

Abstract
Purpose
Elevated levels of transforming-growth-factor (TGF)-β2 in the trabecular meshwork (TM) and aqueous humor are associated with primary open-angle glaucoma (POAG). The underlying mechanism includes alteration of extracellular matrix homeostasis through Smad-dependent and independent signaling. Smad4, an essential co-Smad, upregulates hepcidin, the master regulator of iron homeostasis. Here, we explored whether TGF-β2 upregulates hepcidin, implicating iron in the pathogenesis of POAG.
Methods
Primary human TM cells and human and bovine ex vivo anterior segment organ cultures were exposed to bioactive TGF-β2, hepcidin, heparin (a hepcidin antagonist), or N-acetyl carnosine (an antioxidant), and the change in the expression of hepcidin, ferroportin, ferritin, and TGF-β2 was evaluated by semiquantitative RT-PCR, Western blotting, and immunohistochemistry. Increase in reactive oxygen species (ROS) was quantified with dihydroethidium, an ROS-sensitive dye.
Results
Primary human TM cells and bovine TM tissue synthesize hepcidin locally, which is upregulated by bioactive TGF-β2. Hepcidin downregulates ferroportin, its downstream target, increasing ferritin and iron-catalyzed ROS. This causes reciprocal upregulation of TGF-β2 at the transcriptional and translational levels. Heparin downregulates hepcidin, and reduces TGF-β2-mediated increase in ferritin and ROS. Notably, both heparin and N-acetyl carnosine reduce TGF-β2-mediated reciprocal upregulation of TGF-β2.
Conclusions
The above observations suggest that TGF-β2 and hepcidin form a self-sustained feed-forward loop through iron-catalyzed ROS. This loop is partially disrupted by a hepcidin antagonist and an anti-oxidant, implicating iron and ROS in TGF-β2-mediated POAG. We propose that modification of currently available hepcidin antagonists for ocular use may prove beneficial for the therapeutic management of TGF-β2-associated POAG.
Keywords: hepcidin, TGF-β2, iron, ROS, glaucoma, trabecular meshwork
TGF-β2 is implicated in the pathogenesis of primary open-angle glaucoma (POAG), one of the leading causes of blindness.1 Cumulative evidence suggests that aberrant signaling of TGF-β2 alters extracellular matrix (ECM) homeostasis by inducing cross-linking of actin networks and increased synthesis and deposition of fibrillogenic proteins in the trabecular meshwork (TM), interfering with its pliability and response to mechanical stress. This increases resistance to aqueous humor (AH) outflow and elevates intraocular pressure (IOP), a precipitating factor of glaucomatous pathology.2–5 Thus, pharmacological agents that reduce or neutralize bioactive TGF-β2 or block downstream pathways of TGF-β2 signaling are being explored as possible therapeutic options.6–8 Although partly successful, a better understanding of TGF-β2-mediated signaling and cross-talk with other pathways is necessary to improve the efficacy of existing agents and explore better options.
The principal downstream pathways of TGF-β2 include canonical or Smad-dependent, and noncanonical or Smad-independent signaling.3,9 Smad-dependent signaling involves association of phosphorylated Smad2 and 3 with co-Smad4, and translocation of the complex to the nucleus for transcriptional activation of fibrillogenic proteins that deposit in the ECM. Interestingly, Smad4 is a transcriptional activator of hepcidin,10 a hepatic peptide hormone that maintains serum iron within a narrow range by downregulating ferroportin (Fpn), the only known iron export protein.11,12
In addition to the liver, the heart, kidney, lungs, and other organs synthesize hepcidin locally, suggesting additional regulation of iron possibly through cross-talk with liver hepcidin.13 In the eye, hepcidin is synthesized locally in the neuroretina and ciliary epithelium, suggesting local regulation of iron transport to the neuroretina and the AH respectively.14–16 Expression of hepcidin in the corneal endothelium, lens epithelium, and TM indicate additional regulation of iron exchange between these structures and the AH.15 Although helpful in maintaining stringent regulation of iron, hepcidin is upregulated by inflammatory cytokines as well,17–19 and is likely to create a potentially toxic microenvironment by increasing intracellular iron and iron-catalyzed reactive oxygen species (ROS).20 In this respect, it is notable that TM cells respond to mechanical stress by upregulating TGF-β1 and IL-6,21 cytokines known to upregulate hepcidin,17,22,23 and the limited but noticeable protection of retinal ganglion cells by iron chelators suggests a prominent role of iron in the pathogenesis of POAG.24 Here, we explored whether local hepcidin in the TM is upregulated by TGF-β2, and the role of downstream pathways of hepcidin activation in TGF-β2-associated POAG.
Methods
Experimental Design
Primary human TM cells, human and bovine TM tissue, ex vivo human and bovine whole globe, anterior segment perfusion culture, and anterior cup models from cadaveric human and bovine eyes were used to evaluate the effect of TGF-β2 on hepcidin and vice versa in the TM. For primary human TM cells, recombinant bioactive TGF-β2 peptides or recombinant hepcidin peptides were used as triggers. For experiments with TM tissue, human and bovine anterior segment samples were infused, or cultured with replication-defective adenovirus expressing bioactive TGF-β2 (AdhuTGF-β2) or vector (AdEmpty).25–27 Ferric ammonium citrate (FAC) was used as a positive control to trigger hepcidin upregulation,12 heparin as a hepcidin antagonist,28–30 and N-acetyl-L-Carnosine31 as an antioxidant in different experimental paradigms.
Human and Bovine Eye Globes
Human eye globes were acquired from the Lions Gift of Sight eye bank (St. Paul, MN, USA). Donors ranged in age from 29 to 92 years (Supplementary Table S1). All experiments involving human tissue and cells were performed in compliance with the tenets of the Declaration of Helsinki. Bovine eyes were collected from a local abattoir within 2 hours of euthanization.
Isolation and Culture of Human and Bovine TM Tissue and Cells
TM cells were isolated from human eye globes as described,32 and cultured in DMEM supplemented with 1% FBS. Cultures from passages 2 and 3 at a confluence of 80% to 90% were used for all experiments. Each culture was tested for dexamethasone-induced upregulation of myocilin before use.32,33
Ex Vivo Perfusion of Human Anterior Segment
Human anterior segment ex vivo perfusion culture was established as described25–27 (Supplementary Table S1), and perfused with DMEM containing 1% FBS at a constant flow rate of 2.5 µl/min using micro infusion pumps (Harvard Apparatus, Holliston, MA, USA). After stabilizing the pressure for 12 hours, control and experimental samples were infused intracamerally with AdEmpty or AdhuTGFβ2, respectively.26 The change in pressure was recorded with a computerized system (National Instruments, Austin, TX, USA) and plotted relative to time. Finally, the samples were collected, rinsed, fixed, and processed for immunohistochemistry.
Ex Vivo Culture Model of Bovine Anterior Eye Cup and Whole Globe
The bovine anterior eye cup was cultured as described.34,35 In short, all tissues were removed from the anterior cup except the TM and cornea, and immersed in DMEM supplemented with 1% FBS and 1% penicillin/streptomycin at 37°C in a humidified atmosphere with 5% CO2. After overnight equilibration, samples were subjected to different experimental conditions, and the TM was isolated and analyzed. For experiments with the whole globes, AH was replaced with an equal volume of DMEM as above, supplemented with vehicle or FAC, and agitated gently in a CO2 incubator for 2 hours.36 Finally, control and experimental globes were dissected to isolate the TM.
Chemicals
Dexamethasone (D1756), dihydroethidium (DHE), FAC (F5879), human recombinant TGF-β2 (T2815), heparin (H3149), and H2O2 (H1009) were from Sigma Aldrich, USA. Human hepcidin-25 peptide (4040671) and trifluoroacetic acid were from BACHEM, USA. Reconstitution Buffer-4 was from R&D System, USA. Fluoromount-G (0100-01) was from SouthernBiotech, USA, and N-acetyl-L-Carnosine (18817) was from Cayman Chemicals, USA.
Immunohistochemistry
Immunohistochemistry was performed as described37 (antibody list in Supplementary Table S2). Stained sections were mounted and imaged with Leica inverted microscope (DMi8).
ROS Detection
Release of superoxide anion was analyzed with the fluorescent probe DHE as described.38
SDS-PAGE, Western Blotting, and RT-PCR
Protein lysates were fractionated by SDS-PAGE and analyzed by Western blotting as described.15,16,33,37 Proteins of interest were visualized by probing with specific antibodies (antibody list in Supplementary Table S2). RT-PCR was carried out as described earlier15 (primer list in Supplementary Table S3).
Statistical Analysis
Densitometry of images was performed with UN-SCAN-IT gels (version 6.1) software (Silk Scientific, USA) and Image J software. All data were statistically analyzed by GraphPad Prism (version 5.0) software (GraphPad Software Inc., USA), and are shown as mean ± SEM. Significant differences between control and experimental samples were determined by the Student’s unpaired t-test or 2-way ANOVA. Differences were considered statistically significant starting at P < 0.05.
Results
Hepcidin is Synthesized in the Trabecular Meshwork and Upregulated by Iron
Local synthesis of hepcidin in the TM was determined by subjecting freshly harvested bovine tissue from three eyes to semiquantitative RT-PCR with hepcidin-specific primers. Tissue from bovine retina and liver were analyzed in parallel as positive controls (Fig. 1A). Remarkably, a band co-migrating with retinal and liver hepcidin was amplified from all three samples (Fig. 1A, lanes 1−3 vs. 4−7). Amplified bands were eluted and sequenced to confirm their identity (Supplementary Fig. S2). The β-actin amplified in parallel indicated variable expression of hepcidin in TM and retina samples, although noticeably less than the liver (Fig. 1A, lanes 1−6 vs. 7).
Figure 1.
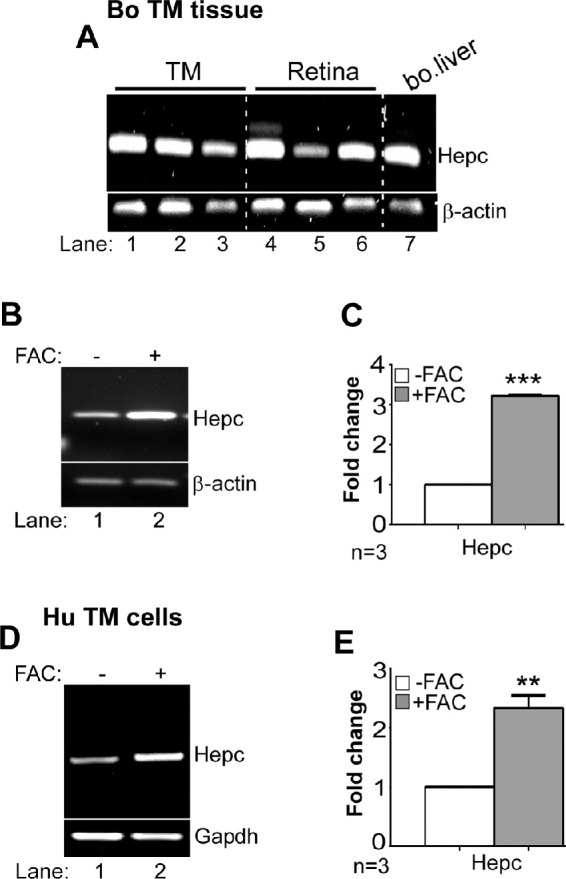
Synthesis of local hepcidin in bovine and human TM. (A) Amplification of hepcidin from bovine TM and retina by RT-PCR shows a band that co-migrates with hepcidin from bovine liver (lanes 1−7). β-Actin was amplified from the same samples for semiquantitative estimation. Full images are shown in Supplementary Figure S1-1A. (B) Amplification of hepcidin from bovine TM exposed to FAC or vehicle by RT-PCR shows upregulation of hepcidin by FAC relative to control (lanes 1 & 2). Full images are shown in Supplementary Figure S1-1B. (C) Densitometry after normalization with β-actin shows three-fold upregulation of hepcidin by FAC relative to controls. Values are mean ± SEM of the indicated n. ***P < 0.001. (D) Amplification of hepcidin from primary human TM cells exposed to FAC or vehicle by RT-PCR shows upregulation of hepcidin by FAC relative to control (lanes 1 & 2). Full images are shown in Supplementary Figure S1-1D. (E) Densitometry after normalization with GAPDH shows 2.3-fold upregulation of hepcidin by FAC relative to controls. Values are mean ± SEM of the indicated n. **P < 0.01.
To evaluate whether hepcidin in the TM responds to exogenous iron as expected, AH from three bovine eye globes was replaced with equal volume of medium containing 1% FBS supplemented with 30 µM FAC or vehicle, and the globes were agitated gently for 2 hours at 37°C. Subsequently, TM was harvested and analyzed. Semiquantitative RT-PCR revealed significant upregulation of hepcidin in samples exposed to FAC relative to controls (Fig. 1B, lanes 1,2; Fig. 1C).
To confirm the above results, primary human TM cells were cultured in the presence of 30 µM of FAC or vehicle for 24 hours, and analyzed as above. Exposure to FAC resulted in significant upregulation of hepcidin relative to controls (Fig. 1D, lanes 1,2; Fig. 1E), supporting the results in bovine TM tissue.
Together, the above results demonstrate local synthesis of hepcidin in human and bovine TM, and transcriptional upregulation by excess iron in the local microenvironment. Subsequent experiments were aimed at evaluating the cross-talk between TGF-β2 and hepcidin, and downstream pathways of hepcidin-mediated regulation of iron in the TM.
TGF-β2 Upregulates Hepcidin and Downstream Proteins in the Trabecular Meshwork
To evaluate whether TGF-β2 upregulates hepcidin, two experimental models were tested; primary human TM cells exposed to recombinant bioactive TGF-β2 peptide at a concentration of 4 to 8 ng/mL diluted in reconstitution buffer-4 (Figs. 2A−D), and ex vivo bovine anterior eye cup model overexpressing human bioactive TGF-β2 (Figs. 2E−H). The dose of TGF-β2, although higher than pathological levels observed in POAG, is within the range that upregulates fibrillogenic proteins in vitro in primary human TM cells5,25,39,40 (Supplementary Fig. S3).
Figure 2.

TGF-β2 upregulates hepcidin in the TM. (A) Amplification of hepcidin from primary human TM cells exposed to TGF-β2 or vehicle shows upregulation of hepcidin by TGF-β2 relative to the control (lanes 1 & 2). Full images are shown in Supplementary Figure S1-2A. (B) Densitometry after normalization with GAPDH shows 3.4-fold upregulation of hepcidin by TGF-β2 relative to controls. Values are mean ± SEM of the indicated n. ***P < 0.001. (C) Western blotting of lysates from primary human TM cells exposed to TGF-β2 or vehicle followed by probing for hepcidin shows increased expression of pro-hepcidin (approximately 12 kDa) by TGF-β2 relative to the control (lanes 1 & 2). Full images are shown in Supplementary Figure S1-2C. (D) Densitometry after normalization with β-actin shows 1.75-fold upregulation of pro-hepcidin by TGF-β2 relative to controls (lanes 1 & 2). Values are mean ± SEM of the indicated n. *P < 0.05. (E) Amplification of hepcidin from bovine TM overexpressing virally encoded human TGF-β2 (AdhuTGF-β2) shows increased expression relative to controls expressing empty vector (AdEmpty) (lanes 3 & 4 vs. 1 & 2). Full images are shown in Supplementary Figure S1-2E. (F) Densitometry after normalization with β-actin shows two-fold upregulation of hepcidin by TGF-β2 relative to controls. Values are mean ± SEM of the indicated n. *P < 0.05. (G) Western blotting of lysates from TM tissue in E above followed by probing for TGF-β2 shows increased expression of bioactive TGF-β2 in samples overexpressing TGF-β2 relative to controls as expected (lanes 3−5 vs. 1 & 2). Reaction for β-actin provides a loading control. Western blot analysis of the same lysates for hepcidin shows increased expression of pro-hepcidin in samples overexpressing TGF-β2 relative to controls (lanes 3−5 vs. 1 & 2). Recombinant hepcidin peptide and lysate from human retina were fractionated as positive controls, and showed hepcidin reactive bands at 3 kDa and approximately 12 kDa respectively (lanes 6 & 7). The membrane was re-probed for β-actin as a loading control. Full images are shown in Supplementary Figure S1-2G. (H) Densitometry after normalization with β-actin shows 4.3-fold increase in TGF-β2 and 2.2-fold increase in hepcidin in samples over-expressing TGF-β2 relative to controls. Values are mean ± SEM of the indicated n. **P < 0.01; ***P < 0.001.
Thus, human TM cells were exposed to 8 ng/mL of TGF-β2 or vehicle for 24 hours, and analyzed. Amplification of hepcidin by RT-PCR revealed significant upregulation by TGF-β2 relative to controls (Fig. 2A, lanes 1,2; Fig. 2B). Western blotting and probing of protein lysates for hepcidin mirrored the above results, and showed significant upregulation of pro-hepcidin migrating at approximately 12 kDa41,42 by TGF-β2 relative to controls (Fig. 2C, lanes 1,2; Fig. 2D). The membrane was re-probed for β-actin as a loading control.
To confirm the above results in TM tissue, ex vivo culture of bovine anterior eye cup was infected with replication defective adenovirus expressing bioactive human TGF-β2 (AdhuTGF-β2).26 Control samples received a similar titer of virus expressing empty vector (AdEmpty). Following an incubation of 2 hours, TM was isolated and processed. Amplification of bovine hepcidin by RT-PCR showed significant upregulation in samples overexpressing TGF-β2 relative to controls (Fig. 2E, lanes 3,4 vs. 1,2; Fig. 2F). Western blotting of protein lysates from the same samples and probing for TGF-β243 confirmed significant increase in expression by AdhuTGF-β2 within 2 hours relative to controls (Fig. 2G upper panel, lanes 3−5 vs. 1,2; Fig. 2H). Re-probing for β-actin provided a loading control. A similar analysis for hepcidin showed significant upregulation in samples overexpressing TGF-β2 relative to vector controls (Fig. 2G lower panel, lanes 3−5 vs. 1,2; Fig. 2H). Recombinant hepcidin peptide migrated at approximately 3 kDa as expected, and samples from human retina showed an approximately 12 kDa band of pro-hepcidin co-migrating with hepcidin from the TM (Fig. 2G lower panel, lanes 6,7; Fig. 2H).44,45 The membrane was re-probed for β-actin as a loading control.
Because hepcidin regulates iron by downregulating Fpn, downstream effects of TGF-β2-mediated upregulation of hepcidin were evaluated by exposing primary human TM cells to 4 and 8 ng/mL of TGF-β2 for 24 hours, and analysis of protein lysates by Western blotting. Parallel cultures were exposed to 30 µM of FAC as a positive control. Probing for Fpn revealed dose-dependent decrease in Fpn by TGF-β2, the effect of 8 ng/mL similar to FAC (Fig. 3A, lanes 2−4 vs. 1; Fig. 3B).
Figure 3.

TGF-β2 downregulates ferroportin in human TM. (A) Western blotting of lysates from primary human TM cells exposed to TGF-β2 or FAC shows decrease in Fpn expression relative to control (lanes 2−4 vs. 1). Full images are shown in Supplementary Figure S1-3A. (B) Densitometry after normalization with β-actin shows a dose dependent decrease in Fpn of 1.2-fold and 2.3-fold with 4 and 8 ng/mL of TGF-β2, respectively, relative to controls. The decrease in Fpn by FAC is equivalent to 8 ng/mL of TGF-β2. Values are mean ± SEM of the indicated n. *P < 0.05, **P< 0 .01. (C) Hematoxylin and eosin staining of fixed tissue from human ex vivo anterior segment model of POAG overexpressing TGF-β2 shows the expected architecture of TM and the Schlemm's canal (SC) (panels 1 & 2). Immunoreaction of serial sections for Fpn shows expression on the plasma membrane in the control sample (panel 3, arrowhead), and internalization and downregulation in TGF-β2-expressing sample that showed increase in IOP with time (panel 4, arrowhead) (Supplementary Figure S4). Serial sections exposed to rabbit IgG and processed in parallel shows no reactivity (panels 5 & 6). Scale bar: 25 µm.
Relevance of the above results to POAG was explored in the ex vivo human anterior segment culture model of glaucoma overexpressing TGF-β2 in the TM.25 Following overnight equilibration, the samples were infused with AdhuTGF-β2 or vector, and change in pressure was monitored over time. As expected, there was a gradual but significant increase in pressure in TGF-β2-expressing sample relative to the control after 6 days (Supplementary Fig. S4), at which time the samples were removed and processed for immunohistochemistry. Staining with hematoxylin and eosin (H & E) confirmed preservation of architecture in both samples with clear demarcation of TM and Schlemm's canal (Fig. 3C, panels 1,2). Immunostaining of serial sections for Fpn showed internalization and downregulation of Fpn in TM cells overexpressing TGF-β2 relative to vector control (Fig. 3C, panels 3,4). Incubation of serial sections with rabbit IgG did not show any reaction, confirming the specificity of Fpn reactivity (Fig. 3C, panels 5,6).
To evaluate whether the TGF-β2-mediated downregulation of Fpn through hepcidin increases intracellular iron and upregulates ferritin, primary human TM cells were exposed to 4 and 8 ng/mL of TGF-β2 for 24 hours as above, and lysates were processed for Western blotting. Probing for ferritin showed significant upregulation in cells exposed to 8 ng/mL of TGF-β2 relative to controls (Fig. 4A, lanes 2,3 vs. 1; Fig. 4B), although less than FAC (Fig. 4A, lane 4; Fig. 4B). Downregulation of Fpn by siRNA also upregulated ferritin, confirming the hepcidin-Fpn-iron axis in these cells (Supplementary Fig. S5).
Figure 4.
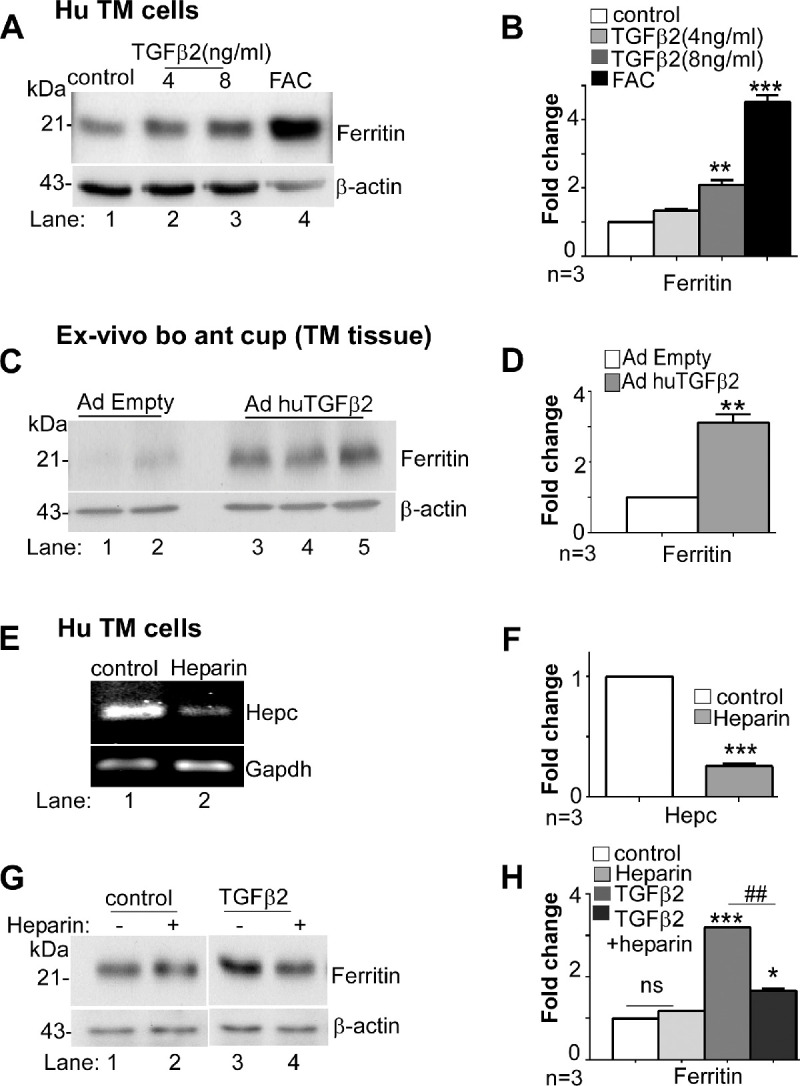
TGF-β2 upregulates ferritin in human TM. (A) Western blotting of lysates from primary human TM cells exposed to TGF-β2 or FAC shows increased expression of ferritin by TGF-β2 and FAC relative to vehicle treated control (lanes 2−4 vs. 1). Full images are shown in Supplementary Figure S1-4A. (B) Densitometry after normalization with β-actin shows two-fold upregulation of ferritin by 8 ng/mL TGF-β2 and 4.2-fold increase by FAC. Values are mean ± SEM of the indicated n. **P < 0.01. (C) Immunoblotting of TM tissue from bovine ex-vivo anterior eye cup overexpressing human TGF-β2 shows increased expression of ferritin relative to vector expressing control (lanes 3−5 vs. 1 & 2). Full images are shown in Supplementary Figure S1-4C. (D) Quantification by densitometry after normalization with β-actin shows three-fold increase in ferritin by TGF-β2 relative to controls. Values are mean ± SEM of the indicated n. **P < 0.01. (E) Treatment of primary human TM cells with heparin followed by amplification of hepcidin by RT-PCR shows significant downregulation relative to vehicle treated control (lane 2 vs. 1). Full images are shown in Supplementary Figure S1-4E. (F) Densitometry after normalization with GAPDH shows reduction in hepcidin mRNA by heparin to one-fifth of the control value. Values are mean ± SEM of the indicated n. ***P < 0.001. (G) Western blotting of lysates from primary human TM cells exposed to TGF-β2 in the absence or presence of heparin shows upregulation of ferritin in the absence of heparin (lane 3 vs. 1) and significant reduction by pre-incubation with heparin (lane 4 vs. 3). Vehicle or heparin treated controls did not show any change (lanes 1 & 2). Full images are shown in Supplementary Figure S1-4G. (H) Densitometry after normalization with β-actin shows three-fold upregulation of ferritin by TGF-β2, and a reduction to ½ the control value with heparin. Values are mean ± SEM of the indicated n. *P < 0.05; ***P < 0.001; ##P < 0.01 (*TGF-β2 vs. control; # TGF-β2 without and with heparin).
A similar evaluation in bovine ex vivo anterior eye cup model infected with AdhuTGF-β2 for 2 hours showed significant upregulation of ferritin relative to vector controls (Fig. 4C, lanes 3−5 vs. 1,2; Fig. 4D). Immunostaining of fixed sections, likewise, showed relatively stronger reactivity for ferritin in TM sections infected with AdhuTGF-β2 relative to AdEmpty controls (Supplementary Fig. 7, panels 1−4). Serial sections reacted with rabbit IgG control showed negative results, confirming the specificity of the reaction (Supplementary Fig. S7, panel 5,6).
To evaluate the specific role of hepcidin in TGF-β2-mediated upregulation of ferritin, primary human TM cells were pre-incubated with heparin (200 µg/mL) for 16 hours, a hepcidin antagonist,29,30 before the addition of 8 ng/mL of TGF-β2 for an additional 24 hours. This dose of heparin did not induce toxicity in primary human TM cells (Supplementary Fig. S6) or influence ferritin levels (Fig. 4G, lanes 1,2; Fig. 4H). Downregulation of hepcidin by heparin was evident when assessed by RT-PCR with hepcidin-specific primers (Fig. 4E, lanes 1,2; Fig. 4F). Evaluation of protein lysates from heparin pretreated samples exposed to TGF-β2 by Western blotting showed significant downregulation of ferritin by heparin relative to untreated controls (Fig. 4G, lane 4 vs. 3; Fig. 4H). These results indicate a prominent role of hepcidin in TGF-β2-mediated upregulation of ferritin.
Reciprocal Upregulation of TGF-β2 by Hepcidin Through ROS
To evaluate whether TGF-β2 increases ROS through hepcidin, triplicate cultures of primary human TM cells were pretreated with 200 µg/mL of heparin for 16 hours followed by 8 ng/mL of TGF-β2 for an additional 24 hours. Additional cultures were incubated with medium supplemented with 0.1% trifluoroacetic acid or 200 µg/mL of heparin as negative controls, and 1 µg/mL of recombinant hepcidin diluted in 0.1% trifluoroacetic acid or 30 µM of FAC as positive controls. One set of cultures were treated with 0.2 mM of H2O2 for the entire duration of the experiment, and considered as a positive control at 100% ROS (standardized in primary human TM cells; Supplementary Fig. S8). None of the treatments exhibited significant cytotoxicity as estimated by the lactate dehydrogenase (LDH) assay (Supplementary Fig. S9).
Control and treated cultures were incubated with DHE for 40 minutes, and fluorescence intensity as a measure of superoxide anion was quantified relative to medium (0%) and H2O2 (100%). TGF-β2 increased ROS to 20%, which was reduced by heparin to 8%. Hepcidin caused the maximum increase in ROS to 88%, followed by FAC at 63% (Fig. 5A). These results suggest that TGF-β2 upregulates ROS through hepcidin.
Figure 5.

Iron and ROS upregulate TGFβ2. (A) Intracellular ROS in primary human TM cells was quantified by recording the change in fluorescence intensity of DHE in triplicate cultures exposed to medium (0%), heparin, TGF-β2, TGF-β2 with heparin, hepcidin, FAC, and 0.2 mM H2O2 (100%). Exposure to hepcidin showed maximum increase in ROS by 88%, followed by 63% by FAC, and 20% by TGF-β2. Pre-incubation with heparin reduced TGF-β2-induced ROS to 8%. Values are mean ± SEM of the indicated n. *P < 0.05, **P < 0.01; #P < 0.05 (*treatment vs. control, #TGFβ2 vs. heparin + TGFβ2). (B) Amplification of TGF-β2 by RT-PCR from TM tissue harvested from ex vivo bovine anterior eye cup exposed to FAC or vehicle shows significant upregulation TGF-β2 relative to controls (lanes 3 & 4 vs. 1 & 2). Full images are shown in Supplementary Figure S1-5B. (C) Quantification by densitometry shows 3.6-fold upregulation of TGF-β2 by FAC relative to controls. Values are mean ± SEM of the indicated n. *P < 0.05. (D) Exposure of primary human TM cells to FAC or hepcidin shows upregulation of TGF-β2 by 24-hour exposure to FAC (lane 1 vs. 3) and hepcidin relative to vehicle treated control (lane 5 vs. 4). Re-probing for ferritin shows increased expression by FAC (lane 3 vs. 1) and hepcidin (lane 5 vs. 4). Full images are shown in Supplementary Figure S1-5D. (E) Densitometry after normalization with β-actin shows 1.8-fold and 2.4-fold increase in TGF-β2, and 2-fold and 1.8-fold increase in ferritin by FAC (24 hours) and hepcidin respectively. Increase in TGF-β2 by hepcidin is significantly more than 24 hour exposure to FAC. Values are mean ± SEM of the indicated n. **P < 0.01, ***P < 0.001; ##P < 0.01 (*TGF-β2 vs. control; #24 hours FAC vs. 4 hours hepcidin). (F) Primary human TM cells were exposed to hepcidin, or pre-incubated with NAC for 1 hour before adding hepcidin. Hepcidin induced significant upregulation of bioactive TGF-β2 (lane 2 vs. 1), which was reduced to control levels by NAC (lanes 1−3). Full images are shown in Supplementary Figure S1-5F. (G) Quantification by densitometry shows 1.5-fold upregulation of bioactive TGF-β2 by hepcidin relative to controls. Pre-treatment with NAC abolishes this effect. Values are mean ± SEM of the indicated n. **P < 0.01; #P < 0.05 (#hepcidin vs. NAC + hepcidin). (H) Amplification of TGF-β2 by RT-PCR from primary human TM cells incubated with bioactive TGF-β2 in the absence or presence of heparin shows significant downregulation of full-length TGF-β2 transcript by heparin (lane 2 vs. 1). Full images are shown in Supplementary Figure S1-5H. (I) Quantification by densitometry shows reduction of TGF-β2 mRNA to ½ of the control by heparin. Values are mean ± SEM of the indicated n. **P < 0.01.
Reciprocal upregulation of certain members of the TGF-β family by iron-catalyzed ROS is known,46 and was evaluated in ex vivo organ cultures of bovine anterior eye cup exposed to 30 µM of FAC for 24 hours. Amplification of bovine TGF-β2 by RT-PCR showed significant upregulation by FAC relative to controls (Fig. 5B, lanes 3,4 vs. 1,2; Fig. 5C).
To evaluate whether primary human TM cells respond similarly to iron, cultures were exposed to 30 µM of FAC for 4 and 24 hours, or 1 µg/mL of recombinant hepcidin peptide for 4 hours. Control cultures received equal volume of diluent. At the indicated times, cell lysates were evaluated by Western blotting. Probing for TGF-β2 revealed significant upregulation of the 25 kDa bioactive form by exposure to FAC for 24 hours and hepcidin for 4 hours (Fig. 5D, lanes 2,3 vs. 1, and lane 5 vs. 4; Fig. 5E). Re-probing for ferritin showed significant increase by 24-hour exposure to FAC and 4 hour treatment with hepcidin relative to matched controls (Fig. 5D, lanes 2,3 vs. 1, and lane 5 vs. 4; Fig. 5E). Surprisingly, upregulation of TGF-β2 by hepcidin was significantly more than FAC although upregulation of ferritin was similar. These results suggest that increased generation of ROS by hepcidin relative to FAC noted in Figure 5A may be responsible for significantly more upregulation of TGF-β2 by the hepcidin.
To evaluate the role of ROS in hepcidin-mediated upregulation of TGF-β2, primary human TM cells were pre-incubated with the antioxidant, N-acetyl-L-Carnosine (NAC; 5 mM) for 1 hour47 before exposure to 1 µg/mL of recombinant hepcidin for 4 hours, and lysates were evaluated by Western blotting. Probing for TGF-β2 revealed significant upregulation of the 25 kDa bioactive form by hepcidin, which was reduced to control levels by pretreatment with NAC (Fig. 5F, lane 2 vs. 1, and lane 3 vs. 2; Fig. 5G).
The contribution of hepcidin in inducing reciprocal upregulation of TGF-β2 in primary human TM cells was evaluated by pre-incubating the cells with 200 µg/mL of heparin for 16 hours before exposure to 8 ng/mL of bioactive TGF-β2 for 24 hours. Amplification of TGF-β2 by RT-PCR showed significant downregulation by heparin treatment, suggesting that bioactive TGF-β2 induces transcriptional activation of TGF-β2 gene through hepcidin (Fig. 5H, lane 2 vs. 1; Fig. 5I).
Together, the above results suggest a self-sustained TGF-β2-hepcidin-TGF-β2 feed-forward loop through iron-catalyzed ROS, which is disrupted by the hepcidin antagonist heparin, and the anti-oxidant NAC (Fig. 6).
Figure 6.

Graphical representation of the TGFβ2-Hepcidin feed-forward loop in the TM. (1) TGF-β2 upregulates transcription of hepcidin by the well-established Smad dependent (canonical) pathway10,56 (Figs. 2A−D; Figs. 2E−H), which (2) results in increased synthesis and secretion of hepcidin peptide. (3) Hepcidin (and TGF-β2) downregulate Fpn (Figs. 3A,B; Fig. 3C), resulting in (4) accumulation of iron in intracellular compartments and compensatory upregulation of ferritin (Figs. 4A-4 D. (5) Downregulation of hepcidin by heparin (Figs. 4E,4F) decreases TGF-β2-mediated upregulation of ferritin (Figs. 4G, 4H), implicating hepcidin in this process. (6) TGF-β2 increases ROS through hepcidin because heparin causes a signification reduction (Fig. 5A). (7) FAC and hepcidin also upregulate TGF-β2 (Figs. 5D,5E) and ferritin, suggesting a role of iron-catalyzed ROS in this process (Figs. 5D,5E). (8) Pre-incubation of TM cells with NAC decreases hepcidin-mediated upregulation of TGF-β2 (Figs. 5F,5G), implicating ROS in this process. (9) TGF-β2 triggers transcriptional activation of TGF-β2 (Figs. 5H,5I) and increased availability of bioactive TGF-β2, forming a self-sustained TGF-β2-hepcidin feed-forward loop.
Discussion
We report upregulation of local hepcidin in human and bovine TM tissue and primary human TM cells by TGF-β2, and reciprocal upregulation of TGF-β2 by hepcidin through iron-catalyzed ROS, forming a self-sustained positive feed-forward loop between TGF-β2 and hepcidin. These observations implicate iron-catalyzed ROS in TGF-β2-associated POAG, providing novel insight into the mechanistic basis of TGF-β2-associated POAG, and therapeutic options aimed at reducing hepcidin and iron-catalyzed ROS in the TM.
Local expression of hepcidin and Fpn in the TM re-inforce similar observations in a recent report,15 and suggest stringent regulation of iron at this site by its autocrine and paracrine activity.11,12,48,49 However, upregulation of hepcidin by TGF-β2 through Smad4, a potent transcriptional activator of hepcidin,10 is likely to increase intracellular iron and the potential for iron-catalyzed ROS. Our observations support this hypothesis, and indicate that TGF-β2 initiates a self-sustained feed-forward loop through hepcidin, implicating iron and ROS in TGF-β2-associated POAG.
As summarized in Figure 6, TGF-β2 upregulates hepcidin in TM cells possibly through Smad4, which downregulates Fpn in the same and adjacent cells by its autocrine and paracrine activity. This results in accumulation of iron in intracellular compartments. Although compensatory upregulation of ferritin stores iron in a relatively nonreactive form, chronic upregulation of hepcidin is likely to increase reactive oxygen and catalyze iron-mediated ROS, triggering the transcriptional activation of TGF-β2, and in addition, the bioactive form of TGF-β2 in the extracellular milieu. Interestingly, heparin, a hepcidin antagonist reduces TGF-β2-mediated increase in ferritin and ROS, and NAC reduces TGF-β2-mediated increase in ROS, supporting a role of hepcidin and ROS in this cycle. This is further supported by upregulation of TGF-β2 by FAC and hepcidin, implicating iron-catalyzed ROS as the proximate cause of increase in TGF-β2 levels. Moreover, bioactive TGF-β2 triggers transcriptional upregulation of TGF-β2, indicating that bioactive TGF-β2 initiates the hepcidin-TGF-β2 feed-forward loop, fueled by iron-catalyzed ROS.
A similar role of ROS in upregulating TGF-β isoforms has been described in other cell types50–52 and TM cells,53 supporting our observations. However, the pathways described above do not exist in isolation, and significant cross-talk with other biochemical pathways, some associated with POAG, is likely to influence the outcome in the in vivo conditions.
The significance of these observations is two-fold; first, this study demonstrates a clear involvement of iron-catalyzed ROS in TGF-β2-associated POAG, and second, the involvement of hepcidin in this process provides an untapped opportunity for using hepcidin antagonists as adjuncts to ROCK inhibitors and other treatment options for POAG.4 A range of hepcidin antagonists are currently undergoing clinical trials for systemic disorders,54,55 and could be modified for ophthalmic use.
Supplementary Material
Acknowledgments
The authors thank Aaron S. Wise and Disha Bhargava for assistance in experimental work.
Supported by R01 NS 092145 (NS). The authors alone are responsible for the content and writing of the paper.
Disclosure: A. Ashok, None; S. Chaudhary, None; A.E. Kritikos, None; M.H. Kang, None; D. McDonald, None; D.J. Rhee, None; N. Singh, None
References
- 1. Fuchshofer R, Tamm ER. The role of TGF-beta in the pathogenesis of primary open-angle glaucoma. Cell Tissue Res. 2012; 347: 279–290. [DOI] [PubMed] [Google Scholar]
- 2. Montecchi-Palmer M, Bermudez JY, Webber HC, Patel GC, Clark AF, Mao W. TGFbeta2 induces the formation of cross-linked actin networks (CLANs) in human trabecular meshwork cells through the Smad and non-Smad dependent pathways. InvestOphthalmolVisSci. 2017; 58: 1288–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pervan CL. Smad-independent TGF-beta2 signaling pathways in human trabecular meshwork cells. Exp Eye Res. 2017; 158: 137–145. [DOI] [PubMed] [Google Scholar]
- 4. Prendes MA, Harris A, Wirostko BM, Gerber AL, Siesky B. The role of transforming growth factor beta in glaucoma and the therapeutic implications. Br J Ophthalmol. 2013; 97: 680–686. [DOI] [PubMed] [Google Scholar]
- 5. Zhao X, Ramsey KE, Stephan DA, Russell P. Gene and protein expression changes in human trabecular meshwork cells treated with transforming growth factor-β. InvestOphthalmolVisSci. 2004; 45: 4023–4034. [DOI] [PubMed] [Google Scholar]
- 6. Van de Velde S, Van Bergen T, Sijnave D, et al.. AMA0076, a novel, locally acting Rho kinase inhibitor, potently lowers intraocular pressure in New Zealand white rabbits with minimal hyperemia. InvestOphthalmolVisSci. 2014; 55: 1006–1016. [DOI] [PubMed] [Google Scholar]
- 7. Tanna AP, Johnson M. Rho kinase inhibitors as a novel treatment for glaucoma and ocular hypertension. Ophthalmology. 2018; 125: 1741–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hsu CR, Chen YH, Liu CP, et al.. A highly selective Rho-kinase inhibitor (ITRI-E-212) potentially treats glaucoma upon topical administration with low incidence of ocular hyperemia. InvestOphthalmolVisSci. 2019; 60: 624–633. [DOI] [PubMed] [Google Scholar]
- 9. Wordinger RJ, Sharma T, Clark AF. The role of TGF-beta2 and bone morphogenetic proteins in the trabecular meshwork and glaucoma. J Ocul Pharmacol Ther. 2014; 30: 154–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang RH, Li C, Xu X, et al.. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab. 2005; 2: 399–409. [DOI] [PubMed] [Google Scholar]
- 11. Ruchala P, Nemeth E. The pathophysiology and pharmacology of hepcidin. Trends Pharmacol Sci. 2014; 35: 155–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sangkhae V, Nemeth E. Regulation of the iron homeostatic hormone hepcidin. Adv Nutr. 2017; 8: 126–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vela D, Vela-Gaxha Z. Differential regulation of hepcidin in cancer and non-cancer tissues and its clinical implications. Exp Mol Med. 2018; 50: e436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gnana-Prakasam JP, Martin PM, Mysona BA, Roon P, Smith SB, Ganapathy V. Hepcidin expression in mouse retina and its regulation via lipopolysaccharide/Toll-like receptor-4 pathway independent of Hfe. Biochem J. 2008; 411: 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ashok A, Chaudhary S, McDonald D, Kritikos A, Bhargava D, Singh N. Local synthesis of hepcidin in the anterior segment of the eye: A novel observation with physiological and pathological implications. ExpEye Res. 2020; 190: 107890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ashok A, Karmakar S, Chandel R, et al.. Prion protein modulates iron transport in the anterior segment: Implications for ocular iron homeostasis and prion transmission. ExpEye Res. 2018; 175: 1−13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Arosio P. New signaling pathways for hepcidin regulation. Blood. 2014; 123: 1433–1434. [DOI] [PubMed] [Google Scholar]
- 18. Langer AL, Ginzburg YZ. Role of hepcidin-ferroportin axis in the pathophysiology, diagnosis, and treatment of anemia of chronic inflammation. Hemodial Int. 2017; 21 suppl 1: S37–S46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Roth MP, Meynard D, Coppin H. Regulators of hepcidin expression. Vitam Horm. 2019; 110: 101–129. [DOI] [PubMed] [Google Scholar]
- 20. Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2014; 10: 9–17. [DOI] [PubMed] [Google Scholar]
- 21. Liton PB, Li G, Luna C, Gonzalez P, Epstein DL. Cross-talk between TGF-β1 and IL-6 in human trabecular meshwork cells. Molecular Vision. 2009; 15: 326. [PMC free article] [PubMed] [Google Scholar]
- 22. Chen S, Feng T, Vujić Spasić M, et al.. Transforming growth factor β1 (TGF-β1) activates hepcidin mRNA expression in hepatocytes. J Biol Chem. 2016; 291: 13160–13174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lehmann C, Islam S, Jarosch S, et al.. The utility of iron chelators in the management of inflammatory disorders. MediatorsInflamm. 2015; 2015: 516740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu P, Zhang M, Shoeb M, et al.. Metal chelator combined with permeability enhancer ameliorates oxidative stress-associated neurodegeneration in rat eyes with elevated intraocular pressure. Free Radic Biol Med. 2014; 69: 289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Oh D-J, Kang MH, Ooi YH, Choi KR, Sage EH, Rhee DJ. Overexpression of SPARC in human trabecular meshwork increases intraocular pressure and alters extracellular matrix. InvestOphthalmolVisSci. 2013; 54: 3309–3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shepard AR, Millar JC, Pang IH, Jacobson N, Wang WH, Clark AF. Adenoviral gene transfer of active human transforming growth factor-{beta}2 elevates intraocular pressure and reduces outflow facility in rodent eyes. InvestOphthalmolVisSci. 2010; 51: 2067–2076. [DOI] [PubMed] [Google Scholar]
- 27. Ethier CR, Wada S, Chan D, Stamer WD. Experimental and numerical studies of adenovirus delivery to outflow tissues of perfused human anterior segments. InvestOphthalmolVisSci. 2004; 45: 1863–1870. [DOI] [PubMed] [Google Scholar]
- 28. Asperti M, Denardo A, Gryzik M, Arosio P, Poli M. The role of heparin, heparanase and heparan sulfates in hepcidin regulation. Vitam Horm. 2019; 110: 157–188. [DOI] [PubMed] [Google Scholar]
- 29. Poli M, Asperti M, Ruzzenenti P, Naggi A, Arosio P. Non-anticoagulant heparins are hepcidin antagonists for the treatment of anemia. Molecules 2017; 22:pii: E598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Poli M, Girelli D, Campostrini N, et al.. Heparin: A potent inhibitor of hepcidin expression in vitro and in vivo. Blood. 2011; 117: 997–1004. [DOI] [PubMed] [Google Scholar]
- 31. Babizhayev MA. Generation of reactive oxygen species in the anterior eye segment. Synergistic codrugs of N-acetylcarnosine lubricant eye drops and mitochondria-targeted antioxidant act as a powerful therapeutic platform for the treatment of cataracts and primary open-angle glaucoma. BBA Clin. 2016; 6: 49–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Keller KE, Bhattacharya SK, Borras T, et al.. Consensus recommendations for trabecular meshwork cell isolation, characterization and culture. Exp Eye Res. 2018; 171: 164–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ashok A, Kang MH, Wise AS, et al.. Prion protein modulates endothelial to mesenchyme-like transition in trabecular meshwork cells: implications for primary open angle glaucoma. SciRep. 2019; 9: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Okumura N, Fujii K, Kagami T, et al.. Activation of the Rho/Rho kinase signaling pathway is involved in cell death of corneal endothelium. InvestOphthalmolVisSci. 2016; 57: 6843–6851. [DOI] [PubMed] [Google Scholar]
- 35. Hu J, Zhang Z, Xie H, et al.. Serine protease inhibitor A3K protects rabbit corneal endothelium from barrier function disruption induced by TNF-α. InvestOphthalmolVisSci. 2013; 54: 5400–5407. [DOI] [PubMed] [Google Scholar]
- 36. Grant WM. Experimental aqueous perfusion in enucleated human eyes. ArchOphthalmol. 1963; 69: 783–801. [DOI] [PubMed] [Google Scholar]
- 37. Ashok A, Singh N. Prion protein modulates glucose homeostasis by altering intracellular iron. SciRep. 2018; 8: 6556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Matesanz N, Park G, McAllister H, et al.. Docosahexaenoic acid improves the nitroso-redox balance and reduces VEGF-mediated angiogenic signaling in microvascular endothelial cells. InvestOphthalmolVisSci. 2010; 51: 6815–6825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hernandez H, Medina-Ortiz WE, Luan T, Clark AF, McDowell CM. Crosstalk between transforming growth factor beta-2 and toll-like receptor 4 in the trabecular meshwork. InvestOphthalmolVisSci. 2017; 58: 1811–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pattabiraman PP, Rao PV. Hic-5 regulates actin cytoskeletal reorganization and expression of fibrogenic markers and myocilin in trabecular meshwork cells. InvestOphthalmolVisSci. 2015; 56: 5656–5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Guillemot J, Canuel M, Essalmani R, Prat A, Seidah NG. Implication of the proprotein convertases in iron homeostasis: proprotein convertase 7 sheds human transferrin receptor 1 and furin activates hepcidin. Hepatology. 2013; 57: 2514–2524. [DOI] [PubMed] [Google Scholar]
- 42. Kulaksiz H, Gehrke S, Janetzko A, et al.. Pro-hepcidin: expression and cell specific localisation in the liver and its regulation in hereditary haemochromatosis, chronic renal insufficiency, and renal anaemia. Gut. 2004; 53: 735–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Khalil N. TGF-β: From latent to active. Microbes and Infection. 1999; 1: 1255–1263. [DOI] [PubMed] [Google Scholar]
- 44. Walker AP, Partridge J, Srai SK, Dooley JS. Hepcidin: What every gastroenterologist should know. Gut. 2004; 53: 624–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hunter HN, Fulton DB, Ganz T, Vogel HJ. The solution structure of human hepcidin, a peptide hormone with antimicrobial activity that is involved in iron uptake and hereditary hemochromatosis. JBiolChem. 2002; 277: 37597–37603. [DOI] [PubMed] [Google Scholar]
- 46. Mehta KJ, Farnaud SJ, Sharp PA. Iron and liver fibrosis: Mechanistic and clinical aspects. World JGastroenterol. 2019; 25: 521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fleisher-Berkovich S, Abramovitch-Dahan C, Ben-Shabat S, Apte R, Beit-Yannai E. Inhibitory effect of carnosine and N-acetyl carnosine on LPS-induced microglial oxidative stress and inflammation. Peptides. 2009; 30: 1306–1312. [DOI] [PubMed] [Google Scholar]
- 48. Loh A, Hadziahmetovic M, Dunaief JL. Iron homeostasis and eye disease. Biochim Biophys Acta. 2009; 1790: 637–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Garcia-Castineiras S. Iron, the retina and the lens: A focused review. Exp Eye Res. 2010; 90: 664–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Krstic J, Trivanovic D, Mojsilovic S, Santibanez JF. Transforming growth factor-beta and oxidative stress interplay: Implications in tumorigenesis and cancer progression. Oxid Med Cell Longev. 2015; 2015: 654594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liu RM, Desai LP. Reciprocal regulation of TGF-beta and reactive oxygen species: A perverse cycle for fibrosis. Redox Biol. 2015; 6: 565–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mehta KJ, Coombes JD, Briones-Orta M, et al.. Iron enhances hepatic fibrogenesis and activates transforming growth factor-beta signaling in murine hepatic stellate cells. Am J Med Sci. 2018; 355: 183–190. [DOI] [PubMed] [Google Scholar]
- 53. Rao V, Lautz J, Kaja S, Foecking E, Lukács E, Stubbs JE. Mitochondrial-targeted antioxidants attenuate TGF-β2 signaling in human trabecular meshwork cells. InvestOphthalmolVisSci. 2019; 60: 3613–3624. [DOI] [PubMed] [Google Scholar]
- 54. Katsarou A, Pantopoulos K. Hepcidin therapeutics. Pharmaceuticals (Basel). 2018; 11: pii: E127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Renders L, Budde K, Rosenberger C, et al.. First-in-human phase I studies of PRS-080#22, a hepcidin antagonist, in healthy volunteers and patients with chronic kidney disease undergoing hemodialysis. PLoS One. 2019; 14: e0212023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Goh JB, Wallace DF, Hong W, Subramaniam VN. Endofin, a novel BMP-SMAD regulator of the iron-regulatory hormone, hepcidin. SciRep. 2015; 5: 13986. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.