null mice confirm that Cyp3a is the dominant enzyme in the formation of M5–M9 (Figure 5). However, M1 formation was only 35% suppressed in liver S9 from Cyp3a-null mice compared to WT counterpart (Figure 5). As multiple enzymes contribute to M1 formation in WT microsomes, including CYP2C and CYP3A4, we expect that CYP2C may play a major role in M1 production in Cyp3a-null mice. Previous studies have shown that mRNA expression levels of Cyp2c29, Cyp2c39, Cyp2c50, Cyp2c55 and Cyp2c66 in Cyp3a-null mouse liver were significantly upregulated with 1.5-, 2.0-, 1.6-, 35- and 3-fold increase, respectively [33, 34]. Cyp2c55 was also highly upregulated at the protein level in the livers of Cyp3a-null mice compared with wild-type mice [34]. It is known that mouse Cyp2c29 has a similar function to human CYP2C8; mouse Cyp2c39, Cyp2c50, Cyp2c55 and Cyp2c66 are similar to human CYP2C19 [35]. Both human enzymes CYP2C8 and CYP2C19 contribute to M1 formation (Table 2). We hypothesize that the formation of M1 in the liver S9 fraction of Cyp3a-null mice was only moderately suppressed compared to wild type because upregulation of Cyp2c-type enzymes associated with the knockout compensated for the loss of Cyp3a.
Together, these findings suggest that using a CYP3A4 inhibitor in wild-type mice might significantly slow JQ1 phase I metabolism and enhance JQ1 pharmacokinetics in vivo. By contrast, our results predict that JQ1 metabolism in Cyp3a-null mice will likely show compensation effects by which other enzymes contribute to rapid JQ1 metabolism. We therefore suggest that researchers seeking to enhance JQ1 half-life to study its effects in mice on BRDT and spermatogenesis (or on other JQ1 targets and activities) should co-administer JQ1 with a CYP3A4 inhibitor (such as ketoconazole or ritonavir). If the CYP3A4 inhibitor increases JQ1 exposure and efficacy in vivo, it would indicate that JQ1 phase I metabolism contributes significantly to loss of efficacy. The availability of multiple CYP3A4 inhibitors would make it possible to achieve such enhancement of JQ1 half-life with different co-administrations, thus controlling for the possibility of a second target being involved. If co-administration were to significantly extend the JQ1 half-life, it could impact the use of JQ1 as a probe for BRDT activity in spermatogenesis and as a probe for bromodomain protein function in other systems.
In summary, the metabolism of JQ1 in human and mouse liver microsomes was comprehensively investigated using LC-MS-based metabolomic approaches. This study distinguished a total of nine phase I metabolites of JQ1 (Table 1 and Figure 6), and CYP3A4 is the primary enzyme contributing to the formation of these JQ1 metabolites in human liver microsomes. CYP3A4 and CYP2C contribute to the formation of the predominant monohydroxylated-JQ1 (M1). These findings will provide useful information for understanding how JQ1 metabolism may modulate its efficacy and for predicting possible drug–JQ1 interactions in future in vivo studies.
Figure 6.
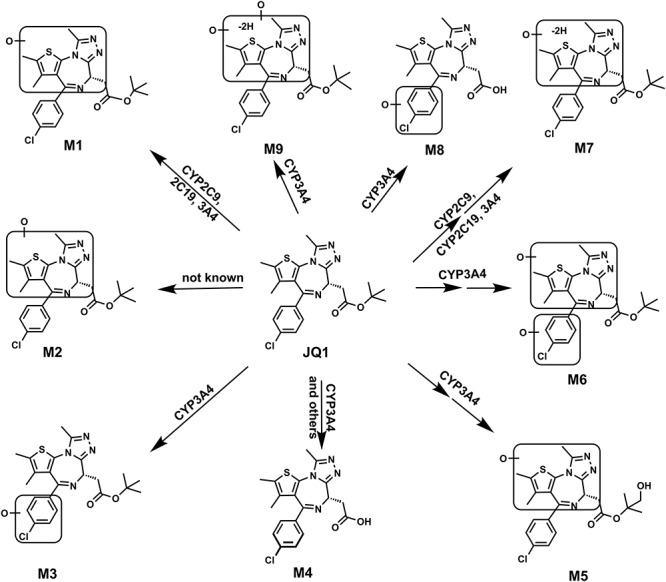
Metabolic map of JQ1 in mice and liver microsomes. All structures were determined based on the exact mass (mass error less than 2 ppm), MS/MS fragments and previous metabolism information of JQ1.
Acknowledgements
We thank Dr. Stacey Kalovidouris for the manuscript review and members of the Center for Drug Discovery for the helpful discussions.
Footnotes
† Grant Support: Cancer Prevention & Research Institute of Texas (RP160805); the Eunice Kennedy Shriver National Institute of Child Health and Human Development (P01 HD087157-01A1); the Welch Foundation (H-Q-0042); the Bill and Melinda Gates Foundation (INV-001902); and the National Institute of Diabetes and Digestive and Kidney (R01 DK121970).
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
- 1. Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S et al. Selective inhibition of BET bromodomains. Nature 2010; 468:1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shi X, Liu C, Liu B, Chen J, Wu X, Gong W. JQ1: a novel potential therapeutic target. Pharmazie 2018; 73:491–493. [DOI] [PubMed] [Google Scholar]
- 3. Belkina AC, Denis GV. BET domain co-regulators in obesity, inflammation and cancer. Nat Rev Cancer 2012; 12:465–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dawson MA, Kouzarides T, Huntly BJ. Targeting epigenetic readers in cancer. N Engl J Med 2012; 367:647–657. [DOI] [PubMed] [Google Scholar]
- 5. Matzuk MM, McKeown MR, Filippakopoulos P, Li Q, Ma L, Agno JE et al. Small-molecule inhibition of BRDT for male contraception. Cell 2012; 150:673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ding N, Hah N, Yu RT, Sherman MH, Benner C, Leblanc M et al. BRD4 is a novel therapeutic target for liver fibrosis. Proc Natl Acad Sci USA 2015; 112:15713–15718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang J, Wang Y, Mei H, Yin Z, Geng Y, Zhang T et al. The BET bromodomain inhibitor JQ1 radiosensitizes non-small cell lung cancer cells by upregulating p21. Cancer Lett 2017; 391:141–151. [DOI] [PubMed] [Google Scholar]
- 8. Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011; 478:524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stathis A, Bertoni F. BET proteins as targets for anticancer treatment. Cancer Discov 2018; 8:24–36. [DOI] [PubMed] [Google Scholar]
- 10. Niu Q, Liu Z, Alamer E, Fan X, Chen H, Endsley J et al. Structure-guided drug design identifies a BRD4-selective small molecule that suppresses HIV. J Clin Invest 2019; 129:3361–3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Trabucco SE, Gerstein RM, Evens AM, Bradner JE, Shultz LD, Greiner DL et al. Inhibition of bromodomain proteins for the treatment of human diffuse large B-cell lymphoma. Clin Cancer Res 2015; 21:113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Moyer MW. First drugs found to inhibit elusive cancer target. Nat Med 2011; 17:1325. [DOI] [PubMed] [Google Scholar]
- 13. Yengi LG, Leung L, Kao J. The evolving role of drug metabolism in drug discovery and development. Pharm Res 2007; 24:842–858. [DOI] [PubMed] [Google Scholar]
- 14. Zhang Z, Tang W. Drug metabolism in drug discovery and development. Acta Pharm Sinica B 2018; 8:721–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kumar GN, Surapaneni S. Role of drug metabolism in drug discovery and development. Med Res Rev 2001; 21:397–411. [DOI] [PubMed] [Google Scholar]
- 16. Zhang Z, Zhu M, Tang W. Metabolite identification and profiling in drug design: current practice and future directions. Curr Pharm Des 2009; 15:2220–2235. [DOI] [PubMed] [Google Scholar]
- 17. Rowley M, Hallett DJ, Goodacre S, Moyes C, Crawforth J, Sparey TJ et al. 3-(4-Fluoropiperidin-3-yl)-2-phenylindoles as high affinity, selective, and orally bioavailable h5-HT(2A) receptor antagonists. J Med Chem 2001; 44:1603–1614. [DOI] [PubMed] [Google Scholar]
- 18. Guengerich FP. Role of cytochrome P450 enzymes in drug-drug interactions. Adv Pharmacol 1997; 43:7–35. [DOI] [PubMed] [Google Scholar]
- 19. Liu X, Lu Y, Guan X, Dong B, Chavan H, Wang J et al. Metabolomics reveals the formation of aldehydes and iminium in gefitinib metabolism. Biochem Pharmacol 2015; 97:111–121. [DOI] [PubMed] [Google Scholar]
- 20. Liu X, Lu YF, Guan X, Zhao M, Wang J, Li F. Characterizing novel metabolic pathways of melatonin receptor agonist agomelatine using metabolomic approaches. Biochem Pharmacol 2016; 109:70–82. [DOI] [PubMed] [Google Scholar]
- 21. Li F, Lu J, Ma X. Metabolomic screening and identification of the bioactivation pathways of ritonavir. Chem Res Toxicol 2011; 24:2109–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li F, Lu J, Ma X. CPY3A4-mediated alpha-hydroxyaldehyde formation in saquinavir metabolism. Drug Metab Dispos 2014; 42:213–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li F, Zhang N, Gorantla S, Gilbertson SR, Pati D. The metabolism of separase inhibitor sepin-1 in human, mouse, and rat liver microsomes. Front Pharmacol 2018; 9:313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lu Y, Zhao X, Hu Z, Wang L, Li F. LC–MS-based metabolomics in the study of drug-induced liver injury. Current Pharmacol Rep 2019; 5:56–67. [Google Scholar]
- 25. Zhao Q, Zhang T, Xiao XR, Huang JF, Wang Y, Gonzalez FJ et al. Impaired clearance of sunitinib leads to metabolic disorders and hepatotoxicity. Br J Pharmacol 2019; 176:2162–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. O'Connell TM, Watkins PB. The application of metabonomics to predict drug-induced liver injury. Clin Pharmacol Ther 2010; 88:394–399. [DOI] [PubMed] [Google Scholar]
- 27. Li F, Lu J, Cheng J, Wang L, Matsubara T, Csanaky IL et al. Human PXR modulates hepatotoxicity associated with rifampicin and isoniazid co-therapy. Nat Med 2013; 19:418–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cazanave SC, Mott JL, Elmi NA, Bronk SF, Werneburg NW, Akazawa Y et al. JNK1-dependent PUMA expression contributes to hepatocyte lipoapoptosis. J Biol Chem 2009; 284:26591–26602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Worley B, Powers R. Multivariate analysis in metabolomics. Curr Metabolomics 2013; 1:92–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Allqvist A, Miura J, Bertilsson L, Mirghani RA. Inhibition of CYP3A4 and CYP3A5 catalyzed metabolism of alprazolam and quinine by ketoconazole as racemate and four different enantiomers. Eur J Clin Pharmacol 2007; 63:173–179. [DOI] [PubMed] [Google Scholar]
- 31. Choi JS, Piao YJ, Kang KW. Effects of quercetin on the bioavailability of doxorubicin in rats: role of CYP3A4 and P-gp inhibition by quercetin. Arch Pharm Res 2011; 34:607–613. [DOI] [PubMed] [Google Scholar]
- 32. Scheer N, Kapelyukh Y, Rode A, Oswald S, Busch D, McLaughlin LA et al. Defining human pathways of drug metabolism in vivo through the development of a multiple humanized mouse model. Drug Metab Dispos 2015; 43:1679–1690. [DOI] [PubMed] [Google Scholar]
- 33. Waterschoot RA, Herwaarden AE, Lagas JS, Sparidans RW, Wagenaar E, Kruijssen CM et al. Midazolam metabolism in cytochrome P450 3A knockout mice can be attributed to up-regulated CYP2C enzymes. Mol Pharmacol 2008; 73:1029–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kumar R, Mota LC, Litoff EJ, Rooney JP, Boswell WT, Courter E et al. Compensatory changes in CYP expression in three different toxicology mouse models: CAR-null, Cyp3a-null, and Cyp2b9/10/13-null mice. PLoS One 2017; 12:e0174355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nelson DR, Zeldin DC, Hoffman SM, Maltais LJ, Wain HM, Nebert DW. Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative-splice variants. Pharmacogenetics 2004; 14:1–18. [DOI] [PubMed] [Google Scholar]