

Abstract
Purpose
Retinitis pigmentosa GTPase regulator (RPGR)-related X-linked retinitis pigmentosa is associated with one of the most severe phenotypes among inherited retinal disease. The aim of this study was to investigate Clustered Regularly Interspaced Short Palindromic Repeat/Cas9-mediated gene editing therapy in a mouse model of Rpgr.
Methods
The Rpgr−/yCas9+/WT male mice were used for this study. At 6 months of age, they received a single subretinal injection of adeno-associated virus vectors carrying sgRNA and donor template separately, and therapeutic effect was examined after 1, 6, and 12 months.
Results
Rpgr knockout mouse showed slow but progressive age-related retinal degeneration, which emulates the disease occurring in humans. Significant photoreceptor preservation was observed in the treated part of the retina, in sharp contrast to the untreated part of the retina in the same eye after 6 and 12 months. It was surprising that precise modification at the target locus as demonstrated by genomic DNA sequencing in the post-mitotic photoreceptor was observed. Moreover, the therapeutic effect lasts for up to 12 months and no off-target effects were shown.
Conclusions
Our study strongly demonstrates that gene editing therapy is a promising therapeutic strategy to treat inherited retinal degeneration.
Keywords: retinitis pigmentosa, gene editing therapy, retinitis pigmentosa GTPase regulator, adeno-associated viral, CRISPR/Cas9
Mutations in the retinitis pigmentosa GTPase regulator (RPGR) gene are associated with X-linked retinitis pigmentosa (XLRP),1 which accounts for 10% to 20% of all retinitis pigmentosa (RP) cases and is one of its most severe forms.2 Approximately 75% of all XLRP cases are caused by mutations in RPGR ORF15,3 which is a highly repetitive and purine-rich region, thus considered a hotspot for mutations.4
Animal models of RPGR-XLRP reported to date include two naturally occurring canine models,5 the naturally occurring retinal degeneration 9 mouse,6 and two genetically engineered Rpgr knockout (Rpgr KO) mouse models,7 which all show a distinct phenotype of RP. Gene replacement therapy has been proven effective in rescuing retinal structure and function in a number of animal models of retinal degeneration8 and in clinical trials for Leber congenital amaurosis and choroideraemia.9 Gene augmentation also showed remarkable preservation of retinal structure and function in Rpgr mutant mice and canine models.10 As both loss-of-function and gain-of-function were evident due to RPGR mutation, gene replacement is not suitable for all kinds of RPGR mutations.
Gene editing therapy using the Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR)/Cas9 system is a promising alternative to tackle RPGR gain-of-function mutations.11 Other gene therapy methods not only add a functional or partially functional copy of a gene to the patient's cells, but also retain the original dysfunctional copy of the gene. Unlike these, the CRISPR/Cas9 system can generate a precise modification at a target locus and entirely eliminate the defective DNA portion.12 CRISPR/Cas9 gene editing system has been demonstrated to be effective in autosomal dominant RP caused by rhodopsin,13 autosomal recessive RP caused by CEP290,14 but has not yet been proven in XLRP animal models with relatively common and currently incurable retinal degeneration, such as RPGR mutations.
As none of the reported animal models of RPGR-XLRP are available for gene editing therapy, we generated a new mouse model with a 5-bp deletion at exon eight of Rpgr. Cre-dependent Cas9 knock-in mice were crossed with Rpgr KO mice to obtain Rpgr−/yCas9+/WT male mice. With this endogenously expressed Cas9 mouse model, we examined the possibility that CRISPR/Cas9-mediated gene editing therapy based on homology-directed repair (HDR) in nondividing photoreceptor cells.
Methods
Animals
All mice were bred and maintained at the Peking University Health Science Center Animal Care Services Facility in Specific Pathogen Free (SPF) conditions under a 12-hour light/12-hour dark cycle. Food and water were available ad libitum. All experiments were performed in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. All animals were maintained in accordance with the guidelines of the Association for the Assessment and Accreditation of Laboratory Animal Care.
Rpgr KO mice (n = 72) used in this study were custom designed and obtained from the Shanghai Model Organisms Center, Inc. (Shanghai, China). The sgRNAs targeting the exon eight of Rpgr gene were designed. The mRNA of in vitro transcribed Cas9 and sgRNA were injected into zygotes of C57BL/6J mice, and the obtained founder mice were validated by PCR and sequencing using the following primer pairs: RPGR-test-F1, 5′- GAGAGTCCGCATAAGAAAGTAACA -3′; RPGR-test-R1, 5′- AACGCCCCCTAGTGGTCAAAG -3′.
Cre-dependent Cas9 knock-in mice (n = 6) were procured from Beijing Biocytogen Co. (Beijing, China). They were generated by inserting a Cas9 transgene expression cassette into the Rosa26 locus, as described previously.15 The transgene was interrupted by a loxP-stop-loxP (LSL) cassette from the CAG promoter and could not express Cas9 without Cre recombinase. The genotype of the Cas9 mice was validated by PCR. Cre-dependent Cas9 knock-in homozygous males were crossed with Rpgr KO female mice to obtain Rpgr−/yCas9+/WT male mice, which were used for the subsequent gene correction study. The wild type (WT) mice used in the study were C57BL/6J (n = 16).
Construction and Production of Adeno-Associated Virus (AAV) Vectors
All AAV vectors were generated by Beijing IDMO (Beijing, China). The sgRNA targeted to the 5-bp deletion in Rpgr KO mice was designed with the sequence GATCCTTAATACGCTCAATT and cloned into the vector pD632-Cre-2A-DsRed-U6. The 5′ and 3′ homology arms were amplified from C57BL/6J mouse genome, after digestion with NotI-AscI (NEB, Ipswich, MA, USA). The RPGR-5′HA and RPGR-3′HA overlap was subcloned into pD632-Cre-2A-DsRed-MCS plasmid. The Cre recombinase was used to delete the LSL cassette and to induce Cas9 expression. The AAV vectors were packaged with serotypes 2/8, which were proven to transduce mouse photoreceptors efficiently and were generated by Beijing FivePlus Molecular Medicine Institute (Beijing, China).
Subretinal Injection
Rpgr −/yCas9+/WT mice (n = 32) were treated on postnatal day 180 (P180) with AAV vectors injected subretinally in the right eye of each mouse as described previously,10 and the left eye for control. Each mouse was injected with 1 µL of AAV vector at a concentration of 1 × 1012 vector genomes per milliliter. Animals with no apparent surgical complications were retained for further evaluation. A total of 26 eyes after treatment met this criterion. Following all injections, 1% atropine eye drops and neomycin-polymyxin B-dexamethasone ophthalmic ointments were applied.
Histology and Immunofluorescence Study
Six or 12 months after treatment, the animals were euthanized, and the eyes were harvested. For immunofluorescence and morphometric studies, a blue dye was used to mark the orientation of the eye before enucleation. The incision point and optic nerve were horizontally aligned to ensure that both treated (nasal quadrant) and untreated areas (temporal quadrant) were in the same section. The eyes were sectioned at a thickness of 7 µm using a cryostat. For histologic analyses, sections were collected at regular intervals from approximately 24 sites per eye, stained with hematoxylin and eosin, and imaged (Nikon, Tokyo, Japan). Images from the nasal and temporal sides of the optic nerve head were montaged digitally and straightened. The outer nuclear layer (ONL) thickness was estimated by outlining the boundaries of the outer plexiform layer and outer limiting membrane, and measuring their distance at regular intervals of approximately 0.2 mm.
As higher formaldehyde concentrations were found to quench the RPGR signal, the in situ detection of RPGR in the connecting cilia was conducted as described previously with some modifications.10 Briefly, eyes were embedded in the optical coherence tomography compound without fixation and quick-frozen in liquid nitrogen. Cryosections were cut at 10 µm and collected without fixation on pretreated glass slides. Sections were stored at −80 °C and used within 2 to 3 days. Just before use, sections were fixed on slides for 2 minutes with 1% formaldehyde in phosphate-buffered saline (PBS) at pH 7.0. Sections were then washed once in PBS, followed by immunofluorescence staining. Immunofluorescence staining was conducted using a previously described method.16 The primary antibodies used in this study were polyclonal rabbit anti-RPGR antibody (1:100 dilution, HPA001593; Sigma-Aldrich Corp., St. Louis, MO, USA), monoclonal mouse anti-protein kinase C α-antibody (1:200 dilution, ab11723; Abcam, Cambridge, MA, USA), biotinylated peanut agglutinin (PNA; 1:200 dilution, B-1075; Vector, Burlingame, CA, USA), polyclonal rabbit anti-M-cone opsin (1:200 dilution, AB5405; Millipore, Burlington, MA, USA), and polyclonal rabbit anti-S-cone opsin (1:200 dilution, AB5407; Millipore). Secondary antibodies included goat anti-rabbit conjugated with Alexa Fluor 488 (Life Technologies, Grand Island, NY, USA), goat anti-mouse conjugated with Alexa Fluor 568 (Life Technologies), and rhodamine (TRITC)-conjugated streptavidin (1:500 dilution, 123126; Jackson ImmunoReseach Laboratories, West Grove, PA, USA) for PNA. Two previously described methods, PNA and M/S-cone, were used to identify cone photoreceptors.17
Electroretinogram (ERGs)
ERGs of treated and controls (n = 3 for each age) were recorded using the Espion E2 recording system (Diagnosis LLC, Lowell, MA, USA) according to the International Society for Clinical Electrophysiology of Vision (ISCEV) standard.18 ERG measurements were recorded after 1, 6, or 12 months of treatment.
Fundus Photography
Mice were anesthetized, pupils were dilated with 1% topical atropine (Alcon Laboratories, Fort Worth, TX, USA), and the cornea was anesthetized with topical Alcaine (0.5% proparacaine hydrochloride; Alcon Laboratories). Methylcellulose solution (2%) was applied to the eyes for corneal hydration. To screen for changes in ocular appearance, a Micro III small animal retinal imaging system from Phoenix Research Laboratories (Pleasanton, CA, USA) was used. Mice were placed on the stage of the imaging system, and fundus images were viewed and photographed using the Micro III software.
DNA Analyses of Rpgr−/yCas9+/WT Mice
After 1, 6, or 12 months of treatment, the mice were euthanized. Genomic DNA of the retinas was isolated using the Blood & Tissue Kit (Qiagen, Frankfurt, Germany) according to the manufacturer's protocol. PCR products containing the homologous arms were amplified with PrimeSTAR HS DNA Polymerase (Takara Bio, Inc., Shiga, Japan), followed by TA cloning using ZT4-Blunt cloning kit (Beijing Zoman Biotechnology Co., Ltd., Beijing, China). PCR products for targeted deep sequencing were prepared as described in HI-TOM Kit19 (Novogene, Beijing, China). Mixed samples were sequenced on the Illumina HiSeq platform (www.hi-tom.net/hi-tom/documentation.html). Genomic region flanking the target site and the 12 off-target sites, which were predicted using the CRISPR Design Tool,20 were PCR-amplified using the primers listed in Supplementary Table S1.
Statistical Analyses
Differences in ONL thickness by genotype were evaluated by two-tailed unpaired Student's t-test, with correction for unequal group variances where relevant. GraphPad Prism 6 (GraphPad Software, La Jolla, CA, USA) was used for statistical analyses.
Results
Generation of Rpgr KO Mouse Model
For the generation of the Rpgr KO mouse model, we designed two sgRNAs targeting exon eight of the Rpgr gene (Fig. 1A). The mRNA of in vitro transcribed Cas9 and sgRNA were injected into zygotes of C57BL/6J mice. Sanger sequencing analyses showed that 10 out of 32 offspring harbored Rpgr mutations (Fig. 1B). Mice with a 5-bp deletion (c.974_978delAAATT; p.K325Nfs*1) in exon eight (Fig. 1C) were chosen for the next study. This mutation, bearing premature translation termination, behaved as a null allele. Expression and localization of Rpgr was subsequently analyzed on WT mice, which showed rod-like immunoreactivity with brighter small spots at the connecting cilium. No staining of Rpgr was detected in Rpgr KO retinas (Fig. 1D). Thus the 5-bp deletion led to inactivation of Rpgr. From a species comparison21 perspective, a 6-month-old mouse approximates to a human in their 30s, which corresponds to the approximate age of the onset of visual acuity decline. A 12- to 13-month-old mouse approximates to a human in their mid-40s, that is, the approximate age of legal blindness induced by RPGR mutation. An 18-month-old mouse is considered to be old-aged. Thus these three ages were chosen for our gene correction study.
Figure 1.

Illustration of Rpgr KO mouse model. (A) Schematic representation of the two Cas9/gRNA-targeting sites in exon eight of mouse Rpgr gene. Targeted genomic sites are indicated in blue. Protospacer adjacent motif (PAM) sequences are marked in red. Exons are indicated by closed boxes. (B) Sanger sequencing analyses of PCR products from DNA isolated from tail biopsies of F0 mice. Number of offspring obtained after transplant into surrogate mothers and mutants generated is indicated. Targeted genomic sites are indicated in red. Inserted nucleotides are shown in blue. (C) Representative Rpgr genomic sequence analyses of PCR products from DNA isolated from tail biopsies of WT mice (upper) and hemizygous mice (lower). (D) Immunostaining of Rpgr in 7-µm fixed retina sections from WT and Rpgr KO mice at 6 months of age. INL, inner nuclear layer; IS, inner segment; OS, outer segment; Scale bar: 50 µm.
Photoreceptor Degeneration in Rpgr KO Mice
Delayed and slow retinal degeneration has been reported in genetically engineered Rpgr KO mice.7,10 A careful phenotypic analysis was conducted in the 5-bp deletion mouse model. The retinal fundi of 3-, 6-, and 12-month-old WT and Rpgr KO mice were imaged using a Micro III fundus camera (Fig. 2A). Until 3 months of age, multiple, uniformly distributed, small yellow-white spots appeared in all quadrants of the retinal fundi of Rpgr KO mice. Accompanying photoreceptor degeneration, these spots become bigger and confluent. By 12 months of age, these spots become sparse, but pigment deposition was apparent.
Figure 2.

Morphological change in the retinae of Rpgr KO mice. (A) Fundus images of Rpgr KO mice at 3, 6, and 12 months and WT controls. Multiple, small yellow-white spots appeared in all quadrants of Rpgr KO mice. Spots became bigger and more confluent with age. (B) Histologic sections of Rpgr KO mice and WT controls. A gradual loss in the number of photoreceptor cells in the ONL was identified with age in Rpgr KO mice. (C) Thickness measurements were taken on sections from nasal to temporal and plotted at defined distances from the optic nerve head (n = 3 animals per group), Scale bar: 50 µm, Error bars represent SD. (D) Representative dark- and light-adapted ERG waveforms of Rpgr KO mice at 3, 6, and 12 months and WT controls. (E) ERG intensity-response of dark-adapted a-wave, b-wave, and light-adapted b-wave in Rpgr KO mice at 6 months (n = 12) compared with age-matched C57 BL/6 mice (n = 6). GCL, ganglion cell layer, INL, inner nuclear layer.
The retinal sections of WT and Rpgr KO mice were evaluated histologically. Until 3 months of age, the retinal morphology of Rpgr KO mice was comparable to that of WT mice, with a subtle aging-related loss of photoreceptor cells in the ONL at 6 months of age. By 12 months of age, the loss of photoreceptor cells became pronounced in the Rpgr KO mice (Fig. 2B). Measurements of ONL thickness across the retina along the horizontal (nasal-temporal) meridian through the optic nerve head for 3-, 6-, and 12-month-old Rpgr KO mice were plotted and are shown in Figure 2C. There were significant differences in the ONL thickness between WT and mutant mice across most of the sampled retinas (Fig. 2C). Concomitant with morphological degeneration, retinal function as measured by ERG also exhibited abnormalities. ERG waveforms for representative WT and Rpgr KO mice at 3, 6, and 12 months of age are shown in Figure 2D. At 3 months, dark-adapted ERGs in the Rpgr KO mice were not remarkably different from those in the WT mice, but the b-wave amplitudes were reduced. At 6 months, ERG amplitudes, including a-wave and b-wave amplitudes, reduced further, with almost no recordable ERG a-wave and b-wave response at 12 months of age (Fig. 2E).
Abnormal Photoreceptor Protein Expression in Rpgr KO Mice
Although retinal cell loss was not apparent in young Rpgr KO retinas, staining for retinal cell markers, which are downregulated during retinal degeneration, indicated that degenerative changes were already underway. At 3 months of age, rhodopsin, M-cone opsin, S-cone opsin, and PNA expression in Rpgr KO mice showed outer-segment labeling similar to that of WT mice, except for the diffused intensity. In 6-month-old Rpgr KO mice, rhodopsin, M-opsin, and S-opsin expression decreased dramatically, and only sparse PNA staining were visible, which was consistent with the decreased outer-segment integrity and protein stability. In 12-month-old Rpgr KO mice, PNA and S-opsin staining were further decreased, and only occasional rhodopsin and M-opsin expression was observed (Fig. 3). The immunohistochemical analysis of phototransduction proteins and PNA expression in Rpgr KO mice showed progressive loss with increasing age.
Figure 3.
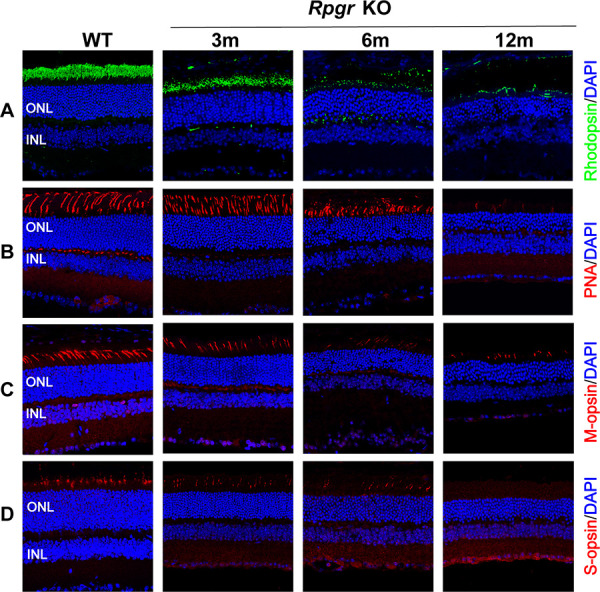
Photoreceptor degeneration in Rpgr KO mice. Immunofluorescent staining of rhodopsin (A), PNA (B), M-cone opsin (C), and S-cone opsin (D) in Rpgr KO mice. All staining was substantially reduced in 12-month-old Rpgr KO mice. PNA, M-cone, and S-cone staining are shown in red. Rhodopsin staining is shown in green. Nuclei are stained blue by DAPI. Scale bar: 50 µm. INL, inner nuclear layer.
Retinal morphology and function of 3-, 6-, 12-, and 24-month-old Rpgr KO mice showed slow but progressive age-related retinal degeneration. Prominent degenerative changes began at 6 months of age, which indicates that this model could emulate the progressive retinal degeneration occurring in humans. Thus this Rpgr KO mouse model provides an excellent animal model system for gene editing therapy study.
Photoreceptor Preservation Following Cas9-Mediated Gene Editing Therapy
Expression cassettes of sgRNA targeted to the mutant Rpgr locus and donor template were delivered to 6-month-old Rpgr−/yCas9+/WT mice by two separate AAV2/8 vectors (Figs. 4A–D). To determine whether the retinal phenotype was altered in treated mice, retinal morphology was assessed 6 months after treatment. Extensive and robust retinal photoreceptor preservation was observed in the treated part of the retina. Up to nine layers of rescued photoreceptors were visible, in sharp contrast to the only four layers of loosely arranged photoreceptor nuclei in the untreated area of the same eye (Figs. 5A, 5B). Photoreceptor rescue was observed exclusively in transfected regions, but not in the untreated regions, with these two areas markedly separated by an injury scar. The ONL of the treated area was markedly thicker than that of the untreated area. The density of photoreceptors in the treated area was 316 nuclei/100 µm, which was 1.5-fold greater than that of the untreated area (128 nuclei/100 µm; Fig. 5C). Immunofluorescence analyses showed that prominent Rpgr expression was observed at the junction between the inner and outer segments of the photoreceptors, indicating staining of the connecting cilia in the treated retina, whereas no staining was identified in the untreated retina. Both PNA and M-opsin expression in the treated retina of the Rpgr KO mice showed outer-segment labeling similar to that of 6-month-old untreated Rpgr KO mice, whereas subtle staining was identified in the untreated retina (Fig. 5D).
Figure 4.

HDR-mediated targeted integration in the retina of Rpgr−/yCas9+/WT mouse. (A) Schematic representation of the Cas9/gRNA-targeting site in exon eight of Rpgr KO mice. Targeted genomic sites are indicated in blue. Protospacer adjacent motif (PAM) sequences are marked in red. Exons are indicated by closed boxes. (B) Schematic overview of HDR-mediated gene editing strategy at the mutant Rpgr locus. (C) Experimental timeline for Rpgr gene correction in Rpgr KO mouse model. (D) Fluorescein shows AAV vectors distribution following unilateral subretinal injection in Rpgr KO mice at 6 months of age. HAL/HAR, left/right homology arm.
Figure 5.

Rescue of retinal structure in Rpgr−/yCas9+/WT mouse 6 months after treatment. Hematoxylin and eosin staining of retinal section of Rpgr KO mouse following vector treatment. The arrow in the left marked the injection site. (B) After treatment for 6 months, extensive and robust retinal photoreceptor preservation was observed in the treated part of the retina, with up to nine layers of rescued photoreceptors, in sharp contrast to the four layers in the untreated area in the same eye. (Upper) Magnified images of the marked areas in (A) are shown. Scale bar: 50 µm, (Lower) Magnified images of the marked areas in (A) are shown. Scale bar: 10 µm. (C) Quantitative analysis of treated and untreated part of ONL thickness from nasal-temporal retinal sections across the optic nerve head. The density of photoreceptors in the treated area was 1.5-fold more than that in the untreated part of the retina. Two-tailed paired t-test was used for statistical analysis. *P < 0.05. Error bars represent SD. INL, inner nuclear layer. (D) Restoration of Rpgr protein expression and higher numbers of PNA and M-opsin expressing cells in Rpgr−/yCas9+/WT mouse following treatment for 6 months. Rpgr are shown in green, with PNA and M-opsin staining shown in red, and nuclei are stained blue.
To determine whether the rescued phenotype could last for a long period, retinal morphology was assessed 12 months after treatment. At this time point, Rpgr staining and prominent PNA staining spanned about three-quarters of the cross-section (Figs. 6A, 6B). The density of photoreceptors in the treated area was 261 nuclei/100 µm, which was three-fold greater than that of the untreated area (87 nuclei/100 µm; Fig. 6C). Both PNA and M-cone expression in the treated area of the Rpgr KO mice were similar to that of 6-month-old untreated Rpgr KO mice, with rhodopsin expression similar to that of 3-month-old untreated Rpgr KO mice (Fig. 6D). However, we did not detect the expression of S-cone in either the treated or the untreated area of the Rpgr KO mice, which can be explained by the phenotype of no staining of S-cone in the 12-month-old untreated Rpgr KO mice. These data suggested that CRISPR/Cas9-mediated Rpgr gene editing therapy prevented photoreceptor degeneration, and that this treatment persisted for a long time.
Figure 6.

Rescue of retinal structure in Rpgr−/yCas9+/WT mouse 12 months after treatment. (A–B) Rpgr and PNA staining of retinal sections of Rpgr KO mouse following 12 months treatment. Rpgr and PNA staining is shown in red, and nuclei are stained blue. (C) Quantitative analysis of treated and untreated part of ONL thickness from nasal-temporal retinal sections. The density of photoreceptors in the treated area was three-fold more than that in the untreated part of the retina. Two-tailed paired t-test was used for statistical analysis. *P < 0.05. Error bars represent SD. INL, inner nuclear layer. (D) More numbers of ID4, PNA, and M-opsin expressing cells in Rpgr−/yCas9+/WT mouse following treatment for 12 months. ID4 was shown in green with PNA, and M-opsin staining shown in red, and nuclei are stained blue.
DNA Level Precise Modification Following Cas9-Mediated Gene Editing Therapy
After treatment for 1 and 6 months, retinas were cut into 4 equal pieces, referred to as L1, L2, L3, L4, and A1, A2, A3, A4, respectively, followed by genomic DNA extraction for Sanger sequencing. Sanger sequencing of TA cloning for PCR products encompassing the sgRNA target site of A3 indicated that 86% (30/35) of all the clones were correctly repaired, whereas the repair proportion after 1 month was 0% (0/19; Figs. 7A, 7B). To determine on-target and potential off-target events, deep sequencing was performed using HI-TOM Kits19 (Novogene, Beijing, China). The efficiency reached up to 96.18% and 94.36% for A3 and A4, showing a higher on-target repair rate 6 months after treatment, compared with those at 1 month after treatment, which showed no correct repair (Supplementary Table S1). We did not find sequence alterations in any of the 12 predicted potential off-target sites (Supplementary Tables S2 and S3). These analyses suggest that the mutant Rpgr DNA was partially corrected via CRISPR/Cas9-mediated gene editing therapy, which subsequently permitted proper Rpgr protein formation and trafficking, and finally aided photoreceptor survival.
Figure 7.

DNA sequencing analyses in Rpgr−/yCas9+/WT mouse 1 and 6 months after treatment. (A) Sequences of genomic DNA of four parts (L1, L2, L3, and L4, treated site located in L3) from treated retina 1 month after treatment. Red line shows the 5-bp deletion site. No correction was noticed. (B) Sequences of the genomic DNA of four parts (A1, A2, A3, and A4, treated site located in A3) from retina following treatment for 12 months. Red rectangle shows the nearly WT sequence in A3 and A4 parts of the retina. Sequencing analyses of clones of A3 part retina showed that 30 out of 35 clones (86%, highlighted in red) were edited correctly. Scale bar: 50 µm. INL, inner nuclear layer.
Discussion
In the present study, we demonstrated the therapeutic efficacy of AAV-CRISPR/Cas9-mediated gene editing therapy via HDR in a RPGR-XLRP mouse model in which SpCas9 is endogenously expressed under the control of Cre. The Rpgr KO mice were treated with subretinal injection of the AAV2/8-donor and AAV2/8-sgRNA, after treatment for 6 or 12 months, prominent rescue of photoreceptors, with thicker ONL and improved expression of markers, were shown in the treated part of the retinas, and mutation was correctly edited as demonstrated by genomic DNA sequencing in the post-mitotic photoreceptor. These observations demonstrate the efficiency of gene editing therapy in the treatment of hereditary retinal degeneration.
Gene replacement therapy is a technology with potential to provide immediate benefits to patients with monogenic retinal dystrophies,10 however, the complexity and particularity of the RPGRORF15 gene hinders its clinical application for the reasons listed next. First, mutations in the ORF15 may translate into truncated proteins, some of which may present a gain-of-function phenotype.22 Second, the RPGRORF15 has poor sequence stability and it may fail to maintain sequence integrity during AAV production, leading to a dominant-negative effect and accelerating the progression of the disease.22 Thus codon-optimized RPGR gene replacement therapy would be a better alternative.23 Finally, the RPGRORF15 gene shows complex post-transcriptional processing24 and inadvertent processing, such as splicing, may occur on the episomal AAV transgene “intronless” primary mRNA transcript. All these factors prompted us to explore gene editing therapy for RPGR.
In vivo genome editing therapy has long been considered an ideal strategy for permanent correction of monogenic disorders and is advancing rapidly since the advent of CRISPR/Cas9 technology.25 One of the key features of precise genome editing, in contrast to viral-vector-based gene replacement, is that endogenous promoters, regulatory elements, and enhancers can be preserved to mediate intrinsic spatiotemporal gene expression. Therapeutic application of CRISPR/Cas9 has shown promising outcomes in animal models of several devastating human diseases, including cystic fibrosis,26 Duchenne muscular dystrophy,12 and hereditary tyrosinemia.27 One of the main challenges observed in the use of CRISPR/Cas9 is the possibility of cleavage at sites in other regions of the genome that share sequence homology with the target locus of interest (off-target effects).28 Inherited retinal diseases could be an ideal target for in vivo CRISPR/Cas9 application, as the retina is easily accessible surgically and is isolated by the blood–retinal barrier.29 In addition, genome modifications can be targeted to specific cell types, and a low number of vectors carrying CRISPR/Cas9 should be sufficient for disease correction. The AAV2/8 vector also specifically targets photoreceptor cells. High cellular levels of Cas9 protein would increase the likelihood for off-target cleavages.30,31 To limit the duration and expression of Cas9, in this study, we adapted a Cre-dependent CRISPR/Cas9 inducible system, which controlled Cas9 expression through Cre recombinase. We found that the ONL in Rpgr−/yCas9+/WT mice following treatment were much thicker than the untreated region after 6 and 12 months, which will benefit clinical translation in the future. When it comes to clinical gene editing therapy, two AAV delivery systems, with one to deliver Cas9 and the other to deliver sgRNA and donor template, should be involved. Delivery of spCas9 via AAV has been reported in 2015,14 to simulate the clinical treatment, we used two AAV systems to deliver sgRNA and HDR template separately in this study and achieved successful gene editing on target cells.
For in vivo AAV-mediated gene editing therapy of retinal disease, the gene editing efficiency differed greatly between different research group, from 4 days13 to approximately 4 to 8 weeks after treatment.14,32,33 In this study, no correction was identified after treatment for 1 month, but significant correction was observed after 6 months. This may be attributed to the low editing efficacy of the HDR method we used, and thus longer treatment will be needed to evaluate the effect. The on-target repair efficiency of A3 and A4 (96.18% and 94.36%) is higher than A1 and A2 (81.6% and 66.14%) after 6-month treatment, the different repair rate is because of the different treated area at one injection. As for the off-target effects, we chose the top 12 off-target sites predicted using the CRISPR Design Tool,20 and found no sequence alterations in any of the predicted potential off-target sites by sanger sequencing. For clinical translation in the future, a stepwise approach should be added to discover and verify the potential off-target site as mentioned in CEP290 gene editing therapy.14
Precise genome editing via CRISPR/Cas9 technology relies on HDR. The CRISPR-mediated gene correction efficacy for eye diseases may occur at a rate that is not sufficiently high for therapeutic benefit, as photoreceptors are in a post-mitotic state and would not be in the correct cell cycle phase for HDR to occur.34 Surprisingly, DNA sequencing revealed that correct genome editing occurred in the photoreceptors, and the Rpgr protein was expressed in the rescued part of the retina. This is consistent with a previous study that showed that genome editing via HDR is possible in mature post-mitotic neurons by combining CRISPR/Cas9 meditated DNA cleavage and the efficient delivery of donor template with AAV.35 New gene editing strategy, such as homology-independent targeted integration33 or “base editing,”36 was also demonstrated recently, which is also a promising alternative to HDR.
Conclusions
In this study, DNA-level precise gene correction was achieved by coinjection of sgRNA and HDR template with AAV2/8 vectors in Rpgr−/yCas9+/WT mice, it can rescue retinal structure and function, and thus ameliorate further degradation in the treated part of the retina. Our studies therefore provide novel insight into precise treatment of retinal degenerative diseases.
Data Availability
The data that support the findings of this study are available from the corresponding author on reasonable request. Next-generation sequences from this study can be found online at https://www.ncbi.nlm.nih.gov/sra/SRP131541.
Supplementary Material
Acknowledgments
The authors thank Hongliang Dou for his helpful insight when developing the project and animal operation, Li Chen for crucial assistance in animal breeding, and Xiaobing Wu for production of AAV vectors.
Supported by the National Natural Science Foundation of China (81470666, 81770966) (LY), and Clinical Key Project of Peking University Third Hospital (BYSY2014004) (LY). The disclosed funders had no role in study design, data collection and analyses, decision to publish, or preparation of the manuscript.
Disclosure: S. Hu, None; J. Du, None; N. Chen, None; R. Jia, None; J. Zhang, None; X. Liu, None; L. Yang, None
References
- 1. Ayyagari R, Demirci FY, Liu J, et al.. X-linked recessive atrophic macular degeneration from RPGR mutation. Genomics. 2002; 80: 166–171. [DOI] [PubMed] [Google Scholar]
- 2. Demirci FYK, Rigatti BW, Wen G, et al.. X-linked cone-rod dystrophy (locus COD1): identification of mutations in RPGR exon ORF15. American J Hum Genet. 2002; 70: 1049–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Iannaccone A, Wang X, Jablonski MM, et al.. Increasing evidence for syndromic phenotypes associated with RPGR mutations. Am J Ophthalmol. 2004; 137: 785–786. [DOI] [PubMed] [Google Scholar]
- 4. Vervoort R, Lennon A, Bird AC, et al.. Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat Genet. 2000; 25: 462–466. [DOI] [PubMed] [Google Scholar]
- 5. Zhang Q, Acland GM, Wu WX, et al.. Different RPGR exon ORF15 mutations in Canids provide insights into photoreceptor cell degeneration. Hum Mol Genet. 2002; 11: 993–1003. [DOI] [PubMed] [Google Scholar]
- 6. Thompson DA, Khan NW, Othman MI, et al.. Rd9 is a naturally occurring mouse model of a common form of retinitis pigmentosa caused by mutations in RPGR-ORF15. PLoS One. 2012; 7: e35865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hong D-H, Pawlyk BS, Shang J, Sandberg MA, Berson EL, Li T. A retinitis pigmentosa GTPase regulator (RPGR)-deficient mouse model for X-linked retinitis pigmentosa (RP3). Proc Natl Acad Sci USA. 2000; 97: 3649–3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boye SE, Boye SL, Lewin AS, Hauswirth WW. A comprehensive review of retinal gene therapy. Mol Ther. 2013; 21: 509–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. MacLaren RE, Groppe M, Barnard AR, et al.. Retinal gene therapy in patients with choroideremia: initial findings from a phase 1/2 clinical trial. Lancet. 2014; 383: 1129–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wu Z, Hiriyanna S, Qian H, et al.. A long-term efficacy study of gene replacement therapy for RPGR-associated retinal degeneration. Hum Mol Genet. 2015;24: 3956–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moreno AM, Mali P. Therapeutic genome engineering via CRISPR-Cas systems. Wiley Interdiscip Rev Syst Biol Med. 2017; 9: e1380. [DOI] [PubMed] [Google Scholar]
- 12. Long C, McAnally JR, Shelton JM, Mireault AA, Bassel-Duby R, Olson EN. Prevention of muscular dystrophy in mice by CRISPR/Cas9–mediated editing of germline DNA. Science. 2014; 345: 1184–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bakondi B, Lv W, Lu B, et al.. In vivo CRISPR/Cas9 gene editing corrects retinal dystrophy in the S334ter-3 rat model of autosomal dominant retinitis pigmentosa. Mol Ther. 2016; 24: 556–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Maeder ML, Stefanidakis M, Wilson CJ, et al.. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat Med. 2019; 25: 229. [DOI] [PubMed] [Google Scholar]
- 15. Platt RJ, Chen S, Zhou Y, et al.. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell. 2014; 159: 440–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Russell S, Bennett J, Wellman JA, et al.. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. 2017; 390: 849–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fu Y, Foden JA, Khayter C, et al.. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013; 31: 822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McCulloch DL, Marmor MF, Brigell MG, et al.. ISCEV standard for full-field clinical electroretinography (2015 update). Doc Ophthalmol. 2015; 130: 1–12. [DOI] [PubMed] [Google Scholar]
- 19. Liu Q, Wang C, Jiao X, et al.. Hi-TOM: a platform for high-throughput tracking of mutations induced by CRISPR/Cas systems. bioRxiv. 2018:235903. [DOI] [PubMed] [Google Scholar]
- 20. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013; 8: 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Flurkey K, Currer JM.. Pitfalls of animal model systems in ageing research. Best Pract Res Clin Endocrinol Metab. 2004; 18: 407–421. [DOI] [PubMed] [Google Scholar]
- 22. Hong D-H, Pawlyk BS, Adamian M, Li T. Dominant, gain-of-function mutant produced by truncation of RPGR . Invest Ophthalmol Vis Sci. 2004; 45: 36–41. [DOI] [PubMed] [Google Scholar]
- 23. Fischer MD, McClements ME, de la Camara CM-F, et al.. Codon-optimized RPGR improves stability and efficacy of AAV8 gene therapy in two mouse models of X-linked retinitis pigmentosa. Mol Ther. 2017; 25: 1854–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Megaw RD, Soares DC, Wright AF. RPGR: its role in photoreceptor physiology, human disease, and future therapies. Exp Eye Res. 2015; 138: 32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cox DB, Platt RJ, Zhang F. Therapeutic genome editing: prospects and challenges. Nat Med. 2015; 21: 121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schwank G, Koo B-K, Sasselli V, et al.. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell. 2013; 13: 653–658. [DOI] [PubMed] [Google Scholar]
- 27. Yin H, Xue W, Chen S, et al.. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat Biotechnol. 2014; 32: 551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cho SW, Kim S, Kim Y, et al.. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014; 24: 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Taylor AW. Ocular immune privilege. Eye. 2009; 23: 1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hsu PD, Scott DA, Weinstein JA, et al.. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013; 31: 827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lin Y, Cradick TJ, Brown MT, et al.. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 2014; 42: 7473–7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ruan GX, Barry E, Yu D, Lukason M, Cheng SH, Scaria A. CRISPR/Cas9-mediated genome editing as a therapeutic approach for Leber congenital amaurosis 10. Mol Ther. 2017; 25: 331–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Suzuki K, Tsunekawa Y, Hernandez-Benitez R, et al.. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature. 2016; 540: 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Heyer W-D, Ehmsen KT, Liu J. Regulation of homologous recombination in eukaryotes. Ann Rev Genet. 2010; 44: 113–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nishiyama J, Mikuni T, Yasuda R. Virus-mediated genome editing via homology-directed repair in mitotic and postmitotic cells in mammalian brain. Neuron. 2017; 96: 755–768.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016; 533: 420. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author on reasonable request. Next-generation sequences from this study can be found online at https://www.ncbi.nlm.nih.gov/sra/SRP131541.