

Abstract
Small cell lung cancer (SCLC) is an aggressive lung cancer subtype with extremely poor prognosis. No targetable genetic driver events have been identified, and the treatment landscape for this disease has remained nearly unchanged for over thirty years. Here, we have taken a CRISPR-based screening approach to identify genetic vulnerabilities in SCLC that may serve as potential therapeutic targets. We used an sgRNA library targeting ~5,000 genes deemed to encode “druggable” proteins to perform loss-of-function genetic screens in a panel of cell lines derived from autochthonous genetically engineered mouse models (GEMMs) of SCLC, lung adenocarcinoma (LUAD), and pancreatic ductal adenocarcinoma (PDAC). Cross-cancer analyses allowed us to identify SCLC-selective vulnerabilities. In particular, we observed enhanced sensitivity of SCLC cells towards disruption of the pyrimidine biosynthesis pathway. Pharmacological inhibition of dihydroorotate dehydrogenase (DHODH), a key enzyme in this pathway, reduced the viability of SCLC cells in vitro and strongly suppressed SCLC tumor growth in human patient-derived xenograft (PDX) models and in an autochthonous mouse model. These results indicate that DHODH inhibition may be an approach to treat SCLC.
One Sentence Summary:
Small cell lung cancer tumors are sensitive to DHODH inhibition, highlighting a potential treatment strategy for this disease.
INTRODUCTION
Small cell lung cancer (SCLC) is an aggressive cancer that is among the deadliest of all solid tumor malignancies. It is characterized by rapid tumor growth and early, widespread metastasis (1), which results in very poor outcomes. Despite decades of research, the combination of platinum and etoposide remains the backbone of SCLC therapy (2). Although initial response rates are high, patients almost invariably relapse. In contrast to the growing number of options for treating non-small cell lung cancer (NSCLC), no new therapies have demonstrated efficacy in SCLC patients (3), highlighting a great need for additional treatments.
Genetic screens have been used to identify and characterize cancer type-specific and genotype-specific vulnerabilities in cancer cells (4). This has provided a useful complementary approach to large-scale cancer genome sequencing studies for identifying new targets for therapy, especially when combined with subsequent functional validation in relevant preclinical models. In this study, we used a focused sgRNA library targeting potentially druggable genes to perform genetic screens in a panel of tumor cell lines derived from autochthonous mouse models of SCLC (5), lung adenocarcinoma (LUAD) (6), and pancreatic ductal adenocarcinoma (PDAC) (7). Through cross-cancer analyses, we identified the de novo pyrimidine biosynthesis pathway as a key vulnerability in SCLC cells. We demonstrate that pharmacological inhibition of DHODH, an enzyme in this pathway, suppresses tumor growth in multiple in vivo models of SCLC, pointing to a potential approach for treating the disease.
RESULTS
SCLC cells are sensitive to disruption of genes involved in de novo pyrimidine synthesis
To identify therapeutically relevant genetic vulnerabilities, we designed an sgRNA library targeting components of the druggable genome (8–10). These include known targets of existing drug compounds (“drugged” genes) as well as genes that belong to gene categories predicted to be druggable (“druggable” genes; Fig. 1, A and B). The library contains 20,160 sgRNAs that target 4,915 mouse genes corresponding to 5,347 human orthologs (Fig. 1C). Cas9-expressing cells were infected with the sgRNA library (fig. S1A) and passaged for 12–15 population doublings (PDs). In this “dropout screen” (11), we focused on genes whose loss was deleterious to cells – cells harboring sgRNAs targeting such genes would be negatively selected and depleted from the final (PD 12–15) cell population compared with the initial (PD 0) cell population.
Fig. 1. SCLC cells are sensitive to disruption of the de novo pyrimidine synthesis pathway.
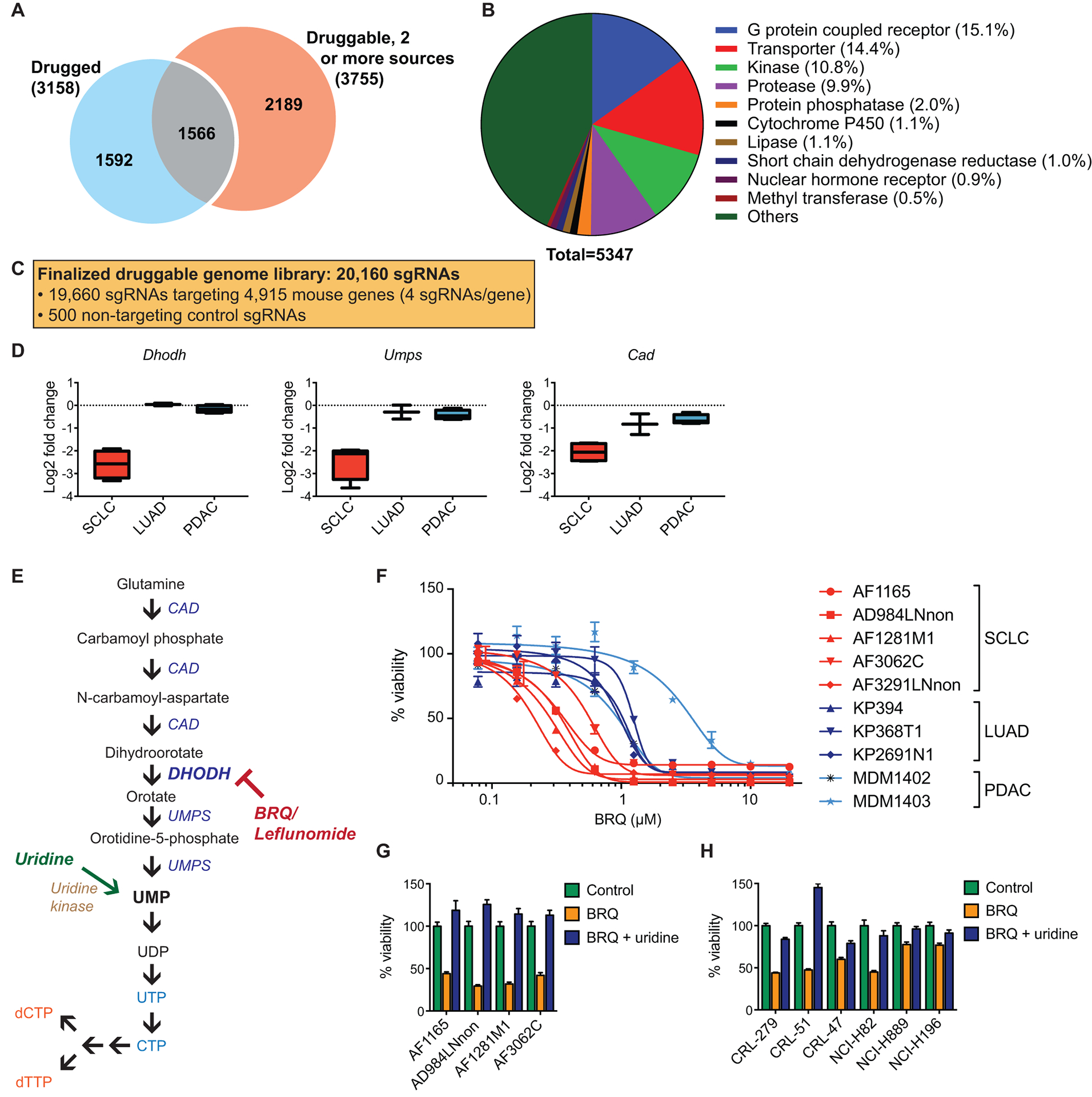
(A) Number of genes in each category in the druggable genome library.
(B) Composition of genes in the druggable genome library by gene category.
(C) Breakdown of the total number of sgRNAs in the druggable genome library.
(D) Gene scores (log2 fold change) for the indicated genes for SCLC (n = 4 biological replicates), LUAD (n = 2 biological replicates), and PDAC (n = 4 biological replicates). Data are presented as median gene scores, with boxes denoting the interquartile range and bars denoting the range.
(E) The pyrimidine nucleotide synthesis pathway.
(F) Dose response curves for brequinar in murine SCLC, LUAD, and PDAC cell lines (n = 4 technical replicates for each sample). Results for each cell line are normalized to control untreated samples. Data are presented as means ± SEM.
(G) Quantification of cell viability in murine SCLC cell lines after treatment with 1 μM brequinar ± 500 μM uridine (n = 4 technical replicates). Data are presented as means ± SEM.
(H) Quantification of cell viability in human SCLC cell lines after treatment with 1 μM brequinar ± 500 μM uridine (n = 4 technical replicates). Data are presented as means ± SEM.
We reasoned that differences in genotype and cell of origin would likely confer different vulnerabilities to different cancer types. Therefore, we performed parallel screens in a panel of cell lines derived from genetically engineered mouse models (GEMMs) of three cancers: SCLC (Trp53−/−; Rb1−/−) (5), LUAD (KrasG12D/+; Trp53−/−) (6), and PDAC (KrasG12D/+; Trp53R172H/−) (7). These models are initiated by well-defined genetic mutations frequently observed in their human counterparts. Notably, loss-of-function mutations in RB1 and TP53 are observed in over 90% of human SCLC tumors (12). To focus on SCLC-specific genetic vulnerabilities, we performed cross-cancer analyses to identify genes whose sgRNAs were preferentially depleted in SCLC cells compared with LUAD and PDAC cells (data file S1). The top candidate gene identified was Dhodh, which encodes dihydroorotate dehydrogenase, an enzyme in the de novo pyrimidine synthesis pathway (Fig. 1D and fig. S1, B and C). In addition, we also observed preferential depletion of sgRNAs targeting Umps and Cad (Fig. 1D), both of which encode multifunctional enzymes catalyzing other steps in de novo pyrimidine synthesis (Fig. 1E). The de novo pyrimidine synthesis pathway generates uridine monophosphate (UMP), which serves as the building block of all pyrimidine ribonucleotides and deoxyribonucleotides required for RNA and DNA synthesis, respectively. UMP is also a precursor for components required for the assembly of various cellular macromolecules, including phospholipids, glycogen, hyaluronic acid, and proteoglycans, as well as for certain post-translational protein modifications (13).
SCLC cells exhibit increased sensitivity to pharmacological inhibition of DHODH
We first sought to validate our finding by pharmacological inhibition of DHODH. Notably, in addition to the de novo pyrimidine synthesis pathway, UMP can also be synthesized by cells through salvage pathways, including phosphorylation of uridine taken up from the environment (Fig. 1E). Standard cell culture medium supplemented with non-dialyzed fetal bovine serum (FBS), which was used in our initial screens, contains variable concentrations of uridine, in some cases approaching the micromolar range (14). Therefore, we performed subsequent experiments in medium supplemented with dialyzed FBS to enable control of uridine concentrations and prevent variable amounts of uridine from confounding interpretation of the results. Using two different inhibitors of DHODH, brequinar (15) and leflunomide (16), we observed that SCLC cell lines displayed higher sensitivity to DHODH inhibition compared with LUAD and PDAC cell lines (Fig. 1F and fig. S1D). The effect of DHODH inhibition was rescued by supplementing the cell culture medium with high concentrations of exogenous uridine (500 μM; Fig. 1G and fig. S1E), arguing that the observed suppression of proliferation results from disruption of the de novo pyrimidine synthesis pathway. Similar results were also observed in a panel of human SCLC cell lines (Fig. 1H).
SCLC cells exhibit lower flux through the de novo pyrimidine synthesis pathway compared with LUAD/PDAC cells
Next, we sought to investigate potential mechanisms underlying SCLC sensitivity towards inhibition of the de novo pyrimidine synthesis pathway. Oncogenic activation of the Rb-E2F pathway has been shown to decrease the concentrations of all rNTPs and dNTPs in cells (17). Consistent with this, we observed smaller UMP pools in the Rb1-null SCLC cells compared with the Rb1-wildtype LUAD/PDAC cells (Fig. 2A). Because increased DHODH activity has been observed in various cancer types (18–22) in which DHODH inhibition by brequinar or leflunomide has demonstrated efficacy, we also assessed the baseline pyrimidine synthesis rate in these cells under regular cell culture conditions using 15N-labeled glutamine tracing. During de novo pyrimidine synthesis, the 15N-amide from 15N-glutamine is incorporated into the pyrimidine ring, so that each molecule of 15N-glutamine gives rise to one molecule of labeled (M+1) UMP. We observed a greater increase in concentrations of newly synthesized (M+1) UMP over time in LUAD/PDAC lines compared with SCLC lines (Fig. 2B), suggesting that SCLC cells exhibit lower flux through the de novo pyrimidine synthesis pathway compared with LUAD/PDAC cells. Collectively, these two factors might explain the increased susceptibility of SCLC cells towards perturbation of the de novo pyrimidine synthesis pathway.
Fig. 2. SCLC cells exhibit lower flux through the de novo pyrimidine synthesis pathway compared with LUAD/PDAC cells.

(A) Baseline uridine monophosphate (UMP) concentrations in untreated SCLC, LUAD, and PDAC cell lines, as measured by liquid chromatography-mass spectrometry (LC/MS). Data are shown as relative amounts normalized to cell number, after absolute quantification using external UMP standards (n = 3 technical replicates per cell line).
(B) Concentrations of newly synthesized UMP (M+1) in untreated SCLC, LUAD, and PDAC cell lines at the indicated time points after the start of 15N-glutamine labeling. Data are shown as relative amounts normalized to pool size/cell (n = 3 technical replicates per condition).
(C, D) Dihydroorotate (C) and N-carbamoyl-aspartate (D) concentrations in SCLC, LUAD, and PDAC cell lines 6 hours after treatment with 1 μM brequinar. Data are normalized to untreated controls and adjusted for cell number (n = 3 technical replicates per cell line).
(E) Fractions of newly synthesized (M+1) and pre-existing (M+0) uridine triphosphate (UTP) in SCLC, LUAD, and PDAC cell lines treated with 1 μM brequinar, at the indicated time points after the start of 15N-glutamine labeling. Data are normalized to total UTP concentrations in each cell line (n = 3 technical replicates per condition).
All data are presented as means ± SEM.
We further examined the effects of brequinar treatment on the different cancer cell lines. After brequinar treatment, metabolite analysis revealed accumulation of dihydroorotate and carbamoyl aspartate, two metabolites upstream of DHODH in the pyrimidine synthesis pathway (Fig. 1E), across all three cancer types (Fig. 2, C and D), confirming on-target inhibition of DHODH by brequinar in both sensitive and insensitive cells. Notably, we observed that the concentrations of both metabolites increased to a greater extent upon brequinar treatment in SCLC cells compared with LUAD and PDAC cells (Fig. 2, C and D), which may suggest more effective inhibition of DHODH by brequinar in SCLC cells compared with LUAD/PDAC cells. In addition, 15N-glutamine tracing in brequinar-treated cells also revealed a greater increase in the fraction of newly synthesized, labeled (M+1) UTP over time in LUAD/PDAC cells compared with SCLC cells (Fig. 2E). Collectively, these data indicate that SCLC cells exhibit lower flux through the de novo pyrimidine synthesis pathway compared with LUAD/PDAC cells, resulting in greater vulnerability of SCLC cells to inhibition of DHODH compared with LUAD/PDAC cells.
LUAD/PDAC cells use alternative pathways to replenish cellular pyrimidine pools
To further validate the greater dependence of SCLC cells on DHODH expression, we performed in vitro competition assays (23) in a subset of the SCLC, LUAD, and PDAC cell lines used in our screen. Cas9-expressing cells were transduced with lentiviral vectors expressing a target sgRNA together with the green fluorescent protein mNeonGreen (fig. S2A), and the percentage of mNeonGreen-positive cells in a mixed population of transduced and untransduced cells was measured over time. In this assay, cells that express sgRNAs targeting genes that are important for cell growth or survival should be rapidly depleted from the mixed population, resulting in a decrease in proportion of mNeonGreen-positive cells over time. We included an sgRNA targeting an intergenic region on chromosome 4 (sgChr4.1) as a negative (neutral) control, and an sgRNA targeting an essential gene, Rpa3 (encoding replication protein A3) (23), as a positive control.
We performed the competition assays in parallel with both dialyzed FBS and regular, non-dialyzed FBS; the latter allowed us to recapitulate the initial screen conditions, as well as to evaluate the effect of more physiological concentrations of uridine on the growth of Dhodh-null cells. All three cell types grew poorly upon Dhodh deletion when cultured in medium with dialyzed FBS (Fig. 3A). Conversely, with regular FBS, Dhodh loss was rapidly selected against in SCLC cell lines, but not in LUAD and PDAC cell lines (fig. S2B), consistent with the results from our initial screens. These results suggest that, while complete inhibition of the de novo pyrimidine synthesis pathway can be deleterious in many proliferating cells, SCLC cells exhibit higher dependence on external salvage of pyrimidines to rescue the effects of DHODH loss compared with LUAD and PDAC cells. This observation led us to investigate whether higher activity of internal pyrimidine nucleotide salvage in LUAD and PDAC cells could explain their lower sensitivity to de novo pyrimidine synthesis pathway inhibition compared with SCLC cells.
Fig. 3. LUAD/PDAC cells use alternative pathways to replenish cellular pyrimidine pools.
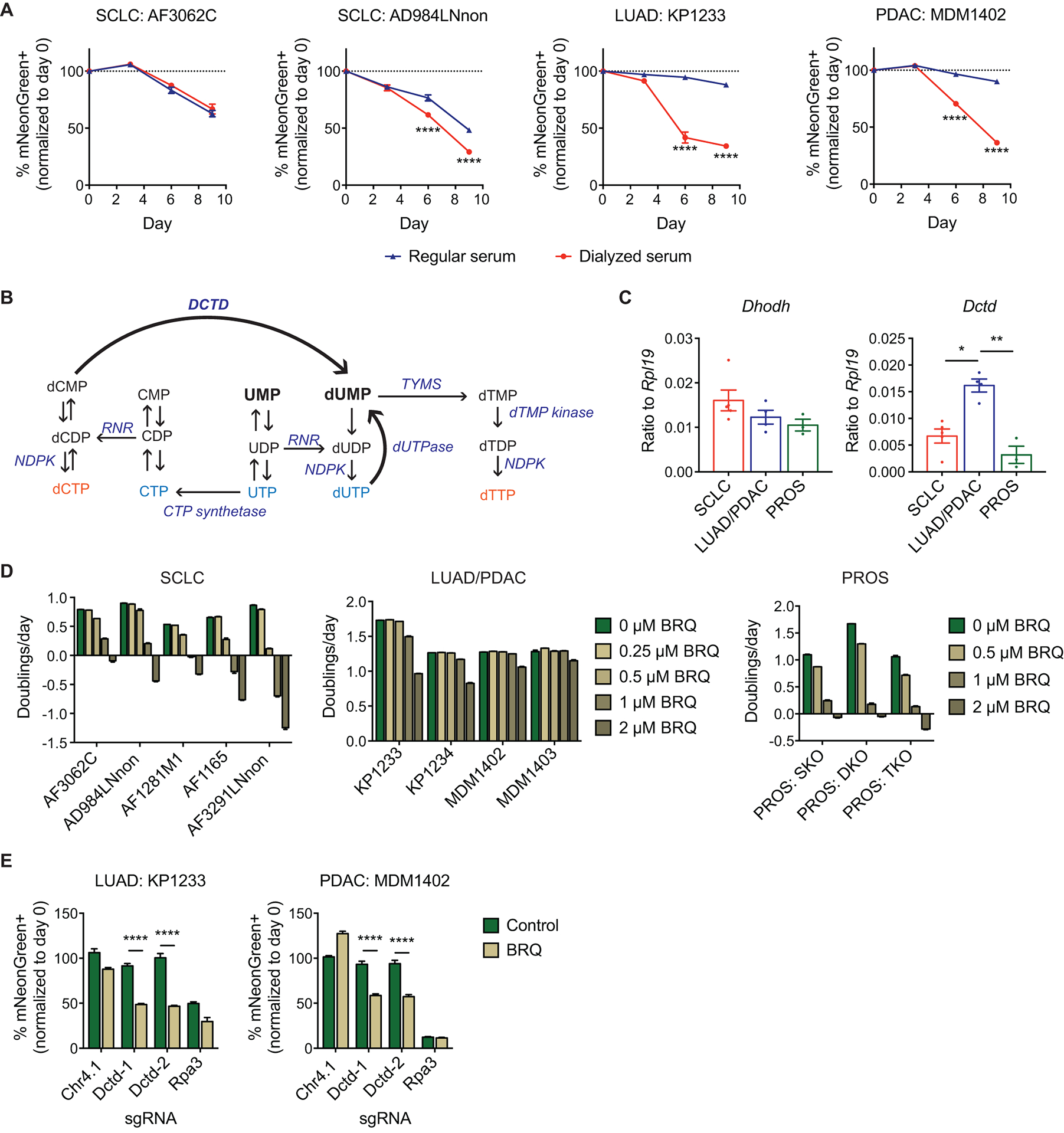
(A) In vitro competition assays for SCLC, LUAD, and PDAC cell lines transduced with sgDhodh, grown in cell culture medium supplemented with either regular (non-dialyzed) serum or dialyzed serum. Data are normalized to the transduction efficiency at day 0 (n = 3 technical replicates for each sample). Data are presented as means ± SEM. **** p<0.0001, 2-way ANOVA with Sidak’s multiple comparison test.
(B) The pyrimidine nucleotide synthesis pathway. RNR: ribonucleotide reductase; TYMS: thymidylate synthetase; NDPK: nucleoside-diphosphate kinase.
(C) RNA expression of Dhodh (left panel) and Dctd (right panel) in SCLC, LUAD/PDAC, and prostate cancer (PROS) cell lines, as assessed by quantitative PCR. Each point represents the mean of 3 technical replicates for one cell line. n = 5 cell lines for SCLC, n = 4 cell lines for LUAD/PDAC (2 each), n = 3 cell lines for PROS. Data are presented as means ± SEM. * p<0.05, ** p<0.01, Mann-Whitney test.
(D) Quantification of population doubling rates for SCLC, LUAD/PDAC, and PROS cell lines treated with the indicated concentrations of brequinar (n = 3 technical replicates per condition). For PROS cell lines, SKO: Pten−/− single knockout prostate adenocarcinoma, DKO: Pten−/−; Rb1−/− double knockout prostate neuroendocrine tumor, TKO: Pten−/−; Rb1−/−; Trp53−/− triple knockout prostate neuroendocrine tumor. Data are presented as means ± SEM.
(E) In vitro competition assays for LUAD (left panel) and PDAC (right panel) cell lines transduced with the indicated sgRNAs, in the absence (green bars) and presence (brown bars) of brequinar (1 μM for KP1233; 2 μM for MDM1402). Data are normalized to the transduction efficiency at day 0 (n = 3 technical replicates for each sample). Control cells were analyzed at day 3, and brequinar-treated cells were analyzed at day 4. Different days were chosen due to the decrease in rate of cell proliferation resulting from brequinar treatment (see main text and fig. S5). Data are presented as means ± SEM. **** p<0.0001, 2-way ANOVA with Sidak’s multiple comparison test.
It has been shown in certain cell types and organisms that an internal salvage pathway serves to replenish intracellular nucleosides and plays a key role in regulating cell growth and proliferation (24, 25). Furthermore, activation of autophagy has been shown to confer resistance to treatment with nucleoside analogues in various cancer types (26–30). Interestingly, in the 15N-glutamine tracing experiments performed with dialyzed FBS and in the absence of brequinar (Fig. 2B), we observed the continued presence of unlabeled UMP/UDP/UTP in LUAD and PDAC cells even after steady state labeling was achieved in these cell lines between 4 and 8 hours after the start of 15N-glutamine labeling (fig. S2C). In addition, after acute brequinar treatment, we observed a transient increase in UMP pools in LUAD and PDAC cell lines (fig. S2D), even though UMP concentrations would be expected to decrease upon inhibition of de novo pyrimidine synthesis. Similar increases were also observed for UDP, CDP, ADP, and GDP in LUAD and PDAC cell lines (fig. S2E), all of which are substrates of ribonucleotide reductase (RNR) for deoxynucleotide synthesis (fig. S2F). Collectively, these results suggest that LUAD and PDAC cells may be more effective at using the internal salvage route as a source of pyrimidine nucleotides, making them less reliant on de novo pyrimidine synthesis.
DCTD expression in cells correlates with sensitivity to DHODH inhibition
In contrast to other dNDPs, dTDP cannot be synthesized directly via RNR, but requires conversion from dUMP through thymidylate synthetase (TYMS) and dTMP kinase (Fig. 3B). We reasoned that this requirement may render dTTP (and hence DNA) synthesis particularly sensitive to decreased pyrimidine production resulting from inhibition of de novo pyrimidine synthesis. Therefore, we focused on deoxycytidylate deaminase (DCTD), an enzyme that supplies the vast majority of dUMP in cells via conversion of dCMP to dUMP (31) (Fig. 3B).
When we queried the Cancer Cell Line Encyclopedia (CCLE) database (32) for the gene expression of several key enzymes involved in de novo pyrimidine deoxynucleotide biogenesis, including CAD, DHODH, UMPS, subunits of RNR (RRM1, RRM2, RRM2B), TYMS, and DCTD, only DCTD exhibited a pattern of gene expression that was consistent with the results from our experiments in murine cell lines (high expression in hLUAD/hPDAC cell lines, low expression in hSCLC cell lines) (fig. S3). In addition, we examined three cell lines derived from prostate cancer GEMMs (PROS) (33), a cancer type that exhibits comparable DCTD expression to SCLC based on the CCLE database (fig. S3B). In these models, Pten loss alone in prostate epithelial cells results in the development of prostate adenocarcinoma (Pten−/− single knockout, or SKO), whereas Pten loss combined with Rb1 loss, with or without Trp53 loss, gives rise to prostate neuroendocrine tumors (Pten−/−; Rb1−/− double knockout, or DKO; Pten−/−; Rb1−/−; Trp53−/− triple knockout, or TKO) (33). Consistent with a potential role of DCTD in determining brequinar sensitivity, all three PROS cell lines expressed low amounts of Dctd (Fig. 3C) and were about as sensitive to brequinar treatment as SCLC cells (Fig. 3D). Interestingly, RB1 mutation status did not affect the sensitivity of the cell lines to brequinar treatment, consistent with what we observed in a panel of RB1-wildtype and RB1-knockout retinal pigment epithelial cells (fig. S4).
To functionally evaluate whether DCTD expression affects sensitivity to de novo pyrimidine synthesis pathway inhibition, we performed competition assays in two LUAD and PDAC cell lines to assess the effect of Dctd loss on cellular fitness, both with and without brequinar treatment. Because brequinar treatment decreased the rate of cell proliferation even in cells transduced with the control sgRNA (fig. S5), we compared the results of the competition assay at different time points for brequinar-treated cells (day 4) versus control cells (day 3). At these respective time points, cells transduced with positive and negative control sgRNAs exhibited comparable percentages of mNeonGreen-positive cells both with or without brequinar treatment (Fig. 3E). Conversely, cells transduced with sgRNAs targeting Dctd did not exhibit any depletion in the absence of brequinar but were depleted from the population in the presence of brequinar (Fig. 3E). Therefore, loss of DCTD appears to sensitize cells to inhibition of the de novo pyrimidine synthesis pathway.
To evaluate DCTD expression across different SCLC tumors, we performed immunohistochemical staining of a human SCLC tissue array (fig. S6A). In contrast to DHODH, which demonstrated a narrow range of expression across different SCLC tumors (fig. S6B), DCTD expression was much more variable across the different tumors (fig. S6C), pointing to the potential for using DCTD expression as a biomarker to identify SCLC tumors that are particularly sensitive to DHODH inhibition.
DHODH inhibition suppresses SCLC tumor progression and extends survival in vivo
DHODH inhibitors are approved for use as immunosuppressive drugs in humans, and these drugs are well-tolerated and have been shown to be effective in some preclinical cancer models, such as acute myeloid leukemia (AML) (34). As shown previously, physiological concentrations of uridine in the serum may limit the effect of DHODH inhibition on cells. Therefore, we assessed the cytotoxic effects of brequinar treatment in medium supplemented with concentrations of uridine spanning the physiological range (35). Consistent with previous experiments, SCLC cells exhibited higher sensitivity to brequinar treatment compared with LUAD and PDAC cells under all conditions tested (fig. S7A).
We next performed preclinical trials of brequinar in an intrasplenic transplant model of SCLC, which causes tumor growth in the liver, a frequent site of metastasis in SCLC patients (fig. S7B). Animals treated with brequinar showed attenuated liver tumor growth (Fig. 4, A to D) and extended survival (Fig. 4E) compared with vehicle-treated animals. To evaluate the potential benefit of combining brequinar treatment with the standard of care for SCLC, we treated animals with a combination of cisplatin and etoposide (fig. S7C). Whereas cisplatin/etoposide alone had little effect on median survival, the combination of brequinar and cisplatin/etoposide further improved survival in these animals compared with brequinar treatment alone (Fig. 4E).
Fig. 4. DHODH inhibition suppresses tumor progression and extends survival in various in vivo models of SCLC.
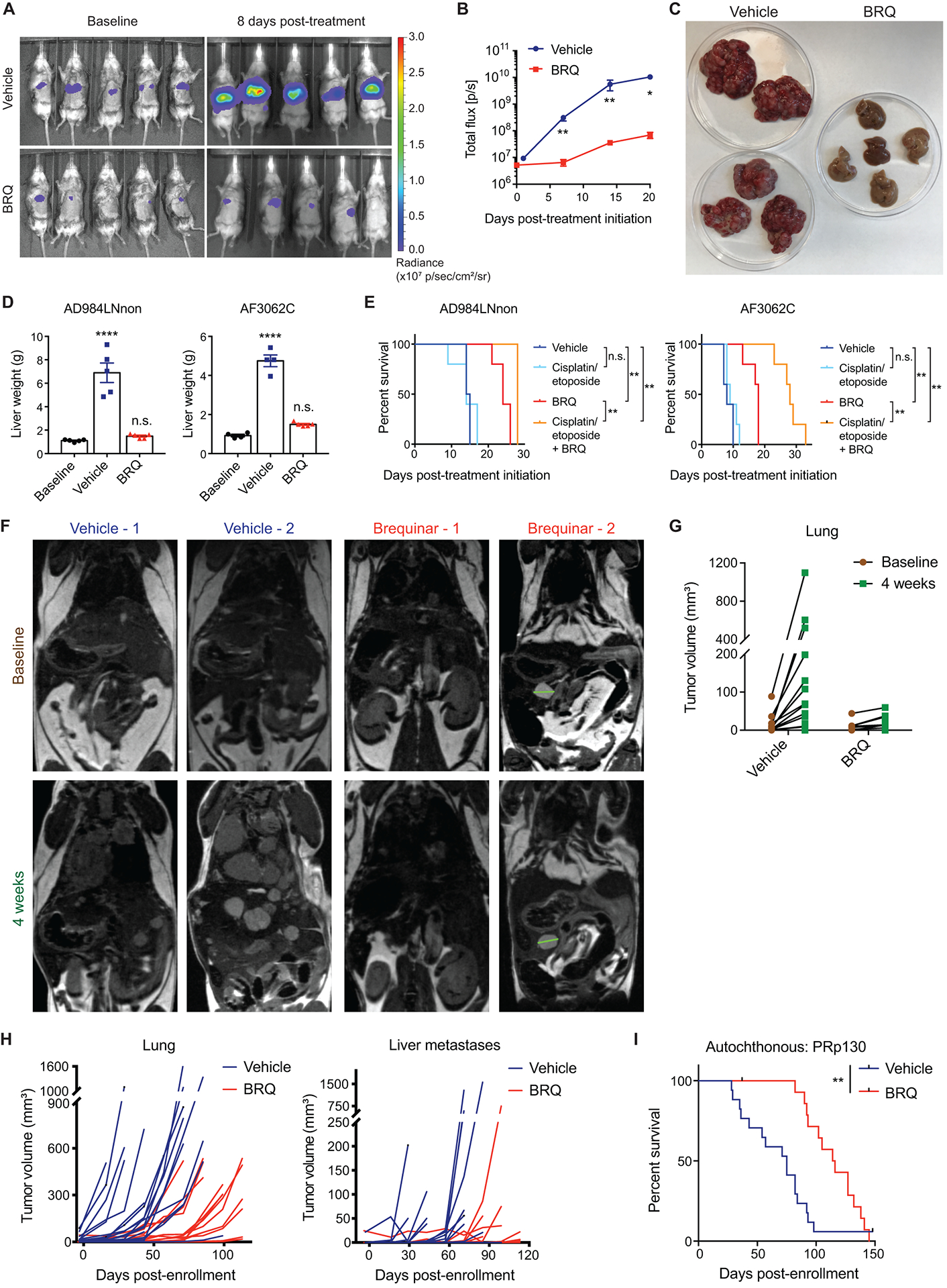
(A) Representative images from in vivo bioluminescence imaging of tumor-bearing animals before the start of drug treatment (left) and after 8 days of treatment with brequinar or vehicle (right).
(B) Quantification of tumor burden (as measured by bioluminescence imaging) at different time points after intrasplenic transplantation of luciferase-expressing AD984LNnon cells (n = 5 for both vehicle-treated and brequinar-treated groups). Data are presented as means ± SEM. * p<0.05, ** p<0.01, two-tailed Student’s t-test.
(C) Representative images of livers harvested from animals 3 weeks after initiation of treatment with brequinar or vehicle. Livers are placed in petri dishes with a diameter of 100 mm. Left: vehicle; right: brequinar.
(D) Quantification of liver tumor burden in animals transplanted with AD984LNnon cells (left panel) or AF3062C cells (right panel) after the indicated treatments, as measured by liver weight at necropsy. For AD984LNnon, n = 5 for all groups. For AF3062C, n = 4 for baseline, n = 4 for vehicle, n = 5 for brequinar. Baseline data were obtained from a separate cohort of animals that was sacrificed 2 weeks after transplantation, before the start of treatment. Treated animals were sacrificed approximately 3 weeks after the start of treatment. Data are presented as means ± SEM. **** p<0.0001, n.s.: not significant, two-tailed Student’s t-test.
(E) Survival analysis in animals transplanted intrasplenically with AD984LNnon cells (left panel) or AF3062C cells (right panel), with the indicated treatments. For AD984LNnon, n = 5 for all treatment groups except for vehicle (n = 4). For AF3062C, n = 5 for all treatment groups. ** p<0.01, log-rank (Mantel-Cox) test.
(F) Representative magnetic resonance imaging (MRI) images of autochthonous PRp130 SCLC animals with detectable tumor burden before treatment (top) and after 4 weeks of treatment (bottom) with vehicle or brequinar.
(G) Quantification of primary tumor burden (as measured by MRI) in autochthonous PRp130 SCLC animals before treatment and after 4 weeks of treatment with vehicle or brequinar (n = 17 for vehicle, n = 15 for brequinar).
(H) Quantification of primary tumor burden (left panel) and metastatic liver tumor burden (right panel) in autochthonous PRp130 SCLC animals throughout the duration of treatment with vehicle or brequinar, as measured by MRI. Each line represents a single animal (n = 17 for vehicle, n = 15 for brequinar). The same cohort of animals was used for survival analysis in (I).
(I) Survival analysis in autochthonous PRp130 SCLC animals with the indicated treatments (n = 17 for vehicle, n = 15 for brequinar). ** p<0.01, log-rank (Mantel-Cox) test.
Next, we tested the efficacy of brequinar treatment in an autochthonous GEMM of SCLC, in which tumors are initiated by conditional deletion of Trp53, Rb1, and p130 in the lung epithelium (36). Assessment of tumor burden in these animals by magnetic resonance imaging (MRI) after treatment initiation revealed attenuated primary tumor progression as well as delayed development of detectable liver metastases in brequinar-treated animals compared with control animals (Fig. 4, F to H). This resulted in an extension of median survival in brequinar-treated animals (115 days) compared with control animals (75 days; Fig. 4I).
Finally, we evaluated the efficacy of brequinar treatment in four human patient-derived xenograft (PDX) models of SCLC, for which responses to chemotherapy (cisplatin/etoposide) have been extensively characterized (37) (Fig. 5A). These four PDX models differed in their expression of DHODH and DCTD (Fig. 5B). Brequinar treatment induced tumor regression and extended time to progression in two of the four models (Fig. 5, C to E). Notably, treatment efficacy in these models appeared to correlate with DCTD expression rather than DHODH expression (Fig. 5B). In addition, the sensitivity of these models to brequinar treatment did not correlate with either chemosensitivity (Fig. 5, C to E; cisplatin/etoposide data were obtained from ref. 37) or the clinical history of chemotherapy exposure (Fig. 5A), suggesting distinct and independent mechanisms of response to brequinar compared with existing SCLC therapies.
Fig. 5. PDX models of SCLC are sensitive to brequinar treatment.
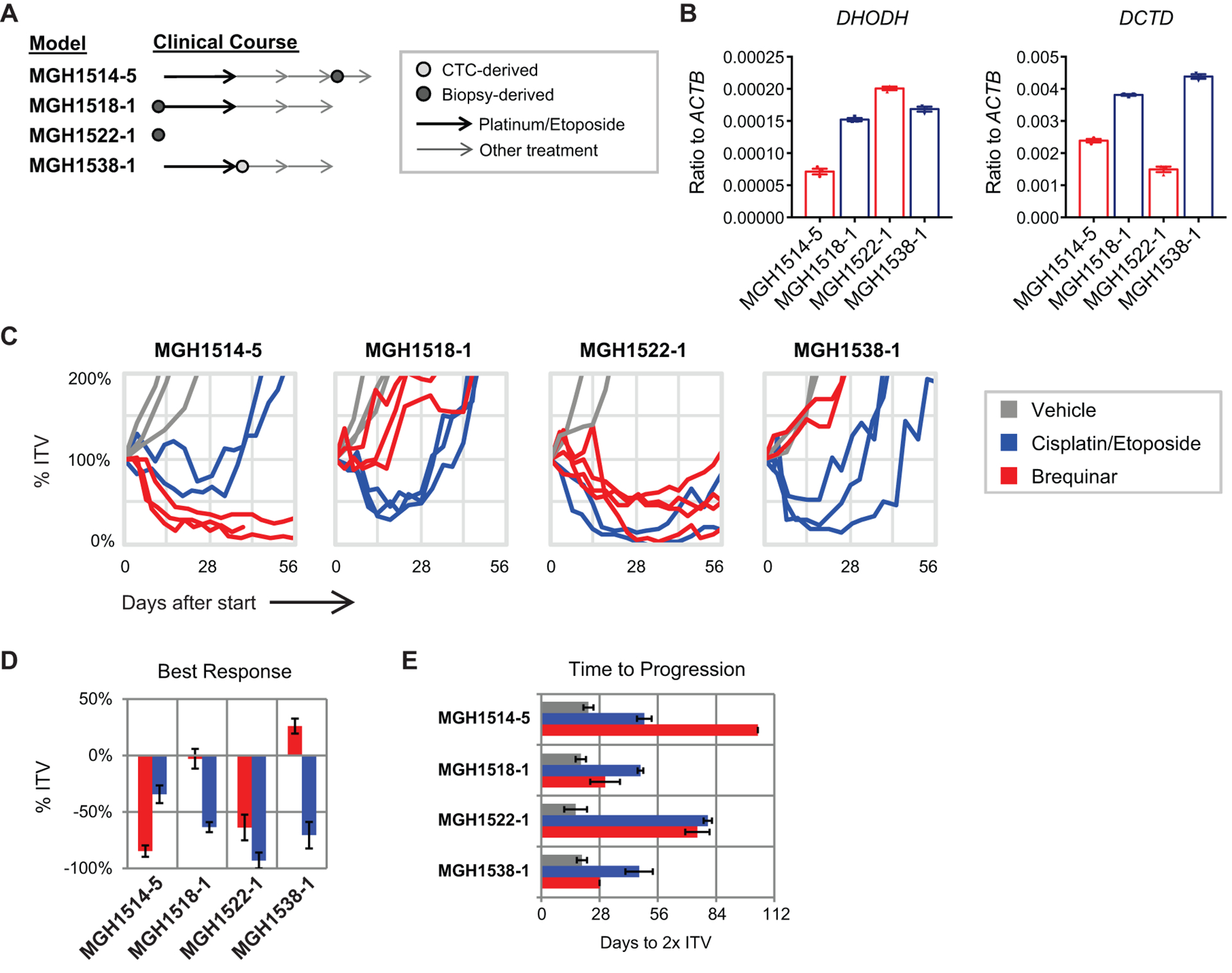
(A) Clinical time point and source of derivation for four SCLC PDX models treated with brequinar. Two models were derived from chemo-naïve patients and two were derived after ≥1 line of therapy.
(B) RNA expression of DHODH (left panel) and DCTD (right panel) in the four PDX models, as assessed by quantitative PCR (n = 3 technical replicates per sample). Data are presented as means ± SEM.
(C) Spider plots of xenograft volume versus time after start of treatment with brequinar, cisplatin/etoposide, or vehicle. Each line represents a single animal. % ITV = % initial tumor volume. Cisplatin/etoposide treatment data were obtained from Drapkin et al., 2018 (37).
(D) Maximum xenograft regression for each model after day +7. Data are presented as means ± SEM.
(E) Time (days) from start of treatment to progression, which is defined as the point at which tumors reach twice of the initial tumor volume (2x ITV). Data are presented as means ± SEM.
DISCUSSION
In the past thirty years, the prognosis for NSCLC patients has greatly improved, largely due to the success of various therapies targeting oncogenic mutations present in NSCLC tumors, such as those in EGFR, ALK, and ROS1 (38). In contrast, SCLC tumors are characterized by inactivating mutations in the tumor suppressor genes TP53 and RB1 (12) and harbor few, if any, actionable oncogenic mutations, thus limiting therapeutic options (39). Although the addition of atezolizumab to carboplatin/etoposide improves overall survival (40), outcomes remain poor, with median overall survival of just over one year, and improved therapeutic strategies for this disease remain a critical need. Here, we applied an unbiased genetic screening approach to identify therapeutic targets for SCLC, uncovering a link between SCLC and pyrimidine metabolism. None of the top candidates identified in our screen exhibited mutation or copy number variation at appreciable frequencies in human SCLC tumors (12), nor were they markedly over- or under-expressed in SCLC compared with other cancer types (32). This highlights the utility of genetic screens for uncovering functional targets that would otherwise not have been identified by genomic or transcriptomic analyses alone.
DCTD-null cells are viable and proliferative in the absence of external salvage (41), suggesting that DCTD is not essential for cell survival. On the other hand, both DCTD enzyme activity (42) and RNA stability (43) increase in rapidly proliferating tissues, including tumors. Our data further suggest that cells with high expression of DCTD are better able to maintain dUMP pools when the de novo pyrimidine synthesis pathway is inhibited (fig. S8). Together with our data demonstrating variability of DCTD expression across different cancer types and different SCLC tumors, these observations collectively point towards the potential utility of DCTD as a biomarker for sensitivity to DHODH inhibition, although this will require future investigation.
RB1 knockout has been shown to decrease nucleotide pool sizes in cells (17). However, we did not observe differences in sensitivity to brequinar treatment as a result of differential RB1 mutation status, whether in a non-cancer cell line (retinal pigment epithelial cells), or in a panel of prostate cancer cell lines (SKO: Rb1-wildtype; DKO, TKO: Rb1-null) (33). Likewise, brequinar sensitivity did not appear to correlate with neuroendocrine differentiation status in the same panel of prostate cancer cell lines (SKO: prostate adenocarcinoma; DKO, TKO: prostate neuroendocrine tumor). These data argue that neither RB1 mutation nor neuroendocrine differentiation status is likely to be the key determinant of brequinar sensitivity.
DHODH inhibition by brequinar or leflunomide has previously demonstrated efficacy in a number of different cancers, either as a monotherapy or in combination with other therapies. These cancer types include AML (34), melanoma (44), triple-negative breast cancer (22), skin tumors (18), mutant KRAS-driven tumors (20), as well as PTEN-deficient breast cancer, glioblastoma, and prostate cancer (21). Our findings demonstrate that SCLC tumors are also selectively sensitive to DHODH inhibition, albeit as a result of unexpected mechanisms, including decreased activity of both the pyrimidine de novo synthesis pathway and the internal salvage route in these tumors, as well as their smaller cellular UMP pools. This provides justification for extending efforts to develop DHODH inhibitors for this indication.
A recent independent study demonstrated selective sensitivity to inhibition of the de novo purine synthesis pathway in the variant subtype of SCLC, which is characterized by low expression of the neuroendocrine transcription factor ASCL1, when compared with the classic ASCL1-high subtype of SCLC investigated in this study (45). It is possible that differential activity of nucleotide synthesis and salvage pathways contributes to the response to purine synthesis inhibitors as well. Whether the variant subtype of SCLC also exhibits sensitivity towards DHODH inhibition, and how differences in molecular subtypes of SCLC may affect their sensitivity to inhibition of different nucleotide synthesis pathways, remain to be further elucidated.
The study presented here uses the DHODH inhibitor brequinar in various in vivo models of SCLC, including an autochthonous mouse model and four PDX models. The 40-day survival benefit conferred by brequinar treatment in the autochthonous SCLC model used here is longer than others reported in such murine SCLC models (46–48). Likewise, we observed that brequinar, as a single agent, matched or outperformed cisplatin and etoposide in two of the four SCLC PDX models tested, even inducing durable tumor regression in a PDX model that is refractory to the current standard of care chemotherapy. Furthermore, the responses of these models to brequinar treatment do not appear to correlate with sensitivity to currently used chemotherapy or a clinical history of chemotherapy exposure. This raises the prospect of using brequinar or other DHODH inhibitors (20, 49, 50) in human SCLC clinical trials, either as a single agent or in combination with standard of care.
We would like to note certain limitations to our study. First, we performed brequinar treatment on a limited set (four) of human PDX models, with brequinar being administered in a first-line, single-agent setting using a previously published dosing schedule optimized for AML (34). Moreover, we have only tested the efficacy of brequinar in the classic subtype of SCLC; whether the variant subtype is sensitive to DHODH inhibition has not been assessed. Therefore, it would be of interest in the future to expand the treatment cohort to a larger panel of human PDX models, including both classic and variant subtypes of SCLC, as well as samples with a wide spectrum of DCTD expression. In addition, it will be important to rigorously examine the efficacy of brequinar or other DHODH inhibitors using different dosing schedules, as well as in combination with the current standard of care, in various preclinical models.
MATERIALS AND METHODS
Study design
The main objective of this study was to identify genetic vulnerabilities in SCLC tumors. We used an sgRNA library to perform genetic screens in four murine SCLC cell lines, two murine LUAD cell lines, and four murine PDAC cell lines. All cell lines were derived from independent tumors from different animals and are therefore biological replicates. Subsequent in vitro validation experiments were also performed in these cell lines, with at least three technical replicates per cell line.
For in vivo validation experiments, sample sizes were determined based on the observed variation in tumor progression from previous studies (36, 37). 4–5 animals per group were used for intrasplenic transplant experiments; at least 10 animals per group were used for experiments involving the autochthonous SCLC model; and 2–4 animals per group were used for experiments involving PDX models. For all experiments, animals were allocated randomly across different treatment groups. For experiments involving the autochthonous SCLC model, this included approximately even distribution based on sex and age. No data were excluded from analyses. For tumor burden quantification by MRI, all image analyses were performed in a blinded fashion, in which the investigator was unaware of the experimental condition while analyzing images for each animal.
Statistical analysis
All statistical analyses were performed using Prism software version 7.02 (GraphPad). P-values for comparisons between two groups were determined by the two-tailed Student’s t-test. P-values for comparisons between multiple groups were determined by 2-way ANOVA with Sidak’s multiple comparison test. P-values for survival analyses were determined by the log-rank (Mantel-Cox) test. For all statistical tests, a p-value of < 0.05 was used to denote statistical significance. All error bars denote the standard error of the mean (SEM), unless otherwise noted in the figure legends.
Supplementary Material
Fig. S1. SCLC cells are preferentially sensitive to loss of DHODH.
Fig. S2. Alternative routes, in addition to de novo pyrimidine synthesis, may contribute to pyrimidine pools in LUAD/PDAC cells.
Fig. S3. Human cancer cell lines exhibit variable expression of key enzymes involved in pyrimidine and deoxynucleotide synthesis.
Fig. S4. RB1 mutation status does not affect sensitivity of RPE cells to brequinar treatment.
Fig. S5. Brequinar treatment decreases cell proliferation in LUAD and PDAC cell lines.
Fig. S6. Human SCLC tumors exhibit variable DCTD expression.
Fig. S7. SCLC cells are sensitive to DHODH inhibition when exposed to physiological concentrations of uridine.
Fig. S8. DCTD expression potentially determines brequinar sensitivity in tumors.
Data file S2. Original data.
Data file S1. Gene scores for all screens performed in this study.
Acknowledgements:
We thank David Sykes, Francisco J. Sánchez-Rivera, Evan Lien, Lucas Sullivan and Alex Muir for helpful scientific discussions and technical advice; Margaret Magendantz for critical reading of the manuscript; the Koch Institute Swanson Biotechnology Center, specifically Wei Huang from the Animal Imaging and Preclinical Testing Core Facility, for in vivo imaging support; the MIT BioMicro Center and the Whitehead Institute Genome Technology Core for sequencing support; and the Whitehead Institute Metabolite Profiling Core Facility for metabolite analysis support.
Funding: This work was supported by the Howard Hughes Medical Institute, the Ludwig Center for Molecular Oncology at MIT, and in part by the Koch Institute Support (core) Grant P30-CA14051 from the National Cancer Institute. L.L. was supported by the Swiss National Science Foundation (SNSF) Early and Advanced Postdoc Mobility Fellowships, and the Hope Funds Postdoctoral Fellowship. S.R.N. was supported by the A*STAR (Agency for Science, Technology and Research, Singapore) National Science Scholarship, and the MIT School of Science Fellowship in Cancer Research. P.P.H. and C.S.N. were supported in part by 2T32CA071345-21A1. A.F.F. and N.J.D. receive NIH grant U01CA220323. M.G.V.H. acknowledges support from SU2C, the MIT Center for Precision Cancer Medicine, R01-CA168653, R01-CA201276, and is also a Howard Hughes Medical Institute Faculty Scholar. T.J. is a Howard Hughes Medical Institute Investigator, the David H. Koch Professor of Biology, and a Daniel K. Ludwig Scholar. M.D.M. is supported by an NCI Mentored Clinical Scientist Research Career Development Award (K08CA208016) and was supported by a KL2/Catalyst Medical Research Investigator Training award (an appointed KL2 award) from Harvard Catalyst | The Harvard Clinical and Translational Science Center (National Center for Research Resources and the National Center for Advancing Translational Sciences) (KL2 TR001100), a Conquer Cancer Foundation-American Society for Clinical Oncology (CCF-ASCO) Young Investigator Award, and the NIH Loan Repayment Program. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Competing interests: T.J. is a member of the Board of Directors of Amgen and Thermo Fisher Scientific. He is also a co-Founder of Dragonfly Therapeutics and T2 Biosystems. T.J. serves on the Scientific Advisory Board of Dragonfly Therapeutics, SQZ Biotech, and Skyhawk Therapeutics. None of these affiliations represent a conflict of interest with respect to the design or execution of this study or interpretation of data presented in this manuscript. Dr. Jacks’s laboratory currently also receives funding from the Johnson & Johnson Lung Cancer Initiative and Calico, but this funding did not support the research described in this manuscript. M.G.V.H. discloses that he is an advisor for Agios Pharmaceuticals, Aeglea Biotherapeutics, and Auron Therapeutics. B.J.D. and N.J.D. receive research funding from Novartis, AstraZeneca, Merck and Abbvie. A.F.F. is consulting for PharmaMar, Abbvie, Loxo, Stemcentrx, Genentech/Roche, Bayer, AstraZeneca, Bristol-Myers Squibb, and Boerhinger Ingelheim, and receives research funding from PharmaMar, AbbVie, AstraZeneca, Bristol-Myers Squibb, Merck, Loxo, Ignyta, Amgen, Genentech/Roche, Bayer and Novartis. P.P.H is a consultant for Auron Therapeutics. M.C.B. is an associate contractor of Anzu Partners. The other authors declare that they have no competing interests.
Data and materials availability: All data associated with this study are available in the main text or the supplementary materials.
REFERENCES AND NOTES
- 1.Califano R, Abidin AZ, Peck R, Faivre-Finn C, Lorigan P, Management of small cell lung cancer: recent developments for optimal care. Drugs 72, 471–490 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Demedts IK, Vermaelen KY, van Meerbeeck JP, Treatment of extensive-stage small cell lung carcinoma: current status and future prospects. Eur Respir J 35, 202–215 (2010). [DOI] [PubMed] [Google Scholar]
- 3.Byers LA, Rudin CM, Small cell lung cancer: where do we go from here? Cancer 121, 664–672 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grimm S, The art and design of genetic screens: mammalian culture cells. Nat Rev Genet 5, 179–189 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Meuwissen R et al. , Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell 4, 181–189 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Jackson EL et al. , Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev 15, 3243–3248 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hingorani SR et al. , Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4, 437–450 (2003). [DOI] [PubMed] [Google Scholar]
- 8.Hopkins AL, Groom CR, The druggable genome. Nat Rev Drug Discov 1, 727–730 (2002). [DOI] [PubMed] [Google Scholar]
- 9.Griffith M et al. , DGIdb: mining the druggable genome. Nat Methods 10, 1209–1210 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wagner AH et al. , DGIdb 2.0: mining clinically relevant drug-gene interactions. Nucleic Acids Res 44, D1036–1044 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lopes R, Korkmaz G, Agami R, Applying CRISPR-Cas9 tools to identify and characterize transcriptional enhancers. Nat Rev Mol Cell Biol 17, 597–604 (2016). [DOI] [PubMed] [Google Scholar]
- 12.George J et al. , Comprehensive genomic profiles of small cell lung cancer. Nature 524, 47–53 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sykes DB, The emergence of dihydroorotate dehydrogenase (DHODH) as a therapeutic target in acute myeloid leukemia. Expert Opin Ther Targets 22, 893–898 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cantor JR et al. , Physiologic Medium Rewires Cellular Metabolism and Reveals Uric Acid as an Endogenous Inhibitor of UMP Synthase. Cell 169, 258–272 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen SF, Ruben RL, Dexter DL, Mechanism of action of the novel anticancer agent 6-fluoro-2-(2’-fluoro-1,1’-biphenyl-4-yl)-3-methyl-4-quinolinecarbo xylic acid sodium salt (NSC 368390): inhibition of de novo pyrimidine nucleotide biosynthesis. Cancer Res 46, 5014–5019 (1986). [PubMed] [Google Scholar]
- 16.Davis JP, Cain GA, Pitts WJ, Magolda RL, Copeland RA, The immunosuppressive metabolite of leflunomide is a potent inhibitor of human dihydroorotate dehydrogenase. Biochemistry 35, 1270–1273 (1996). [DOI] [PubMed] [Google Scholar]
- 17.Bester AC et al. , Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 145, 435–446 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hosseini M et al. , Energy Metabolism Rewiring Precedes UVB-Induced Primary Skin Tumor Formation. Cell Rep 23, 3621–3634 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Bajzikova M et al. , Reactivation of Dihydroorotate Dehydrogenase-Driven Pyrimidine Biosynthesis Restores Tumor Growth of Respiration-Deficient Cancer Cells. Cell Metab 29, 399–416 e310 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koundinya M et al. , Dependence on the Pyrimidine Biosynthetic Enzyme DHODH Is a Synthetic Lethal Vulnerability in Mutant KRAS-Driven Cancers. Cell Chem Biol 25, 705–717 e711 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Mathur D et al. , PTEN Regulates Glutamine Flux to Pyrimidine Synthesis and Sensitivity to Dihydroorotate Dehydrogenase Inhibition. Cancer Discov 7, 380–390 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown KK, Spinelli JB, Asara JM, Toker A, Adaptive Reprogramming of De Novo Pyrimidine Synthesis Is a Metabolic Vulnerability in Triple-Negative Breast Cancer. Cancer Discov 7, 391–399 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zuber J et al. , Toolkit for evaluating genes required for proliferation and survival using tetracycline-regulated RNAi. Nat Biotechnol 29, 79–83 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Y et al. , Autophagy-dependent ribosomal RNA degradation is essential for maintaining nucleotide homeostasis during C. elegans development. Elife 7, e36588 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wei CW et al. , Equilibrative Nucleoside Transporter 3 Regulates T Cell Homeostasis by Coordinating Lysosomal Function with Nucleoside Availability. Cell Rep 23, 2330–2341 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Wu HM, Shao LJ, Jiang ZF, Liu RY, Gemcitabine-Induced Autophagy Protects Human Lung Cancer Cells from Apoptotic Death. Lung 194, 959–966 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Yin H et al. , HMGB1-mediated autophagy attenuates gemcitabine-induced apoptosis in bladder cancer cells involving JNK and ERK activation. Oncotarget 8, 71642–71656 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu J et al. , Gemcitabine induces apoptosis and autophagy via the AMPK/mTOR signaling pathway in pancreatic cancer cells. Biotechnol Appl Biochem 65, 665–671 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Hashimoto D et al. , Autophagy is needed for the growth of pancreatic adenocarcinoma and has a cytoprotective effect against anticancer drugs. Eur J Cancer 50, 1382–1390 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Chen M et al. , The cytoprotective role of gemcitabine-induced autophagy associated with apoptosis inhibition in triple-negative MDA-MB-231 breast cancer cells. Int J Mol Med 34, 276–282 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Reichard P, Interactions between deoxyribonucleotide and DNA synthesis. Annu Rev Biochem 57, 349–374 (1988). [DOI] [PubMed] [Google Scholar]
- 32.Ghandi M et al. , Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ku SY et al. , Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 355, 78–83 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sykes DB et al. , Inhibition of Dihydroorotate Dehydrogenase Overcomes Differentiation Blockade in Acute Myeloid Leukemia. Cell 167, 171–186 e115 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deng Y et al. , An adipo-biliary-uridine axis that regulates energy homeostasis. Science 355, eaaf5375 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schaffer BE et al. , Loss of p130 accelerates tumor development in a mouse model for human small-cell lung carcinoma. Cancer Res 70, 3877–3883 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Drapkin BJ et al. , Genomic and Functional Fidelity of Small Cell Lung Cancer Patient-Derived Xenografts. Cancer Discov 8, 600–615 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thomas A, Liu SV, Subramaniam DS, Giaccone G, Refining the treatment of NSCLC according to histological and molecular subtypes. Nat Rev Clin Oncol 12, 511–526 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Mamdani H, Induru R, Jalal SI, Novel therapies in small cell lung cancer. Transl Lung Cancer Res 4, 533–544 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Horn L et al. , First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N Engl J Med 379, 2220–2229 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Bianchi V, Pontis E, Reichard P, Regulation of pyrimidine deoxyribonucleotide metabolism by substrate cycles in dCMP deaminase-deficient V79 hamster cells. Mol Cell Biol 7, 4218–4224 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maley F, Maley GF, Nucleotide interconversions. II. Elevation of deoxycytidylate deaminase and thymidylate synthetase in regenerating rat liver. J Biol Chem 235, 2968–2970 (1960). [PubMed] [Google Scholar]
- 43.Bernengo MG, Pegoraro L, Deoxycytidylate deaminase activity and its template stability in human acute lymphoblastic and myeloblastic leukaemic cells. Eur J Cancer 12, 611–616 (1976). [DOI] [PubMed] [Google Scholar]
- 44.White RM et al. , DHODH modulates transcriptional elongation in the neural crest and melanoma. Nature 471, 518–522 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang F et al. , Inosine Monophosphate Dehydrogenase Dependence in a Subset of Small Cell Lung Cancers. Cell Metab 28, 369–382 e365 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mollaoglu G et al. , MYC Drives Progression of Small Cell Lung Cancer to a Variant Neuroendocrine Subtype with Vulnerability to Aurora Kinase Inhibition. Cancer Cell 31, 270–285 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meder L et al. , Combined VEGF and PD-L1 Blockade Displays Synergistic Treatment Effects in an Autochthonous Mouse Model of Small Cell Lung Cancer. Cancer Res 78, 4270–4281 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Jahchan NS et al. , Identification and Targeting of Long-Term Tumor-Propagating Cells in Small Cell Lung Cancer. Cell Rep 16, 644–656 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shih KC, Lee CC, Tsai CN, Lin YS, Tang CY, Development of a human dihydroorotate dehydrogenase (hDHODH) pharma-similarity index approach with scaffold-hopping strategy for the design of novel potential inhibitors. PLoS One 9, e87960 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ladds M et al. , A DHODH inhibitor increases p53 synthesis and enhances tumor cell killing by p53 degradation blockage. Nat Commun 9, 1107 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eyre TA, Wright MW, Lush MJ, Bruford EA, HCOP: a searchable database of human orthology predictions. Brief Bioinform 8, 2–5 (2007). [DOI] [PubMed] [Google Scholar]
- 52.Gray KA, Yates B, Seal RL, Wright MW, Bruford EA, Genenames.org: the HGNC resources in 2015. Nucleic Acids Res 43, D1079–1085 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wright MW, Eyre TA, Lush MJ, Povey S, Bruford EA, HCOP: the HGNC comparison of orthology predictions search tool. Mamm Genome 16, 827–828 (2005). [DOI] [PubMed] [Google Scholar]
- 54.Doench JG et al. , Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 34, 184–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sanjana NE, Shalem O, Zhang F, Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11, 783–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Joung J et al. , Genome-scale activation screen identifies a lncRNA locus regulating a gene neighbourhood. Nature 548, 343–346 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dooley AL et al. , Nuclear factor I/B is an oncogene in small cell lung cancer. Genes Dev 25, 1470–1475 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dimitrova N et al. , Stromal Expression of miR-143/145 Promotes Neoangiogenesis in Lung Cancer Development. Cancer Discov 6, 188–201 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Muzumdar MD et al. , Survival of pancreatic cancer cells lacking KRAS function. Nat Commun 8, 1090 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Akama-Garren EH et al. , A Modular Assembly Platform for Rapid Generation of DNA Constructs. Sci Rep 6, 16836 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen S et al. , Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell 160, 1246–1260 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang T, Wei JJ, Sabatini DM, Lander ES, Genetic screens in human cells using the CRISPR-Cas9 system. Science 343, 80–84 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sanchez-Rivera FJ et al. , Rapid modelling of cooperating genetic events in cancer through somatic genome editing. Nature 516, 428–431 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Madisen L et al. , A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13, 133–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sutherland KD et al. , Cell of origin of small cell lung cancer: inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell 19, 754–764 (2011). [DOI] [PubMed] [Google Scholar]
- 66.DuPage M, Dooley AL, Jacks T, Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat Protoc 4, 1064–1072 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Faber AC et al. , Assessment of ABT-263 activity across a cancer cell line collection leads to a potent combination therapy for small-cell lung cancer. Proc Natl Acad Sci U S A 112, E1288–1296 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. SCLC cells are preferentially sensitive to loss of DHODH.
Fig. S2. Alternative routes, in addition to de novo pyrimidine synthesis, may contribute to pyrimidine pools in LUAD/PDAC cells.
Fig. S3. Human cancer cell lines exhibit variable expression of key enzymes involved in pyrimidine and deoxynucleotide synthesis.
Fig. S4. RB1 mutation status does not affect sensitivity of RPE cells to brequinar treatment.
Fig. S5. Brequinar treatment decreases cell proliferation in LUAD and PDAC cell lines.
Fig. S6. Human SCLC tumors exhibit variable DCTD expression.
Fig. S7. SCLC cells are sensitive to DHODH inhibition when exposed to physiological concentrations of uridine.
Fig. S8. DCTD expression potentially determines brequinar sensitivity in tumors.
Data file S2. Original data.
Data file S1. Gene scores for all screens performed in this study.