

Abstract
Purpose
Activating the cell survival modulator sigma 1 receptor (Sig1R) delays cone photoreceptor cell loss in Pde6βrd10/J (rd10) mice, a model of retinitis pigmentosa. Beneficial effects are abrogated in rd10 mice lacking NRF2, implicating NRF2 as essential to Sig1R-mediated cone neuroprotection. Here we asked whether activation of NRF2 alone is sufficient to rescue cones in rd10 mice.
Methods
Expression of antioxidant genes was evaluated in 661W cells and in mouse retinas after treatment with monomethylfumarate (MMF), a potent NRF2 activator. Rd10 mice were administered MMF (50 mg/kg) or the Sig1R ligand (+)-pentazocine (PTZ; 0.5 mg/kg) intraperitoneally (every other day, P14-42). Mice were evaluated for visual acuity (optokinetic tracking response), retinal function (electroretinography) and architecture (SD-OCT); histologic retinal sections were evaluated morphometrically.
Results
MMF treatment increased Nrf2, Nqo1, Cat, Sod1, and Hmox1 expression in vitro and in vivo. Visual acuity of (+)-PTZ–treated rd10 mice was similar to wild-type mice; however, MMF treatment did not alter acuity compared with nontreated rd10 mice. Cone electroretinography b-wave amplitudes were greater in PTZ-treated than nontreated or MMF-treated rd10 mice. SD-OCT assessment of retinal thickness was greater in (+)-PTZ–treated mice versus nontreated or MMF-treated rd10 mice. Morphometric assessment of the outer nuclear layer revealed approximately 18 cells/100 µm retinal length in (+)-PTZ–treated rd10 mice, but only approximately 10 to 12 cells/100 µm in MMF-treated and nontreated rd10 retinas.
Conclusions
Activation of NRF2 using MMF, at least at our dosing regimen, is insufficient to attenuate catastrophic photoreceptor damage characteristic of rd10 mice. The data prompt investigation of additional mechanisms involved in Sig1R-mediated retinal neuroprotection.
Keywords: retina, oxidative stress, rd10 mouse, retinal neuroprotection, NRF2-KEAP1, NRF2
Although cataract (or opacity of the ocular lens) is the leading cause of blindness worldwide, the remarkable improvements in synthetic lens development have provided ophthalmologists excellent tools to manage this form of blindness. In contrast, degenerative diseases of the retina present major therapeutic hurdles to clinicians and constitute the major cause of untreatable blindness.1 Retinal disease affects millions of individuals worldwide and includes macular degeneration and diabetic retinopathy. Inherited retinal degenerative diseases, such as retinitis pigmentosa (RP), affect more than 1.5 million individuals worldwide. RP compromises the function of retinal photoreceptor cells and typically manifests during the first few decades of life progressing to severely limited vision (and often blindness). It is associated with more than 3000 mutations in more than 50 genes.2
Such genetic heterogeneity presents significant impediments to the treatment of retinal degenerations and has prompted researchers to investigate common disease mechanisms that might then offer reasonable therapeutic intervention targets. Of the myriad causes proposed for retinal degenerations (including inflammation, vascular alterations, and cellular demise), oxidative stress has emerged as a major factor underlying these diseases.3 In RP, rod photoreceptor cells frequently are affected initially. For example, mutations of rhodopsin can lead to protein misfolding, mislocalization, disrupted intracellular traffic, and protein instability.4 However, it is the subsequent loss of cones that is most debilitating because these cells are responsible for vision in bright light. There is considerable evidence that cones die in RP owing to the hyperoxic environment that ensues after the death of rod cells.5
A modulator of cellular oxidative stress that has emerged as a novel target for a number of degenerative diseases is sigma 1 receptor (Sig1R).6 Sig1Rs are nonopioid binding sites that are distinct from other known neurotransmitters or hormone receptors and are ubiquitously expressed in different tissues.7 They modulate intracellular Ca2+ 8 by interacting with L-type voltage gated Ca2+ channels,9 increased synthesis of inositol triphosphate and by interacting with the C-terminal of ankyrin-B 220.10 The production of inositol triphosphate and release of Ca2+ from the endoplasmic reticulum occur when cells are exposed to oxidative stress. Many reports show that Sig1R attenuates oxidative stress.11–16
Our laboratory has investigated Sig1R as a novel therapeutic target for retinal degenerative disease.17 In a recent study, we discovered that activation of Sig1R, using (+)-pentazocine ((+)-PTZ), a high-affinity, high-specificity Sig1R ligand, yielded robust rescue of cone structure and function in a rodent model of RP.18 When we administered (+)-PTZ to Pde6βrd10/J (rd10) mice, we observed retention of cone function, determined by photopic flash electroretinography (ERG) and a natural luminance noise test, at an age when rd10 cone function is typically nondetectable.18 This benefit was abrogated in rd10/Sig1R−/− mice demonstrating the specificity of (+)-PTZ in targeting Sig1R. Analysis of lipid peroxidation, protein carbonylation and other measures of oxidative stress, demonstrated that Sig1R activation attenuated this stress in retinas of rd10 mice and importantly normalized levels of NRF2.18
Nuclear factor erythroid 2-related factor 2 (NRF2) is a transcription factor that regulates more than 500 antioxidant and cytoprotective genes.19 When stress is minimal, NRF2 is retained at low levels in the cytosol by Kelch ECH associated protein 1 (KEAP1); however, under stress NRF2 translocates to the nucleus and binds to antioxidant response elements that are present in many antioxidant proteins and detoxification enzymes.20 Owing to our observation that Sig1R activation altered NRF2 levels in rd10 mice,18 we used several assay to evaluate whether (+)-PTZ might actually be an activator of NRF2. The results indicated unequivocally that (+)-PTZ does not disrupt KEAP1–NRF2 binding, and thus it is not an activator of NRF2.21 We performed a subsequent studies to ask whether the beneficial effects of Sig1R activation (via (+)-PTZ) would persist in the rd10 mouse if NRF2 was absent.21 We reasoned that if NRF2 is a factor underlying cone rescue owing to Sig1R activation, then (+)-PTZ treated rd10/nrf2−/– mice would show minimal improvement in cone function or survival, which is what we observed. Indeed, (+)-PTZ treatment of rd10/nrf2−/− mice did not improve retinal structural or functional outcomes compared with nontreated rd10/nrf2−/− mice.21 Thus, in the absence of NRF2, activation of Sig1R does not mediate robust rescue of cone cells in rd10 mice. These findings implicate NRF2 as an important factor in Sig1R-medicated cone neuroprotection. What was not determined in the study, however, was whether activation of NRF2 alone is sufficient to rescue cones in this severe retinal degeneration model. The present study addressed this question by administering a potent NRF2 activator, monomethylfumarate (MMF), to rd10 mice and comparing retinal structure and function with rd10 mice administered the Sig1R activator (+)-PTZ). MMF is a thioreactive electrophile that has been shown to activate NRF2 in a concentration-dependent manner.22 The binding of MMF to cysteine residues of KEAP1 induces a conformational change that dissociates NRF2 from KEAP1, permitting it to enter the nucleus. Additional studies have analyzed MMF activation using SH-SY5Y cells expressing the Neh2-luc reporter.23 The cells are a valuable tool to measure the disruption of KEAP1-NRF2, which leads to expression of transcriptionally active NRF2. Studies from the Thomas laboratory using these cells showed that MMF activates the NRF2 pathway by S-alkylation of KEAP1 (as well as facilitating the nuclear exit of the NRF2 repressor protein, BTB domain and CNC homolog 1.
Studies from the Duh laboratory reported that MMF (administered systemically at a concentration of 50 mg/kg), exerted neuronal protection via the NRF2 pathway in retinas of mice subjected to ischemia–reperfusion (I/R) injury.24 In the present study, we administered MMF to rd10 mice and evaluated retinal function and structure compared with rd10 mice administered (+)-PTZ. Interestingly, our data show that activation of NRF2 by MMF does not yield the retinal benefit conferred by activation of Sig1R and suggest that, although NRF2 is important in the mechanism of Sig1R neuroprotection, its activation alone may not account completely for the retinal neuroprotective effects of Sig1R.
Methods
Evaluation of MMF Treatment on Upregulation of Antioxidant Genes
MMF has been reported as a potent inducer of the NRF2 antioxidant pathway.22,24 To confirm the role of MMF in increasing expression of genes known to be regulated by NRF2 (Nqo1, Cat, Sod1, Hmox1), we performed an in vitro assay in a cone photoreceptor cell line (661W) and evaluated gene expression following MMF treatment compared with tert-butylhydroquinone (tBHQ) (Sigma-Aldrich, St Louis, MO) a known activator of Nrf2.25 The 661W cells were obtained from Dr M. Al-Ubaidi (University of Houston). These cells express blue and green cone pigments, transducin and cone arrestin characteristic of cone photoreceptor cells.26 They were cultured in Dulbecco's Modified Eagle's Medium (with 4.5 g/L glucose, L-glutamine, and sodium pyruvate, Cat. # 10-013-CV, Corning Cellgro, ThermoFisher Scientific, Waltham, MA) supplemented with 5% fetal bovine serum for regular culture or 1% fetal bovine serum in treatment, and 100 U/mL penicillin, 100 µg/mL streptomycin. The cells were treated for 8 hours in the presence or absence of MMF (400 µM), a concentration that approximates the dosage administered in the in vivo studies described elsewhere in this article. tBHQ (20 µM) was used as the positive control. Expression levels of mRNA transcripts specific for Nrf2, Nqo1, Sod1, Cat, and Hmox-1 were determined. Total RNA was purified from the cells using TRIzol (Invitrogen, Carlsbad, CA) and was reverse-transcribed using the iScript Synthesis kit (Bio-Rad, Hercules, CA). cDNAs were amplified for 40 cycles using SsoAdvanced SYBR Green Supermix (Bio-Rad) and gene-specific primers (Table 1) in a CFX96 Touch Real-Time PCR Detection System (Bio-Rad). Expression levels were calculated by comparing cycle threshold (Ct) values (ΔΔCt).27 In additional studies, expression levels of mRNA transcripts specific for Nrf2, Nqo1, Sod1, Cat, and Hmox-1 were examined in retinas of mice used in the study. Total RNA purified from isolated neural retinas using Trizol (Invitrogen) was reverse-transcribed using the iScript Synthesis kit (Bio-Rad). cDNAs were amplified 40 cycles using SsoAdvanced SYBR Green Supermix (Bio-Rad) and gene-specific primers (Table 1) in a CFX96 Touch Real-Time PCR Detection System (Bio-Rad). Expression levels were calculated by comparison of Ct values (ΔΔ Ct).27
Table 1.
Sequences of Primers Used for Real-Time Quantitative Reverse Transcription Plus PCR
| Gene | NCBI Accession Number | Primer Sequence | Product Size (bp) | |
|---|---|---|---|---|
| Sod1 | NM_011434 | Forward | 5′-AACCAGTTGTGTTGTCAGGAC-3′ | |
| Reverse | 5′-CCACCATGTTTCTTAGAGTGAGG-3′ | 139 | ||
| Cat | NM_009804 | Forward | 5′-AGCGACCAGATGAAGCAGTG-3′ | |
| Reverse | 5′-TCCGCTCTCTGTCAAAGTGTG-3′ | 181 | ||
| Nqo1 | NM_008706 | Forward | 5′-AGGATGGGAGGTACTCGAATC-3′ | |
| Reverse | 5′-AGGCGTCCTTCCTTATATGCTA-3′ | 144 | ||
| Hmox1 | NM_010442 | Forward | 5′-AAGCCGAGAATGCTGAGTTCA-3′ | |
| Reverse | 5′-GCCGTGTAGATATGGTACAAGGA-3′ | 100 | ||
| Nrf2 | NM_010902 | Forward | 5′-TAGATGACCATGAGTCGCTTGC-3′ | |
| Reverse | 5′-GCCAAACTTGCTCCATGTCC-3′ | 153 | ||
| Gapdh | NM_008084 | Forward | 5′-AGGTCGGTGTGAACGGATTTG-3′ | |
| Reverse | 5′-TGTAGACCATGTAGTTGAGGTCA-3′ | 123 |
Cat, catalase; Gapdh, glyceraldehyde 3-phosphate dehydrogenase; Hmox1, heme oxygenase 1; Nqo1, nicotinamide adenine dinucleotide phosphate (NAD(P)H)-quinone dehydrogenase 1; Sod1, superoxide dismutase.
Animals
Breeding pairs of B6.CXBI-Pde6βrd10/J (rd10) mice were purchased from Jackson Laboratories (Bar Harbor, ME). Genotyping was confirmed as described.18 We also confirmed that the Crb1rd8/rd8 mutation, which causes focal retinal disruption in certain mouse strains,28 was not present in mice used in the study. Teklad Irradiated Rodent diets 8904 and 2918 (Teklad, Madison, WI) were provided for breeding and maintenance, respectively. Mice were maintained on a standard 12-hour light:dark cycle. We adhered to institutional guidelines for humane treatment of animals and to the ARVO statement for Use of Animals in Ophthalmic and Vision Research. Three groups of mice were evaluated over a period of 42 days: rd10 mice (nontreated), rd10+MMF, and rd10+PTZ mice. The mice in the treatment groups received an intraperitoneal injection on alternate days beginning at P14 of either MMF (Sigma-Aldrich; 50 mg/kg, dissolved in dimethylsufoxide and 0.01 M PBS) or (+)-PTZ (Sigma-Aldrich; 0.5 mg/kg, dissolved in dimethylsufoxide and 0.01 M PBS). The dosage of MMF was based on previously published studies demonstrating neuroprotection in mice with retinal ischemia-reperfusion injury.24 The dosage of (+)-PTZ was based on our previously published studies.18,21,29 Table 2 provides information on the numbers of mice used in the study.
Table 2.
Number of Animals Used in the Study
| Mouse Group and Analysis | n | Age (Postnatal Days) |
|---|---|---|
| OptoMotry analysis of vision acuity | ||
| WT | 7 | 21, 28, 41 |
| rd10 | 5 | 21, 28, 41 |
| rd10 (50 mg kg−1 MMF) | 8 | 21, 28, 41 |
| rd10 (0.5 mg kg−1 (+)-PTZ) | 4 | 21, 28, 41 |
| ERG analysis of retinal function | ||
| rd10 | 6 | 35 |
| rd10 (50 mg kg−1 MMF) | 7 | 35 |
| rd10 (0.5 mg kg−1 (+)-PTZ) | 6 | 35 |
| OCT analysis of retinal structure | ||
| WT | 5 | 42 |
| rd10 | 4 | 42 |
| rd10 (50 mg kg−1 MMF) | 12 | 42 |
| rd10 (0.5 mg kg−1 (+)-PTZ) | 6 | 42 |
| Histologic analysis and peanut agglutinin labeling of cone photoreceptor cells | ||
| WT | 3 | 42 |
| rd10 | 3 | 42 |
| rd10 (50 mg kg−1 MMF) | 3 | 42 |
| rd10 (0.5 mg kg−1 (+)-PTZ) | 3 | 42 |
| Gene expression analyses | ||
| WT | 4 | 42 |
| rd10 | 3 | 42 |
| rd10 (50 mg kg−1 MMF) | 9 | 42 |
| rd10 (0.5 mg kg−1 (+)-PTZ) | 3 | 42 |
Visual Acuity Assessment
Visual acuity was evaluated in rd10-nontreated, rd10+MMF mice, and rd10+PTZ mice ages P21, P28 and P41; a cohort of wild-type (WT) C57Bl/6J mice (Jackson Laboratories) were also included in the assessment for comparison. Acuity was measured as described previously30 following the method of Prusky et al.31 Spatial thresholds for optokinetic tracking of sine-wave gratings were measured using the OptoMotry system (CerebralMechanics, Medicine Hat, Alberta, Canada). Mice were placed unrestrained on a pedestal and were presented vertical sine-wave gratings moving at 12°/s or gray of the same mean luminance within the OptoMotry device, which functions as a virtual cylinder. The cylinder hub was continually centered between the mouse's eyes to establish the spatial frequency of the grating at the mouse's viewing position as it shifted its position. Gray color was projected while the mouse was moving; when movement ceased, the gray was replaced with the grating. Grating rotation under these circumstances elicited reflexive tracking, which was scored via live video using a method of limits procedure with a yes/no criterion as recommended by the manufacturer. A measure of spatial resolution was taken as the asymptote of a staircase procedure. The two eyes were tested in an interleaved fashion.
Electroretinography
The rd10-nontreated, rd10+MMF mic and rd10+PTZ mice were dark-adapted overnight. Before testing on day P35, mice were anesthetized using isoflurane and electrophysiologic function was assessed as described.18,29 Briefly, dark-adapted ERGs were performed using silver-coated nylon fibers joined to flexible wires placed on the cornea, the electrical contact of which was enhanced with hypromellose. The 1-mm diameter optic fibers were positioned in front of the pupil through which highly controllable illumination was delivered to eyes using a 5500° white LED. Rod function was assessed using a series of tests with 5-ms flashes of increasing luminance, followed by assessment of cone function using photopic testing with 5-ms flashes above a pedestal. Additionally, a photopic "natural noise" stimulus was presented. This stimulus changes luminance pseudorandomly over time, with the amplitudes of those changes inversely proportional to temporal frequency (as has been described for human subjects32), and phase being random. This process produces relatively slow, continuous changes in luminance, rather than flashes, and is natural in the sense that real-world visual stimuli similarly change slowly. Responses to noise stimuli are also random, but responses are correlated with stimuli to generate kernels that describe how the retina transforms arbitrary stimuli into ERG responses.
Spectral Domain Optical Coherence Tomography
Rd10-nontreated, rd10+MMF mice, and rd10+PTZ mice were anesthetized with ketamine/xylazine as described.18,29 Retinal structure was evaluated in vivo using a Bioptigen Spectral Domain Ophthalmic Imaging System (SDOIS; Bioptigen Envisu R2200, Durham, NC) in mice at P42. The imaging protocol included averaged single B scan and volume intensity scans with images centered on the optic nerve head. The imaging protocol included averaged single B scan and volume intensity scans with images centered on the optic nerve head. There is substantial disruption of the outer retina in the rd10 mouse retina, thus it is preferable to use the manual caliper feature to measure retinal layers versus autosegmentation post-imaging analysis (a feature of the InVivoVue Diver 2.4 software [Bioptigen]). We used this feature to acquire inner, outer, and total retinal thickness measurements. The inner retina was measured from the upper edge of the inner limiting membrane to the lower edge of the inner nuclear layer. Outer retina was measured from the lower inner nuclear layer edge to the inferior boundary of the RPE layer; if there was separation of photoreceptors from RPE, we obtained outer retinal thickness by adding the RPE layer thickness and the distance from the lower edge of the inner nuclear layer to the edge of neuronal retina as described.29,33
Microscopic Evaluation, Morphometric Analysis, and Immunofluorescence Studies
Eyes were harvested from euthanized rd10-nontreated, rd10+MMF, mice and rd10+PTZ mice (P42). One eye was immersion fixed in 2% paraformaldehyde/2% glutaraldehyde in 0.1 M cacodylate buffer and processed for embedding in JB-4 methacrylate (Electron Microscopy Sciences, Hatfield, PA). Sections (2 µm thickness) were stained with hematoxylin and eosin. The contralateral eye was enucleated and prepared for cryosectioning. Eyes were flash frozen in liquid nitrogen and embedded in optimal cutting temperature compound (Elkhart, IN). The 10 µm thick cryosections were fixed for 10 minutes in 4% paraformaldehyde and blocked with 10% goat serum in 0.1%Triton-X100/phosphate-buffered saline for 1 hour at room temperature.
Plastic-embedded, hematoxylin and eosin–stained retinal sections were imaged using a Zeiss Axio Imager D2 microscope (Carl Zeiss, Göttingen, Germany) equipped with a high-resolution camera and processed using Zeiss Zen23pro software. Given the extent of outer nuclear layer (ONL) loss in the rd10 mouse, we not only measured the thickness of this layer in micrometers, but also counted the total number of photoreceptor cells along the full length of retina (from the temporal to nasal ora serrata) in three areas of three separate retinal sections per mouse. Photoreceptor cell counts were averaged per mouse and then the average was calculated per treatment group.
To determine whether the photoreceptor cells were cones, the retinal cryosections were incubated with FITC-conjugated peanut agglutinin, a marker for cone photoreceptors. Retinal sections were viewed by epifluorescence using the Zeiss Axio Imager D2 microscope.
Data Analysis
Statistical analysis used GraphPad Prism analytical program (La Jolla, CA). Data were analyzed by one-way ANOVA; Tukey's test was used as the post hoc test. For the ERG results, we used Igor Pro (WaveMetrics, Lake Oswego, OR). Significance for all analyses was P < 0.05.
Results
MMF Induces Upregulation of Antioxidant Genes Regulated by NRF2
To evaluate effects of MMF in inducing NRF2-regulated genes including Nqo1, Cat, Sod1, Hmox1, we treated the 661W photoreceptor cell line with MMF at a concentration that approximated the dosage we intended to use in the in vivo study with rd10 mice. We isolated RNA and evaluated gene expression using qRT-PCR and observed a highly significant increase in Sod1, Nqo1, Cat, and Hmox1 levels within 8 hours of MMF exposure (Fig. 1). Nrf2 and Sod1 levels were approximately 1.5-fold greater when cells were treated with MMF compared with nontreated cells, which was comparable with the effects of tBHQ treatment (Fig. 1A). For analysis of Nqo1 and Cat, treatment with MMF increased expression levels by seven- and four-fold, respectively compared with nontreated cells (Fig. 1B). Expression of Hmox1 increased by 20-fold after MMF treatment (Fig. 1C). Thus, MMF treatment resulted in a highly significant increase in expression of antioxidant genes that are regulated by NRF2. We therefore proceeded with our assessment of MMF on the retinal phenotype of rd10 mice. After completing the functional analyses described elsewhere in this article, retinas were isolated from mice and were evaluated for expression of these same genes. There was a significant increase in expression of Nrf2, Sod1, Nqo1, Cat (Fig. 1D) and Hmox1 (Fig. 1E) as a consequence of MMF treatment in rd10 mice compared with nontreated rd10 and WT mice (that received no treatment). Thus, the administration of MMF activated NRF2, as well as several antioxidant genes, known to be regulated by NRF2. The significant increase in expression of these genes in rd10 mice is expected since oxidative stress, which is considerable in the rd10 retina,18 is itself an activator of NRF2. Treatment of rd10 mice with (+)-PTZ resulted in increased retinal expression of Nrf2, Cat, and Hmox1 compared with nontreated rd10 and WT mice, although it did not alter retinal expression of these genes compared with MMF-treated rd10 mice, except in the case of Hmox1 (Fig. 1E).
Figure 1.

Evaluation of antioxidant gene expression in 661W cells treated with MMF. The 661W cells were incubated with MMF [400 µM] for 8 hours. RNA was isolated and subjected to qRT-PCR to analyze expression of (A) Nrf2, Sod1, (B) Nqo1, Cat, and (C) Hmox1. Primer pairs used for analysis are listed in Table 1. Data are mean ± SEM of three independent assays. Significant differences (***P < 0.001; ****P < 0.0001) are in reference to the control (nontreated cells). The positive control used to induce Nrf2 was tBHQ (20 µM). RNA was isolated from neural retinas of WT, rd10, rd10+MMF, rd10+PTZ mice and subjected to qRT-PCR analysis of (D) Nrf2, Sod1, Nqo1, Cat, and (E) Hmox1 using the primer pairs listed in Table 1. Data are the mean ± SEM of three to four assays. Significantly different from gene expression in WT mice (**P < 0.01; ****P < 0.0001).
Assessment of Visual Acuity
Our earlier studies reported improved visual acuity in rd10 mice when Sig1R was activated using (+)-PTZ.29 Additional work indicated that Sig1R-mediated cone rescue in rd10 mice required NRF2,21 but whether activation of NRF2 alone would improve visual acuity in rd10 mice had not been evaluated. Here we analyzed the effects on visual acuity in rd10 mice treated with MMF, a known activator of NRF2, compared with (+)-PTZ. Visual acuity was assessed by measuring the optokinetic tracking response using the OptoMotry system. The optokinetic tracking response measured the reflex response for clockwise and counterclockwise movement of gratings drifting at 12°/s. Data were recorded as the asymptotic convergence of a staircase procedure that estimates spatial resolution, or acuity, in units of cycles per degree (c/d). The visual acuity in WT mice at P28 and P42 was 0.4 c/d (Fig. 2A), whereas for rd10-nontreated mice it was approximately 0.30 c/d at P21 and approximately 0.35 c/d at P28, but decreased rapidly to 0.10 to 0.15 c/d by P42 (Fig. 2B). (The visual acuity observed at P28 in rd10 mice reflects function of cones that are still present at this age,34,35 despite the near absence of rods.) In the rd10 + MMF mice, the visual acuity was approximately 0.35 at P28, but decreased significantly by P42 to approximately 0.15 (Fig. 2C), which was quite similar to nontreated rd10 mice. In contrast, rd10 mice treated with (+)-PTZ retained visual acuity through P42 (the median visual acuity was approximately 0.35 c/d at P21, approximately 0.34 c/d at P28, and approximately 0.32 c/d at P42) (Fig. 2D). The data support previously reported observations,29 indicating that (+)-PTZ treatment delays the marked decline in visual acuity observed in nontreated rd10 mice, whereas MMF treatment does not preserve visual acuity significantly beyond that of nontreated rd10 mice. The averaged values for the visual acuity data are presented for comparisons between rd10-non, rd10+MMF, and rd10-PTZ mice at P21, P28, and P42 (Fig. 2E).
Figure 2.
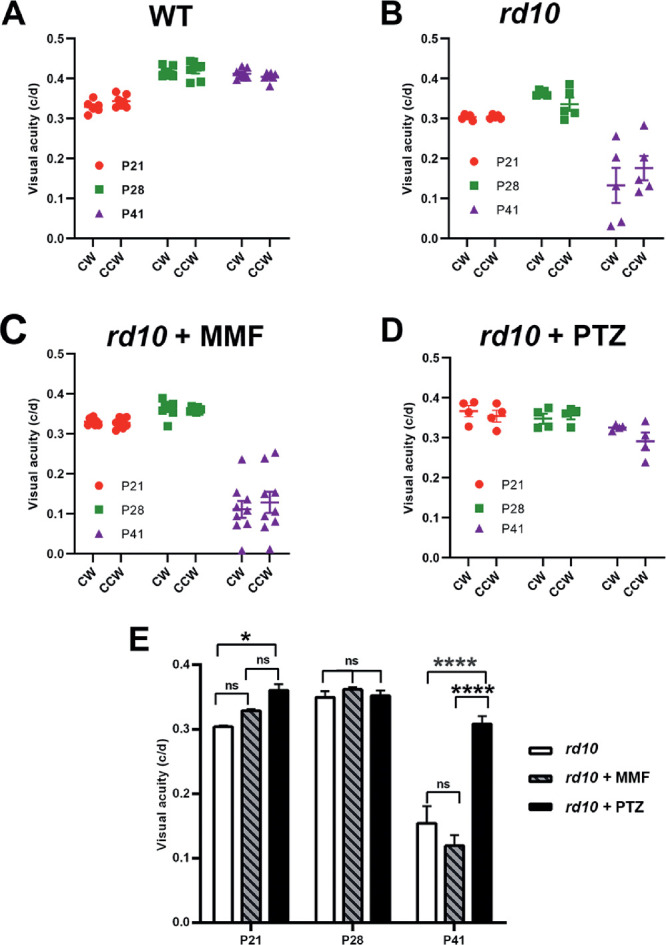
Assessment of visual acuity in rd10 mice treated with MMF compared with nontreated and PTZ-treated. The rd10 mice were administered MMF or (+)-PTZ every other day beginning at P14 and were compared with nontreated rd10 mice. The optokinetic tracking response (OKR) was measured using the OptoMotry system to assess visual acuity at P21, P28, and P41. Data are expressed as cycles/degree (c/d) for (A) WT mice, (B) nontreated rd10 mice, (C) rd10+MMF mice, and (D) rd10 +PTZ mice. The data were summarized for comparison (E). Significance is depicted as *P < 0.05, ****P < 0.0001. CW, left eye; CCW, right eye; ns, not significant.
ERG Assessment of Retinal Function
We then evaluated the effects of MMF treatment on cone and rod photoreceptor cell responses. Mice, administered either MMF or (+)-PTZ on alternate days beginning at P14, were subjected to comprehensive ERG analysis at P35 to assess cone (photopic) and rod (scotopic) function. Responses were compared with age-matched, rd10-nontreated mice. Regarding cone responses, the b-wave amplitude measured in the photopic ERG response was similar between rd10 nontreated and rd10 MMF-treated mice, but was greater in the rd10+PTZ group. Representative tracings at increasing light intensities are shown (Fig. 3A–C). The photopic flash response is summarized in Figure 3D, with the rd10+PTZ mice having the most robust response. Cone function was tested also with a stimulus that changed more slowly in time. This natural stimulus (green noise) detected responses in rd10+PTZ mice that were significantly greater than nontreated rd10 or rd10+MMF at P42 (Fig. 3E). Regarding rod activity, there was minimal detection of function at low light intensities, but detectable responses, albeit low, at the highest intensities (Fig. 3F). At higher luminous intensities, the average b-wave amplitude for (+)-PTZ–treated rd10 mice were greater than rd10-nontreated or MMF-treated rd10 mice. The data indicate that PRC function is more robust in (+)-PTZ–treated rd10 mice compared with MMF-treated animals.
Figure 3.
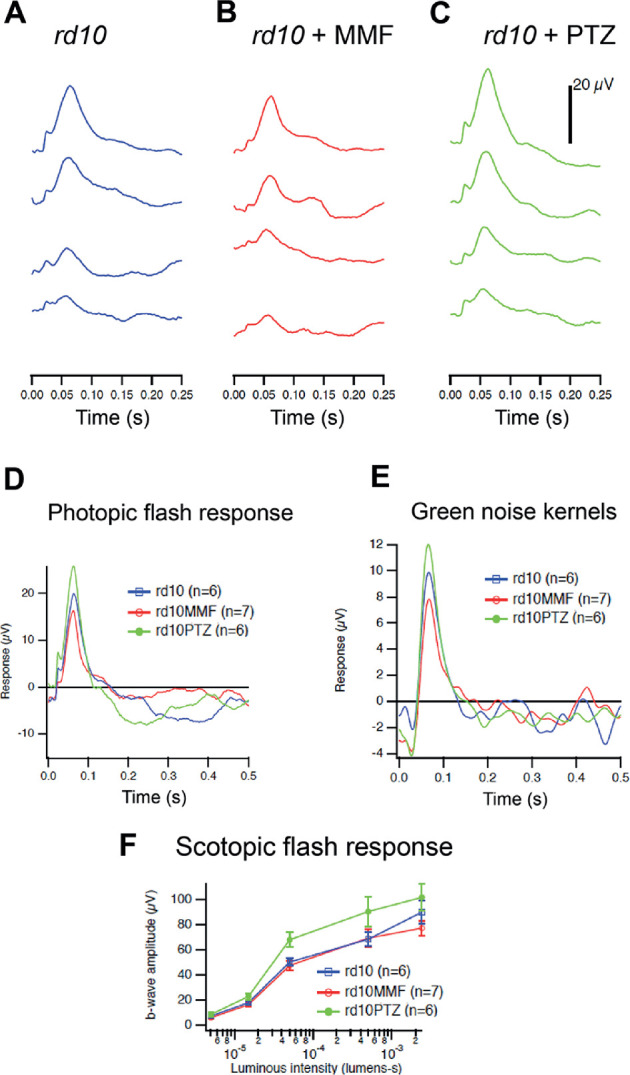
Assessment of retinal function by photopic and scotopic ERG in rd10 mice treated with MMF compared with nontreated and PTZ-treated mice. Rd10 mice were administered MMF or (+)-PTZ every other day beginning at P14 and were compared with nontreated rd10 mice. Photopic and scotopic ERG responses were performed on rd10 mice, rd10+MMF mice, and rd10+PTZ mice at P35. Averaged photopic responses to 5ms flashes at a series of intensities were provided for (A) rd10, (B) rd10+MMF, and (C) rd10+PTZ mice at P35. (D) Averaged responses to the highest intensity photopic flashes. (E) Averaged kernels derived from responses to natural noise stimuli. (F) Averaged scotopic ERG responses to 5-ms flashes at a series of intensities for rd10, rd10+MMF and rd10+PTZ. Intensities were in units of candela-seconds per meter squared.
SD-OCT Assessment of Retinal Architecture
We visualized retinal structure in living mice using SD-OCT. Mice, administered either MMF or (+)-PTZ on alternate days beginning at P14, were subjected to SD-OCT at P42. Representative OCT images are shown (Fig. 4A–C). We used the calipers feature of the OCT software to determine thickness of the total retina, the inner retina and the outer retina. At P42, the total retinal thickness in rd10 was approximately 108 µm thick. In rd10+MMF mice, it was approximately 117, which was significantly greater than nontreated rd10 mice. In the (+)-PTZ–treated animals, the total retinal thickness averaged 123 µm, which was significantly thicker than rd10+MMF mice, as well as nontreated rd10 mice (Fig. 4D). Within the inner retina, there was no significant difference in the measurement of the nontreated rd10 versus the rd10+MMF retina, but there was a significant increase in inner retinal thickness in the rd10+PTZ mice versus the other two groups (Fig. 4E). Regarding the outer retina, there was a significant difference in the measurement of the nontreated rd10 versus the rd10+MMF retina, but an even more significant increase in the rd10+PTZ mice versus the other two groups (Fig. 4F). Thus, in terms of retinal architecture, there was an improvement in the MMF-treated rd10 mice, albeit not as marked as observed in the rd10+PTZ mice.
Figure 4.

Assessment of retinal structure in vivo using SD-OCT in rd10 mice treated with MMF compared with nontreated and PTZ-treated mice. Rd10 mice were administered MMF or (+)-PTZ every other day beginning at P14 and were compared with nontreated rd10 mice. SD-OCT was performed on mice at P42. Representative SD-OCT images are shown for (A) rd10 (nontreated) mice, (B) rd10+MMF mice, and (C) rd10+PTZ mice. Data from segmentation analysis for (D) total retinal thickness, (E) thickness of the inner retina, and (F) thickness of the outer retina. Significance is depicted as *P < 0.05; **P < 0.01; ****P < 0.0001. ns, not significant. Data are the mean ± SEM of analyses in 5 to 15 mice per group.
Histologic Assessment of Retinal Structure
After the in vivo functional and structural analyses (Figs. 123–4), mice were euthanized and one eye per mouse was processed for embedding in JB-4 methacrylate to permit histologic evaluation of retinas. Representative histologic sections of rd10, rd10+MMF, and rd10+PTZ are shown in Figure 5A–C. By P42, the rd10 retina (Fig. 5A) is less than one-half the thickness of the WT mouse retina (Fig. 5D, inset). The marked disruption is reflected in the severely compromised ONL, which typically consists of 10 to 12 rows of photoreceptor cell nuclei, but is reduced to approximately 1 row in the mutant. In the rd10 mice treated with MMF, the ONL is similar to the nontreated mice (Fig. 5B), whereas the rd10+PTZ has more cells in this row (Fig. 5C). To quantify the differences among groups, we measured the thickness of the ONL and observed no difference between nontreated and MMF-treated rd10 mice, but a small, albeit significant, increase in the (+)-PTZ–treated mice (Fig. 5E). A more precise indicator of the integrity of the ONL in rd10 mice is the number of photoreceptor cell nuclei expressed per retinal length. For nontreated rd10 mice, there were 12.2 ± 1.7 photoreceptor cells per 100 µm retinal length, which was similar to the rd10+MMF mice (12.6 ± 1.3 cells/100 µm) (Fig. 5F). In the rd10+PTZ group, the number of cells was 18.9 ± 1.9 per 100 µm retinal length, which was significantly greater than either nontreated or MMF-treated rd10 mice (Fig. 5F).
Figure 5.

Assessment of retinal histologic structure and detection of cone photoreceptor cells in rd10 mice treated with MMF compared with nontreated and PTZ-treated. Rd10 mice were administered MMF or (+)-PTZ every other day beginning at P14 and were compared with nontreated rd10 mice. After the functional tests, mice were euthanized at P42 and eyes taken for plastic embedding or frozen sections. The plastic sections were stained with hematoxylin and eosin and viewed by light microscopy. Representative retinas are provided for (A) rd10 (nontreated), (B) rd10 + MMF, (C) rd10+PTZ. (D) A representative retinal section of an age-matched WT mouse. Morphometric analysis was performed on the retinal sections; data are presented as mean ± SEM for (None;) the measurement of ONL thickness and (F) the number of photoreceptor cell nuclei in the ONL expressed per 100 µm retinal length. Significance *P < 0.05. ns, not significant. Frozen sections were used for immunodetection of cone photoreceptor cells using FITC-peanut agglutinin (PNA) shown at low and high magnification, respectively for WT (G, H), rd10-nontreated (I, J), rd10 + MMF (K, L), and rd10+PTZ (M, N). Green fluorescence is associated with FITC-PNA; nuclei fluoresce blue owing to labeling using 4′,6-diamidino-2-phenylindole (DAPI) staining. gcl, ganglion cell layer; ipl, inner plexiform layer; inl, inner nuclear layer; opl, outer plexiform layer; onl, outer nuclear layer; is, inner segment; os, outer segment. Calibration bar: 100 µm.
In addition to plastic embedding of one eye (per mouse) for histologic assessment, the contralateral eye was processed for cryosectioning to permit immunolabeling of the inner and outer segments of cone photoreceptor cells using peanut agglutinin. The WT mouse has an abundance of peanut agglutinin labeling in retinas (Fig. 5G, H). In the rd10 mice, many of the cells remaining in the ONL were cones as shown for rd10-nontreated retinas (Fig. 5I, J), rd10+MMF mice (Fig. 5K, L) and rd10+PTZ retinas (Fig. 5M, N).
Discussion
Work published from our laboratory reporting robust cone protection in the rd10 mouse18,21,29 is encouraging because it identifies Sig1R as a potential target protein that might prove beneficial for retinal disease. Elucidating the mechanism(s) by which Sig1R activation mediates neuroprotection is critical if such therapeutic strategies are to move into the realm of clinical studies in patients.
To that end, we have been investigating the role of NRF2 in Sig1R-mediated retinal neuroprotection. The rationale for this line of investigation began more than a decade ago when we observed increased [3H]-(+)–PTZ binding activity to Sig1R in primary retinal Müller glial cells when they were exposed to nitric oxide or reactive oxygen species (ROS) donors.12 Subsequently, we discovered that if Sig1R was absent in Müller cells (i.e., Müller cells harvested from Sig1R−/− mice), endogenous levels of ROS were significantly higher than Müller cells isolated from WT mice.16 The Sig1R−/– cells had perturbed antioxidant balance reflected in reduced levels of numerous antioxidant genes, whose transcription was regulated by NRF2, and these cells showed reduced NRF2 expression and decreased NRF2 binding activity to antioxidant response elements. Subsequent work demonstrating that Sig1R activation in the rd10 mouse led to reduced oxidative stress and normalization of NRF2 levels accompanied by rescue of cone photoreceptors18 prompted us to test directly whether (+)-PTZ modulated NRF2 levels and whether treating rd10/Nrf2−/– mice with (+)-PTZ would afford neuroprotection.21 In those studies, we found that treating cone photoreceptor cells in vitro with (+)-PTZ increased NRF2 at the gene and protein level, whereas silencing Sig1R reduced NRF2 expression. In addition, (+)-PTZ increased the binding activity of NRF2 to antioxidant response elements. Importantly, (+)-PTZ treatment did not improve cone function or retinal architecture in rd10/Nrf2−/– mice, but did so in rd10/Nrf2+/+ mice. Those data implicate NRF2 as a factor in Sig1R-mediated retinal neuroprotection, raising the question: is activation of NRF2—alone—sufficient to yield cone photoreceptor protection in rd10 mice that is comparable with the robust neuroprotection attributable to Sig1R activation?
The present study compared an activator of NRF2 (MMF) and an activator of Sig1R ((+)-PTZ) for their effects in attenuating the profound retinal photoreceptor loss characteristic of rd10 mice. This murine model of RP has a rapid loss of photoreceptor cells such that by P25 nearly all rod photoreceptor cells are lost and by P35 cone function is barely detectable.34,35 The degeneration is due to a spontaneous, missense point mutation in the gene encoding the visual cycle protein, cGMP-phosphodiesterase, producing a devastating retinopathy. Our initial analyses in this mutant focused on functional responses to MMF and (+)-PTZ, specifically measuring visual acuity and electrophysiologic function. We anticipated that because RP and, in particular the rd10 mouse, are characterized by retinal oxidative stress, activation of NRF2 using MMF would slow the rate of decline in visual acuity, as we have shown previously for (+)-PTZ,29 but that was not the result we obtained. The visual acuity in the rd10+MMF mice was not improved; instead it was quite similar to nontreated rd10 mice, whereas the (+)-PTZ–treated animals showed retention of visual acuity that was approximately 75% that of WT mice. Similarly photopic ERG responses were better in rd10+PTZ mice compared with nontreated rd10 mice as well as the MMF-treated mutants. The structural evaluation of retinas, using the powerful in vivo imaging tool SD-OCT, demonstrated a significant increase in retinal thickness, including the thickness of the outer retina in MMF-treated rd10 mice; however, the increased thickness compared with rd10-nontreated mice was not as robust as in (+)-PTZ–treated rd10 mice. Histologic examination of the retinas showed significantly more photoreceptor cells in the ONL of the rd10+PTZ mice compared with either the rd10-nontreated or the rd10+MMF mice. Immunodetection methods confirmed that the cells within this layer in all three mouse groups were largely cone photoreceptors. The data suggest that activation of NRF2 in this severe retinopathy (at least using MMF) was not sufficient to delay the marked degeneration characteristic of this mouse, as has been reported for (+)-PTZ-treatment of rd10 mice.18,29
Although our data do not show MMF-mediated neuroprotection against the rapid loss of photoreceptor cells in the rd10 mouse, the findings do not negate the key role that NRF2 (or MMF) play in modulating retinal oxidative stress. For example, Chen et al.36 exposed 661W cells to blue light significantly increasing ROS levels, expression of Nrf2 and several NRF2-regulated genes; however, when they silenced Nrf2 they observed even greater ROS levels and cell death, indicating that NRF2 is a key endogenous protective factor mitigating oxidative stress in photoreceptor cells.36 Recent studies from the Pennesi laboratory tested the neuroprotective effects of MMF in a murine model of induced-photoreceptor degeneration.37 Specifically, they used a light-induced retinopathy model in Balb/c mice and observed rapid decline of ERG, decreased retinal thickness (by OCT), microglial activation, and increased oxidative stress and inflammation in nontreated mice, but a highly significant protection against these manifestations of disease when mice were pretreated with MMF. Interestingly, if MMF was administered following the light exposure (either systemically or intravitreally), significant protection of the light-damaged retina was not observed.37 The authors deduce that MMF can prevent, but not restore, photoreceptor cell death owing to excess light. It is noteworthy, that in that study, the authors examined levels of hydroxyl-carboxylic acid receptor 2 (HCAR2), because MMF is also an agonist of the HCAR2 receptor. They found that, under light-induced retinopathy conditions, HCAR2 mRNA levels were increased, whereas they were attenuated by MMF treatment. Compelling studies from the Duh laboratory showed that NRF2 activation in the I/R mouse model decreased neuronal loss of cells in the retinal ganglion cell layer and improved the retinal function measured by ERG.24 The compound that they selected for the NRF2 activation was MMF and the dosage used was 50 mg/kg, the same as we used in our study. There are, however, important differences in their study versus ours. One is that the I/R model is induced in WT mice allowing the investigators the opportunity to administer MMF several times (2 days, 1 day, and day 0) before the induction of I/R. Thus, the study by Cho et al, like the MMF light-induced retinopathy study37 may have primed the system with respect to cytoprotective protein availability at the onset of disease. Interestingly, it is well-established that caloric restriction is beneficial in models of I/R-induced retinopathy preserving ganglion cells38 and caloric restriction has been shown to modulate the NRF2 pathway.39 Finally, a study from the Martin laboratory demonstrated that MMF can attenuate the retinopathy characteristic of sickle cell disease.40 That study validated the Townes humanized sickle cell disease mouse as a model of Sickle retinopathy and reported that administration of MMF (in drinking water) beginning at 1 month improved the sickle retinopathy as evidenced by functional and structural retinal assessments. It is noteworthy that the onset of detectable retinal alterations in the sickle cell disease model was considerably later (4–7 months) and the severity significantly less than the rapid and fulminant photoreceptor cell loss observed in rd10 mice.34,35 Taken collectively, data amassed from the aforementioned studies support the notion that activation of NRF2 may have therapeutic value in more slowly progressing retinopathies, whereas it may have less efficacy in retinal diseases in which cellular stress is extreme. Indeed, mouse models of RP that have a slower progression, such as the P23H opsin mutation characterized by the Palczewski laboratory,41 may be more amenable to NRF2 activation as a treatment modality.
Regarding the scientific premise, which launched the current study, it seems that activation of NRF2, at least using MMF, may not be sufficient to delay the catastrophic photoreceptor damage characteristic of the rd10 mouse. The findings are relevant to our working hypothesis that NRF2 is central to Sig1R-mediated retinal photoreceptor rescue. Although earlier studies suggested that NRF2 is essential (because (+)-PTZ does not rescue cones in rd10/Nrf2−/– mice), the current work suggests that NRF2 may not be sufficient since a potent activator of this transcription factor (MMF) did not yield the same level of rescue as reported for (+)-PTZ. It is possible that a different dosing regimen of MMF (other than that used in this study) or alternative activators of NRF2 might yield retinal protection that was not observed in the present work. Future studies to address this issue could explore a wider range of MMF dosages or alternative NRF2 activators as well as additional mechanisms by which Sig1R mediates retinal neuroprotection.
Acknowledgments
The authors thank members of the Augusta University EM/Histology core for the expert assistance.
Supported by NIH/NEI (R01EY028103) and the Foundation Fighting Blindness (TA-NMT-0617-0721-AUG). The Culver Vision Discovery Institute and Augusta University provided financial support in acquiring the OptoMotry device for visual acuity studies.
Disclosure: H. Xiao, None; J. Wang, None; A. Saul, None; S.B. Smith, None
References
- 1. Duncan JL, Pierce EA, Laster AM, Daiger SP, Birch DG, Ash JD, et al., the Foundation Fighting Blindness Scientific Advisory Board. Inherited retinal degenerations: current landscape and knowledge gaps. Transl Vis Sci Technol. 2018; 7: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Daiger SP, Sullivan LS, Bowne SJ. Genes and mutations causing retinitis pigmentosa. Clin Genet. 2013; 84: 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hernández-Rabaza V, López-Pedrajas R, Almansa I. Progesterone, lipoic acid, and sulforaphane as promising antioxidants for retinal diseases: a review. Antioxidants (Basel). 2019; 8(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Athanasiou D, Aguila M, Bellingham J, Li W, McCulley C, Reeves PJ, et al.. The molecular and cellular basis of rhodopsin retinitis pigmentosa reveals potential strategies for therapy. Prog Retin Eye Res. 2018; 62: 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Campochiaro PA, Mir TA. The mechanism of cone cell death in retinitis pigmentosa. Prog Retin Eye Res. 2018; 62: 24–37. [DOI] [PubMed] [Google Scholar]
- 6. Nguyen L, Lucke-Wold BP, Mookerjee S, Kaushal N, Matsumoto RR. Sigma-1 receptors and neurodegenerative diseases: towards a hypothesis of sigma-1 receptors as amplifiers of neurodegeneration and neuroprotection. Adv Exp Med Biol. 2017; 964: 133–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hayashi T, Su TP. An update on the development of drugs for neuropsychiatric disorders: focusing on the sigma 1 receptor ligand. Expert Opin Ther Targets. 2008; 12: 45–58. [DOI] [PubMed] [Google Scholar]
- 8. Novakova M, Ela C, Bowen WD, Hasin Y, Eilam Y. Highly selective sigma receptor ligands elevate inositol 1,4,5-trisphosphate production in rat cardiac myocytes. Eur J Pharmacol. 1998; 353: 315–327. [DOI] [PubMed] [Google Scholar]
- 9. Tchedre KT, Huang RQ, Dibas A, Krishnamoorthy RR, Dillon GH, Yorio T. Sigma-1 receptor regulation of voltage-gated calcium channels involves a direct interaction. Invest Ophthalmol Vis Sci. 2008; 49: 4993–5002. [DOI] [PubMed] [Google Scholar]
- 10. Wu Z, Bowen WD. Role of sigma-1 receptor C-terminal segment in inositol 1,4,5-trisphosphate receptor activation: constitutive enhancement of calcium signaling in MCF-7 tumor cells. J Biol Chem. 2008; 283: 28198–28215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Whang WK, Park HS, Ham IH, Oh M, Namkoong H, Kim HK, et al.. Methyl gallate and chemicals structurally related to methyl gallate protect human umbilical vein endothelial cells from oxidative stress. Exp Mol Med. 2005; 37: 343–352. [DOI] [PubMed] [Google Scholar]
- 12. Jiang G, Mysona B, Dun Y, Gnana-Prakasam JP, Pabla N, Li W, et al.. Expression, subcellular localization, and regulation of sigma receptor in retinal Müller cells. Invest Ophthalmol Vis Sci. 2006; 47: 5576–5582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hayashi T, Su TP. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell. 2007; 131: 596–610. [DOI] [PubMed] [Google Scholar]
- 14. Yang ZJ, Carter EL, Torbey MT, Martin LJ, Koehler RC. Sigma receptor ligand 4-phenyl-1-(4-phenylbutyl)-piperidine modulates neuronal nitric oxide synthase/postsynaptic density-95 coupling mechanisms and protects against neonatal ischemic degeneration of striatal neurons. Exp Neurol. 2010; 221: 166–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pal A, Fontanilla D, Gopalakrishnan A, Chae YK, Markley JL, Ruoho AE. The sigma-1 receptor protects against cellular oxidative stress and activates antioxidant response elements. Eur J Pharmacol. 2012; 682: 12–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang J, Shanmugam A, Markand S, Zorrilla E, Ganapathy V, Smith SB. Sigma 1 receptor regulates the oxidative stress response in primary retinal Müller glial cells via NRF2 signaling and system xc(-), the Na(+)-independent glutamate-cystine exchanger. Free Radic Biol Med. 2015; 86: 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Smith SB, Wang J, Cui X, Mysona B, Zhao J, Bollinger KE. Sigma 1 Receptor as a therapeutic target in retinal disease. ProgRetinalEye Res. 67;2018: 130–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang J, Saul A, Roon P, Smith SB. Activation of the molecular chaperone, sigma 1 receptor, preserves cone function in a murine model of inherited retinal degeneration. Proc Natl Acad Sci USA. 2016; 113: E3764–3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yamamoto M, Kensler TW, Motohashi H. The KEAP1-NRF2 system: a thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol Rev. 2018; 98: 1169–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Raghunath A, Sundarraj K, Nagarajan R, Arfuso F, Bian J, Kumar AP, et al.. Antioxidant response elements: discovery, classes, regulation and potential applications. Redox Biol. 2018; 17: 297–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang J, Zhao J, Cui X, Mysona BA, Navneet S, Saul A, et al.. The molecular chaperone sigma 1 receptor mediates rescue of retinal cone photoreceptor cells via modulation of NRF2. Free Radic Biol Med. 2019; 134: 604–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Linker RA, Lee DH, Ryan S, van Dam AM, Conrad R, Bista P, et al.. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain. 2011; 134: 678–692. [DOI] [PubMed] [Google Scholar]
- 23. Ahuja M, Ammal Kaidery N, Yang L, Calingasan N, Smirnova N, Gaisin A, et al.. Distinct Nrf2 signaling mechanisms of fumaric acid esters and their role in neuroprotection against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced experimental Parkinson's-like disease. J Neurosci. 2016; 36: 6332–6351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cho H, Hartsock MJ, Xu Z, He M, Duh EJ. Monomethyl fumarate promotes Nrf2-dependent neuroprotection in retinal ischemia-reperfusion. J Neuroinflammation. 2015; 12: 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zagorski JW, Turley AE, Freeborn RA, VanDenBerg KR, Dover HE, Kardell BR, et al.. Differential effects of the Nrf2 activators tBHQ and CDDO-Im on the early events of T cell activation. Biochem Pharmacol. 2018; 147: 67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tan E, Ding XQ, Saadi A, Agarwal N, Naash MI, Al-Ubaidi MR. Expression of cone-photoreceptor-specific antigens in a cell line derived from retinal tumors in transgenic mice. Invest Ophthalmol Vis Sci. 2004; 45: 764–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008; 3: 1101–1108. [DOI] [PubMed] [Google Scholar]
- 28. Chang B, Hurd R, Wang J, Nishina P. Survey of common eye diseases in laboratory mouse strains. Invest Ophthalmol Vis Sci. 2013; 54: 4974–4981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang J, Saul A, Smith SB. Activation of Sigma 1 Receptor extends survival of cones and improves visual acuity in a murine model of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2019; 60: 4397–4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Navneet S, Zhao J, Wang J, Mysona B, Barwick S, Ammal Kaidery N, et al.. Hyperhomocysteinemia-induced death of retinal ganglion cells: the role of Müller glial cells and NRF2. Redox Biol. 2019; 24: 101199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Prusky GT, Alam NM, Beekman S, Douglas RM. Rapid quantification of adult and developing mouse spatial vision using a virtual optomotor system. Invest Ophthalmol Vis Sci. 2004; 45: 4611–4616. [DOI] [PubMed] [Google Scholar]
- 32. Saul AB, Still AE. Multifocal Electroretinography in the presence of temporal and spatial correlations and eye movements. Vision. 2016; 1: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mezu-Ndubuisi OJ, Taylor LK, Schoephoerster JA. Simultaneous fluorescein angiography and spectral domain optical coherence tomography correlate retinal thickness changes to vascular abnormalities in an in vivo mouse model of retinopathy of prematurity. J Ophthalmol. 2017;9620876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gargini C, Terzibasi E, Mazzoni F, Strettoi E. Retinal organization in the retinal degeneration 10 (rd10) mutant mouse: a morphological and ERG study. J Comp Neurol. 2007; 500(2): 222–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang J, Saul A, Cui X, Roon P, Smith SB. Absence of sigma 1 receptor accelerates photoreceptor cell death in a murine model of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2017; 58(11): 4545–4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen WJ, Wu C, Xu Z, Kuse Y, Hara H, Duh EJ. Nrf2 protects photoreceptor cells from photo-oxidative stress induced by blue light. Exp Eye Res. 2017; 154: 151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jiang D, Ryals RC, Huang SJ, Weller KK, Titus HE, Robb BM, et al.. Monomethyl fumarate protects the retina from light-induced retinopathy. Invest Ophthalmol Vis Sci. 2019; 60: 1275–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kawai SI, Vora S, Das S, Gachie E, Becker B, Neufeld AH. Modeling of risk factors for the degeneration of retinal ganglion cells after ischemia/reperfusion in rats: effects of age, caloric restriction, diabetes, pigmentation, and glaucoma. FASEB J. 2001; 15: 1285–1287. [DOI] [PubMed] [Google Scholar]
- 39. Izuta Y, Imada T, Hisamura R, Oonishi E, Nakamura S, Inagaki E, et al.. Ketone body 3-hydroxybutyrate mimics calorie restriction via the Nrf2 activator, fumarate, in the retina. Aging Cell. 2018; 17(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Promsote W, Powell FL, Veean S, Thounaojam M, Markand S, Saul A, et al.. Oral monomethyl fumarate therapy ameliorates retinopathy in a humanized mouse model of sickle cell disease. Antioxid Redox Signal. 2016; 25: 921–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sakami S, Maeda T, Bereta G, Okano K, Golczak M, Sumaroka A, et al.. Probing mechanisms of photoreceptor degeneration in a new mouse model of the common form of autosomal dominant retinitis pigmentosa due to P23H opsin mutations. J Biol Chem. 2011; 286: 10551–10567. [DOI] [PMC free article] [PubMed] [Google Scholar]