

Abstract
Purpose
Age-related macular degeneration (AMD) is the leading cause of blindness in Western populations. While an overactive complement system has been linked to pathogenesis, mechanisms contributing to its activation are largely unknown. In aged and AMD eyes, loss of the elastin layer (EL) of Bruch's membrane (BrM) has been reported. Elastin antibodies are elevated in patients with AMD, the pathogenic significance of which is unclear. Here we assess the role of elastin antibodies using a mouse model of smoke-induced ocular pathology (SIOP), which similarly demonstrates EL loss.
Methods
C57BL/6J mice were immunized with elastin or elastin peptide oxidatively modified by cigarette smoke (ox-elastin). Mice were then exposed to cigarette smoke or air for 6 months. Visual function was assessed by optokinetic response, retinal morphology by spectral-domain optical coherence tomography and electron microscopy, and complement activation and antibody deposition by Western blot.
Results
Ox-elastin IgG and IgM antibodies were elevated in ox-elastin immunized mice following 6 months of smoke, whereas elastin immunization had a smaller effect. Ox-elastin immunization exacerbated smoke-induced vision loss, with thicker BrM and more damaged retinal pigment epithelium (RPE) mitochondria compared with mice immunized with elastin or nonimmunized controls. These changes were correlated with increased levels of IgM, IgG2, IgG3, and complement activation products in RPE/choroid.
Conclusions
These data demonstrate that SIOP mice generate elastin-specific antibodies and that immunization with ox-elastin exacerbates ocular pathology. Elastin antibodies represented complement fixing isotypes that, together with the increased presence of complement activation seen in immunized mice, suggest that elastin antibodies exert pathogenic effects through mediating complement activation.
Keywords: smoke-induced ocular pathology, elastin, immune response, complement activation, electron microscopy
Age-related macular degeneration (AMD) is characterized by progressive loss of central vision resulting from damage to macular photoreceptors. AMD occurs in two forms, wet and dry,1 and both forms are associated with pathology at the retinal pigment epithelium (RPE)/choroid interface. As a disease, AMD is considered a complex multifactorial disease, with age representing the primary risk factor. The genetic risk is driven largely by variations in two loci, one on chromosome 1 (1q32; CFH) and the other on chromosome 10 (10q26; ARMS2),2 and complement activation has been proposed as an initiator of AMD pathology in patients with both genetic risk factors.3–6 Finally, smoking is the only environmental risk factor unequivocally linked to AMD.7
The RPE, Bruch's membrane (BrM), and choriocapillaris all undergo degenerative changes in AMD. All three appear to be targets of complement activation,8 and these surfaces undergo oxidative stress modifications.9,10 BrM, an extracellular matrix compartment, is made up of the RPE basement membrane, the inner collagenous layer, the middle elastic layer (EL), the outer collagenous zone, and the choriocapillaris basement membrane. BrM undergoes age-related changes, including thickening with aging likely due to lipid buildup11 and the development of basal laminar deposits (BLamD; localized between the RPE basement membrane and its plasma membrane)12 and basal linear deposits (BLinD; localized between the RPE basement membrane and the inner collagenous zone).13 These changes impact conductivity across the BrM.14 While the accumulation of lipoprotein-like particles during aging occurs in the peripheral and the macular BrM, the time course appears to be faster in the macula.15 In contrast, there are regional differences in the structure of the middle EL that are augmented by aging and disease. The middle EL is made up of stacked layers of linear elastin fibers in addition to collagen VI, fibronectin, and other proteins.16 Importantly, the middle EL is thinner, with less structural integrity in the macula than in the periphery, a discrepancy that is more severe in eyes with early AMD and active choroidal neovascularization (CNV).17 This reduction in elastin fibers, which are important for BrM's biomechanical properties and for maintaining a physical barrier to the pathologic invasion of blood vessels, might provide some rationale why CNV occurs in the macula.17 In connection with this observation, patients with AMD have elevated concentrations of elastin-peptide in serum18; control subjects have the lowest concentration, and those with early AMD with intermediate and neovascular AMD have the highest concentrations. Correspondingly, elevated levels of elastin IgG autoantibodies are found in neovascular AMD compared with controls, and elevated levels of elastin IgM autoantibodies are found in neovascular AMD when compared with controls or dry AMD.19 Antielastin B- and T-cell immunity has also been observed in other diseases such as chronic obstructive pulmonary disease.20 These observations suggest that abnormalities in elastin homeostasis may play a role in AMD.
Autoantibodies are pathologically produced by the immune system in response to protein epitopes found in an organism's own tissues. IgGs are generated randomly or in response to foreign substances. Those IgGs that recognize self-epitopes are typically eliminated by clonal deletion, but some IgGs escape this screening mechanism and generate autoantibodies that attack the “self”, leading to inflammation and damage. Studies have suggested that autoantibodies might play a role in AMD pathogenesis. Not only do patients with AMD have elevated levels of IgG autoantibodies when compared with controls,21 but their ligands include many “self” proteins such as glial fibrillary acidic protein (GFAP),22 α-crystallin, α-enolase,22 annexin II,5 cardiolipin,23 and elastin.19 Proof of concept that autoantibodies generate disease was provided by immunizing mice with Carboxyethyl-pyrrole (CEP) adducted mouse serum albumin, which led to pathologies similar to dry AMD.24 Finally, IgGs and/or IgMs bound to ligands on cell surfaces, basement membranes, or extracellular matrices can participate in inflammation via two distinct mechanisms: (1) IgGs/IgMs can directly activate complement via the classical or lectin pathway of complement, leading to an inflammatory environment by generating anaphylatoxins or membrane-attack-complex formation and direct cell injury (complement-dependent cytotoxicity [CDC]), or (2) target-bound antibodies (IgG, IgA, or IgE) can bind to their specific receptors (Fcγ-receptors; FcγR) to trigger antibody-dependent cell-mediated cytotoxicity (ADCC).25 Both complement fixation and immune complex formation are rapid, yet CDC and ADCC can participate in chronic, slowly progressing diseases.26,27 In CDC, lytic/destructive activation of “self-cells” is prevented due to the presence of membrane-bound complement inhibitors, while over time, elevated C3a and/or C5a signaling may enhance vascular permeability,28 mediate chronic inflammation, and result in increased production of reactive oxygen and nitrogen species,29 overall generating a toxic microenvironment. In ADCC, it has been speculated that high concentrations of immune complexes may have a FcγR-blocking effect, resulting in immune abnormalities present in certain chronic diseases.27
Here we characterize the response of RPE/BrM in a mouse model of ocular damage with features of human dry AMD, using a smoke-induced ocular pathology (SIOP) model.30 The SIOP model has revealed that long-term smoke exposure in C57BL/6J mice leads to a reduction in retinal function, concomitant with RPE/BrM alterations that include mitochondrial swelling, thickening of BrM, and severe loss of the EL and lipid deposition in the area of BrM in a complement-dependent manner.30–32 In other pathologies associated with cigarette smoke–induced injury, such as emphysema, the presence of autoantibodies and autoantibodies directed against oxidatively modified extracellular matrix components has been shown to be associated with disease severity.33 Therefore, here to mechanistically assess the impact of elastin autoantibodies in SIOP pathology, we immunized mice with elastin or cigarette smoke–modified elastin (ox-elastin) peptides to determine whether amplify autoantibodies exacerbated SIOP. We demonstrate that immunization amplifies autoreactive antibodies to both elastin and ox-elastin antibodies, increases complement activation, and exacerbates SIOP.
Materials and Methods
Animals
C57BL/6J mice were purchased (Jackson Laboratory, Bar Harbor, ME, USA) and maintained as breeding colonies. Mice were housed under a 12:12-hour, light-dark cycle with access to food and water ad libitum.
To investigate the role of autoantibodies in smoke-induced ocular pathology, mice were immunized as previously reported by Brandsma et al.34 In brief, mice were injected intraperitoneally with 10 µg mouse lung elastin peptides (Elastin Products Company, Owensville, MO, USA) or 10 µg cigarette smoke–modified elastin peptides mixed 1:1 with 100 µL Sigma Adjuvant (Sigma-Aldrich, St. Louis, MO, USA), followed by an immunization booster 3 weeks later. Cigarette smoke–modified elastin peptides (termed oxidized elastin, or ox-elastin) were generated by reconstituting mouse lung elastin peptides at 1 mg/mL in PBS (pH 6.4) and incubating the peptides in 10% cigarette smoke extract31 for 24 hours at 37°C (see smoke exposure of animals for further information), followed by dialysis (Tube-O-DIALYZER, Micro 1K MWCO; G-Biosciences, St Louis, MO, USA) for 24 hours in prechilled PBS (pH 7.2) at 4°C.
Additional schedules are outlined in the Results section. All experiments were approved by the Medical University of South Carolina Institutional Animal Care and Use Committee and performed in accordance with the Association for Research in Vision and Ophthalmology statement for the use of animals in ophthalmic and vision research.
Exposure to Cigarette Smoke
Cigarette smoke exposure was carried out according to our published protocol.30 In short, mice were exposed to cigarette smoke using the Teague TE-10 total body smoke exposure system (Teague Enterprises, Woodland, CA, USA) for 5 hours per day, 5 days per week for 6 months, using 3R4F reference cigarettes (University of Kentucky, Louisville, KY, USA).
Optokinetic Response Test
Visual acuity and contrast sensitivity of mice were measured by observing their optomotor responses to moving sine-wave gratings (OptoMotry, Cerebral Mechanics, Lethbridge, AB, CA) as previously described by us.30 In short, mice were placed individually on the central elevated pedestal surrounded by a square array of computer monitors that display stimulus gratings and allow monitoring of mice via an overhead closed-circuit TV camera. All tests were conducted under photopic conditions with a mean luminance of 52 candela (cd) m−2. Since visual acuity does not change in response to smoke exposure,30,32 we only assessed contrast sensitivity. Contrast sensitivity was determined by taking the reciprocal of the contrast threshold at a fixed spatial frequency (0.131 cycles per degree) and speed (12 deg/s). Contrast of the pattern was decreased systematically in a staircase manner until the animal stopped responding (considered the threshold response). For the time course analysis, the immunized mice were compared against each other; for the 6-month analysis, values from animals raised in room air and animals each raised in smoke that were uninjected obtained from previous studies30,32 were included.
Elastin ELISA
Microtiter (Immulon2; Dynatech Laboratories, Chatilly, VA, USA) plates were coated with 25 µg/mL mouse lung elastin peptides or cigarette smoke–modified elastin peptides overnight at 4°C. The plates were then blocked for 1.5 hours at 37 degree in 0.2% milk in PBS and then washed 3 times with PBST, followed by exposure to increasing doses of mouse serum (1:100 to neat) for 2 hours at 37°C. The plates were again washed and incubated with biotinylated anti-mouse secondary antibodies (anti-IgG and anti-IgM) followed by streptavidin (1:1000) and color development using Turbo-TMB ELISA (Pierce; Thermo Scientific, Rockford, IL, USA).
Western Analysis
Mouse RPE/choroid/sclera (hereafter referred to as RPE/choroid fraction) preparations were prepared from eyecups and protein was extracted by solubilizing in RIPA buffer (10 mM Tris-HCl [pH 7.5], 300 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1% SDS, and 0.1% sodium deoxycholate; ThermoFisher Scientific, Waltham, MA, USA) containing protease inhibitor cocktail (Sigma-Aldrich). For Western blot analysis, equal amounts of protein were added to Laemmli sample buffer and boiled. For some blots, mouse lung elastin peptides or cigarette smoke–modified elastin peptides (10 µg per lane) were loaded. Samples were separated by electrophoresis on 4% to 20% Criterion TGX Precast Gels (Bio-Rad Laboratories, Inc., Hercules, CA, USA) and proteins transferred to a polyvinylidene fluoride (PVDF) membrane. Membranes to identify the presence of complement activation products in the RPE/choroid fraction were incubated with primary antibody against C3d (reference) followed by appropriate secondary antibody coupled to peroxidase, whereas those to identify the presence of Igs were incubated with horseradish peroxidase–conjugated secondary antibodies (anti-mouse IgG and IgM [Sigma-Aldrich], anti-mouse IgG1, IgG2a,b and IgG3 [Abcam, Cambridge MA, USA]). In addition, membranes holding the elastin peptides (15 µg each) were probed with mouse serum, followed by incubation with horseradish peroxidase–conjugated secondary antibodies (anti-mouse IgG and IgM; Santa Cruz Biotechnology). Following secondary antibody incubation, all blots were incubated with Clarity Western ECL Blotting Substrate (Bio-Rad Laboratories, Inc.) and chemiluminescent detection. All blots were normalized to β-actin (Cell Signaling Technology, Danvers MA, USA). Protein bands were scanned and densities quantified using ImageJ software (National Institutes of Health, Bethesda, MD, USA). Control and oxidized elastin peptides run on SDS polyacrylamide gels were used for silver stain to identify the elastin fragments. The procedure was carried out in accordance with the manufacturer’s instructions (Pierce Silver Stain Kit 24612, ThermoFisher). Briefly, gels were fixed in 30% ethanol/10% acetic acid solution; incubated in sensitizer working solution, followed by incubation in staining working solution for 30 minutes; and then immersed in developer working solution to visualize the bands. Once the desired band intensity was reached, the reaction was stopped by 5% acetic acid solution. Finally, comparable transfer of mouse lung elastin peptides when compared with cigarette smoke–modified elastin peptides to PVDF membranes was confirmed using Ponceau S staining (0.1% w/v Ponceau S in 5% v/v acetic acid) according to the manufacturer's recommendations (Tocris Bioscience, Minneapolis, MN, USA).
Electronmicroscopy
Tissue Preparation
The eyes were enucleated, and a slit was cut into the cornea to allow for rapid influx of fixative. Eyes were fixed overnight in 2.5% glutaraldehyde, 1% formaldehyde, 3% sucrose, and 1 mM MgSO4 in 0.1 molar (M) phosphate buffer, pH 7.4. The eyes were then dissected and small central, nasal portions were osmicated for 60 minutes in 0.5% OsO4 in 0.1 M phosphate buffer, processed in maleate buffer for en bloc staining with uranyl acetate, dehydrated in graded ethanols, and processed for resin embedding as published.30 Serial sections were cut at 90 nm on a Leica Ultramicrotome, Leica Biosystems Division of Leica Microsystems Inc. Buffalo Grove, IL, USA, onto carbon-coated Formvar films, Ted Pella, Inc. Redding, CA, USA, supported by nickel slot-grids.
Ultrastructural Analysis
Electron microscopy images were captured using a JEOL USA Inc., Peabody MA, USA, JEM 1400 transmission electron microscope using SerialEM software (https://bio3d.colorado.edu/SerialEM/) to automate image capture overnight with 1200–1500 images captured per section, yielding data sets that were then processed with the NCR Toolset (Scientific Computing and Imaging Institute, Salt Lake City, UT, USA) to generate image mosaics with corrections for image aberrations induced by electron microscopy. Images were evaluated using Adobe Photoshop (Adobe Systems, San Jose, CA, USA) and ImageJ software (National Institutes of Health) as published previously. Each image was downsampled by 4 and set on a global scale with ImageJ to 0.000219 µm/pixel. BrM thickness was determined by analyzing ∼24-µm-length sections of damaged BrM areas of each sample. Areas of each BrM sample were outlined measuring 1.22 µm away from the choroidal intercapillary pillars, using the basement membrane of the RPE and choriocapillaris as boundaries. Based on a normal BrM in age-matched room air–exposed mice of consistently 0.22 ± 0.04 µm, thickness exceeding 0.28 µm was considered damaged. For all additional readouts, two RPE cells per sample, four to six animals per condition each, above an area of thickened BrM and randomly chosen, were examined. The basal lamina was measured within individual RPE cells, using the RPE basement membrane and basal infoldings as borders. The membrane infoldings and inner space enclosed by them were calculated as their respective area. Mitochondria were analyzed by manually generating a masking layer for all mitochondria present within an RPE cell (i.e., each mitochondrion was circled in Adobe Photoshop) that was created to calculate mitochondrial number (on average 450 mitochondria per condition), shape, size, and localization. To determine mitochondrial localization, the centroid coordinates for each mitochondrion were identified based on normalized x-y coordinates (1:1 ratio size). After plotting the data, we identified percent mitochondria localized in the center of the cells (central: 0.20 ≤ x-value ≤0.80 and 0.33 ≤ y-value <0.66), as mislocalization of mitochondria is a sign of RPE impairment in SIOP.30 Overall, this approach, which we have used before,30,32 has high statistical power as it analyzes multiple copies of cells per retina, and within each cell, there are many mitochondria.
Statistics
Data consisting of multiple groups and/or repeated measures were analyzed with post hoc ANOVA using Bonferroni correction (P < 0.05; Statview; SAS Institute, Inc., Cary, NC, USA).
Results
Antielastin Antibody Production
Two-month-old C57BL/6J were immunized with two doses of ox-elastin or elastin, followed by exposure to 6 months of smoke. A blood draw revealed that IgG and IgM antibodies were increased ∼10- to 13-fold (Fig. 1 in response to ox-elastin when compared with control elastin (Fig. 1) after cigarette smoke exposure.
Figure 1.
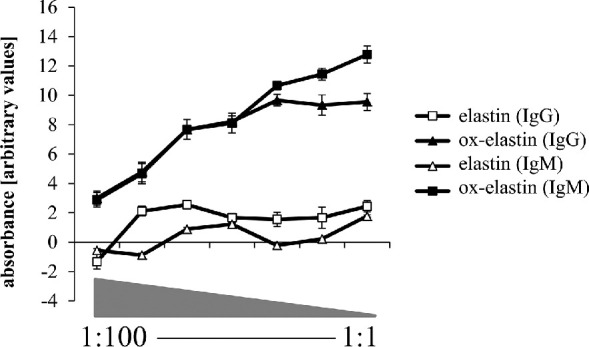
Antibody production in response to immunization with elastin or oxidized elastin. ELISA analysis was performed, coating plates with elastin or oxidized elastin peptide. Serum at different concentrations (1:100 to neat) from animals immunized with elastin or oxidized elastin was used to probe for binding, which was visualized with corresponding anti-mouse IgG and IgM secondary antibodies. Values were background subtracted and averaged (n = 3). After immunization and smoke exposure, a significant immune response against ox-elastin could be detected, based on IgG and IgM binding, whereas the response against control elastin was more modest.
Elastin is a large insoluble and durable complex made by crosslinking small soluble precursor tropoelastin protein molecules performed by the enzyme lysyl oxidase. A silver stain of the elastin and ox-elastin peptides revealed elastin fragments between 250 and 15 kDa (Fig. 2). Serum samples from ox-elastin immunized mice recognized a prominent band at ∼72 kDa when revealed with anti-mouse IgG, and the corresponding elastin band was not recognized. Staining with the anti-mouse IgM was not as clear, identifying multiple bands, including a 72- and the 250-kDa band.
Figure 2.
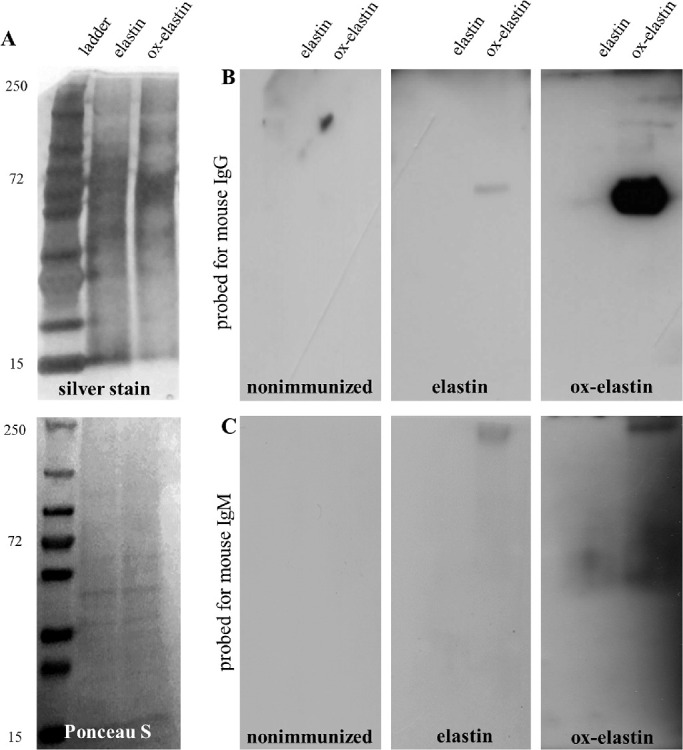
Antibodies generated after immunization recognized specific elastin fragments. Elastin and ox-elastin peptides (15 µg/lane) were loaded per lane and visualized by silver stain or transferred to PVDF membranes and used for Wester blotting. Serum samples from control animals (no immunization, no smoke exposure) or serum samples from smoke-exposed animals immunized with control elastin or oxidized elastin were used. (A) Silver stain identified distinct fragments between 1 and 250 kDa, and Ponceau S staining confirmed comparable transfer of control elastin or oxidized elastin peptides. (B) When blots were probed with serum samples from animals and developed with secondary antibodies against mouse IgG, a distinct ∼72-kDa band was identified with serum samples from oxidized elastin immunized mice. (C) Blots were probed with serum samples from animals and developed with secondary antibodies against mouse IgM, which identified full-length elastin as well as a smear in the ∼72-kDa range.
Immunization with Ox-Elastin Does Augment Smoke-Induced Ocular Pathology in Mouse
Using the SIOP model, we have shown previously that the mouse elastin layer, which in C57B/6J mice raised in room air exhibits a regular banding pattern, is fragmented or is no longer recognizable in electron microscopy (EM) images in mice exposed to 6 months of cigarette smoke.30 Additional changes included a reduction in retinal function, swelling of RPE mitochondria, thickening of BrM, and loss of basolaminar infoldings, all dependent on an alternative pathway of complement activation.30–32
Following immunization, mice were placed into the smoke chamber, and visual function was assessed in monthly intervals. As with nonimmunized mice,30 spatial acuity did not change over the 6-month period (data not shown). Contrast sensitivity, on the other hand, was affected by smoke inhalation in an antigen-specific manner. A repeated-measures ANOVA identified a treatment (P < 0.01) and treatment-by-time effect (P = 0.008) for the ox-elastin versus elastin mice over time (Fig. 3A). At the final time point (6 months), when compared with control mice exposed to room air, a robust decrease in contrast sensitivity (more contrast was required) was observed for both the elastin-immunized and the ox-elastin-immunized smoke-exposed mice (P < 0.01) as well as the nonimmunized smoke-exposed mice (P < 0.01). However, when compared with nonimmunized smoke-exposed mice, vision loss was only significantly greater in ox-elastin-immunized mice (P < 0.01) but not in elastin-immunized mice (P = 0.2) (Fig. 3B).
Figure 3.
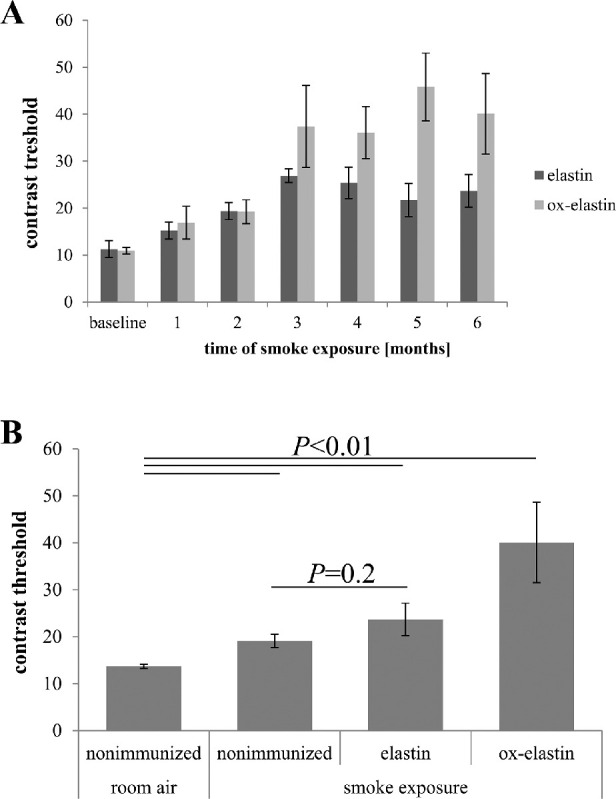
Immunization with oxidized elastin impairs contrast sensitivity. (A) Optomotor responses were analyzed in C57BL/6 mice over 6 months. Immunized elastin or oxidized elastin was exposed to 6 months of cigarette smoke. Contrast sensitivity was determined by measuring the contrast threshold at a fixed spatial frequency (0.131 cycles per degree) and speed (12 deg/sec) and expressed as threshold (percent contrast required for perception). We previously determined that this spatial frequency falls within the range of maximal contrast sensitivity for 9-month-old wild-type mice (data not shown). Smoke-exposed mice showed a significant reduction in contrast sensitivity compared with controls raised in room air, which was augmented in mice immunized with oxidized elastin (repeated-measures ANOVA: P = 0.01). (B) Contrast sensitivity of mice from panel A at 6 months was compared with nonimmunized room air–raised and nonimmunized smoke-exposed mice. Contrast sensitivity was affected by smoke exposure and immunization, with the effect of control elastin (P < 0.05) being less severe than that of oxidized elastin (P < 0.0001). Data are expressed as mean ± SEM (n = 5–9 per condition).
Given the functional deficits, ultrastructural differences were analyzed by EM (Fig. 4) and evaluated on multiple criteria, including BrM thickening, changes to the basolaminar infoldings, and mitochondrial morphology. The masks used to identify these features are indicated (Fig. 5). As reported previously, smoke exposure leads to a thickening of BrM,30,32 albeit not uniformly, but the RPE and choroid associated with these areas show the most pathologic changes. Hence, for all additional analyses (basal infoldings and mitochondria), cells above thickened BrM were analyzed. As the control (room air–exposed) mice show signs of aging by 9 months (time when all animals were analyzed), we focused analysis on those tissues also on cells above thickened BrM. The extent of thickened BrM increased with smoke exposure and immunization (Fig. 6A). Specifically, the percent thickened BrM increased to 25% to 35% in smoke-exposed mice whether uninjected or immunized with elastin, compared with ∼41% in smoke-exposed mice immunized with oxidized elastin (Table). While BrM thickness of the elastin-immunized, smoke-exposed group increased, it was not significant, whereas the ox-elastin-immunized mice were significantly worse when compared with the control mice (P < 0.0001) or the elastin-immunized mice (P < 0.0001). No difference in RPE cell height (as measured from BrM to the apical microvilli) could be identified among the four conditions (data not shown). The basal side of the RPE is highly infolded (basal infoldings) and regulates the exchange of nutrients, waste, and signaling molecules between the RPE and the choroid. One of the age-related changes on the mouse RPE is the presence of extended and enlarged basal infoldings,35 similar to that observed by us in the animals exposed to smoke.30,32 Density was measured as the overall area occupied by the membrane infoldings, as opposed to the open space between the infoldings. When comparing uninjected smoke-exposed mice to those immunized with control elastin, the infoldings and their open space were increased significantly (P < 0.05), but a treatment effect could not be established (P = 0.25) (Fig. 6B; Table). Mitochondrial swelling and mislocalization also have been observed in mice-exposed smoke.30,32 Mitochondrial swelling is typically considered a hallmark of mitochondrial dysfunction, and mitochondrial mislocalization has been shown to be associated with cell damage. Mitochondrial swelling was assessed as the overall area that mitochondria occupy (Fig. 7A, Table) and found to be associated with smoke exposure in untreated, control elastin and oxidized elastin-immunized mice (P < 0.05), but no treatment effect could be established (P = 0.7). As an additional damage feature of mitochondria, the number of mitochondria present in the center of the cell, rather than along the basal or basolateral region where they are required for cellular functions, was determined (Fig. 7B, Table). The percent mitochondria that are mislocalized occurred in response to smoke (P < 0.05), and an ox-elastin treatment effect could be established (P < 0.01). Finally, if we bin mitochondria based on shape (form factor between 0 and 1, with 1 representing a perfect circle), mitochondria in all four groups have the majority of their mitochondria in bin 7/8 (0.629 ± 0.15); however, more mitochondria are oblong in the control elastin (P < 0.05) and ox-elastin group (P < 0.05) (Fig. 7C, Table).
Figure 4.
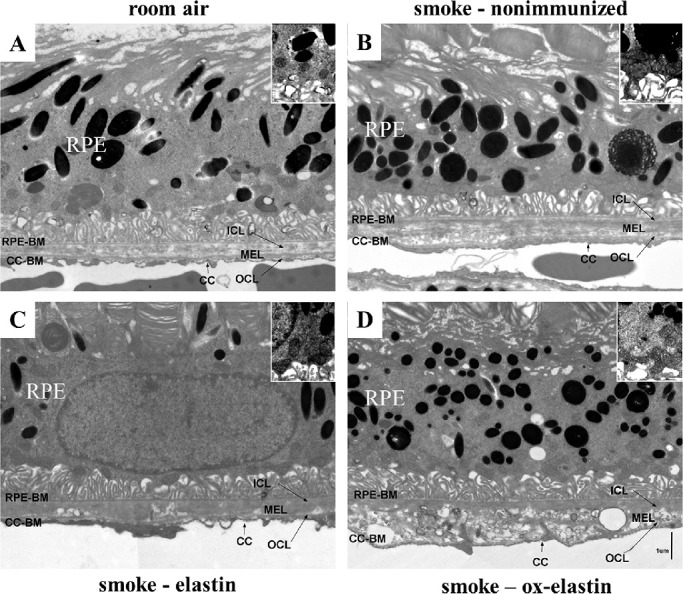
Ultrastructural changes in mice following smoke exposure and elastin immunization. Electron micrographs of the RPE/BrM/choriocapillaris complex (RPE/BrM/CC) obtained from C57BL/6J mice exposed to 6 months of room air were compared with those exposed to 6 months of smoke in the absence (smoke – untreated) and presence of elastin immunization (smoke – elastin; smoke – ox-elastin). (A) In a control animal raised in room air, BrM exhibits an organized pentalaminar structure, consisting of RPE-BM (RPE basement membrane), ICL (inner collagenous layer), MEL (middle elastic layer), OCL (outer collagenous layer), and CC-BM (choriocapillaris basement membrane). (B) The RPE/BrM/CC in animals exposed to smoke exhibit pathologic changes, including a thickening of BrM, which becomes disorganized, losing its pentalaminar structure, at its thicker points. (C) The RPE/BrM/CC is similarly affected in mice immunized with control elastin when compared with mice that are smoke exposed but not immunized. (D) The RPE/BrM/CC in animals exposed to smoke exhibit and immunized with oxidized elastin exhibit more severe pathologic changes, including larger areas of BrM being disorganized, losing its pentalaminar structure and the middle elastic layer. Insets highlight some of the morphologic features of mitochondria with degraded outer membranes and disorganized cristae in mice exposed to smoke and oblong mitochondria, particularly in the ox-elastin group.
Figure 5.

Masks overlying the morphologic features to be analyzed. Electron micrographs described in Figure 4 were used for analysis. Adobe Photoshop was used to generate masks for BrM, basal infolding, and mitochondria for further analysis in ImageJ. Masks highlight the differences in RPE/BrM obtained from C57BL/6J mice exposed to 6 months of room air when compared with those exposed to 6 months of smoke in the absence (smoke – untreated) and presence of elastin immunization (smoke – elastin; smoke – ox-elastin).
Figure 6.

Morphologic alterations in Bruch's membrane thickness and RPE basal infoldings in response to smoke and elastin immunization. Summary of alterations in Bruch's membrane thickness and basal infoldings obtained from EM images described in Figure 4. (A) BrM thickness was determined using the masks outlined in Figure 5, and the percent BrM along a given RPE cell that is damaged (exceed the normal thickness of BrM in age-matched room air–exposed mice) is established. A smoke and immunization effect could be established. (B) Basal infoldings were measured for both the lamellar portion and the open space in between. However, no consistent differences were observable. Data are expressed as mean ± SEM (n = 4–6 retinas per condition, representing 8–12 cells).
Table.
| Criteria | Room Air | Smoke Uninjected | Smoke Elastin | Smoke Ox-elastin |
|---|---|---|---|---|
| mitochondria number | 72±4.65 | 81±3.46 | 151±14.5## | 107±4.41† |
| mitochondria size % | 3.83±0.24 | 5.32±0.26* | 6.46±0.36 | 6.73±0.83† |
| mitochondria localization % | 4.17±0.71 | 8.26±1.31* | 6.03±0.53 | 12.84±1.58†† |
| BrM thickness % | 22.84±1.73 | 34.66±1.42** | 25.74±1.86# | 40.78±3.22†† |
| basal infoldings (plasma membrane) % | 0.61 ± 0.03 | 0.55 ±0.05 | 0.70±0.07 | 0.61±0.01 |
| basal infoldings (open area) % | 0.39 ±0.03 | 0.46±0.06 | 0.30±0.07 | 0.39±0.01 |
*, ** P<0.05 and P<0.001. Comparison between room air and smoke PBS.
#, ## P<0.05 and P<0.001. Comparison between smoke (uninjected) and smoke + elastin.
†, †† P<0.05 and P<0.001. Comparison between smoke + elastin and smoke + ox-elastin.
Figure 7.
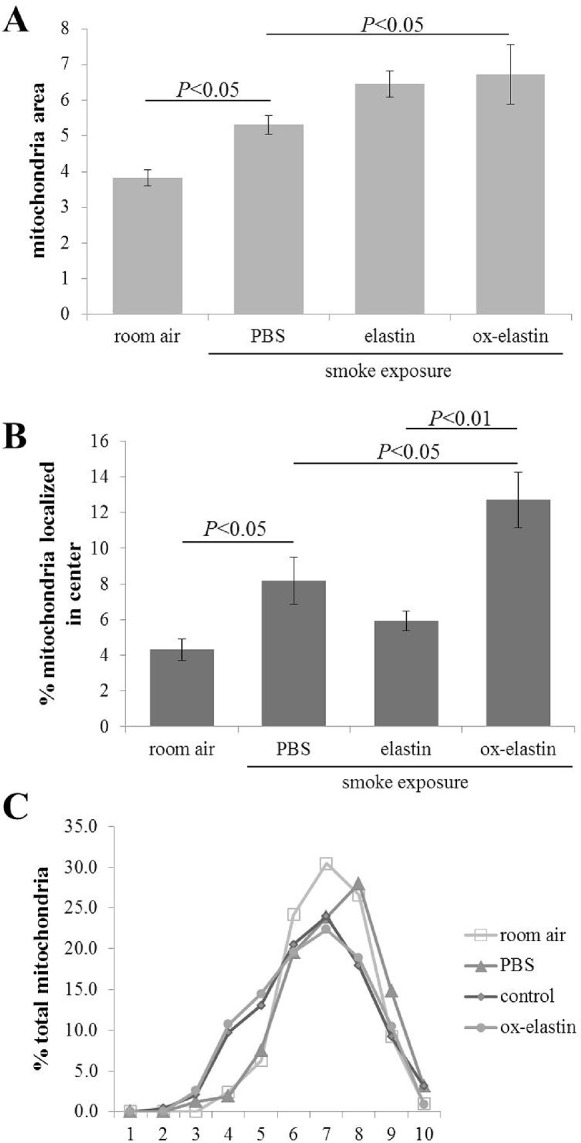
Morphologic alterations in mitochondria in response to smoke and elastin immunization. Summary of alterations in mitochondrial features obtained from EM images described in Figure 4. (A) Mitochondrial area (area of RPE cells occupied by mitochondria) was determined from electron micrographs, indicative of mitochondrial swelling and biogenesis. (B) Mitochondrial position was determined from electron micrographs by determining their centroid coordinates as a percentage of the corresponding RPE length and thickness, respectively. Each centroid was subsequently assigned to one of four bins (basal, apical, basolateral, and central). Based on our previous publication demonstrating that mitochondria exhibit an apical shift from the basal to central compartment in response to smoke, only the central bin is depicted here, demonstrating a smoke and immunization dependent shift of mitochondria. (C) Mitochondria shape was assessed in ImageJ, binning shapes from 0 to 1 into 10 bins, with 1 being a perfect circle. The normalized total mitochondrial distribution across the 10 bins demonstrates a shift to more oblong mitochondria in control elastin and oxidized elastin-immunized mice. Data are expressed as mean ± SEM (n = ∼450 mitochondria per condition).
Immunization with Ox-Elastin Increased Ocular Complement Activation and IgG/IgM Deposition Upon Smoke Exposure
To quantify complement activation in RPE/choroid of immunized mice, protein samples were analyzed by quantitative Western blotting for the presence of C3 breakdown products C3a, C3b, C3d, and C3dg based on their molecular weights (Fig. 8A). Overall, when analyzing the four parameters together, using a repeated-measures ANOVA, complement activation products by treatment effect could be confirmed (P < 0.0001). Specifically, with the exception of C3a, all other C3 activation products (C3b, C3d, and C3dg) were significantly increased by smoke in the control elastin-immunized mice (P < 0.05), and all four readouts were significantly increased by ox-elastin immunization (P < 0.05) (Figs. 8B–8D).
Figure 8.

Analysis of complement products in response to smoke and elastin immunization. (A) Equal amounts of RPE/choroid extracts (15 µg/lane) were loaded per lane, probed for C3 (Comptech), and band intensities quantified. Arbitrary values were established based on normalization with β-actin. Age-matched animals exposed to room air were compared with those raised in smoke and immunized with control or oxidized elastin. (B) C3a levels were detectable in the majority of animals exposed to smoke and immunized with oxidized elastin. (C) C3d and (D) C3dg levels were elevated and showed additivity by smoke and immunogen. (E) C3b levels were also elevated, but additivity between smoke and immunogen could not be established due to a larger variation between samples in the smoke + ox-elastin group and the difficulty of quantifying the bands accurately (partial overlap of C3b and C3dg). Data are expressed as mean ± SEM (n = 2–6 independent samples per condition).
The increase of complement activation products in the RPE/choroid in the context of immunization suggests that complement is activated in an IgG- and/or IgM-dependent manner.25 The same RPE/choroid samples were probed for the presence of IgG and IgM antibodies using quantitative Western blotting (Fig. 9A). Smoke exposure increased both IgG and IgM levels in the RPE choroid fraction when compared with room air (P < 0.05), and both levels were elevated when comparing control elastin and ox-elastin (P < 0.05) (Figs. 9B, 9C). To identify the IgG subclasses responsible for driving this difference, RPE/choroid samples were probed for the presence of IgG1, IgG2a, IgG2b, and IgG3 levels (Fig. 10, top row). While all four subclasses were increased in response to smoke, only IgG2b and IgG3 levels were further increased by ox-elastin immunization (P < 0.005 and P < 0.001, respectively) (Fig. 10, bottom row).
Figure 9.

Analysis of IgG and IgM binding in RPE/choroid in response to smoke and elastin immunization. (A) Equal amounts of RPE/choroid extracts (15 µg/lane) were loaded per lane, probed for mouse IgG (top blot) and IgM (middle blot), and band intensities quantified. Arbitrary values were established based on normalization with β-actin (bottom blot). Age-matched animals exposed to room air were compared with those raised in smoke and immunized with control or oxidized elastin. (B) IgG and (C) IgM levels were elevated and showed additivity by smoke and immunogen. Data are expressed as mean ± SEM (n = 2–3 independent samples per condition).
Figure 10.

Analysis of binding of IgG subtypes in RPE/choroid in response to smoke and elastin immunization. (A) Equal amounts of RPE/choroid extracts (15 µg/lane) were loaded per lane; probed for mouse IgG1, 2a, 2b, and 3; and band intensities quantified. Arbitrary values were established based on normalization with β-actin. For each set of gel images, the respective top blot represents the IgG subclass and the bottom blot the respective β-actin. Age-matched animals exposed to room air were compared with those raised in smoke and immunized with control or oxidized elastin. (B) IgG2a and IgG3 levels were elevated and showed additivity by smoke and immunogen, whereas IgG1 and IgG2b were elevated by smoke but were unaffected by the type of immunogen exposure. Data are expressed as mean ± SEM (n = 2–3 independent samples per condition).
Discussion
The main results of the current study are as follows: (1) immunization with a cigarette smoke–modified form of elastin (ox-elastin) led to the generation of IgG and IgM antibodies, whereas the self-version (elastin) had a smaller effect. (2) Ox-elastin immunization exacerbated SIOP. Specifically, after 6 months of smoke, mice immunized with ox-elastin exhibited more pronounced vision loss, thicker BrM, and more damaged RPE mitochondria when compared with mice immunized with elastin or no immunization. (3) Protein analysis revealed that the RPE/choroid fraction of these smoke-exposed mice immunized with ox-elastin contained significantly higher levels of IgM, IgG3, and IgG2b together with C3 activation or breakdown products when compared with mice immunized with elastin or controls. (4) Finally, a single peptide was identified as the antigen for the serum-derived IgGs generated in response to immunization in smoke-exposed mice. (5) In summary, our results suggest that in the SIOP model, antibodies generated de novo against ox-elastin (IgG) bound to ox-elastin generated by smoke in BrM might generate cytotoxicity and inflammation. Inflammation might be generated by antibodies activating complement via the classical or lectin pathway leading to complement-dependent cytotoxicity. Alternatively, these ox-elastin-bound antibodies could engage Fcγ receptors on effector cells, eliciting antibody-dependent cell-mediated cytotoxicity. Additional experiments are under way to distinguish between these two potential mechanisms of action.
AMD has been found to be associated with elevated levels of IgG Abs,21 but unlike in posterior uveitis, which is associated with the prevalence of serum antiretinal antibodies,36 the antigens in AMD appear to be more ubiquitous, including GFAP,22 CEP,37 α-crystallin and α-enolase,22 annexin II,5 cardiolipin,23 and elastin.19 What these antigens appear to have in common is that they tend to mostly be associated with cell stress, cell death, or matrix remodeling.
Smoking has been shown to be associated with autoantibody development in a variety of diseases, including systemic lupus erythematosus,38 Graves ophthalmopathy, 39 and chronic obstructive pulmonary disease.40 While the association between smoking, autoantibodies, and AMD has not yet been studied, there is ample evidence that smoking and AMD are. Specifically, smoking has been shown to increase the risk of developing AMD two- to fourfold41 and significantly accelerates the progression of atrophic AMD to wet AMD.42,43 However, the underlying mechanisms for this correlation are unclear. The main hypothesis is that components in cigarette smoke generate free radicals44 and deplete the system of antioxidants,45–50 overall contributing to oxidative stress. Oxidative stress, leading to endoplasmic reticulum (ER) stress31 and dysfunctional autophagy,51 coupled with the vasoconstrictive activity of nicotine-reducing choroidal blood flow,52 might be involved in inefficient debris clearance and accumulation of deposits.53 Finally, as of yet unidentified components in cigarette smoke can modify C3 that reduces its ability to bind CFH, overall leading to an increase in complement activation.54 Here we explore the concept that cigarette smoke exposure and the respective chemicals in cigarettes generate neoepitopes on membranes (eg, Wang et al.55), which are then recognized by their respective antibodies, leading to cell damage, potentially in a complement-dependent manner. We have previously confirmed this concept in RPE monolayers. Specifically, it was shown that a specific neoepitope (malondialdehyde) generated under oxidative stress conditions (H2O2 exposure) and recognized by its respective IgM antibody can activate the complement cascade, leading to RPE dysfunction.10 In unpublished experiments, we showed that smoke exposure also leads to the presentation of neoepitopes on RPE cells that are recognized by natural antibodies.
To further explore the concept of antibody-mediated damage in AMD, we focused on elastin as a neoepitope. The rationale for that is fourfold. First, there are elevated levels of elastin-peptide18 and α-elastin antibodies in AMD19; second, the thickness and integrity of the EL are impaired in early AMD and active CNV17; third, in the mouse SIOP model, EL degradation is observed, concomitant with an increase in complement activation in BrM30; and finally fourth, HTRA1, potentially one of the risk factors associated with AMD, has recently been found to possess elastase activity.56 Elastin per se is not antigenic, with the exception of autoimmune disorders in which abnormal levels of α-tropoelastin and α-elastin have been reported in serum. However, elastin peptides modified by smoke extract are highly immunogenic, as has been shown previously by Kirkham and colleagues33 as well as by us here, resulting in both the selection and amplification of IgM and the de novo generation of IgG antibodies (Figs. 1, 2).
We tested whether these ox-elastin antibodies might augment pathology in an animal model relevant to AMD, SIOP. In the SIOP model, in which the antigen used to generate the immune response is predicted to be similar to the neoepitopes generated during the disease, an additive effect could be identified. In other words, when ox-elastin generated by smoke exposure was used to immunize mice, ocular pathology in response to long-term smoke exposure of the animals was observed. When contrast sensitivity was used as a readout of retinal function, a deviation in sensitivity between elastin- and ox-elastin-immunized mice was observed starting at 3 months after the onset of smoke, reaching significance by 6 months. This loss in contrast sensitivity was correlated with changes previously reported,30 which included a thicker BrM covering a larger portion of the eye, in addition to RPE basal infoldings with lesser surface area and RPE mitochondria with altered shapes and localization within the cell, when compared with mice immunized with elastin or no immunization. Some of these observed changes occur with aging. For example, the presence of extended and enlarged basal infoldings has been reported in the aged mouse eye,35 disorganization of the basal infoldings has been shown in the aged eye,57 elongated mitochondria are present in the macular RPE of aging monkeys,58 and mitochondrial fragmentation, together with a loss of cristae and matrix density, was found in aged RPE.59 In AMD, these mitochondrial changes are augmented.59 Overall, these observations suggest that AMD might be viewed as accelerated aging,60 and the SIOP model might be a good model for that.
Our results demonstrate pathologic differences between immunization with elastin and ox-elastin peptides, which raises interesting questions about the relevance of the reported human data on elastin antibodies as biomarkers. While patients with AMD have antielastin antibodies,19 as do smokers,20 it has not yet been investigated whether the two are additive or whether the differences between affected and controls would be even greater if testing were done for antibodies against ox-elastin instead of elastin. Similarly, differential testing for both elastin and ox-elastin antibodies might explain this unexpected observation that elevated levels of antielastin IgG antibodies are only found in neovascular but not in dry AMD.19 It is plausible that a rapid breakdown of BrM in wet AMD generates antibodies against elastin, whereas the slow breakdown of BrM in dry AMD in the presence of oxidative stress might generate antibodies against modified elastin. Future experiments might shed light on these questions.
Our study has a number of limitations. We did not include adjuvant only–treated smoke-exposed mice or tested the effects of immunization in room air–only mice. Adjuvant has short-term and possibly longer-term consequences independent of the antigen,61,62 but Brandsma and colleagues34 found no negative impact of adjuvant only after 6 months on lung morphology in their model. Immunization against elastin in the absence of smoke is not expected to be damaging to the eye due to the lack of antigens present in the ocular tissues in the absence of stress, although Brandsma and colleagues34 did find that immunization against elastin itself resulted in increased macrophage numbers in the lung. A definitive experiment to demonstrate that the anti-ox-elastin antibodies are indeed pathogenic is still outstanding. In important follow-up studies, we will be using passive transfer of serum from immunized mice to constant smoke-exposed mice to answer this question. Finally, our experiments do not address the question of signaling by soluble elastin. Skeie and colleagues63 treated 7-month-old mice with human skin elastin-derived peptides (EDPs: 200 ng, three times weekly for 4 weeks) to identify a potential role of elastin in neovascular AMD. They reported that “a trend toward increasing basal lamina thickness was observed in some EDP treated eyes, but this was not consistently observed.” The influence of an immune response toward the human peptide was not determined or discussed; rather, it was suggested that “EDPs may play a role in neovascular AMD by binding to and inducing neovascular phenotypes in choroidal endothelial cells through their receptor, GLB1” (Galactosidase, β 1).
In two previous studies, we have shown that SIOP is dependent on complement activation. Specifically, we have shown that in mice in which complement factor B is eliminated systemically, most of the smoke-induced changes (loss in contrast sensitivity, BrM thickening, and mitochondrial changes) can be prevented,30 and systemic injection of a targeted inhibitor of the alternative pathway (CR2-fH) accelerates structural and functional recovery from SIOP.32 Here we confirmed our previous finding that the RPE/choroid fraction of smoke-exposed mice contained significantly higher levels of C330 and a readout of complement activation, an effect that was significantly increased in mice immunized with ox-elastin. Here we also showed that the increase in pathology in smoke-exposed mice immunized with ox-elastin was correlated with higher levels of IgM, IgG3, and IgG2b but not IgG1 and IgG2. It is presumed, therefore, that the difference in response to smoke is mediated by IgM, IgG3, and IgG2b. In general, both IgM and IgG can activate the lectin and classical pathway of the complement cascade.64 However, the mouse IgG isotypes differ in their capacity to either activate complement or interact with Fcγ receptors. For example, for complement-dependent opsonophagocytosis, the hierarchy of IgG3 > IgG2b = IgG2a >> IgG1 has been established.65 The mouse has three activating Fcγ receptors, R1, RIII, and RIV, with RIII being the main receptor. FcγRIII is expressed on most myeloid cells, including monocytes, macrophages, and dendritic cells and natural killer cells. FcγRIII is activated by mouse IgG1, IgG2a, and IgG2b.66 Hence, the effect of ox-elastin antibodies on SIOP pathology could be driven by a mixed effect, mediated by IgM, IgG3, and IgG2b. IgM and IgG3 antibodies might be activating complement, leading to CDC, whereas IgG2b could engage FcγRΙΙΙ receptors on effector cells, eliciting ADCC. Additional experiments using FcγRIII−/− mice and complement inhibitors might be able to further distinguish between these two pathways of antibody-dependent cell damage. In this context, we observed that the elastin + smoke-challenged mice had a thinner BrM and fewer mislocalized mitochondria than the mice exposed to smoke alone. It is plausible that the small amount of antibodies against elastin is protective, triggering IgG/C1q-mediated removal of debris,67 whereas the large amount of anti-ox-elastin is damaging, triggering CDC or ADCC.
Taken together, a growing body of evidence links oxidative stress, smoking, and complement activation as well as autoimmunity to AMD pathogenesis. Our new data provided here show that functional and morphologic defects in the retina, RPE, and BrM generated by smoke exposure are potentiated by immunization with elastin peptides modified by smoke. Our experiments showing differential responses in binding of subclasses of IgG antibodies suggest that both complement activation and Fcγ receptor-mediated cytotoxicity might be required for these functional and structural alterations to occur. These results may open up additional avenues for anti-complement-based therapies, as well as immunotherapies, in dry AMD.
Acknowledgments
Supported by the National Institutes of Health R01EY019320 (BR), R01HL091944 (CA), R01EY015128 (BJ), R01EY028927 (BJ), and P30EY014800 (BJ); the Department of Veterans Affairs RX000444 and BX003050 (BR); the South Carolina SmartState Endowment (BR); and an Unrestricted Research Grant from Research to Prevent Blindness, New York, to the Department of Ophthalmology & Visual Sciences, University of Utah.
Disclosure: B. Annamalai, None; C. Nicholson, None; N. Parsons, None; S. Stephenson, None; C. Atkinson, None; B. Jones, None; B. Rohrer, None
References
- 1. Brown MM, Brown GC, Stein JD, Roth Z, Campanella J, Beauchamp GR. Age-related macular degeneration: economic burden and value-based medicine analysis. Can J Ophthalmol. 2005; 40: 277–287. [DOI] [PubMed] [Google Scholar]
- 2. Grassmann F, Heid IM, Weber BH. Recombinant haplotypes narrow the ARMS2/HTRA1 association signal for age-related macular degeneration. Genetics. 2017; 205: 919–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Johnson LV, Leitner WP, Staples MK, Anderson DH. Complement activation and inflammatory processes in Drusen formation and age related macular degeneration. Exp Eye Res. 2001; 73: 887–896. [DOI] [PubMed] [Google Scholar]
- 4. Klein RJ, Zeiss C, Chew EY, et al.. Complement factor H polymorphism in age-related macular degeneration. Science. 2005; 308: 385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Umeda S, Suzuki MT, Okamoto H, et al.. Molecular composition of drusen and possible involvement of anti-retinal autoimmunity in two different forms of macular degeneration in cynomolgus monkey (Macaca fascicularis). Faseb J. 2005; 19: 1683–1685. [DOI] [PubMed] [Google Scholar]
- 6. Mullins RF, Dewald AD, Streb LM, Wang K, Kuehn MH, Stone EM. Elevated membrane attack complex in human choroid with high risk complement factor H genotypes. Exp Eye Res. 2011; 93: 565–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schmidt S, Hauser MA, Scott WK, et al.. Cigarette smoking strongly modifies the association of LOC387715 and age-related macular degeneration. Am J Hum Genet. 2006; 78: 852–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sparrow JR, Ueda K, Zhou J. Complement dysregulation in AMD: RPE-Bruch's membrane-choroid. Mol Aspects Med. 2012; 33: 436–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Weismann D, Hartvigsen K, Lauer N, et al.. Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature. 2011; 478: 76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Joseph K, Kulik L, Coughlin B, et al.. Oxidative stress sensitizes RPE cells to complement-mediated injury in a natural antibody-, lectin pathway- and phospholipid epitope-dependent manner. J Biol Chem. 2013; 288: 12753–12765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Curcio CA, Johnson M, Rudolf M, Huang JD. The oil spill in ageing Bruch membrane. Br J Ophthalmol. 2011; 95: 1638–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sarks SH. Ageing and degeneration in the macular region: a clinico-pathological study. Br J Ophthalmol. 1976; 60: 324–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sarks SH, Van Driel D, Maxwell L, Killingsworth M. Softening of drusen and subretinal neovascularization. Trans Ophthalmol Soc U K. 1980; 100: 414–422. [PubMed] [Google Scholar]
- 14. Starita C, Hussain AA, Pagliarini S, Marshall J. Hydrodynamics of ageing Bruch's membrane: implications for macular disease. Exp Eye Res. 1996; 62: 565–572. [DOI] [PubMed] [Google Scholar]
- 15. Johnson M, Dabholkar A, Huang JD, Presley JB, Chimento MF, Curcio CA. Comparison of morphology of human macular and peripheral Bruch's membrane in older eyes. Curr Eye Res. 2007; 32: 465–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Curcio CA, Johnson M. Structure, Function, and Pathology of Bruch's Membrane. London, UK: Elsevier; 2013: 465–481. [Google Scholar]
- 17. Chong NH, Keonin J, Luthert PJ, et al.. Decreased thickness and integrity of the macular elastic layer of Bruch's membrane correspond to the distribution of lesions associated with age-related macular degeneration. Am J Pathol. 2005; 166: 241–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sivaprasad S, Chong NV, Bailey TA. Serum elastin-derived peptides in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2005; 46: 3046–3051. [DOI] [PubMed] [Google Scholar]
- 19. Morohoshi K, Patel N, Ohbayashi M, et al.. Serum autoantibody biomarkers for age-related macular degeneration and possible regulators of neovascularization. Exp Mol Pathol. 2012; 92: 64–73. [DOI] [PubMed] [Google Scholar]
- 20. Rinaldi M, Lehouck A, Heulens N, et al.. Antielastin B-cell and T-cell immunity in patients with chronic obstructive pulmonary disease. Thorax. 2012; 67: 694–700. [DOI] [PubMed] [Google Scholar]
- 21. Patel N, Ohbayashi M, Nugent AK, et al.. Circulating anti-retinal antibodies as immune markers in age-related macular degeneration. Immunology. 2005; 115: 422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Joachim SC, Bruns K, Lackner KJ, Pfeiffer N, Grus FH. Analysis of IgG antibody patterns against retinal antigens and antibodies to alpha-crystallin, GFAP, and alpha-enolase in sera of patients with “wet” age-related macular degeneration. Graefes Arch Clin Exp Ophthalmol. 2007; 245: 619–626. [DOI] [PubMed] [Google Scholar]
- 23. Ozkan B, Karabas LV, Altintas O, Tamer GS, Yuksel N, Caglar Y. Plasma antiphospholipid antibody levels in age-related macular degeneration. Can J Ophthalmol. 2012; 47: 264–268. [DOI] [PubMed] [Google Scholar]
- 24. Hollyfield JG, Perez VL, Salomon RG. A hapten generated from an oxidation fragment of docosahexaenoic acid is sufficient to initiate age-related macular degeneration. Mol Neurobiol. 2010; 41: 290–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Saeed AF, Wang R, Ling S, Wang S. Antibody engineering for pursuing a healthier future. Front Microbiol. 2017; 8: 495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Natoli R, Mason E, Jiao H, et al.. Dynamic interplay of innate and adaptive immunity during sterile retinal inflammation: insights from the transcriptome. Front Immunol. 2018; 9: 1666. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27. Wang TT, Ravetch JV. Immune complexes: not just an innocent bystander in chronic viral infection. Immunity. 2015; 42: 213–215. [DOI] [PubMed] [Google Scholar]
- 28. Hugli TE. Structure and function of the anaphylatoxins. Springer Semin Immunopathol. 1984; 7: 193–219. [DOI] [PubMed] [Google Scholar]
- 29. Klos A, Tenner AJ, Johswich KO, Ager RR, Reis ES, Kohl J. The role of the anaphylatoxins in health and disease. Mol Immunol. 2009; 46: 2753–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Woodell A, Coughlin B, Kunchithapautham K, et al.. Alternative complement pathway deficiency ameliorates chronic smoke-induced functional and morphological ocular injury. PLoS ONE. 2013; 8: e67894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kunchithapautham K, Atkinson C, Rohrer B. Smoke exposure causes endoplasmic reticulum stress and lipid accumulation in retinal pigment epithelium through oxidative stress and complement activation. J Biol Chem. 2014; 289: 14534–14546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Woodell A, Jones BW, Williamson T, et al.. A targeted inhibitor of the alternative complement pathway accelerates recovery from smoke-induced ocular injury. Invest Ophthalmol Vis Sci. 2016; 57: 1728–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kirkham PA, Caramori G, Casolari P, et al.. Oxidative stress-induced antibodies to carbonyl-modified protein correlate with severity of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011; 184: 796–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brandsma CA, Timens W, Geerlings M, et al.. Induction of autoantibodies against lung matrix proteins and smoke-induced inflammation in mice. BMC Pulm Med. 2010; 10: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mishima H, Kondo K. Ultrastructure of age changes in the basal infoldings of aged mouse retinal pigment epithelium. Exp Eye Res. 1981; 33: 75–84. [DOI] [PubMed] [Google Scholar]
- 36. Ten Berge JC, Schreurs MW, Vermeer J, Meester-Smoor MA, Rothova A. Prevalence and clinical impact of antiretinal antibodies in uveitis. Acta Ophthalmol. 2016; 94: 282–288. [DOI] [PubMed] [Google Scholar]
- 37. Gu X, Meer SG, Miyagi M, et al.. Carboxyethylpyrrole protein adducts and autoantibodies, biomarkers for age-related macular degeneration. J Biol Chem. 2003; 278: 42027–42035. [DOI] [PubMed] [Google Scholar]
- 38. Frostegard J. Prediction and management of cardiovascular outcomes in systemic lupus erythematosus. Expert Rev Clin Immunol. 2015; 11: 247–253. [DOI] [PubMed] [Google Scholar]
- 39. Hiromatsu Y, Eguchi H, Tani J, Kasaoka M, Teshima Y. Graves' ophthalmopathy: epidemiology and natural history. Intern Med. 2014; 53: 353–360. [DOI] [PubMed] [Google Scholar]
- 40. Daffa NI, Tighe PJ, Corne JM, Fairclough LC, Todd I. Natural and disease-specific autoantibodies in chronic obstructive pulmonary disease. Clin Exp Immunol. 2015; 180: 155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lois N, Abdelkader E, Reglitz K, Garden C, Ayres JG. Environmental tobacco smoke exposure and eye disease. Br J Ophthalmol. 2008; 92: 1304–1310. [DOI] [PubMed] [Google Scholar]
- 42. Chakravarthy U, Augood C, Bentham GC, et al.. Cigarette smoking and age-related macular degeneration in the EUREYE Study. Ophthalmology. 2007; 114: 1157–1163. [DOI] [PubMed] [Google Scholar]
- 43. Mitchell P, Wang JJ, Smith W, Leeder SR. Smoking and the 5-year incidence of age-related maculopathy: the Blue Mountains Eye Study. Arch Ophthalmol. 2002; 120: 1357–1363. [DOI] [PubMed] [Google Scholar]
- 44. Church DF, Pryor WA. Free-radical chemistry of cigarette smoke and its toxicological implications. Environ Health Perspect. 1985; 64: 111–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Alberg A. The influence of cigarette smoking on circulating concentrations of antioxidant micronutrients. Toxicology. 2002; 180: 121–137. [DOI] [PubMed] [Google Scholar]
- 46. Bruno RS, Traber MG. Vitamin E biokinetics, oxidative stress and cigarette smoking. Pathophysiology. 2006; 13: 143–149. [DOI] [PubMed] [Google Scholar]
- 47. Chow CK, Thacker RR, Changchit C, et al.. Lower levels of vitamin C and carotenes in plasma of cigarette smokers. J Am Coll Nutr. 1986; 5: 305–312. [DOI] [PubMed] [Google Scholar]
- 48. Panda K, Chattopadhyay R, Ghosh MK, Chattopadhyay DJ, Chatterjee IB. Vitamin C prevents cigarette smoke induced oxidative damage of proteins and increased proteolysis. Free Radic Biol Med. 1999; 27: 1064–1079. [DOI] [PubMed] [Google Scholar]
- 49. Moriarty SE, Shah JH, Lynn M, et al.. Oxidation of glutathione and cysteine in human plasma associated with smoking. Free Radic Biol Med. 2003; 35: 1582–1588. [DOI] [PubMed] [Google Scholar]
- 50. van der Vaart H, Postma DS, Timens W, ten Hacken NH. Acute effects of cigarette smoke on inflammation and oxidative stress: a review. Thorax. 2004; 59: 713–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Golestaneh N, Chu Y, Xiao YY, Stoleru GL, Theos AC. Dysfunctional autophagy in RPE, a contributing factor in age-related macular degeneration. Cell Death Dis. 2017; 8: e2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhu BQ, Parmley WW. Hemodynamic and vascular effects of active and passive smoking. Am Heart J. 1995; 130: 1270–1275. [DOI] [PubMed] [Google Scholar]
- 53. Friedman E. Update of the vascular model of AMD. Br J Ophthalmol. 2004; 88: 161–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kew RR, Ghebrehiwet B, Janoff A. Cigarette smoke can activate the alternative pathway of complement in vitro by modifying the third component of complement. J Clin Invest. 1985; 75: 1000–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang D, Nasto LA, Roughley P, et al.. Spine degeneration in a murine model of chronic human tobacco smokers. Osteoarthritis Cartilage. 2012; 20: 896–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jones A, Kumar S, Zhang N, et al.. Increased expression of multifunctional serine protease, HTRA1, in retinal pigment epithelium induces polypoidal choroidal vasculopathy in mice. Proc Natl Acad Sci USA. 2011; 108: 14578–14583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bonilha VL. Age and disease-related structural changes in the retinal pigment epithelium. Clin Ophthalmol. 2008; 2: 413–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gouras P, Ivert L, Neuringer M, Nagasaki T. Mitochondrial elongation in the macular RPE of aging monkeys, evidence of metabolic stress. Graefes Arch Clin Exp Ophthalmol. 2016; 254: 1221–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Feher J, Kovacs I, Artico M, Cavallotti C, Papale A, Balacco Gabrieli C. Mitochondrial alterations of retinal pigment epithelium in age-related macular degeneration. Neurobiol Aging. 2006; 27: 983–993. [DOI] [PubMed] [Google Scholar]
- 60. Alavi MV. Aging and vision. Adv Exp Med Biol. 2016; 854: 393–399. [DOI] [PubMed] [Google Scholar]
- 61. Leenaars PP, Koedam MA, Wester PW, Baumans V, Claassen E, Hendriksen CF. Assessment of side effects induced by injection of different adjuvant/antigen combinations in rabbits and mice. Lab Anim. 1998; 32: 387–406. [DOI] [PubMed] [Google Scholar]
- 62. Stills HF., Jr Adjuvants and antibody production: dispelling the myths associated with Freund's complete and other adjuvants. ILAR J. 2005; 46: 280–293. [DOI] [PubMed] [Google Scholar]
- 63. Skeie JM, Hernandez J, Hinek A, Mullins RF. Molecular responses of choroidal endothelial cells to elastin derived peptides through the elastin-binding protein (GLB1). Matrix Biol. 2012; 31: 113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Muller-Eberhard HJ. Molecular organization and function of the complement system. Annu Rev Biochem. 1988; 57: 321–347. [DOI] [PubMed] [Google Scholar]
- 65. Michaelsen TE, Kolberg J, Aase A, Herstad TK, Hoiby EA. The four mouse IgG isotypes differ extensively in bactericidal and opsonophagocytic activity when reacting with the P1.16 epitope on the outer membrane PorA protein of Neisseria meningitidis. Scand J Immunol. 2004; 59: 34–39. [DOI] [PubMed] [Google Scholar]
- 66. Ding JW, Zhou T, Zeng H, et al.. Hyperacute rejection by anti-Gal IgG1, IgG2a, and IgG2b is dependent on complement and Fc-gamma receptors. J Immunol. 2008; 180: 261–268. [DOI] [PubMed] [Google Scholar]
- 67. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010; 11: 785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]