

Abstract
Through whole-exome/genome sequencing, human geneticists identify rare variants that segregate with disease phenotypes. To asess if a specific variant may be pathogenic, one must query many databases to determine whether the gene of interest is linked to a genetic disease, whether the specific variant has been reported before, and what functional data is available in model organism databases that may provide clues about the gene’s function in human. MARRVEL (Model organism Aggregated Resources for Rare Variant ExpLoration) is a one-stop data collection tool for human genes and variants and their orthologous genes in seven model organisms including in mouse, rat, zebrafish, fruit fly, nematode worm, fission yeast and budding yeast. In this Protocol, we provide an overview of what MARRVEL can be used for and discuss how different data sets can be used to assess whether a variant of unknown significance (VUS) in a known disease -causing gene or a variant in a gene of uncertain significance (GUS) may be pathogenic. This protocol will guide a user through searching multiple human databases simultaneously starting with a human gene with or without a variant of interest. We also discuss how to utilize data from OMIM, ExAC/gnomAD, ClinVar, Geno2MP, DGV and DECHIPHER. Moreover, we illustrate how to interpret a list of ortholog candidate genes, expression patterns, and GO terms in model organisms associated with each human gene. Furthermore, we discuss the value protein structural domain annotations provided and explain how to use the multiple species protein alignment feature to assess whether a variant of interest affects an evolutionarily conserved domain or amino acid. Finally, we will discuss three different use-cases of this website. MARRVEL is an easily accessible open access website designed for both clinical and basic researchers and also serves as starting point to design experiments for functional studies.
Keywords: Human genomics, variant prioritization, model organisms, genetics, rare and undiagnosed diseases, functional genomics, database integration, translational research, medical diagnosis, variant of unknown significance (VUS), gene of uncertain significance (GUS), web-based tool
SUMMARY:
Here, we present a protocol to access and analyze many human and model organism databases efficiently. This protocol demonstrates the use of MARRVEL to analyze human disease candidate variants identified from next-generation sequencing efforts.
INTRODUCTION:
The use of next generation sequencing technology is expanding in both research and clinical genetic laboratories1. Whole-exome (WES) and whole-genome sequencing (WGS) analyses reveal numerous rare variants of unknown significance (VUS) in known disease -causing genes as well as variants in genes that are yet to be associated with a Mendelian disease (GUS: genes of uncertain significance). Presented with a list of genes and variants in a clinical sequence report, medical geneticists must manually visit multiple online resources to obtain more information to assess which variant maybe responsible for a certain phenotype seen in the patient of interest. This process is time consuming and its efficacy is highly dependent on the expertise of the individual. Although several guideline papers have been published2,3, interpretation of WES and WGS requires manual curation since there is yet to be a standardized methodology for variant analysis. For the interpretation of VUS, knowledge on the previously reported genotype-phenotype relationship, mode of inheritance, and allele frequencies in the general population become valuable. In addition, knowledge on whether the variant affects a critical protein domain or an evolutionarily conserved residue may increase or decrease the likelihood of pathogenicity. To gather all of this information, one typically needs to navigate through 10–20 human and model organism databases since the information is scattered through the World Wide Web.
Similarly, model organism scientists who work on specific genes and pathways are often interested in connecting their findings to human disease mechanisms and wish to take advantage of the knowledge that are being generated in the human genomics field. However, due to rapid expansion and evolution of data sets regarding the human genome, it has been challenging to identify databases that provide useful information. In addition, since most model organism databases are designed for researchers who work with the specific organism on a daily basis, it is very difficult, for example, for a mouse researcher to search for specific information in a Drosophila database and vice versa. Similar to variant interpretation searches performed by medical geneticists, identifying useful human and other model organism information is time consuming and heavily dependent on the background of the model organism researcher. MARRVEL (Model organism Aggregated Resources for Rare Variant ExpLoration)4 is a tool designed for both groups of users to streamline their workflow.
MARRVEL (http://marrvel.org) was designed as a centralized search engine that collects data systematically in an efficient and consistent manner for clinicians and researchers. With information from 20 or more publicly available databases, this program allows users to quickly gather information and access a large number of human and model organism databases without reiterative searches. The search result pages also contain hyperlinks to the original sources of information, allowing individuals to access the raw data and gather additional information provided by the sources.
In contrast to many of the variant prioritization tools that require large sequencing data input in the form of VCF or BAM files and installations of often proprietary/commercial software, MARRVEL operates on any web-browser. It can be used at no cost and compatible with portable devices (e.g. smartphones, tablets) as long as one is connected to the internet. We chose this format since many clinicians and researchers typically need to search one or a few genes and variants at a time. Note that we are developing batch-download and API (application programming interface) features for MARRVEL to eventually allow users to curate hundreds of genes and variants at a time through customized query tools if necessary.
Due to the wide range of applications, this Protocol will describe a broadly encompassing approach on how to navigate through different datasets that MARRVEL displays. More targeted examples that are tailored towards a specific users’ needs will be described in Representative Results. It is important to note that the output of MARRVEL still requires a certain level of background knowledge in either human genetics or model organisms to extract valuable information. We refer the readers to the table that lists primary papers that describe the function of each of the original databases that are curated by MARRVEL (Table 1).
Table 1. List of Data Sources for MARRVEL.
All databases where MARRVEL obtains data from are listed in this table. For each database, we list the type of database, URL/Link, rationale for including in MARRVEL, and primary references.
| Type of database | Name of Database | URL/Link to Database | Rationale for Inclusion | Reference |
|---|---|---|---|---|
| Human Genetics | OMIM | https://omim.org | The three main pieces of information that we draw from OMIM are: gene function, associated phenotypes, and reported alleles. It is helpful to know if a gene is a part of a known Mendelian phenotype whose molecular basis is known (#entries). Genes without this knowledge are candidates for novel gene discovery and for genes that are this category if the patient’s phenotype does not match the reported disease and phenotype as well as those of the patients in the literature, then this increases the opportunity to provide a phenotypic expansion. | PMID: 28654725 |
| Human Genetics | ExAC | exac.broadinstitute.org/ | ExAC contains more than 60,000 exomes and is, other than gnomAD (http://gnomad.broadinstitute.org/), the largest public collection of exomes that have been selected against individuals with severe early-onset Mendelian phenotypes. For MARRVEL’s purposes, ExAC serves as the best control population minor allele frequency. We are interested in two sets of outputs from ExAC. The first output is the gene-centric overview of the expected versus observed number of missense and loss of function alleles. A metric called pLI (probability of Loss of function intolerance) ranges between 0 and 1 is likely related to how essential both copies of a gene are before reproductive age. A pLI score of 1 means that this gene is very intolerant of any loss of function variants and is under selective constraint. The second output is data from ExAC that pertains to the specific variant. | PMID: 27535533 |
| Human Genetics | gnomAD | http://gnomad.broadinstitute.org | gnomAd contains a total of 123,136 exome sequences and 15,496 whole-genome sequences from unrelated individuals sequenced as part of various disease-specific and population genetic studies. In MARRVEL we display the population frequencies that pertains to specific variant. | PMID: 27535533 |
| Human Genetics | ClinVar | https://www.ncbi.nlm.nih.gov/clinvar/ | ClinVar is a public archive of reports of the relationships among human variations and phenotypes, with supporting evidence. Variants with interpretations reported by researchers and clinicians are valuable for analyzing how likely a variant is pathogenic. | PMID: 29165669 |
| Human Genetics | Geno2MP | http://geno2mp.gs.washington.edu/Geno2MP/ | Geno2MP is a collection of samples from the University of Washington Center for Mendelian Genetics. It contains ~9,650 exomes of affected individuals and unaffected relatives. This database links the phenotypic as well as mode of inheritance information to specific alleles. For phenotype, we focus on comparing the affected organ system of the patient to the affected individuals in Geno2MP. A match in allele, mode of inheritance, and phenotype provides an increased probability that the variant likely pathogenic. However, due to small sample size a negative association does not necessarily decrease a variant’s pathogenic priority. | http://geno2mp.gs.washington.edu/Geno2MP/#/ |
| Human Genetics | DGV | http://dgv.tcag.ca/dgv/app/home | To our knowledge, DGV is the largest public-access collection of structural variants from more than 54,000 individuals. The database includes samples of reportedly healthy individuals, at the time of ascertainment, from up to 72 different studies. Possible limitations to this data include variation in source and method of the data acquired the lack of information regarding incomplete penetrance of pathogenic CNVs, and whether individuals will develop associated diseases subsequent to data collection. | PMID: 24174537 |
| Human Genetics | DECIPHER | https://decipher.sanger.ac.uk/ | The data displayed on MARRVEL includes common variants from the control population. The data displayed includes structural variants that cover the genomic location of the input variant. DECIPHER also contains variant and phenotypic information for affected individuals but can only be accessed on their database. | PMID: 19344873 |
| Integration | DIOPT | https://www.flyrnai.org/cgi-bin/DRSC_orthologs.pl | DIOPT provided multiple protein sequence alignment of the best predicted orthologs in six model organisms against the protein sequence of the human gene of interest. The alignment will provide information on the conservation of specific amino acids as well as functional protein domains. | PMID: 21880147 |
| Gene Function | GO Central | http://www.geneontology.org/ | MARRVEL displays only gene ontology terms (Molecular Function, Cellular Component, and Biological Process) derived from experimental evidence for each gene. They are filtered by “experimental evidence codes” and GO terms based on “computational analysis evidence codes” and “electronic annotation evidence codes” (predictions) are avoided. | PMID: 10802651, 25428369 |
| Model Organism | SGD | https://www.yeastgenome.org/ | We collected data from multiple model organism databases and provide a summary of the biological and genetic functions of the predicted orthologs derived by DIOPT. | PMID: 22110037 |
| Model Organism | PomBae | https://www.pombase.org/ | PMID:22039153 | |
| Model Organism | WormBase | http://wormbase.org | PMID:26578572 | |
| Model Organism | FlyBase | http://flybase.org | PMID:26467478 | |
| Model Organism | ZFIN | https://zfin.org/ | PMID:26097180 | |
| Model Organism | MGI | http://www.informatics.jax.org/ | PMID:25348401 | |
| Model Organism | RGD | https://rgd.mcw.edu/ | PMID:25355511 | |
| Model Organism | GTEx | https://gtexportal.org/home/ | MARRVEL displays both mRNA and protein expression pattern in human tissues of each gene. The expression pattern can add insight into the phenotypes observed in patients and/or model organisms. | PMID: 29019975, 23715323 |
| Model Organism | The Human Protein Atlas | https://www.proteinatlas.org/ | PMID: 21752111 | |
| Gene Function | IMPC | http://www.mousephenotype.org/ | MARRVEL provides a link to the mouse gene page on IMPC. If there has been a knock out mouse made by IMPC, an exhaustive list of assays and their results are made available publicly and can provide insight into the phenotype when a gene is lost. | PMID: 27626380 |
| Gene Function | Monarch Initiative | https://monarchinitiative.org/ | MARRVEL provides a link to the Phenogrid of a human gene on Monarch Initiative. This grid provides comparisons between the phenotype of model organisms and known human diseases. | PMID: 27899636 |
| Integration | Ensembl | https://useast.ensembl.org/index.html | Ensembl gene IDs are used to link the different databases. | PMID: 29155950 |
| Integration | HGNC | https://www.genenames.org/ | HGNC official gene symbols are used for MARRVEL searches. | PMID: 27799471 |
| Integration | Mutalyzer | https://mutalyzer.nl/ | MARRVEL uses Mutalyzer’s API to convert different variant nomenclatures to genomic location. | PMID: 18000842 |
PROTOCOL:
The following Protocol is divided into three sections: (1) How to begin a search, (2) how to interpret MARRVEL human genetics outputs, and (3) how to make use of model organism data in MARRVEL. In the Representative Results section, more focused and specific approaches are described.
MARRVEL is being actively updated so please refer to the current website’s FAQ page for details about data sources. We strongly recommend the users of MARRVEL to sign up in order to receive update notifications through the e-mail submission form located at the bottom of the MARREL home page (http://marrvel.org/)
1. How to begin a search
1.1 For human gene and variant-based search go to 1.1.1.–1.1.2., for human gene-based search (no variant input), go to 1.2., and for model organism gene-based search, refer to 1.3.1.–1.3.2.
1.1.1. Go to the home page of MARRVEL4 at http://marrvel.org/. Start by entering a human gene symbol. Candidate gene names should be listed below the input box with each character entry. If the search comes back negative, make sure the gene symbol used is up to date using the HUGO Gene Nomenclature Committee website5 (HGNC; https://www.genenames.org/).
1.1.2. Enter a human variant. The search bar is compatible with two types of variant nomenclature: genome location similar to how variants are displayed on ExAC and GnomAD6 and transcript-based nomenclature according to HGVS guidelines. Examples of such formats are shown in grey text within the search box. For genomic location nomenclature, use the coordinates according to hg19/GRCh37. Proceed to step 2.
NOTE: If a search returns an error, the most common problems are either the gene symbol is not up to date or the variant nomenclature is incorrect. In those cases, the HGNC (https://www.genenames.org/), Mutalyzer7 (https://www.mutalyzer.nl/), and TransVar8 (https://bioinformatics.mdanderson.org/transvar/) websites are great resources to correct the error. HGNC provides official gene symbols and their aliases for all human genes. When users still encounter error messages after confirming the gene name is up to date, one can use Mutalyzer and TransVar to check and convert variant nomenclature. In some situations such as a very recent gene symbol change in HGNC, MARRVEL may not provide the correct information due a lag in data update. In this circumstance, try using a synonym for the gene and please contact the MARRVEL operating team using the “Feedback” tab so we can update our source data.
1.2. Enter a human gene symbol and leave the human variant search bar blank. If an error is encountered, go to HGNC (https://www.genenames.org/) to check for official gene symbol or try an older gene symbol.
1.3.1 Click on “Model Organisms Search” tab on the top banner (Figure 1) or go to http://marrvel.org/model. Select the model organism of choice and enter a model organism gene symbol. Click on the gene symbol as the name is autocompleted and then click search. If the search result is negative, check the official gene symbol that is used in model organism databases (Table 1).
Figure 1. Representative output from a MARRVEL search.

This specific example is showing a gene/variant search for “TBX2/17:59477596 G>A” (http://marrvel.org/search/pair/TBX2/17:59477596%20G%3EA). Sidebar on the left supports navigations through the data output. Note the “external link” signs here provide links to the appropriate pages of the UCSC genome browser (https://genome.ucsc.edu/). The tabs on the top allow one to perform model organism gene based searches, obtain additional information about MARRVEL and provide user feedbacks. The ‘Search Results’ panels display gene and variant information from the sources indicated in the image.
NOTE: If the search result is still negative, it is possible that there are no good predicted orthologs for the gene of interest. In this scenario, users should access DIOPT (DRSC Integrative Ortholog Prediction Tool, https://www.flyrnai.org/cgi-bin/DRSC_orthologs.pl) and HCOP (https://www.genenames.org/tools/hcop/) to assess if this is the case. DIOPT is an ortholog prediction search engine run by the DRSC (Drosophila RNAi Screening Center) and HCOP is a similar suite developed by HGNC. Additional searches using BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi) may allow users to find orthologs that may be missed by prediction algorithms used in DIOPT and HCOP.
1.3.2. Click on the “MARRVEL it” bottom for the predicted human ortholog of choice. The “DIOPT score9” and “Best score from Human gene to model organism?” helps the selection of the human gene. Proceed to Step 2.
NOTE: “DIOPT score”9 (https://www.flyrnai.org/cgi-bin/DRSC_orthologs.pl) is a value of how many ortholog prediction algorithms predict a pair of genes in two organisms to be orthologous to one another. For more information about these values and the specific algorithms used to calculate this score, refer to Hu et al9. When “Best score from Human gene to model organism?” is “Yes,” it indicates that the human gene is more likely to be a true human orthologs of the gene of interest but there could be exceptions, especially when multiple human genes are orthologous to multiple model organism genes due to gene duplication events during evolution. If the gene of interest is a member of a complex gene family that have undergone divergent evolution in multiple species, users should identify a publication that has performed an extensive phylogenetic analysis of the gene family of interest to identify the most likely ortholog candidate gene.
2. How to interpret MARRVEL human genetics outputs for a gene and variant search
On the results page, there are seven human databases that are displayed (Table 1, Figure 1). For each output box, there is an “external link” button (small box with a diagonal arrow)” on the upper right hand corner that will link to the original database for more details.
2.1. OMIM (Online Mendelian Inheritance in Man, https://www.omim.org/)10 is the first database that is displayed. OMIM is a manually curated database that aggregates and summarizes information on genetic diseases and traits in human.
2.1.1. Use the “Human Gene Description” box from OMIM for a short summary of what is known about the gene and gene product.
2.1.2. Use the “Gene-Phenotype Relationships” box to determine if this gene is a known disease-causing gene or not. This box provides manually curated known disease or phenotype associations with the gene of interest.
2.1.3. Use the “Reported Alleles From OMIM” box to get a list of pathogenic variants curated by OMIM.
NOTE: Since manual curation of a publication regarding a new disease gene discovery is necessary for any gene-disease association to appear in OMIM, some time lag and/or missed publications may lead to misconception. We recommend that users perform PubMed (https://www.ncbi.nlm.nih.gov/pubmed/) searches to look into recent literature as well (See 4.1.2.). For additional information curated in OMIM, refer to Amberger et al 2017 and 201810,11.
2.2. ExAC (Exome Aggregation Consortium, http://exac.broadinstitute.org/)6 and gnomAD (genome Aggregation Database, http://gnomad.broadinstitute.org/) are large population genomics databases based on WES and WGS of people who are selected to exclude severe pediatric diseases. ExAC contains ~60,000 WES whereas gnomAD contains ~120,000 WES and ~15,000 WGS. Both ExAC and gnomAD can be used as a “control population database”, especially for severe pediatric disorders, but its interpretation requires some degree of caution.
NOTE: In general, gnomAD can be considered as an updated and expanded version of ExAC since most cohorts that are included in ExAC is also included in gnomAD. However since there are some exceptions (see cohort information in http://exac.broadinstitute.org/about and http://gnomad.broadinstitute.org/about, respectively), MARRVEL displays data from both sources.
2.2.1. Use the “Control Population Gene Summary” box to obtain gene-level statistics such as probability of finding loss of function (LOF) alleles in the general population. This is called the pLI (probability of LOF Intolerance) score in ExAC and can be used to infer how likely a single copy of a LOF allele for a specific gene may cause a dominant disease through haploinsufficient mechanisms.
NOTE: Looking at the pLI score of a gene has value, especially when dealing with dominant disorders that present as severe pediatric diseases associated with de novo variants. If a gene has a pLI score of 0.00, it means it is highly tolerant of LOF variants thus the gene unlikely cause a disease via a dominant haploinsufficiency mechanism. This does not, however, necessarily rule out other dominant gain of function (GOF) or dominant negative mediated mechanisms may cause disease. In addition, genes that cause recessive diseases may have low pLI scores since careers are expected to be found in the general population. On the other hand if a gene has a pLI score of 1.00, it is possible that the loss of one copy of this gene is detrimental for human health. Additional searches in websites such as DOMINO ( https://wwwfbm.unil.ch/domino/) may also be used in combination to assess the likelihood of a variant in a specific gene causing a dominant disorder.
2.2.2. Use the next two boxes to obtain the allele frequencies of the variant of interest in ExAC and gnomAD, respectively. This may help interpret whether or not the variant may be pathogenic depending on if the patient has dominant or recessive disease. This box will only be displayed when the user inputs variant information when initiating the search.
NOTE: If one hypothesizes a recessive disease scenario and the pLI score of the gene of interest is low, one should pay attention to the allele frequency listed here. Some geneticists may establish a cut-off point of 0.005 to 0.0001 as the maximum allele frequency for pathogenic variants that can cause severe recessively inherited disease2. On the other hand, if one hypothesizes a dominant disease scenario, it is less likely to find the identical or similar variant in a “control” population. Again, this requires caution because individuals with late-onset disorders, diseases with mild presentation, psychiatric disorders or diseases not screened by the ExAC/gnomAD researchers may be still included and the variant may still be a dominant pathogenic variant. Also, there have been some instances of variants linked to pediatric conditions found in a few individuals in these databases12–14, potentially due to incomplete penetrance or somatic mosaicism13,15,16. In addition, although ExAC and gnomAD will display variants that are found in a homozygous state, it will not indicate whether any of the variants are found in a compound heterozygous state. Finally, some variants found in these databases are tagged as “low confidence” due to technical challenges in sequencing (e.g. low sequence coverage, repetitive sequence). To look more carefully into these data sets, users are recommended to use the “external link” button to visit the original ExAC and gnomAD websites to gain additional information.
2.3. Geno2MP (Genotype to Mendelian Phenotype Browser, http://geno2mp.gs.washington.edu/Geno2MP/) is a collection of WES-based data from the University of Washington Center for Mendelian Genetics. It contains about 9,600 exomes (as of 1/18/2019) of affected individuals and unaffected relatives with some phenotypic descriptions (Figure 1).
2.3.1. Use the “Disease population” box to obtain the allele frequency of the variant of interest in this cohort.
2.3.2. Use the “Gene-Phenotype Relationships” box to obtain HPO (human phenotype ontology)17 terms for the individuals with the variant of interest. This is one of many ways for one to look for patients that may have the same disease.
NOTE: If a gene of interest is suspected to be associated with a patient’s disease and there are matches found in Geno2MP, additional important information may be present in the data source beyond what is displayed. Click the “external link” button to the gene-specific page on Geno2MP, filter for mutations that are similar to those of the patient (e.g. missense, LOF), and carefully review the lists of variants. Take note of the variants with high CADD18 scores and click into the HPO profiles. For example, CADD scores higher than 20 are within the top 1% of all variants predicted to be deleterious, CADD scores that are higher than 10 are within the top 10%. HPO terms provides standardized description of human phenotypes. Here, make sure to check if the variant was identified in an affected individual or in a relative. If variants are found in patients that are affected in the same organ system as your patient, consider using the e-mail form to contact the physician that submitted these cases to Geno2MP using the feature provided on the Geno2MP website. Note that not all physicians respond to such queries, so one should explore other avenues of patient matchmaking. Other ways to gather a cohort of patients affected by the same diseases is to use tools such as GeneMatcher19 (https://www.genematcher.org/) and other databases that are part of the Matchmaker Exchange19,20 (https://www.matchmakerexchange.org/). See accompanying JoVE article for more information on matchmaking21.
2.4. ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/)22 is a database supported by the National Institutes of Health (NIH) where researchers and clinicians submit variants with or without determination of pathogenicity. This may include single nucleotide variants (SNV), small indels and larger copy number variations (CNV).
2.4.1. Use the top row to review a summary of the number of each type of variants reported in ClinVar (Figure 1).
2.4.2. Check the list of variants below in the box “Reported Alleles From ClinVar.” Note that if a variant was included in the initial search, the highlighted variants in teal are all variants that include the genomic location of the variant of interest [including large CNVs, which are often labeled as; genomic coordinate…x1 (deletion) and …x3 (duplication)].
2.5. DGV23 (Database of Genomic Variants, http://dgv.tcag.ca/dgv/app/home) and DECIPHER24 (DatabasE of genomiC varIation and Phenotype in Humans using Ensembl Resources, https://decipher.sanger.ac.uk/) are both collections of CNVs. DGV is the largest public-access collection of structural variants from more than 54,000 individuals. This database includes samples of reportedly healthy individuals, at the time of ascertainment, from up to 72 different studies. Similarly, the data displayed from DECIPHER includes common variants from the control population.
NOTE : Since MARRVEL does not have permission to display patient derived data from DECIPHIER, users are encouraged to directly visit the DECIPHER website to access potentially pathogenic CNV information.
2.5.1. Use the “Copy Number Variation In Control Population (DGV Database)” box to obtain variants that contain the gene of interest. Information such as the size, subtype, and reference of the copy number variation can be found in the same box.
2.5.2. Use the “Common Copy Number Variants (DECIPHER Database)” box to obtain variants that contain the genomic location of the variant of interest. This information may help determine if the gene is duplicated or deleted in “control” individuals.
NOTE: If the gene of interest is deleted in many individuals in the control population, it means that this gene is likely to be highly tolerant of LOF variants. Similar to low pLI scores, this suggests that a single copy loss of this gene is less likely to cause a severe disease via a haploinsufficiency mechanism. This does not, however, necessarily rule out other dominant gain of function or dominant negative mechanisms (e.g. antimorphic, hypermorphic and neomorphic alleles) caused by specific missense and truncation alleles. Possible limitations to these data include variation in source and method of the data acquired, lack of information regarding incomplete penetrance of pathogenic CNVs, and whether individuals developed certain diseases subsequent to data collection.
3. How to use model organism data in MARRVEL
3.1. The “Gene Function Table” provides the following information for eight model organism including human (human, rat, mouse, zebrafish, Drosophila, C elegans, budding yeast and fission yeast):
3.1.1. Gene name: Each gene name is hyperlinked to gene pages on respective model organism databases. Click on these links to find out more about the phenotypic information and resources available for each model organism. For example on FlyBase25 (http://flybase.org/), there will be a list of all alleles that have been generated, their respective phenotypes and the availability of each allele from public stock centers.
3.1.2. PubMed link: Click on the PubMed link to go to a list of publications that relates to the gene of interest in each organism. Without using these links, searching for the human gene directly in PubMed may lead to missing some publications that used an old gene alias to refer to the human gene. Similarly, model organism gene names may have fluctuated historically.
3.1.3. DIOPT9 score: Check this column for a score of how many ortholog prediction algorithms predict the gene is likely to be an ortholog of the human gene of interest. One may use a DIOPT score of 3 or above as a reasonable cut-off to identify solid ortholog candidates. However, there are cases where genuine orthologs only have a DIOPT score of 1 due to limited homology. At the top of the gene function table, un-check the “Show only best DIOPT score gene” box to display all candidates that typically include homologous genes that are not necessarily orthologs.
3.1.4. Expression: Check this column for the list of the tissues where the gene or protein of interest has been reported to be expressed in human or model organism databases. Human gene and protein expression data are from GTEx26 (https://gtexportal.org/) and Human Protein Atlas27 (https://www.proteinatlas.org/), respectively. Some have a button with a pop-up links, such as for human and for fly that display the expression pattern using a heat map, whereas others are hyperlinked to respective model organism databases pages.
3.1.5. Gene Ontology28 (GO) terms – these are filtered by “experimental evidence codes” and obtained from respective human or model organism databases. GO terms based on “computational analysis evidence codes” and “electronic annotation evidence codes” (predictions) are not displayed. Please visit each model organism website to gather these information if necessary.
3.1.6. Other links such as Monarch Initiative29 (https://monarchinitiative.org/) and IMPC30 (http://www.mousephenotype.org/): The “Monarch Initiative” hyperlink brings the user to the Phenogrid page for the specific human gene, a chart that provides a quick comparison between the phenotypes associated with the gene of interest to known human diseases and model organism mutants that have phenotypic overlaps. If a mouse gene has a knockout mouse made or planned by the International Mouse Phenotyping Consortium (IMPC), the “IMPC” links to the page that details the phenotype of the knockout mouse and its availability from public stock centers.
3.2. Human Protein Domains
Use the “human gene protein domains” box to obtain predicted protein domains of the human gene. The data are derived from DIOPT, which uses Pfam (https://pfam.xfam.org/) and CCD (Conserved Domains Database, https://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml). A single residue maybe annotated more than once due to some overlap in domains annotated in the two sources.
3.3. Use the “Multiple Protein Alignment” box to obtain the amino acid multiple alignment generated by DIOPT9 which includes human (hs), rat (rn), mouse (mm), zebrafish (dr), fruit fly (dm), worm (ce), and yeasts (sc and sp). To highlight the amino acid of interest, scroll down to the bottom of the box and enter the amino acid numbers below and the amino acids of interest will be highlighted in teal. The alignment is provided by DIOPT and uses MAFFT aligner (Multiple alignment program for amino acid or nucleotide sequences, https://mafft.cbrc.jp/alignment/software/31).
NOTE: If the amino acid that is highlighted based on the number is not the one expected, it may be due to different splicing isoforms used for the alignment. In principal, DIOPT uses the longest isoform to display in this box. Also, for segments of genes that are not well conserved, alignment of multi-species sequences using default parameters may not be optimal. We recommend using other websites and software like Clustal Omega and ClustalW/X (http://www.clustal.org/)32 to optimize the alignment parameters and matrices accordingly.
REPRESENTATIVE RESULTS:
Human geneticists and model organism scientists each use MARRVEL in distinct ways, each with different desired outcomes. Below are three vignettes of possible uses for MARRVEL.
1. Evaluating pathogenicity of a variant in a dominant disease
Most of the users that visit MARRVEL use this website to analyze the likelihood that a rare human variant may cause a certain disease. For example, a missense (17:59477596 G>A, p.R20Q) variant in TBX2 was found to segregate in an autosomal dominant manner in a small family with dysmorphic features and cleft palate, cardiac defects, skeletal and digit abnormalities, thyroid-related phenotypes and immune defects12. The mother and two children affected with these symptoms carried the variant, whereas the father did not. The 9 year old son had the most severe phenotype, whereas the 36 year old mother and the 6 year old daughter had milder forms of this disease. To assess whether this variant is likely pathogenic, one can start a MARRVEL search by entering the gene and variants on the starting page on http://MARRVEL.org. Note that the variant search bar requires the removal of “Chr” in front of the variant if this is listed in the original clinical report to indicate “Chromosome”. At the time of the original study, the results page showed that there is no OMIM phenotype associated with this gene, and this variant is found only once in gnomAD but not in ExAC, ClinVar, or Geno 2MP. One may think this identification of one individual may be evidence against p.R20Q being a pathogenic variant, but it is important to note that the mother of the family exhibited mild form of the disease. A variant found in 1/~150,000 individual is indeed a very rare variant and the identification of an individual with the identical variant maybe explained by reduced expressivity or penetrance. In the Gene Function table, it is often helpful to check if the gene is expressed in relevant tissues in humans (via GTEx and Protein Atlas) in reference to the phenotypes of the patient. In this case, the expression pattern matches since the patient has phenotypes in multiple tissues and the gene is also widely expressed, including cardiac, and immune related organs.
Based on model organism information displayed in MARRVEL, one can quickly see that the gene is conserved from C. elegans and Drosophila to human and the amino acid of interest, p.R20 is also highly conserved throughout evolution as shown in Figure 2 (note that rat Tbx2 does not align well in this region, likely due to the transcript that is used for alignment). Phenotypic information in mouse and zebrafish indicates that this gene affects development or function of a number of tissues including cardiovascular system, craniofacial/palate and digits. In sum, these data suggest that this variant is possibly pathogenic and further functional study is valuable. Considering that the gene and variant is conserved in organisms like C. elegans and Drosophila, functional studies in invertebrate animals will be faster and cheaper compared to performing the same experiment in vertebrate model organisms such as zebrafish, mouse and rat. Please see the accompanying article by Harnish et al.21 regarding how we designed and performed functional assays for this case12. The involvement of this gene/variant in this family’s disease was further strengthened by identification of unrelated 8 year old male patient with overlapping phenotypes with a de novo missense variant in the same gene using GeneMatcher. The variants in the two families were both found to be functional using experiments in Drosophila, further supporting the pathogenicity of the rare variants in TBX2. The disease has recently been curated as ‘Vertebral anomalies and variable Endocrine and T-cell Dysfunction (VETD, OMIM #618223)’ in OMIM.
Figure 2. Summary of the model organism ortholog table and multi-species alignment for TBX2.
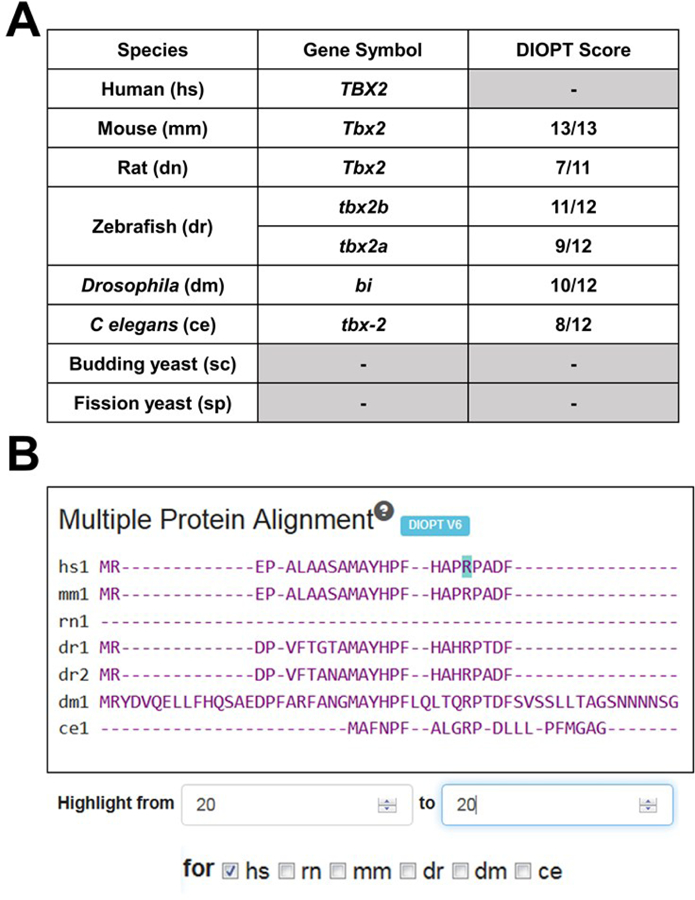
A) MARRVEL selects the top ortholog candidate for each species based on the DIOPT tool. For example, a DIOPT score of 10/12 shown for the Drosophila bi gene means 10 out of 12 orthology prediction programs used by DIOPT predicted that bi is the most likely fly ortholog of human TBX2. Since 25% of genes are duplicated in zebrafish compared to human, MARRVEL displays two paralogous genes (in this case tbx2a and tbx2b) when this is applicable. B) Snapshot of the multi-species alignment window. By selecting a specific organism [in this case human (hs)] and entering the amino acid of interest, one can highlight the specific amino acid in teal. In this example, p.R20 of human TBX2 seems to be conserved in mouse (mm1), both zebrafish orthologs (dr1 and dr2), Drosophila (dm1) and C. elegans (ce1). Rat Tbx2 does not seem to align well compared to other species, most likely due to the isoform used by the DIOPT to perform the multi-species alignment.
2. Evaluating pathogenicity of a variant in a recessive disease
There are significant differences between analyzing human variants in dominant and recessive diseases. For example, pLI score, minor allele frequency, and presence of deletions in the control population become less important because two alleles are necessary to reveal any phenotype.
One example of analysis of a recessive disease is detailed in Yoon et al33 and Wang et al4 which is summarized here. A 15-year-old girl exhibited developmental delay, microcephaly, ataxia, motor impairment, hypotonia, language impairments, brain abnormalities, and hypoplasia of the corpus callosum33. The proband, her unaffected parents, and an unaffected sibling received WES. After filtering for variants that were both unique to the proband and rare in the population, variants in 13 different genes remained. Manual filtering and analysis of the 13 candidates by following the protocol described here resulted in the prioritization of one specific variant in OGDHL as a good candidate for functional studies. The key pieces of information that led to prioritizing p.S778L in OGDHL (10:50946295 G>A) over other variants include: (1) no previous disease association in OMIM, (2) variant not found in control populations, (3) gene ontology associated with microtubule and mitochondria, two systems that have many links to neurological disorders34,35, (4) highly expressed in human cerebellum, a tissue severely affected in this patient, and (5) the variant of interest affecting a highly conserved amino acid (from yeast to human) and located within the catalytic domain4. pLI score for this gene is 0.00 but this doesn’t affect the prioritization of this variant/gene for this case since we are suspecting a recessive mode of inheritance and that carriers of deleterious variants in this gene can present in the general population.
Model organism studies performed in parallel showed that loss of Ogdh ( also referred to as Nc73EF), the Drosophila ortholog of OGDHL, in the nervous system exhibits a neurodegenerative phenotype consistent with the proband’s neurological disorder33. Functional studies in Drosophila showed that the variant of interest (p.S778L) affects protein function, making this a strong candidate gene for this disease. Since then, this information about a potential pathogenic variant in OGDHL linked to a novel neurological disorder has been incorporated into OMIM (https://www.omim.org/entry/617513) very recently but have not yet been assigned a disease-phenotype number because only one case has been reported as of January 2019.
3. Is the human ortholog of a model organism gene of interest associated with genetic diseases?
Many model organism researcher may be interested to see whether the human ortholog of their gene of interest may have links to genetic diseases. In this example, we will search whether the human ortholog(s) of the fly Notch (N) gene has any relevance to genetic diseases. To do this, we will start with performing a “Model Organisms Search (1.3.1.–1.3.2.)” and select “Drosophila melanogaster” as the species name and “N” as the model organism gene name. The four predicted human orthologs for this fly gene will be displayed in the results window as NOTCH1, NOTCH2, NOTCH3, and NOTCH4. The four genes have different DIOPT scores (10/12 for NOTCH1, 8/12 for NOTCH2 and NOTCH3, 5/12 for NOTCH4) due to the degree of homology between fly N and each human gene. Considering the “Best score from Human gene to Fly” is listed as “Yes” for all four genes, the reverse search from each human gene picks up the fly N gene as the most likely ortholog candidate. Indeed, the four human NOTCH genes are thought to have arisen from a single Notch gene during the two rounds of whole genome duplication events that happened in the vertebrate lineage after splitting from the invertebrate lineage36. By clicking the “MARRVEL it” buttons for each human gene, one can obtain the human gene-based outputs for NOTCH1-4. On the results page of each gene, the top boxes for OMIM indicate that while NOTCH1, 2, and 3 are associated with genetic diseases, NOTCH4 is currently not associated with any human diseases. Note that there have been debates on whether variants in NOTCH4 are associated with schizophrenia based on genome -wide association studies (GWAS)37,38. Since OMIM generally does not curate GWAS data with some exceptions (e.g. APOE, PTPN22), this information is not available from the OMIM window. Similarly, since OMIM does not generally curate cancer-associated somatic mutation information, information on whether somatic mutations in these genes are associated with certain cancer types will not be listed with a few exceptions (e.g. TP53, RB1, BRCA1). By clicking the “PubMed” or “Monarch” box, one can identify some disease related papers that are not curated in OMIM.
DISCUSSION:
Critical steps in this protocol include the initial input (Steps 1.1–1.3) and subsequent interpretation of the output. The most common reason why search results are negative is because of the many ways that a gene and/or variant can be described. While MARRVEL is updated on a scheduled basis, these updates may cause disconnects between the different databases that MARRVEL links to. Thus, the first step in troubleshooting is invariably checking to see if alternative names of the gene or variant will lead to a successful search result. If it still cannot be resolved, please send a message to the development team using the feedback form in http://marrvel.org/message.
One limitation to MARRVEL is that it does not yet include all the useful databases necessary for gene and variant analysis. For example, pathogenicity prediction algorithms such as CADD18 are not currently provided. Similarly, protein structure information and protein-protein interaction information that may also provide structural and functional links to known disease causing variants in genes are not currently displayed in MARRVEL. In our next major update, we plan to integrate this information into MARRVEL, in addition to incorporating more phenotypic information from model organism websites, IMPC, Monarch Initiative and Alliance of Genome Resources (AGR, https://www.alliancegenome.org/). Since MARRVEL was designed to facilitate rare disease research, the program currently focuses on germline variants and does not provide access to somatic variant information. No cancer genetics related databases are integrated as of publication of this protocol. As MARRVEL is actively being developed and upgraded, we highly appreciate feedback, and strongly encourage the existing users to sign up for newsletters on http://marrvel.org/message for any future additional databases that become integrated.
Although data from MARRVEL can be used to prioritize variants that maybe pathogenic. However in order to demonstrate pathogenicity, one will need to identify other patients with similar genotypes and phenotypes, or perform functional studies to provide solid evidence that the variant of interest have functional consequences that are relevant to the disease condition. For more information on additional information outside of MARRVEL that maybe useful to judge if a variant is worth experimentally investigating in model organism, please refer to the accompanying JoVE article Harnish et al21. In order to take the next steps in using model organisms to study human variants, human geneticists and model organism researchers must be able to connect and collaborate. GeneMatcher and other genomic consortia that are part of the Matchmaker Exchange consortium are resources that facilitate this next step. If the users reside in Canada, one can also register in the Rare Disease Models and Mechanisms Network (RDMM, http://www.rare-diseases-catalyst-network.ca/) to identify clinicians and/or model organism researchers that are willing to collaborate (REF). Japan (J-RDMM, https://irudbeyond.nig.ac.jp/en/index.html), Europe (RDMM-Europe, http://solve-rd.eu/rdmm-europe/), and Australia (Australian Functional Genomics Network: https://www.functionalgenomics.org.au/) have recently adopted the Canadian RDMM model to facilitate similar collaborations within their countries/regions. Furthermore, by using tools such as BioLitMine (https://www.flyrnai.org/tools/biolitmine/web/) one can search for potential collaborators among Principal Investigators who have previously worked on the gene of interest.
Lastly, in addition to MARRVEL, there are a number of other cross-species datamining tools available including Gene2Function39 (http://www.gene2function.org/), Monarch Initiative29 (https://monarchinitiative.org/) and Alliance of Genome Resources (AGR, https://www.alliancegenome.org/). While Gene2Function provides access to cross-species data and Monarch Initiative provide phenotypic comparisons, MARRVEL has a larger emphasis on human variants and linking human genomic data with model organisms. AGR is an initiative that involves six model organism databases and the Gene Ontology Consortium that integrates data from different database in a uniform way to increase the accessibility of data accumulated by each database. These resources are complementary and users should understand the strengths of each database to navigate the vast amount of knowledge that has been accumulated by researchers in the communities. As MARRVEL development continues, we plan to include more databases that are relevant to studying human variants in model organisms. The overarching goal of MARRVEL is to provide an easily accessible way for clinicians and researchers alike to analyze human genes and variants for further study by integrating useful information while keeping the interface as simple as we can.
Supplementary Material
ACKNOWLEDGMENTS:
We thank Drs. Rami Al-Ouran, Seon-Young Kim, Yanhui (Claire) Hu, Ying-Wooi Wan, Naveen Manoharan, Sasidhar Pasupuleti, Aram Comjean, Dongxue Mao, Michael Wangler, Hsiao-Tuan Chao, Stephanie Mohr, and Norbert Perrimon for their support in the development and maintenance of MARRVEL. We are grateful to Samantha L. Deal and J. Michael Harnish for their input on this manuscript. The initial development of MARRVEL was supported in part by the Undiagnosed Diseases Network Model Organisms Screening Center through the NIH Commonfund (U54NS093793) and through the NIH Office of Research Infrastructure Programs (ORIP) (R24OD022005). JW is supported by the NIH Eunice Kennedy Shriver National Institute of Child Health & Human Development (F30HD094503) and The Robert and Janice McNair Foundation McNair MD/PhD Student Scholar Program at BCM. HJB is further supported by the NIH National Institute of General Medical Sciences (R01GM067858) and is an Investigator of the Howard Hughes Medical Institute. ZL is supported by the NIH National Institute of General Medical Science (R01GM120033), National Institute of Aging (R01AG057339), and the Huffington Foundation. SY received additional support from the NIH National Institute on Deafness and other Communication Disorders (R01DC014932), the Simons Foundation (SFARI Award: 368479), the Alzheimer’s Association (New Investigator Research Grant: 15-364099), Naman Family Fund for Basic Research and Caroline Wiess Law Fund for Research in Molecular Medicine.
Footnotes
Members of the Undiagnosed Diseases Network is provided in Supplemental Table 1.
DISCLOSURES:
The authors have nothing to disclose.
REFERENCES
- 1.Yang Y et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 369 (16), 1502–1511, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Richards S et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17 (5), 405–424, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.MacArthur DG et al. Guidelines for investigating causality of sequence variants in human disease. Nature. 508 (7497), 469–476, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang J et al. MARRVEL: Integration of Human and Model Organism Genetic Resources to Facilitate Functional Annotation of the Human Genome. Am J Hum Genet 100 (6), 843–853, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Povey S et al. The HUGO Gene Nomenclature Committee (HGNC). Hum Genet 109 (6), 678–680, (2001). [DOI] [PubMed] [Google Scholar]
- 6.Lek M et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 536 (7616), 285–291, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wildeman M, van Ophuizen E, den Dunnen JT & Taschner PE Improving sequence variant descriptions in mutation databases and literature using the Mutalyzer sequence variation nomenclature checker. Hum Mutat 29 (1), 6–13, (2008). [DOI] [PubMed] [Google Scholar]
- 8.Zhou W et al. TransVar: a multilevel variant annotator for precision genomics. Nat Methods. 12 (11), 1002–1003, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu Y et al. An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinformatics. 12 357, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amberger JS & Hamosh A Searching Online Mendelian Inheritance in Man (OMIM): A Knowledgebase of Human Genes and Genetic Phenotypes. Curr Protoc Bioinformatics. 58 1 2 1–1 2 12, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amberger JS, Bocchini CA, Scott AF & Hamosh A OMIM.org: leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res 10.1093/nar/gky1151, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu N et al. Functional variants in TBX2 are associated with a syndromic cardiovascular and skeletal developmental disorder. Hum Mol Genet 27 (14), 2454–2465, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ropers HH & Wienker T Penetrance of pathogenic mutations in haploinsufficient genes for intellectual disability and related disorders. Eur J Med Genet 58 (12), 715–718, (2015). [DOI] [PubMed] [Google Scholar]
- 14.Shashi V et al. De Novo Truncating Variants in ASXL2 Are Associated with a Unique and Recognizable Clinical Phenotype. Am J Hum Genet 100 (1), 179, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen R et al. Analysis of 589,306 genomes identifies individuals resilient to severe Mendelian childhood diseases. Nat Biotechnol 34 (5), 531–538, (2016). [DOI] [PubMed] [Google Scholar]
- 16.Halvorsen M et al. Mosaic mutations in early-onset genetic diseases. Genet Med 18 (7), 746–749, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kohler S et al. The Human Phenotype Ontology in 2017. Nucleic Acids Res 45 (D1), D865–D876, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rentzsch P, Witten D, Cooper GM, Shendure J & Kircher M CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 10.1093/nar/gky1016, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sobreira N, Schiettecatte F, Valle D & Hamosh A GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat 36 (10), 928–930, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sobreira NLM et al. Matchmaker Exchange. Curr Protoc Hum Genet 95 9 31 31–39 31 15, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harnish M, Deal S, Wangler M & Yamamoto S In vivo functional study of disease-associated rare human variants using Drosophila. Journal of Visualized Experiments., (2019). [DOI] [PMC free article] [PubMed]
- 22.Harrison SM et al. Using ClinVar as a Resource to Support Variant Interpretation. Curr Protoc Hum Genet 89 8 16 11–18 16 23, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.MacDonald JR, Ziman R, Yuen RK, Feuk L & Scherer SW The Database of Genomic Variants: a curated collection of structural variation in the human genome. Nucleic Acids Res 42 (Database issue), D986–992, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Firth HV et al. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am J Hum Genet 84 (4), 524–533, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thurmond J et al. FlyBase 2.0: the next generation. Nucleic Acids Res 10.1093/nar/gky1003, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Consortium GT Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 348 (6235), 648–660, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ponten F, Jirstrom K & Uhlen M The Human Protein Atlas--a tool for pathology. J Pathol 216 (4), 387–393, (2008). [DOI] [PubMed] [Google Scholar]
- 28.The Gene Ontology, C. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res 10.1093/nar/gky1055, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mungall CJ et al. The Monarch Initiative: an integrative data and analytic platform connecting phenotypes to genotypes across species. Nucleic Acids Res 45 (D1), D712–D722, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meehan TF et al. Disease model discovery from 3,328 gene knockouts by The International Mouse Phenotyping Consortium. Nat Genet 49 (8), 1231–1238, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Katoh K, Rozewicki J & Yamada KD MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform 10.1093/bib/bbx108, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sievers F & Higgins DG Clustal Omega for making accurate alignments of many protein sequences. Protein Sci 27 (1), 135–145, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoon WH et al. Loss of Nardilysin, a Mitochondrial Co-chaperone for alpha-Ketoglutarate Dehydrogenase, Promotes mTORC1 Activation and Neurodegeneration. Neuron 93 (1), 115–131, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deal S & Yamamoto S Unraveling novel mechanisms of neurodegeneration through a large-scale forward genetic screen in Drosophila (In Press). Frontiers in Genetics. (2019). [DOI] [PMC free article] [PubMed]
- 35.Matamoros AJ & Baas PW Microtubules in health and degenerative disease of the nervous system. Brain Res Bull 126 (Pt 3), 217–225, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Theodosiou A, Arhondakis S, Baumann M & Kossida S Evolutionary scenarios of Notch proteins. Mol Biol Evol 26 (7), 1631–1640, (2009). [DOI] [PubMed] [Google Scholar]
- 37.Shayevitz C, Cohen OS, Faraone SV & Glatt SJ A re-review of the association between the NOTCH4 locus and schizophrenia. Am J Med Genet B Neuropsychiatr Genet. 159B (5), 477–483, (2012). [DOI] [PubMed] [Google Scholar]
- 38.Wang Z et al. A review and re-evaluation of an association between the NOTCH4 locus and schizophrenia. Am J Med Genet B Neuropsychiatr Genet 141B (8), 902–906, (2006). [DOI] [PubMed] [Google Scholar]
- 39.Hu Y, Comjean A, Mohr SE, FlyBase C & Perrimon N Gene2Function: An Integrated Online Resource for Gene Function Discovery. G3 (Bethesda) 7 (8), 2855–2858, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.