

Abstract
Purpose
Severing corneal nerves during orthotopic corneal transplantation elicits the elaboration of the neuropeptide substance P (SP), which induces the generation of CD11c+ contrasuppressor (CS) cells. CS cells disable T regulatory cells (Tregs) that are induced when antigens enter the anterior chamber (AC), either by direct injection or by orthotopic corneal transplantation. This study examined the crucial cell surface molecules on Tregs that are adversely affected by CS cells that are generated by severing corneal nerves.
Methods
CS cells were induced by producing shallow 2.0-mm circular incisions in the corneal epithelium in BALB/c mice. CD8+ Tregs were generated by injecting ovalbumin into the AC. The effects of CS cells and SP on the expression and function of two cell surface molecules (CD103 and the receptor of interferon-γ) that are crucial for the induction and function of CD8+ Tregs were analyzed.
Results
SP converted CD11c+, but not CD11c−, dendritic cells (DCs) to CS cells. Severing corneal nerves resulted in a 66% reduction in the expression of CD103 on CD8+ AC-associated immune deviation (ACAID) Tregs, and a 50% reduction in the interferon-γ receptor (IFN-γR). These effects could be mimicked in vitro by coculturing CS cells with CD8+ ACAID Tregs.
Conclusions
The elaboration of SP in response to corneal nerve ablation converts CD11c+ DCs to CS cells. CS cells disable CD8+ ACAID Tregs by downregulating two crucial cell surface molecules, CD103 and IFN-γR, by an SP-dependent pathway. Blocking this pathway may provide a means of restoring ocular immune privilege in corneas subjected to corneal nerve injury.
Keywords: anterior chamber, contrasuppressor cells, immune privilege, substance P, Tregs
Ocular immune privilege prevents immune-mediated injury to tissues that have little or no regenerative properties. Immune privilege is the sum total of multiple anatomic, physiological, and regulatory processes that block the induction and expression of both innate and adaptive immune responses in the eye.1 The aqueous humor is a cocktail of soluble factors that promote immune privilege by dampening inflammatory responses and suppressing T cell-activity within the eye.2–6 The cells lining the eye are decorated with molecules, such as FasL, PD-L1, and tumor-necrosis factor-related apoptosis-inducing ligand, that either induce apoptosis or suppress activation of cells of the adaptive and innate immune systems.7–10 Dynamic regulatory processes also preserve immune privilege within the eye. Antigens entering the anterior chamber (AC) elicit the generation of T regulatory cells (Tregs) that suppress T cell–mediated immunity and promote corneal allograft survival.11,12 This dynamic systemic immunoregulatory phenomenon has been termed AC-associated immune deviation (ACAID) and occurs after antigens, such as viral peptides, soluble proteins, tumor antigens, or alloantigens, enter the AC.1,13 ACAID is associated with corneal allograft survival. Mice bearing long-standing corneal allografts display an antigen-specific suppression of T cell–mediated alloimmune responses that closely resembles the ACAID phenotype.14,15 Moreover, maneuvers that abolish ACAID (e.g., splenectomy or anti-IFN-γ antibody treatment) invariably lead to the immune rejection of corneal allografts.16,17
Immune privilege is not absolute and is terminated under a variety of conditions. In rare circumstances, penetrating injuries to the eye can elicit a condition called sympathetic ophthalmia (SO) in which trauma to one eye elicits inflammation in the opposite “sympathizing” eye. This curious phenomenon was recognized by the ancient Greeks and was even discussed by Hippocrates.18 SO remains poorly understood but current thinking posits that trauma to the eye, especially penetrating injuries, causes a release of uveal and retina antigens that elicit an inflammatory reaction in both eyes.18
We recently discovered that application of a corneal transplant to one eye caused a steep increase in the incidence of immune rejection of subsequent corneal allografts placed into the opposite eye—even corneal allografts from donors that were genetically different from the donors of the first transplant.19 Further analysis revealed that simply severing the corneal nerves in one eye produced a similar effect and robbed the opposite eye of its immune privilege, and led to a dramatic increase in the incidence and tempo of immune rejection of corneal allografts placed into the opposite eye. This sympathetic loss of immune privilege (SLIP) is reminiscent of SO but is distinctly different in the sense that SLIP is not antigen-specific and is not related to the release of ocular antigens. SLIP not only affects corneal allograft survival but it also prevents the induction of ACAID in the opposite eye that was not subjected to nerve injury.20 The underlying mechanisms responsible for SLIP are associated with the generation of antigen nonspecific CD11c+ contrasuppressor (CS) cells that disable Tregs that are induced either by orthotopic corneal allografts or by AC injection of antigens (i.e., ACAID).21 The present study explored the mechanisms utilized by SLIP CS cells to disable ACAID Tregs.
Materials and Methods
Animals
BALB/c (H-2d) mice were purchased from the University of Texas Southwestern Mouse Breeding Facility. All mice were maintained in a pathogen-free environment and were cared for in accordance with the guidelines of the Institutional Animal Care and Use Committee of the University of Texas Southwestern Medical Center and the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
Induction of ACAID
ACAID was induced as described previously using microinjection of antigen into the AC of the eye.22 A Hamilton automatic dispensing apparatus was used to inject 6 µL of the 16.67 mg/mL ovalbumin (OVA) (Sigma-Aldrich, St. Louis, MO, USA) in PBS (100 µg of OVA) into the AC. Seven days after the AC injection, animals were immunized subcutaneously (SC) with 200 µg of OVA in an equal volume of Complete Freud's Adjuvant (CFA; Sigma-Aldrich). Ears were challenged 7 days after SC immunization by injecting OVA (400 µg in 20 µL of PBS). The opposite ear was injected with 20 µL of PBS as a negative control. Ear swelling was measured 24 hours later to assess the delayed-type hypersensitivity (DTH) response to OVA.
Isolation of ACAID CD8+ Tregs
Magnetic microbeads conjugated with anti-CD8 (Ly-2) monoclonal antibody were used to isolate CD8+ Tregs from spleen cells of BALB/c mice 3 to 4 days following AC injection of OVA. The cell suspension was then loaded on to a MACS Column (Miltenyi Biotec; Santa Barbara, CA), which was placed in a magnetic field of the MACS separator. The magnetically labeled CD8+ Tregs were retained within the column and the unlabeled cells removed. The column was removed from the magnetic field, and the retained CD8+ cells were eluted as the positively selected cell fraction. We have previously found that this enrichment technique yields >95% CD8+ T cells.20
Antibodies and Flow Cytometry
CD8+ ACAID Tregs were interrogated for their cell surface expression of the IFN-γ receptor and the CD103 molecule. The antibodies used for these flow cytometric analyses were PE-CD 103, PE-IFN-γ receptor, FITC-CD8, and 7- AAD (BD Biosciences, San Jose, CA, USA), and isotype control monoclonal antibodies. All flow cytometric analyses were performed on an Attune NxT acoustic focusing cytometer (Applied Biosystems; Life Technologies, Grand Island, NY, USA). The data from the flow cytometer were analyzed using FlowJo version 10 software (Tree Star, Ashland, OR, USA).
DTH Assay
DTH was measured using a conventional ear-swelling assay.22 An eliciting dose of 400 µg of OVA (Sigma-Aldrich) in 20 µL of Hanks’ balanced salt solution (HBSS) was injected into the SC tissue of the right ear. The left ear served as a negative control and was injected with 20 µL HBSS without cells. Results were expressed as antigen-specific ear-swelling response = (experimental ear 24 hour measurement – experimental ear 0 hour measurement) – (negative control ear 24 hour measurement – negative ear 0 hour measurement).
Isolation of CS Cells
We have previously reported that 2.0-mm circular incisions of the corneal epithelium induce the generation of CS cells that express the CD11c surface marker.20 Corneas of BALB/c mice were trephined as described previously, and CD11c+ spleen cells were isolated 4 days later using a Miltenyl Biotec (Santa Barbara, CA) pan dendritic cell (DC) isolation kit.20 The CD11c+ cells from trephined mice and untreated mice were used for detecting regulatory cell activity in vitro.20
Neuropeptide Receptor Antagonists
The receptor of substance P (SP), NK1-R, was blocked in vitro with Spantide II (Sigma-Aldrich).19,20 The melanocortin receptor 4 receptor antagonist, HS014 (Bachem, Torrance, CA, USA), was used to block α-melanocyte stimulating hormone (α-MSH) receptors.23,24
Transwell Coculture Assays
CS cells and CD8+ Tregs were cultured in Transwell culture plates (Millipore; St. Louis, MO). CS cells were placed in the top chamber (1 × 106 cells), and 1 × 106 CD8+ Tregs were placed in the bottom chamber. A semipermeable membrane separates the two chambers and permits free movement of macromolecules between chambers but prevents both cell populations from entering the other chamber. Cells were cultured for 20 hours at 37°C, washed in HBSS, and the CD8+ Treg cell suspensions were examined by flow cytometry for CD103 expression.
Statistical Analyses
Results for DTH assays and flow cytometry analyses were evaluated by the Student's t-test. Results are expressed as mean ± SEM. P values in all experiments were considered to be statistically significant if the P values were <0.05.
Results
SP Converts CD11c+ DCs to CS Cells
Previous studies revealed that the severing corneal nerves that occurs during penetrating keratoplasty to one eye induces a steep upregulation in the neuropeptide SP in the anterior segment of both eyes, and leads to a commensurate increase in the expression of the SP receptor NK1-R.19 Moreover, in vitro treatment of naive CD11c+ DCs with SP endows these cells with CS activity.21 We wished to determine if the SP effect on CS cell activity was restricted to CD11c+ DCs or if other DCs not expressing the CD11c marker would also be converted to CS cells. Accordingly, splenic CD11c+ and CD11c− DCs were isolated and enriched using a Miltenyl Biotech pan DC isolation kit. Both cell populations were incubated for 1 hour in HBSS alone or HBSS containing 1.0 pg/mL of SP. Cells were washed in HBSS and injected intravenously (IV) (1 × 106 cells/mouse) into naive mice. One day later, OVA was injected into the AC, and mice were immunized SC with OVA emulsified in CFA 7 days after AC injection.
Ear swelling responses were measured 10 days after SC immunization. As expected, ACAID was not induced in mice that had received SP-conditioned CD11c+ DCs indicating that these cells blocked either the induction or the suppressive activity of Tregs (Fig. 1). By contrast, the induction and expression of ACAID was unaffected in recipients of either CD11c− cells conditioned in vitro with SP, or mice that received CD11c+ DCs that were treated with HBSS before adoptive transfer.
Figure 1.
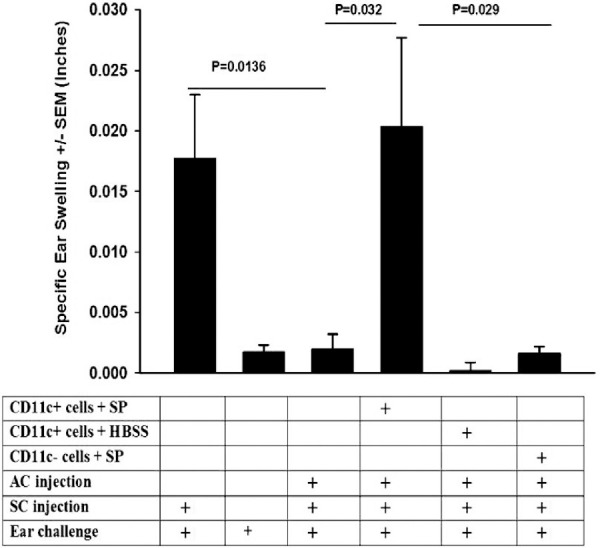
SP convers CD11c+ DCs to CS cells. CD11c+ and CD11c− spleen cells were isolated from naive BALB/c mice and incubated in either HBSS or SP (1.0 pg/mL) for 1 hour prior to IV injection into BALB/c mice. One day later OVA was injected into the AC of mice who were immunized SC with OVA 7 days later. DTH responses were assessed 10 days after SC immunization. There were five mice per group. This experiment was performed twice with similar results. Results are expressed as mean ± SEM.
Corneal Nerve Ablation Results in Downregulation of CD103 on ACAID Tregs
CD103 is a marker that is expressed on CD4+ CD25+ Tregs and CD8+ ACAID Tregs.25-29 Keino et al.26 performed DNA microarray analysis on 12,000 genes expressed in CD8+ ACAID Tregs and found an 85-fold increase in the expression of CD103. Moreover, these investigators found that ACAID could not be induced in CD103 knockout mice, thereby confirming the key role of CD103 in ocular immune privilege. Accordingly, we entertained the hypothesis that corneal nerve ablation adversely affected the expression of CD103 on ACAID Tregs. To test this, the corneas of mice were subjected to shallow circular incisions produced by trephining one eye. One day later, mice received AC injections of OVA introduced into the opposite eye. CD8+ Tregs were isolated 4, 9, or 15 days later and were interrogated for CD103 expression by flow cytometry using a gating procedure described previously30 and shown schematically in Figure 2. Trephining resulted in a 66% reduction in CD103 expression on CD8+ cells on day 4 (P = 0.00009), and a 20% reduction (P > 0.05) on day 9 following AC injection of OVA (Figs. 3A, 3B, 3D, 3E). However, CD103 expression on splenic CD8+ Tregs returned to normal levels by day 14 (Figs. 3C, 3F). To establish a link between the downregulation of CD103 on CD8+ Tregs and CS cells, we cocultured CD11c+ CS cells from trephined donor mice with CD8+ Tregs isolated from mice with ACAID and assessed CD103 expression on the ACAID Tregs 20 hours later. We found that CD103 expression on CD8+ Tregs cocultured with CS cells was reduced by over 30% (Figs. 4A, 4B).
Figure 2.

Flow cytometry gating strategy for dual staining CD8 and CD103 on T cells from ACAID mice. (A) Live cells were distinguished from dead cells based on forward scatter (FSC) versus side scatter (SSC). Surface markers CD8 (FITC) and CD103 (PE) or CD8 and CD119 (PE) were dual stained, and the results were depicted as cells that expressed CD103 or CD119 for Fluorescence-activated cell sorting (FACS) profile figures. (B) In subsequent figures the results represented in bar graphs are expressed as “mean fluorescence intensity” and represent the intensity of staining for either CD103 or CD119. FACS profile figures represent the mean number of cells staining for either CD103 or CD119 and are labeled as “counts.”
Figure 3.

Corneal nerve ablation results in downregulation of CD103 on ACAID Tregs. The right eyes of BALB/c mice were trephined, and 1 day later ACAID was induced by injecting OVA into the left eyes. CD8+ putative Tregs were isolated from the spleens at 4 (A, D), 9 (B, E), or 14 (C, F) days later and were interrogated by flow cytometry for CD103 expression. (A–C) Y-axis represents mean fluorescence intensity (MFI) of CD103+ cells. (D, E, F) Y-axis represents “counts” (% of cells that were CD103+). There were five animals per group. These experiments were performed twice with similar results. Results are expressed as mean ± SEM. P values were calculated based on either (A) MFI or (B) number of CD8+ CD103+ cells.
Figure 4.

Trephine-induced CS cells or SP alone downregulates CD103 on ACAID Tregs. CD8+ ACAID Tregs were induced by AC injection of OVA. Four days later, CD8+ ACAID Tregs were isolated from the spleens and were cocultured for 20 hours with (A, B) CD11c+ and CD11c− CS cells or (C, D) with SP alone (0.1 pg/mL). (E) CD11c+ CS cells or CD11c+ CS cells in the presence of either Spantide II (36 ug/mL) or HSO14 (36 ug/mL). CD103 expression on CD8+ T cells was evaluated by flow cytometry 24 hours later. (A,C, E) Y-axis label represents mean fluorescence intensity (MFI), which is a reflection of the density of CD103 expression. (B, D) Y-axis label represents “counts” or % of cells that were CD103+. There were five animals per group. Each experiment was performed twice with similar results. Results are expressed as mean ± SEM. P values were determined based on either MFI (A, C, E) or number of CD103+ cells (B, D).
We have previously shown that CS cells produce SP, and that in vivo blockade of the SP receptor NK1-R prevents the loss of immune privilege by corneal nerve ablation.19 Accordingly, we tested the hypothesis that SP produced by CS cells downregulates CD103 expression on ACAID Tregs. To test this, CD8+ ACAID Tregs were isolated from the spleens of mice 4 days following AC injection of OVA and were incubated for 20 hours in either normal culture medium or culture medium containing SP (0.01 pg/mL/1 × 106 cells). Cells were washed and examined by flow cytometry for CD103 expression. CD103 expression was reduced by approximately 75% in CD8+ Tregs exposed to SP compared with untreated cells (Figs. 4C, 4D).
These results suggested that CS cells induced by trephining the cornea were directly responsible for the downregulation of CD103 on ACAID Tregs. This hypothesis was tested in vitro by coculturing CD11c+ cells from trephined donors with ACAID Tregs in the presence or absence of Spantide II, an SP receptor (NK1-R) antagonist. For comparison, CD11c− cells from trephined donors were similarly tested in vitro. The results indicated that CD11c− cells from trephined donors had no effect on CD103 expression on CD8+ ACAID Tregs, either in the presence or absence of Spantide II (Fig. 4E). By contrast, CD11c+ cells from trephined donors downregulated CD103 expression on ACAID Tregs. Moreover, addition of Spantide II significantly relieved the downregulation of CD103 expression on ACAID Tregs, even though CD103 expression was not completely restored to the levels found in ACAID Tregs cocultured with CD11c− spleen cells. To confirm that the restoration of CD103 expression by the NK1-R, the antagonist Spantide II was specific for the SP receptor (NK1-R), additional experiments evaluated the effect of blocking the receptor for an unrelated neuropeptide, α-MSH, using the melanocortin receptor (MC4R) antagonist HS014. Blockade of the MC4R failed to affect the expression of CD103 on CD8+ Tregs cocultured with CD11c+ CS cells, thereby confirming that SP was responsible for the modulation of CD103 on CD8+ ACAID Tregs (Fig. 4E).
Additional experiments were performed to determine if the downregulation of CD103 on CD8+ACAID Tregs that was produced by CD11c+ CS cells was contact-dependent. This hypothesis was based on published reports indicating that antigen-presenting DCs produce and respond to SP.31–36 Accordingly, the right eyes of naive BALB/c mice were trephined, and 4 days later CD11c+ cells were isolated from spleens and were placed into the top wells of Transwell culture plates. CD8+ ACAID Tregs were isolated from mice primed in the AC with OVA 5 days earlier and were placed either into the top or bottom wells of Transwell culture plates. CD8+ ACAID Tregs were removed from the both top and bottom wells 20 hours later and were interrogated by flow cytometry for CD103 expression. The results suggested that the previously observed downregulation CD103 expression by CD11c+ CS cells was contact-dependent because placing a membrane barrier (i.e., Transwell culture) between these two cell populations prevented the downregulation of CD103 on CD8+ ACAID Tregs, whereas CD8+ Tregs in direct contact with CD11c+ CS expressed significantly less CD103 (Figs. 5A, 5B). These findings support the notion that CS cells must be in direct contact with CD8+ ACAID Tregs for the stimulation of SP secretion and the ensuing downregulation of CD103 on CD8+ Tregs. However, we subsequently learned that many neuropeptides stick to membranes, such as those used in Transwell culture plates, and that the putative contact-dependency that we observed might be due to SP adhering to the Transwell membrane and not reaching the bottom chamber of the culture plates. This suspicion was tested by adding SP to the top chamber of Transwell culture plates that did not contain cells in either chamber. SP concentrations in the top and bottom chambers were assessed by ELISA 24 hours later. The results revealed that the Transwell membrane completely blocked the diffusion of 1.0 pg/mL of SP into the bottom chamber (Fig. 5C). The effect of membrane incarceration of SP on downregulation of CD103 on CD8+ Tregs was confirmed by placing SP into the top chambers of other Transwell culture plates, and adding CD8+ Tregs into the bottom chamber. Assessment of CD103 expression 24 hours later revealed that the previously observed downregulation of CD103 by SP was blocked by the Transwell membrane (Fig. 5D). Thus the conclusion that the downregulation of CD103 by CD11c+ CS cells is contact-dependent cannot be validated in this model, and we can only speculate as to whether direct contact between CD11c+ DC and CD8+ Tregs is required for downregulation of CD103.
Figure 5.

Is downregulation of CD103 on CD8+ ACAID Tregs contact-dependent? (A, B) CD11c+ cells from trephined donors were cocultured in the top chambers with equal numbers of CD8+ ACAID Tregs, and a separate suspension of CD8+ ACAID Tregs were cultured in the bottom chamber of the same Transwell culture plate. (A) Y-axis label represents mean fluorescence insensitivity (MFI) for CD103 staining. (B) Y-axis label represents counts or % of cells that were CD103+. (C) To determine if the Transwell membrane impeded diffusion of SP, medium containing either 1.0, 5.0, or 10.0 pg of SP was placed into the top chambers of Transwell culture plates, and medium was placed into the bottom chamber. SP levels were determined by ELISA 24 hours later. According to the manufacturer, the sensitivity of the SP ELISA kit is 1.0 to 1000.0 pg/mL. ND = not detected. (D) SP (0.1 pg/mL or 0.01 pg/mL) was placed into the top chamber of a Transwell culture plate, and CD8+ ACAID Tregs were placed into the bottom chamber. Parallel experiments were performed using a culture plate without a membrane barrier. These experiments were performed twice with similar results. Results are expressed as mean ± SEM. P values for fluorescence-activated cell sorting (FACS) data were based on either (A) MFI or (B) number of CD103+ cells.
Effect of Corneal Nerve Ablation on IFN-γ Receptor Expression on ACAID Tregs
It was previously demonstrated that IFN-γ was needed for the development of ACAID.37 For CD8+ ACAID Tregs to suppress DTH responses they must first respond to IFN-γ through engagement of their IFN-γ receptor, which licenses them to express suppressive properties.37,38 Thus expression of both CD103 and the IFN-γ receptor is required for CD8+ ACAID Tregs to suppress DTH responses. With this in mind, we tested the hypothesis that in addition to downregulating CD103 on ACAID Tregs, CS cells also downregulate the expression of the IFN-γ receptor. This was tested by trephining the right eyes of BALB/c mice 1 day prior to injecting OVA into the AC of the left eye. CD8+ T cells were isolated from the spleens of trephined mice and nontrephined mice 4 days later. CD8+ cells were examined by flow cytometry for their expression of the IFN-γ receptor. The results revealed that expression of the IFN-γ receptor on CD8+ cells was reduced by approximately 50% in trephined mice compared with mice not subjected to trephining (Figs. 6A, 6B). Coculturing CD11c+ CS cells from trephined donors with CD8+ ACAID Tregs for 20 hours resulted in a significant reduction in the expression of the IFN-γ receptor (Figs. 6C, 6D).
Figure 6.

Effect of corneal nerve ablation on IFN-γ receptor expression on CD8+ ACAID Tregs. (A, B) The right eyes of BALB/c mice were trephined 1 day prior to injecting OVA into the AC of the left eye. CD8+ T cells were isolated from the spleens of trephined mice and nontrephined mice 4 days later. CD8+ cells were examined by flow cytometry for their expression of the IFN-γ receptor (CD119). (C, D) CD8+ ACAID Tregs were isolated from the spleens and were cocultured with CD11c+ and CD11c− cells that were isolated from the spleens of mice 4 days after trephining. Expression of IFN-γ receptor (CD119) on CD8+ ACAID Tregs was assessed by flow cytometry 20 hours later. (E, F) CD8+ ACAID Tregs were cultured in the absence or presence of SP (0.01 pg/mL/106 cells) and were assessed for the expression of the IFN-γ receptor 20 hours later. (A,C, E) Y-axis represents mean fluorescence intensity (MFI) of CD119 cells. (B, D, F) Y-axis represents “counts” (% of cells that were CD119+). These experiments were performed twice with similar results. Results are expressed as mean ± SEM. P values were based on either MFI (A, C, E) or number of CD8+ CD119+ cells (B, D, F).
The role of SP in this downregulation of the IFN-γ receptor was then examined in vitro. CD8+ ACAID Tregs were cultured in the absence or presence of SP (0.01 pg/mL/106 cells) and were assessed for the expression of the IFN-γ receptor 20 hours later. SP treatment resulted in a 36% reduction in the expression of the IFN-γ receptor on CD8+ ACAID Tregs (Figs. 6E, 6F).
Discussion
We have previously reported that severing corneal nerves—either by shallow circular incisions or by the application of an orthotopic corneal transplant—abolishes immune privilege and prevents the induction of ACAID in the opposite eye.20 A similar phenomenon was subsequently reported by Guzman et al.39 who found that either semicircular corneal incisions or alkali corneal burns in one eye prevented the induction of immune tolerance by topical application of OVA in the opposite eye. Earlier studies by Lucas et al.40 found that retinal laser burns (RLB) to one eye prevented the induction of ACAID in the other eye. RLB also induced a steep upregulation of the SP receptor (NK1-R) in the anterior segment of both eyes. These effects could be mimicked by simply injecting SP IV into naive mice, which resulted in the abrogation of ACAID.20 Thus SP and the NK1-R play pivotal roles in the abrogation of ACAID in the induction of SLIP. With this in mind, we directed our attention to the ocular cells that express the NK1-R. NK1-R is widely distributed among various bone marrow–derived cells, including DCs .32,41,42 Stimulation of DCs via their NK1-R inhibits their production of IL-10 and tilts the immune response toward a Th1 pathway.32 Moreover, antigen-presenting DCs produce and respond to SP.31–35 This, along with the observations that SP and its receptor (NK1-R) are upregulated in the anterior segment following corneal nerve ablation, and that ocular surface DCs reside in the precise location were trephining occurs, led us to suspect that these cells were the precursors to CS cells induced by corneal nerve ablation. It is noteworthy that unilateral alkali burns to the cornea of one eye leads to a steep increase in the number of CD11c+ DCs in both the ipsilateral and contralateral draining lymph nodes and prevents the induction of immune tolerance by topical application of OVA.39 Our current and previous findings indicate that SP released in response to corneal nerve ablation converts ocular surface CD11c+ DCs to CS cells. However, CD11c– cells exposed to SP do not display any detectable CS cell activity. Corneal nerve injury also leads to a transient, albeit steep, reduction in CD103 expression of CD8+ ACAID Tregs. In vitro analysis revealed that the downregulation of CD103 on CD8+ ACAID Tregs could be blocked using an SP receptor antagonist, Spantide II. Previous in vitro analyses indicated that CS cells elaborated SP, which downregulated CD103.21 Here we show that the SP-mediated downregulation of CD103 required the participation of CD11c+ CS cells.
Like CD103, interferon-γ receptor (IFN-γR) expression on CD8+ ACAID Tregs is required for their capacity to suppress DTH responses. Our findings indicate that CD11c+ CS cells downregulated IFN-γR by a process that was SP-dependent. As mentioned earlier, CD103 and IFN-γ are crucial for the induction and expression of ACAID Tregs. CD103 is a cell-adhesion molecule that binds to E-cadherin, which is expressed on multiple cells including antigen-presenting cells (APCs).43–45 It is noteworthy that TGF-β2 is crucial for the generation of ACAID APCs and coincidentally upregulates the expression of E-cadherin APCs.46 We are attracted to the hypothesis offered by Keino et al.,26 which suggests that CD103/E-cadherin interactions are enhanced by signaling via the T-cell receptor complex.47 This in turn, would stabilize the CD8+ Tregs and effector T cells at the site of antigenic challenge in DTH responses. This is consistent with the finding that expression of CD103 on ACAID Tregs is required for suppressing the efferent phase of DTH.26 We have previously shown that CD11c+ CS cells produce SP,21 and here we show that the release of SP by CD11c+ CS cells downregulates CD103 on CD8+ ACAID Tregs, and disables their suppressive activity.
We propose that corneal nerve ablation results in the elaboration of SP and a coincidental upregulation of the SP receptor (NK1-R) on CD11c+ DCs residing at the site of injury. SP converts these cells to CS cells, which in turn induce the downregulation CD103 and the IFN-γR on splenic CD8+ ACAID Tregs. Thus both CD103 and the IFN-γR are required for the induction and expression of suppressive activity by CD8+ ACAID Tregs.37,38
We and others have proposed that ACAID protects the eye from injury inflicted by immune-mediated inflammation. AC injection of retinal S-antigen induces ACAID and dampens inflammation directed against retinal antigens in a mouse model of experimental autoimmune uveitis.48 Ferguson et al.49,50 reported that exposure to light is required for the induction of ACAID. Mice reared in the dark cannot develop ACAID and are presumably unable to maintain normal circadian rhythm. We have proposed that in the absence of light, ACAID is unnecessary as vision is limited or absent. Likewise, photoentrainment is absent in animals residing in the total absence of light (i.e., troglodytes). We hypothesize that this leads to a condition in which the retinal elements that are normally required for photoentrainment are unnecessary and as a result, so is ACAID. One wonders if the circumvention of ACAD by rearing mice in the dark is because of the local production of SP.
At first blush, it would seem counterintuitive that conditions that abolish immune privilege in one eye would also affect the opposite unmanipulated eye. If ocular immune privilege is designed to shield the eye from immune-mediated injury, what is the advantage of robbing the opposite eye of this protection? We propose that SLIP is a safeguard to defend the host from life-threatening ocular infections. The dense corneal innervation serves as an early warning system for detecting the presence of infectious agents, which in some cases can be lethal. Two of the major causes of infectious keratitis, Pseudomonas aeruginosa and herpes simplex virus (HSV), are associated with activation of SP.51–54 We propose that infectious agents that deliver potent “danger signals” via cornea nerve stimulation evoke the release of SP, which in turn leads to the termination of immune privilege in both eyes. This is consistent with a recent report indicating that pain signals transmitted via TRPV1+ sensory nerves in the skin represent danger signals that activate innate Th17 immune responses in the skin of the affected area, as well as unaffected adjacent cutaneous tissues.55 Moreover, the authors proposed that stimulation of TRPV+ sensory nerves in the skin activates “anticipatory immunity” that prepares neighboring previously unaffected tissues against second cutaneous infections with Staphylococcus aureus and Candida albicans.55
Conclusions
It stands to reason that infection of the ocular surface in one eye would have a high likelihood to also occur in the opposite eye. Therefore terminating immune privilege would lead to the full expression of immunity in both eyes in anticipation of an imminent infection in the opposite eye, and thereby prevent the development of potentially lethal infections. HSV keratitis provides support for this hypothesis. Corneal HSV infections in T cell–deficient nude mice, which lack an adaptive immune response, culminate in fatal viral encephalitis.56 By contrast, similar corneal infections in immunocompetent mice induce a robust T-cell response and cornea inflammation; however, the hosts survive even though they are blinded by the intense inflammatory response. Thus we propose that the termination of immune privilege that occurs in both eyes due to SLIP is an adaptation to preserve life, even if the cost is blindness.
Acknowledgments
The authors thank Andrew Taylor (Boston University) for helpful advice regarding α-MSH receptor antagonists.
Supported by the National Institutes of Health Grants EY007641 and EY030413, and an unrestricted grant from Research to Prevent Blindness.
Disclosure: S. Neelam, None; J.Y. Niederkorn, None
References
- 1. Niederkorn JY. See no evil, hear no evil, do no evil: the lessons of immune privilege. Nat Immunol. 2006; 7: 354–359. [DOI] [PubMed] [Google Scholar]
- 2. Taylor AW, Alard P, Yee DG, Streilein JW. Aqueous humor induces transforming growth factor-beta (TGF-beta)- producing regulatory T-cells. Curr Eye Res. 1997; 16: 900–908. [DOI] [PubMed] [Google Scholar]
- 3. Taylor AW, Streilein JW, Cousins SW. Alpha-melanocyte-stimulating hormone suppresses antigen-stimulated T cell production of gamma-interferon. Neuroimmunomodulation. 1994; 1: 188–194. [DOI] [PubMed] [Google Scholar]
- 4. Taylor AW, Streilein JW, Cousins SW. Immunoreactive vasoactive intestinal peptide contributes to the immunosuppressive activity of normal aqueous humor. J Immunol. 1994; 153: 1080–1086. [PubMed] [Google Scholar]
- 5. Taylor AW, Yee DG.. Somatostatin is an immunosuppressive factor in aqueous humor. Invest Ophthalmol Vis Sci. 2003; 44: 2644–2649. [DOI] [PubMed] [Google Scholar]
- 6. Taylor AW, Yee DG, Streilein JW. Suppression of nitric oxide generated by inflammatory macrophages by calcitonin gene-related peptide in aqueous humor. Invest Ophthalmol Vis Sci. 1998; 39: 1372–1378. [PubMed] [Google Scholar]
- 7. Griffith TS, Brunner T, Fletcher SM, Green DR, Ferguson TA. Fas ligand-induced apoptosis as a mechanism of immune privilege. Science. 1995; 270: 1189–1192. [DOI] [PubMed] [Google Scholar]
- 8. Hori J, Wang M, Miyashita M, et al.. B7-H1-induced apoptosis as a mechanism of immune privilege of corneal allografts. J Immunol. 2006; 177: 5928–5935. [DOI] [PubMed] [Google Scholar]
- 9. Shen L, Jin Y, Freeman GJ, Sharpe AH, Dana MR. The function of donor versus recipient programmed death-ligand 1 in corneal allograft survival. J Immunol. 2007; 179: 3672–3679. [DOI] [PubMed] [Google Scholar]
- 10. Wang S, Boonman ZF, Li HC, et al.. Role of TRAIL and IFN-gamma in CD4+ T cell-dependent tumor rejection in the anterior chamber of the eye. J Immunol. 2003; 171: 2789–2796. [DOI] [PubMed] [Google Scholar]
- 11. Chauhan SK, Saban DR, Lee HK, Dana R. Levels of Foxp3 in regulatory T cells reflect their functional status in transplantation. J Immunol. 2009; 182: 148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cunnusamy K, Chen PW, Niederkorn JY. IL-17A-dependent CD4+CD25+ regulatory T cells promote immune privilege of corneal allografts. J Immunol. 2011; 186: 6737–6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Niederkorn JY. Anterior chamber-associated immune deviation and its impact on corneal allograft survival. Curr Opin Organ Transplant. 2006; 11: 360–365. [Google Scholar]
- 14. Niederkorn JY. The immune privilege of corneal grafts. J Leukoc Biol. 2003; 74: 167–171. [DOI] [PubMed] [Google Scholar]
- 15. Niederkorn JY, Larkin DF.. Immune privilege of corneal allografts. Ocul Immunol Inflamm. 2010; 18: 162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Niederkorn JY. High-risk corneal allografts and why they lose their immune privilege. Curr Opin Allergy Clin Immunol. 2010; 10: 493–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Niederkorn JY. Cornea: window to ocular immunology. Curr Immunol Rev. 2011; 7: 328–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Castiblanco CP, Adelman RA. Sympathetic ophthalmia. Graefes Arch Clin Exp Ophthalmol. 2009; 247: 289–302. [DOI] [PubMed] [Google Scholar]
- 19. Paunicka KJ, Mellon J, Robertson D, Petroll M, Brown JR, Niederkorn JY. Severing corneal nerves in one eye induces sympathetic loss of immune privilege and promotes rejection of future corneal allografts placed in either eye. Am J Transplant. 2015; 15: 1490–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mo J, Neelam S, Mellon J, Brown JR, Niederkorn JY. Effect of corneal nerve ablation on immune tolerance induced by corneal allografts, oral immunization, or anterior chamber injection of antigens. Invest Ophthalmol Vis Sci. 2017; 58: 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Neelam S, Mellon J, Wilkerson A, Niederkorn JY. Induction of contrasuppressor cells and loss of immune privilege produced by corneal nerve ablation. Invest Ophthalmol Vis Sci. 2018; 59: 4738–4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. D'Orazio TJ, Niederkorn JY.. A novel role for TGF-beta and IL-10 in the induction of immune privilege. J Immunol. 1998; 160: 2089–2098.9498745 [Google Scholar]
- 23. Clemson CM, Yost J, Taylor AW. The role of alpha-MSH as a modulator of ocular immunobiology exemplifies mechanistic differences between melanocortins and steroids. Ocul Immunol Inflamm. 2017; 25: 179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Taylor AW, Lee DJ.. The alpha-melanocyte stimulating hormone induces conversion of effector T cells into Treg cells. J Transplant. 2011; 2011: 246856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Andrew DP, Rott LS, Kilshaw PJ, Butcher EC. Distribution of alpha 4 beta 7 and alpha E beta 7 integrins on thymocytes, intestinal epithelial lymphocytes and peripheral lymphocytes. Eur J Immunol. 1996; 26: 897–905. [DOI] [PubMed] [Google Scholar]
- 26. Keino H, Masli S, Sasaki S, Streilein JW, Stein-Streilein J. CD8+ T regulatory cells use a novel genetic program that includes CD103 to suppress Th1 immunity in eye-derived tolerance. Invest Ophthalmol Vis Sci. 2006; 47: 1533–1542. [DOI] [PubMed] [Google Scholar]
- 27. Myers L, Croft M, Kwon BS, Mittler RS, Vella AT. Peptide-specific CD8 T regulatory cells use IFN-gamma to elaborate TGF-beta-based suppression. J Immunol. 2005; 174: 7625–7632. [DOI] [PubMed] [Google Scholar]
- 28. Suffia I, Reckling SK, Salay G, Belkaid Y. A role for CD103 in the retention of CD4+CD25+ Treg and control of Leishmania major infection. J Immunol. 2005; 174: 5444–5455. [DOI] [PubMed] [Google Scholar]
- 29. Uss E, Rowshani AT, Hooibrink B, Lardy NM, van Lier RA, Berge IJ. CD103 is a marker for alloantigen-induced regulatory CD8+ T cells. J Immunol. 2006; 177: 2775–2783. [DOI] [PubMed] [Google Scholar]
- 30. Bourdin B, Segura E, Tetreault MP, Lesage S, Parent L. Determination of the relative cell surface and total expression of recombinant ion channels using flow cytometry. J Vis Exp. 2016: 54732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ho WZ, Lai JP, Zhu XH, Uvaydova M, Douglas SD. Human monocytes and macrophages express substance P and neurokinin-1 receptor. J Immunol. 1997; 159: 5654–5660. [PubMed] [Google Scholar]
- 32. Janelsins BM, Sumpter TL, Tkacheva OA, et al.. Neurokinin-1 receptor agonists bias therapeutic dendritic cells to induce type 1 immunity by licensing host dendritic cells to produce IL-12. Blood. 2013; 121: 2923–2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lai JP, Douglas SD, Ho WZ. Human lymphocytes express substance P and its receptor. J Neuroimmunol. 1998; 86: 80–86. [DOI] [PubMed] [Google Scholar]
- 34. Lambrecht BN, Germonpre PR, Everaert EG, et al.. Endogenously produced substance P contributes to lymphocyte proliferation induced by dendritic cells and direct TCR ligation. Eur J Immunol. 1999; 29: 3815–3825. [DOI] [PubMed] [Google Scholar]
- 35. Marriott I, Bost KL. Substance P receptor mediated macrophage responses. Adv Exp Med Biol. 2001; 493: 247–254. [DOI] [PubMed] [Google Scholar]
- 36. Simeonidis S, Castagliuolo I, Pan A, et al.. Regulation of the NK-1 receptor gene expression in human macrophage cells via an NF-kappa B site on its promoter. Proc Natl Acad Sci U S A. 2003; 100: 2957–2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cone R, Chattopadhyay S, O'Rourke J. Control of delayed-type hypersensitivity by ocular-induced CD8+ regulatory T cells. Chem Immunol Allergy. 2008; 94: 138–149. [DOI] [PubMed] [Google Scholar]
- 38. Paunicka K, Chen PW, Niederkorn JY. Role of IFN-gamma in the establishment of anterior chamber-associated immune deviation (ACAID)-induced CD8+ T regulatory cells. J Leukoc Biol. 2012; 91: 475–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Guzman M, Miglio MS, Zgajnar NR, et al.. The mucosal surfaces of both eyes are immunologically linked by a neurogenic inflammatory reflex involving TRPV1 and substance P. Mucosal Immunol. 2018; 11: 1441–1453. [DOI] [PubMed] [Google Scholar]
- 40. Lucas K, Karamichos D, Mathew R, Zieske JD, Stein-Streilein J. Retinal laser burn-induced neuropathy leads to substance P-dependent loss of ocular immune privilege. J Immunol. 2012; 189: 1237–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ansel JC, Kaynard AH, Armstrong CA, Olerud J, Bunnett N, Payan D. Skin-nervous system interactions. J Invest Dermatol. 1996; 106: 198–204. [DOI] [PubMed] [Google Scholar]
- 42. Bowden JJ, Baluk P, Lefevre PM, Vigna SR, McDonald DM. Substance P (NK1) receptor immunoreactivity on endothelial cells of the rat tracheal mucosa. Am J Physiol. 1996; 270: L404–L414. [DOI] [PubMed] [Google Scholar]
- 43. Geiger B, Ayalon O. Cadherins. Annu Rev Cell Biol. 1992; 8: 307–332. [DOI] [PubMed] [Google Scholar]
- 44. Hermiston ML, Gordon JI.. In vivo analysis of cadherin function in the mouse intestinal epithelium: essential roles in adhesion, maintenance of differentiation, and regulation of programmed cell death. J Cell Biol. 1995; 129: 489–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tang A, Amagai M, Granger LG, Stanley JR, Udey MC. Adhesion of epidermal Langerhans cells to keratinocytes mediated by E-cadherin. Nature. 1993; 361: 82–85. [DOI] [PubMed] [Google Scholar]
- 46. Wilbanks GA, Streilein JW.. Studies on the induction of anterior chamber-associated immune deviation (ACAID). 1. Evidence that an antigen-specific, ACAID- inducing, cell-associated signal exists in the peripheral blood. J Immunol. 1991; 146: 2610–2617. [PubMed] [Google Scholar]
- 47. Higgins JM, Mandlebrot DA, Shaw SK, et al.. Direct and regulated interaction of integrin alphaEbeta7 with E-cadherin. J Cell Biol. 1998; 140: 197–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mizuno K, Altman NF, Clark AF, Streilein JW. Histopathologic analysis of experimental autoimmune uveitis attenuated by intracameral injection of S-antigen. Curr Eye Res. 1989; 8: 113–121. [DOI] [PubMed] [Google Scholar]
- 49. Ferguson TA, Hayashi JD, Kaplan HJ. Regulation of the systemic immune response by visible light and the eye. FASEB J. 1988; 2: 3017–3021. [DOI] [PubMed] [Google Scholar]
- 50. Ferguson TA, Mahendra SL, Hooper P, Kaplan HJ. The wavelength of light governing intraocular immune reactions. Invest Ophthalmol Vis Sci. 1992; 33: 1788–1795. [PubMed] [Google Scholar]
- 51. Foldenauer ME, McClellan SA, Barrett RP, Zhang Y, Hazlett LD. Substance P affects growth factors in Pseudomonas aeruginosa-infected mouse cornea. Cornea. 2012; 31: 1176–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hazlett LD, McClellan SA, Barrett RP, Liu J, Zhang Y, Lighvani S. Spantide I decreases type I cytokines, enhances IL-10, and reduces corneal perforation in susceptible mice after Pseudomonas aeruginosa infection. Invest Ophthalmol Vis Sci. 2007; 48: 797–807. [DOI] [PubMed] [Google Scholar]
- 53. McClellan SA, Zhang Y, Barrett RP, Hazlett LD. Substance P promotes susceptibility to Pseudomonas aeruginosa keratitis in resistant mice: anti-inflammatory mediators downregulated. Invest Ophthalmol Vis Sci. 2008; 49: 1502–1511. [DOI] [PubMed] [Google Scholar]
- 54. Twardy BS, Channappanavar R, Suvas S. Substance P in the corneal stroma regulates the severity of herpetic stromal keratitis lesions. Invest Ophthalmol Vis Sci. 2011; 52: 8604–8613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cohen JA, Edwards TN, Liu AW, et al.. Cutaneous TRPV1(+) neurons trigger protective innate type 17 anticipatory immunity. Cell. 2019; 178: 919–932.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Metcalf JF, Hamilton DS, Reichert RW. Herpetic keratitis in athymic (nude) mice. Infect Immun. 1979; 26: 1164–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]