

Abstract
Purpose
To study cone structure and function in patients with retinitis pigmentosa (RP) owing to mutations in rhodopsin (RHO), expressed in rod outer segments, and mutations in the RP-GTPase regulator (RPGR) gene, expressed in the connecting cilium of rods and cones.
Methods
Four eyes of 4 patients with RHO mutations, 5 eyes of 5 patients with RPGR mutations, and 4 eyes of 4 normal subjects were studied. Cone structure was studied with confocal and split-detector adaptive optics scanning laser ophthalmoscopy (AOSLO) and spectral-domain optical coherence tomography. Retinal function was measured using a 543-nm AOSLO-mediated adaptive optics microperimetry (AOMP) stimulus. The ratio of sensitivity to cone density was compared between groups using the Wilcoxon rank-sum test.
Results
AOMP sensitivity/cone density in patients with RPGR mutations was significantly lower than normal (P < 0.001) and lower than patients with RHO mutations (P < 0.015), whereas patients with RHO mutations were similar to normal (P > 0.9).
Conclusions
Retinal sensitivity/cone density was lower in patients with RPGR mutations than normal and lower than patients with RHO mutations, perhaps because cones express RPGR and degenerate primarily, whereas cones in eyes with RHO mutations die secondary to rod degeneration. High-resolution microperimetry can reveal differences in cone degeneration in patients with different forms of RP.
Keywords: adaptive optics scanning laser ophthalmoscopy, microperimetry, retinitis pigmentosa, cones
Retinitis pigmentosa (RP) refers to a heterogeneous group of inherited diseases caused by mutations in at least 87 genes (https://www.omim.org/phenotypicSeries/PS268000) causing progressive, relentless vision loss due to retinal degeneration. Some of the first symptoms patients with RP notice are night blindness and peripheral visual field loss. As the disease progresses, patients also experience reduced visual acuity and eventual blindness because not only rod but also cone photoreceptor cells die. RP is the leading cause of hereditary blindness in developed countries, and its worldwide prevalence is 1:3000, although this may be an underestimation for individual populations.1,2
RP is most commonly inherited in three ways: autosomal dominant (30%–40% cases), autosomal recessive (50%–60% cases), or X-linked (5%–15% cases), although mitochondrial inheritance also occurs.2 Mutations in the genes most commonly associated with RP include RP-GTPase regulator (RPGR; 10%–20% of cases) and rhodopsin (RHO; 8%–10% of cases).3–6 Mutations in these genes typically cause rod, then cone, photoreceptor loss. For patients with RHO mutations, there are two classes of disease expression: class A patients have severe night blindness early in life, whereas class B patients can have little or no night vision loss, and relatively preserved rod thresholds and electroretinographic responses.7 Patients with RPGR mutations experience early onset night blindness, visual field loss onset is evident earlier, at approximately age 10 years, and significant visual impairment occurs at approximately age 30 to 40 years.8
The mechanism of degeneration responsible for these two forms of RP is different. Rhodopsin localizes to the outer segments (OS) of rods and RHO mutations often cause protein misfolding and retention in the endoplasmic reticulum, leading to cellular stress and eventual cell death.6 In patients with RHO mutations, even though rhodopsin is expressed in rods only, the cones eventually also die, perhaps due to intercellular connections,9 nutrient deficiency,10 oxidative stress,11 or loss of neurotrophic factors produced by rods, such as rod-derived cone viability factor.12,13 RPGR is localized to the connecting cilium of both rods and cones. The connecting cilium is crucial for regulating the flow of proteins from the inner segment (IS) to the OS, and RPGR mutations affect intracellular protein trafficking, which compromises photoreceptor function and survival.6
The current study tests the hypothesis that macular cone structure and function are significantly different in patients with mutations in RHO compared with patients with mutations in RPGR at similar stages of disease progression assessed by visual field and optical coherence tomography (OCT) images of retinal structure. The goal of this study was to compare cone structure and function in patients with RHO mutations and patients with RPGR mutations using high-resolution measures of photoreceptor structure and function.
Methods
Study Design
Research procedures followed the tenets of the Declaration of Helsinki. Informed consent was obtained from all subjects. The study protocol was approved by the institutional review boards of the University of California, San Francisco and the University of California, Berkeley.
Patients with RP owing to mutations in RHO or RPGR were compared with age-similar normal subjects. Subjects were excluded if they had conditions that could affect imaging, including unsteady fixation, cataract, amblyopia, and foveal cystoid macular edema. Genetic testing was performed on patients with X-linked RP and autosomal-dominant RP through the eyeGENE research consortium,14 or using a next-generation sequencing panel testing between 181 and 266 genes associated with retinal dystrophy through the genetic testing study of My Retina Tracker, an online registry for patients with inherited retinal degenerations (NCT02435940).
Clinical Examination
Refractive error and best-corrected visual acuity were measured according to the Early Treatment of Diabetic Retinopathy Study (ETDRS) protocol15 using a standard illuminated eye chart. Axial length was measured using partial coherence interferometry (IOL Master; Carl Zeiss Meditec, Dublin, CA, USA). After visual acuity and axial length measurements, pupils were dilated with tropicamide 1% and phenylephrine 2.5%.
Structural Measures
Spectral-Domain (SD) OCT
SD-OCT (Spectralis HRA+OCT system; Heidelberg Engineering, Vista, CA, USA) images were acquired, including 15, 20, or 30 degree horizontal cross section B-scans through the fovea. The manufacturer's automated retinal tracking feature was used to acquire and average 100 B-scans to increase the signal-to-noise ratio of the horizontal B-scan through the fovea. Manual segmentation was performed using custom software to measure OS and IS thickness lengths.16–21 The OS thickness was measured from the OS/RPE band to the IS/OS junction, and the IS thickness was between the IS/OS band and the external limiting membrane. An example of a segmented SD-OCT B-scan using this method for a patient eye with retinal degeneration can be found in Figure 1 and in a previous work.22
Figure 1.

AOSLO split-detector and confocal images with AOMP test locations superimposed and SD-OCT B-scans beneath each AOSLO/AOMP image. Images are from a normal (40154) subject (A), a patient (40095) with a RHO mutation (B), and a patient (40064) with a RPGR mutation (C). Retinal sensitivity values in color-coded circles are shaded from green (normal) to red (stimulus not seen) based on the sensitivity measured at each location; color scale bar at left of panel; scale bar: 200 µm. The inner black circle represents the actual stimulus size, and the bigger colored circle is 10 times larger to increase its visibility in the figure. The B-scans show interpolated boundaries based on manual segmentation of the RPE/Bruch's membrane, outer segment-RPE (OS/RPE), IS/OS, external limiting membrane, inner nuclear layer/outer plexiform layer, inner plexiform layer/inner nuclear layer, retinal nerve fiber layer/retinal ganglion cell, and vitreous/nerve fiber layer borders.17,18,20,21,44
Adaptive Optics Scanning Laser Ophthalmoscopy (AOSLO)
A noninvasive, high resolution AOSLO imaging system was used to acquire simultaneous confocal22–27 and nonconfocal images28 as have been described previously. The specific system employed a 940-nm light for wavefront sensing and a 840-nm light for imaging and tracking the retina. All light sources were drawn from a supercontinuum light source (SuperK EXTREME; NKT Photonics, Birkerod, Denmark) and coupled into single-mode fibers using a custom-built fiber-coupling optical system. Wavefront measurements were made with a custom-built Shack Hartman wavefront sensor. Aberration correction was done with a 97 actuator continuous membrane deformable mirror (DM97; ALPAO, Montbonnot-Saint-Martin, France).
The benefit of a combined confocal and nonconfocal AOSLO system is that it allows for confocal imaging of direct backscattered light from waveguiding cones (generally a combination of light scattered at the IS/OS junction and from the cone OS tips29,30) and nonconfocal imaging of multiply scattered and refracted light from the IS28,31 at the same location simultaneously. Nonconfocal split-detection allows the scattered light from cone IS to be observed even in cones in which the OSs are nonwaveguiding.28,32
Videos at 512 × 512 pixels were acquired at 30 frames per second over a 1.2 × 1.2 degree field of view on the retina (approximately 360 × 360 micrometer field). High signal-to-noise images were generated from each video after correcting for eye movements in each video frame.33 Images from a series of overlapping locations along the horizontal meridian were stitched together into a single montage using custom software (Automontage; https://github.com/BrainardLab/AOAutomontaging).34
Functional Measures
Adaptive Optics Microperimetry
To assess cone function, stimuli were delivered via AOSLO by modulating the scanning light source in a technique called adaptive optics microperimetry (AOMP) (Fig. 1). A combination of high speed fundus imaging and tracking with an infrared beam and visible stimulus delivery in real-time was used to measure sensitivity at select locations with the AOSLO system.35 This approach allowed for imaging and delivering light stimuli to small groups of cone photoreceptors to test localized regions in the retina and measure sensitivity thresholds. AOMP has a delivery error of 0.89 arcmin, which is less than the cone-to-cone spacing at all eccentricities beyond 1 degree, and is approximately 5.5 times better than tracking errors of standard fundus-guided microperimeters.35 A wavelength of 543 nm was chosen to measure cone photoreceptor sensitivity thresholds because it is equally sensed by both long- and medium-wavelength-sensitive cones.36 The tests were made against a background light comprised of the AOSLO imaging wavelength of 840 nm, the wavefront sensing wavelength of 910 nm, and a small amount of 543-nm light, which leaked through the acousto-optic modulator that was used to modulate the visible stimulus power. Stimuli were presented over a dynamic range of 0 to 1 arbitrary units (au) on a 1000-step linear scale (30 dB dynamic range) above a fixed background. Stimuli were presented over six frames for an approximate duration of 200 msec. The highest intensity for the 543-nm stimulus was 1 au = ∼3.5 log Trolands (Td), and the total background luminance was ∼1.65 log Td. The background intensity was greater than rod saturation, which ensured that the sensitivity tests were mediated by cones only. A 3.45 arcmin diameter stimulus (50% of the diameter of a Goldmann I stimulus) was used, with 30 trials tested twice at each test location using a yes-no adaptive (QUEST) staircase algorithm thresholding procedure.37 The retina was tested at approximately 2° intervals and the temporal meridian was chosen to minimize blood vessel interaction.
Data Syntheses and Analyses
Images and data from all modalities (AOSLO, SD-OCT, en face, and B-scans) were all carefully coregistered manually using commercial software using the optic nerve and retinal vascular landmarks (Adobe Illustrator; Adobe, Inc., San Jose, CA, USA). This enabled the direct selection of locations to report and/or compute structure/function metrics (cone density, retinal thickness, sensitivity) at common retinal locations.
Cone Density Analysis
Regions of interest (ROIs) were identified using AOSLO images and selected based on areas with unambiguous cones as close to the locations imaged with cross-sectional SD-OCT scans as possible. Cone density was measured using custom software (Translational Imaging Innovations, Hickory, NC, USA) as previously described.38 Specifically, the software computed bound cone density using Voronoi tessellation analysis for metric calculations at each ROI. Density was thereby defined as a ratio of number of bound Voronoi cells to the total area of the bound Voronoi cells.38
This method has previously been used to characterize cones in RP patients.39 A combination of confocal and split-detector images were used for analysis for locations where scattered and refracted light provided the most clear signals, approximately <2° and approximately >2°, respectively. ROIs were ∼0.17° x ∼0.17° (72 pixels2) in size and their locations were measured as eccentricity in degrees from the preferred retinal locus (PRL). The PRL was identified with a recorded 10-second video of the patient observing a target created by modulating the scanning raster of the AOSLO as described previously.40,41 The PRL in eyes with normal central vision is generally within approximately 0.10° of the location of maximum cone density.42,43
Structure-Function Comparisons and Statistical Analysis
Correlations between cone density, IS and OS thickness, and AOMP sensitivity were assessed with Spearman correlation coefficients using bootstrap analyses clustered by person and excluding zero values. The Wilcoxon rank-sum test were used to compare the ratio of AOMP sensitivity/cone density between normal, RHO, and RPGR patients. P values reflect clustering by patient.
Results
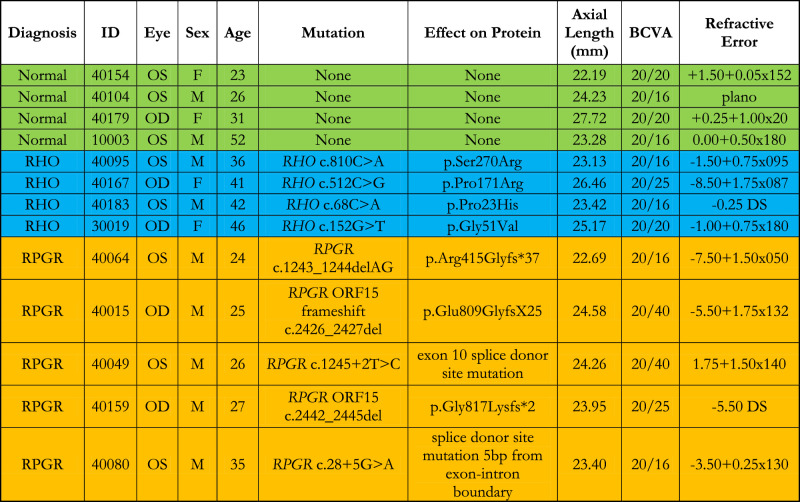
One eye from each of four patients with mutations in RHO (two female, two male; mean age 41.3 ± 3.8 years), five patients with mutations in RPGR (all male; mean age 27.3 ± 4.5 years), and four healthy subjects with normal eye examinations (two female, two male; mean age 32.9 ± 13.2 years), all from unrelated families, were recruited for the study. Normal subjects and patients with RHO mutations were not significantly different in age (P = 0.27), and normal subjects were not significantly different in age from patients with RPGR mutations (P = 0.40), but patients with RHO mutations were slightly, but significantly, older than patients with RPGR mutations (P = 0.002). Characteristics of the patients are shown in the Table.
Table.
Clinical characteristics of healthy normal subjects and patients with RP. OD, right eye; OS, left eye; M, male; F, female; BCVA, best corrected visual acuity; DS, diopter sphere. “None” indicates the normal subjects did not undergo genetic testing.
|
Figure 1 shows confocal and split-detector AOSLO image montages along with coregistered SD-OCT B-scans across the horizontal meridian of a representative subject from each cohort. AOMP tested locations and the corresponding thresholds are also indicated on the figure.
Figure 2 shows an example of split-detector AOSLO images at similar locations in a patient with a RHO mutation (left panel), and a patient with a mutation in the RPGR gene (right panel). The region tested using AOMP is shown with a small black circle near the ROI in which cones were counted. The two selected locations have similar cone densities, yet the threshold in the RPGR patient was higher (indicating lower sensitivity) than in the RHO patient. This result indicates dissociation of function and structure in the RPGR-related RP retina, whereas the RHO-related RP retina shows reduced sensitivity that likely corresponds to reduced cone density. The finding in the patient with RPGR-related RP is novel; reduced sensitivity in eyes with retinal disease has been attributed to reductions in cone density in prior studies.45–48
Figure 2.

AOSLO split-detection image with AOMP sensitivity-threshold values superimposed as color-coded circle, black circle indicating actual size of stimulus, ROIs outlined with black box, and magnified view of ROI with red marks indicating positions of cones used to assess cone density. (A) Image from patient 40095 with RHO mutation, ROIs at 3.6 degrees eccentricity; (B) image from patient 40159 with RPGR mutation, ROIs at 1.1 degrees eccentricity. Retinal sensitivity-threshold values in color-coded circles are shaded from green (normal) to red (stimulus not seen) based on the thresholds measured at each location; color scale bar at left of panel; scale bar: 0.17 degrees.
To control for density-dependent sensitivity differences, we divided the sensitivity measured with AOMP by cone density (cones/degree2) at each ROI in which sensitivity was tested, and cones could be unambiguously identified and quantified. Figure 3A shows AOMP sensitivity divided by density at each location for normal controls, patients with RHO mutations, and patients with RPGR mutations.
Figure 3.

Ratios of retinal sensitivity, retinal thickness, and cone density. (A) AOMP/cone density ratio. Normal subjects are shown with green bars, patients with RHO mutations are shown with blue bars, patients with RPGR mutations are shown with orange bars. Error bars indicate the standard error of the mean, and asterisks represent statistical significance. The number of measurements represented by each bar depended on the number of locations at which it was possible to compute density, perform AOMP, and which had measurable OCT thickness; zero values were excluded: (A), normal = 20 measurements, RHO = 13 measurements, and RPGR = 11 measurements; (B) OS thickness/density ratio; (C) IS thickness/density ratio. The number of measurements represented by each bar depended on the number of locations at which it was possible to compute cone density; (B), normal = 36 measurements, RHO = 19 measures, and RPGR = 12 measures; (C), normal = 36 measurements, RHO = 22 measures, and RPGR = 14 measures. (D) AOMP/OS thickness; (E) AOMP/IS thickness. The number of measurements represented by each bar depended on the number of locations at which it was possible to measure AOMP; (D), normal = 20 measurements, RHO = 13 measures, and RPGR = 11 measures; (E), normal = 20 measurements, RHO = 14 measures, and RPGR = 13 measures.
Figure 3A shows lower sensitivity per cone density in the patients with RPGR mutations compared with both normal subjects and patients with RHO mutations. Sensitivity in patients with RHO mutations was not significantly lower than normal eyes when measured using AOMP (P > 0.9). However, sensitivity in patients with RPGR-related RP was significantly lower than normal when measured using AOMP (P < 0.001), and was also lower than in patients with RHO mutations when measured using AOMP (P = 0.0015).
To determine if patients with RPGR mutations showed lower sensitivity/density ratios because the cone IS or OS (as measured using segmented SD-OCT images) were shorter than normal and/or patients with RHO mutations, we measured the ratio of OS length/cone density and IS length/cone density for the three cohorts. OS thickness per cone density in patients with RHO mutations was not significantly lower than normal eyes (P = 0.48) (Fig. 3B). However, OS thickness per cone density in patients with RPGR mutations was significantly lower than normal (P < 0.001) and was also lower than patients with RHO mutations (P = 0.032). IS thickness per cone density was significantly lower than normal both in patients with RHO mutations (P = 0.013) and RPGR mutations (P = 0.002) (Fig. 3C), but there was no statistically significant difference between the two patient groups (P = 0.42).
To determine whether OS or IS length is a likely structural factor that can explain the sensitivity loss in RPGR patients compared with RHO patients, we plotted the ratio of AOMP to OS and IS thickness for each cohort (Figs. 3D–E). The ratio of AOMP/OS thickness was not significantly different from normal in patients with RHO mutations (P = 0.39) or RPGR mutations (P = 0.79), and there was no difference between patients with RHO and RPGR mutations (P = 0.47) (Fig. 3D).
The ratio of AOMP/IS thickness was significantly different from normal in patients with RHO mutations (P = 0.04) and RPGR mutations (P =0.0024), and patients with RHO mutations were significantly different from those with RPGR mutations (P = 0.0004). Sensitivity/OS thickness and IS thickness was slightly greater in the patients with RHO mutations than normal subjects, (Figs. 3D–E), and significantly lower in the patients with RPGR mutations than both patients with RHO mutations and normal subjects for IS thickness only (Figs. 3D–E).
Discussion
High-resolution retinal imaging demonstrated significant differences in the structure and function of cones and function of rods in patients with RHO mutations, as well as patients with RPGR mutations. Patients with RP due to RHO mutations have normal sensitivity/cone density, indicating that visual losses are due to cone death. Patients with RP due to RPGR mutations demonstrate abnormal sensitivity/cone density, likely because cones express RPGR intrinsically, and potentially demonstrating a dissociation between loss of cone structure and function in patients with RPGR-related RP for the first time of which we are aware. The difference in the two patient types has implications for therapy in which gene augmentation with RPGR might be expected to improve visual function of remaining cones, whereas therapy for RHO mutations might delay cone death but would be unlikely to improve visual function of the remaining cones.
Patients with RHO mutations showed greater sensitivity than patients with RPGR mutations at a given cone density. These trends observed are likely a result of the different mechanisms of disease between the two groups. Patients with RHO mutations included in the present study had the class B phenotype, which progresses more slowly than patients with the class A phenotype,7,49 and which shows loss of cone function only after 75% of rod OS have been lost.7 RPGR-related retinal degeneration is phenotypically heterogeneous; patients have a range of disease onset and severity even within the same family, phenotype expression is not consistently related to location or type of mutation, and families with cone-rod dystrophy or maculopathy rather than RP have been reported.50 Some patients with RPGR-ORF15-related RP showed greater cone sensitivity loss, and regional variation was typical among patients with a range of disease severity caused by different mutations.50 Two patients in the current study had mutations in the ORF15 region, whereas the other three had mutations in the region homologous to the regulator of chromosome condensation 1 (RCC1)-like domain in the N-terminal region of the protein.51 However, to our knowledge, prior studies have not reported a dissociation between photoreceptor structure and function as we report in the current manuscript, in which patients with similar cone density at similar retinal locations show greater loss of visual function in RPGR compared with RHO-related RP.
Patients in the two groups that were included in the study were in stages of disease progression, which retained well-preserved macular outer retinal structure. Patients with RPGR mutations, who typically express more severe clinical manifestations than the class B phenotype patients with RHO mutations, were enrolled at earlier stages of disease progression to include patients with preserved macular photoreceptors.52 Thus in this study, patients with RHO mutations were significantly older than the patients with RPGR mutations, which is a result of the increased rate of disease progression in RPGR.
Photoreceptor OS thickness is lost earliest in RP patients, followed by IS, then outer nuclear layer thickness.53 In the present study, the IS thickness/density ratios (Fig. 3C) were more similar between patients with RHO mutations and patients with RPGR mutations than the OS thickness/density ratios (Fig. 3B), perhaps because cone OS degenerated earlier in patients with RPGR mutations. Patients with RPGR mutations had shorter OS for a given density (Fig. 3B) and correspondingly lower sensitivity (Fig. 3A).
We chose to characterize sensitivity/cone density to assess the impact of the two different types of mutation on macular function. Because neither RHO nor RPGR encode proteins that are involved in cone phototransduction, it was not clear whether macular function would be affected more by mutations in one or the other gene. The current study was conducted to see if loss of sensitivity was attributable simply to fewer cones surviving versus reduced sensitivity of the cones that persist. Retinal degenerations with dissociation between loss of photoreceptor structure and function have been particularly amenable to therapeutic intervention.54 Characterizing cone function in eyes with similar degrees of cone loss owing to mutations in two genes commonly associated with retinal degeneration could demonstrate if either of these diseases might be more likely to respond to treatment with visual improvement.
The sensitivity/cone density metric in the current study indicated that patients with RP due to RHO mutations had normal sensitivity/cone density, indicating that visual loss was due to cone death. Patients with RP due to RPGR mutations demonstrated abnormal sensitivity/cone density, likely because cones express RPGR intrinsically. This result demonstrated a dissociation between loss of cone structure and function in patients with RPGR-related RP. The difference in the two patient types has implications for therapy, in which gene augmentation with RPGR might be expected to improve visual function of remaining cones, whereas therapy for RHO mutations might delay cone death but would be unlikely to improve visual function of the remaining cones.
Prior studies have shown enhanced rod bipolar function in rat eyes with transgenic P23H RHO mutations that compensate for some of the rod loss.55 The greater sensitivity/IS thickness and sensitivity/OS thickness ratios observed in the RHO patients compared with normal (Figs. 3D–E) in the present study could in part be explained by this phenomenon, in which compensatory mechanisms increase sensitivity per retinal region as photoreceptors are lost.
In prior studies of patients with retinal degenerations, AOSLO-mediated acuity measures were not significantly better than ETDRS acuity in patients with retinal degenerations, perhaps because acuity was limited by reduced cone densities near the fovea.41 Until AOMP becomes widely available, clinical microperimetry could be a useful tool to characterize macular function in RP patients, and may identify abnormal function in regions with relatively preserved outer retinal structure. However, if precise stimulus localization is required for disease assessment, AOMP can be useful to provide exact placement of stimuli on particular retinal areas on interest.56
The present study has limitations that go beyond the small number of subjects assessed with each genetic form of RP studied. Longitudinal studies with greater numbers of patients who have RP associated with mutations in RHO and RPGR would provide a more comprehensive assessment of the relationships reported. In addition, future studies should compare structure and function of rods in addition to cones in eyes with RP because rods are affected earliest in patients with RP. To test rod function with AOMP, the AOSLO system would need to be adapted to prevent bleaching of the rods with the imaging system. With a modified scotopic AOSLO, one could elucidate pressing research questions, such as how rods are affected in the macula of RP patients who retain central cones; evaluate the threshold of rod loss before which cone spacing becomes abnormal; and whether rod loss is different in eyes with RP caused by mutations expressed only in rods compared with mutations expressed in rods and cones. A deeper understanding of rod loss in patients with RP is essential to advance developments of therapies for such a relentless, progressive disease affecting all photoreceptors.
Conclusions
Sensitivity/cone density in patients with RHO mutations was greater than in patients with RPGR mutations (Figs. 2, 3). This is likely because cones express RPGR but not RHO, and degenerate intrinsically in patients with RPGR mutations, which causes the cones to have shorter OS (Fig. 3B). Patients with RHO mutations exhibited cone function that was more similar to normal, with a trend toward increased sensitivity despite shorter than normal IS and OS, suggesting the possibility of compensatory increased function in response to degeneration.55,57,58
AOSLO and AOMP as part of a multimodal approach are vital to understanding relationships between structure and function and genotype/phenotype variations. This study provides a foundation for investigating RP disease progression, as well as a measurement for potential treatment efficacy using AOSLO imaging techniques. The high resolution measures used here to visualize structure and measure function have the capacity to deliver specific, precise, objective, and sensitive measures of health and survival of cone photoreceptors. Thus this approach may facilitate improved understanding of disease mechanism in different forms of retinal degeneration. Furthermore, the present study highlights fundamental differences in retinal structure and function in patients with RP due to different genetic mutations. The results support the need for natural history trials of genetically characterized patients with RP to identify the most sensitive outcome measures of disease progression in patients with different genetic forms of RP.
Acknowledgments
Supported by the National Institutes of Health Grants R01EY023591, P30EY002162, and T32EY007043; Foundation Fighting Blindness; Research to Prevent Blindness RP Nelson Trust Award and Unrestricted funds; Claire Giannini Foundation; That Man May See, Inc.; and the Minnie Flaura Turner Memorial Fund for Impaired Vision Research Award.
Disclosure: K.G. Foote, Carl Zeiss Meditec, Inc. (E); J.J. Wong, None; A.E. Boehm, None; E. Bensinger, C. Light Technologies (C); T.C. Porco, None; A. Roorda, USPTO#7,118,216, USPTO#6,890,076 (University of Rochester, University of Houston [P]), C. Light Technologies (I); J.L. Duncan, Vedere Bio (C), 4D Therapeutics (C), Applied Genetics Technology Corporation (C), California Institute for Regenerative Medicine (C), Editas Medicine (C), Eloxx (C), Foundation Fighting Blindness (C), Biogen/Nightstar Therapeutics (C), ProQR Therapeutics (C), SparingVision (C), Spark Therapeutics (C)
References
- 1. Zhang Q. Retinitis pigmentosa: progress and perspective. Asia Pac J Ophthalmol (Phila). 2016; 5: 265–271. [DOI] [PubMed] [Google Scholar]
- 2. Fahim A. Retinitis pigmentosa: recent advances and future directions in diagnosis and management. Curr Opin Pediatr. 2018; 30: 725–733. [DOI] [PubMed] [Google Scholar]
- 3. Breuer DK, Yashar BM, Filippova E, et al.. A comprehensive mutation analysis of RP2 and RPGR in a North American cohort of families with X-linked retinitis pigmentosa. Am J Hum Genet. 2002; 70: 1545–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Briscoe AD, Gaur C, Kumar S. The spectrum of human rhodopsin disease mutations through the lens of interspecific variation. Gene. 2004; 332: 107–118. [DOI] [PubMed] [Google Scholar]
- 5. Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006; 368: 1795–1809. [DOI] [PubMed] [Google Scholar]
- 6. Wright AF, Chakarova CF, Abd El-Aziz MM, Bhattacharya SS. Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nat Rev Genet. 2010; 11: 273–284. [DOI] [PubMed] [Google Scholar]
- 7. Cideciyan AV, Hood DC, Huang Y, et al.. Disease sequence from mutant rhodopsin allele to rod and cone photoreceptor degeneration in man. Proc Natl Acad Sci USA. 1998; 95: 7103–7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tee JJ, Smith AJ, Hardcastle AJ, Michaelides M. RPGR-associated retinopathy: clinical features, molecular genetics, animal models and therapeutic options. Br J Ophthalmol. 2016; 100: 1022–1027. [DOI] [PubMed] [Google Scholar]
- 9. Ripps H. Cell death in retinitis pigmentosa: gap junctions and the ‘bystander’ effect. Exp Eye Res. 2002; 74: 327–336. [DOI] [PubMed] [Google Scholar]
- 10. Punzo C, Kornacker K, Cepko CL. Stimulation of the insulin/mTOR pathway delays cone death in a mouse model of retinitis pigmentosa. Nat Neurosci. 2009; 12: 44–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shen J, Yang X, Dong A, et al.. Oxidative damage is a potential cause of cone cell death in retinitis pigmentosa. J Cell Physiol. 2005; 203: 457–464. [DOI] [PubMed] [Google Scholar]
- 12. Leveillard T, Mohand-Said S, Lorentz O, et al.. Identification and characterization of rod-derived cone viability factor. Nat Genet. 2004; 36: 755–759. [DOI] [PubMed] [Google Scholar]
- 13. Narayan DS, Wood JP, Chidlow G, Casson RJ. A review of the mechanisms of cone degeneration in retinitis pigmentosa. Acta Ophthalmol. 2016; 94: 748–754. [DOI] [PubMed] [Google Scholar]
- 14. Sullivan LS, Bowne SJ, Reeves MJ, et al.. Prevalence of mutations in eyeGENE probands with a diagnosis of autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2013; 54: 6255–6261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ferris FL 3rd Kassoff A, Bresnick GH, Bailey I. New visual acuity charts for clinical research. Am J Ophthalmol. 1982; 94: 91–96. [PubMed] [Google Scholar]
- 16. Aizawa S, Mitamura Y, Baba T, Hagiwara A, Ogata K, Yamamoto S. Correlation between visual function and photoreceptor inner/outer segment junction in patients with retinitis pigmentosa. Eye (London). 2009; 23: 304–308. [DOI] [PubMed] [Google Scholar]
- 17. Birch DG, Wen Y, Locke K, Hood DC. Rod sensitivity, cone sensitivity, and photoreceptor layer thickness in retinal degenerative diseases. Invest Ophthalmol Vis Sci. 2011; 52: 7141–7147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hood DC, Cho J, Raza AS, Dale EA, Wang M. Reliability of a computer-aided manual procedure for segmenting optical coherence tomography scans. Optom Vis Sci. 2011; 88: 113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hood DC, Lin CE, Lazow MA, Locke KG, Zhang X, Birch DG. Thickness of receptor and post-receptor retinal layers in patients with retinitis pigmentosa measured with frequency-domain optical coherence tomography. Invest Ophthalmol Vis Sci. 2009; 50: 2328–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wen Y, Klein M, Hood DC, Birch DG. Relationships among multifocal electroretinogram amplitude, visual field sensitivity, and SD-OCT receptor layer thicknesses in patients with retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2012; 53: 833–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wen Y, Locke KG, Klein M, et al.. Phenotypic characterization of 3 families with autosomal dominant retinitis pigmentosa due to mutations in KLHL7. Arch Ophthalmol. 2011; 129: 1475–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Foote KG, De la Huerta I, Gustafson K, et al.. Cone spacing correlates with retinal thickness and microperimetry in patients with inherited retinal degenerations. Invest Ophthalmol Vis Sci. 2019; 60: 1234–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Roorda A, Metha AB, Lennie P, Williams DR. Packing arrangement of the three cone classes in primate retina. Vision Res. 2001; 41: 1291–1306. [DOI] [PubMed] [Google Scholar]
- 24. Duncan JL, Zhang Y, Gandhi J, et al.. High-resolution imaging with adaptive optics in patients with inherited retinal degeneration. Invest Ophthalmol Vis Sci. 2007; 48: 3283–3291. [DOI] [PubMed] [Google Scholar]
- 25. Merino D, Dainty C, Bradu A, Podoleanu AG. Adaptive optics enhanced simultaneous en-face optical coherence tomography and scanning laser ophthalmoscopy. Opt Express. 2006; 14: 3345–3353. [DOI] [PubMed] [Google Scholar]
- 26. Merino D, Duncan JL, Tiruveedhula P, Roorda A. Observation of cone and rod photoreceptors in normal subjects and patients using a new generation adaptive optics scanning laser ophthalmoscope. Biomed Opt Express. 2011; 2: 2189–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Talcott KE, Ratnam K, Sundquist SM, et al.. Longitudinal study of cone photoreceptors during retinal degeneration and in response to ciliary neurotrophic factor treatment. Invest Ophthalmol Vis Sci. 2011; 52: 2219–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Scoles D, Sulai YN, Langlo CS, et al.. In vivo imaging of human cone photoreceptor inner segments. Invest Ophthalmol Vis Sci. 2014; 55: 4244–4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jonnal RS, Kocaoglu OP, Zawadzki RJ, Lee SH, Werner JS, Miller DT. The cellular origins of the outer retinal bands in optical coherence tomography images. Invest Ophthalmol Vis Sci. 2014; 55: 7904–7918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Roorda A, Duncan JL. Adaptive optics ophthalmoscopy. Annu Rev Vis Sci. 2015; 1: 19–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guevara-Torres A, Williams DR, Schallek JB. Origin of cell contrast in offset aperture adaptive optics ophthalmoscopy. Opt Lett. 2020; 45: 840–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Scoles D, Sulai YN, Cooper RF, et al.. Photoreceptor inner segment morphology in best vitelliform macular dystrophy. Retina. 2017; 37: 741–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stevenson SB, Roorda A, Kumar G. Eye tracking with the adaptive optics scanning laser ophthalmoscope. In: Spencer SN, ed. Proceedings of the 2010 Symposium on Eye-Tracking Research and Applications. New York: Association for Computed Machinery; 2010: 195–198. [Google Scholar]
- 34. Chen M, Cooper RF, Han GK, Gee J, Brainard DH, Morgan JI. Multi-modal automatic montaging of adaptive optics retinal images. Biomed Opt Express. 2016; 7: 4899–4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tuten WS, Tiruveedhula P, Roorda A. Adaptive optics scanning laser ophthalmoscope-based microperimetry. Optom Vis Sci. 2012; 89: 563–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Harmening WM, Tuten WS, Roorda A, Sincich LC. Mapping the perceptual grain of the human retina. J Neurosci. 2014; 34: 5667–5677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Watson AB, Pelli DG. QUEST: a Bayesian adaptive psychometric method. Percept Psychophys. 1983; 33: 113–120. [DOI] [PubMed] [Google Scholar]
- 38. Cooper RF, Wilk MA, Tarima S, Carroll J. Evaluating descriptive metrics of the human cone mosaic. Invest Ophthalmol Vis Sci. 2016; 57: 2992–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sun LW, Johnson RD, Langlo CS, et al.. Assessing photoreceptor structure in retinitis pigmentosa and usher syndrome. Invest Ophthalmol Vis Sci. 2016; 57: 2428–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rossi EA, Roorda A.. The relationship between visual resolution and cone spacing in the human fovea. Nat Neurosci. 2010; 13: 156–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Foote KG, Loumou P, Griffin S, et al.. Relationship between foveal cone structure and visual acuity measured with adaptive optics scanning laser ophthalmoscopy in retinal degeneration. Invest Ophthalmol Vis Sci. 2018; 59: 3385–3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Putnam NM, Hofer HJ, Doble N, Chen L, Carroll J, Williams DR. The locus of fixation and the foveal cone mosaic. J Vis. 2005; 5: 632–639. [DOI] [PubMed] [Google Scholar]
- 43. Wang Y, Bensaid N, Tiruveedhula P, Ma J, Ravikumar S, Roorda A. Human foveal cone photoreceptor topography and its dependence on eye length. eLife. 2019; 8: e47148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hood DC, Lazow MA, Locke KG, Greenstein VC, Birch DG. The transition zone between healthy and diseased retina in patients with retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2011; 52: 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Choi SS, Doble N, Hardy JL, et al.. In vivo imaging of the photoreceptor mosaic in retinal dystrophies and correlations with visual function. Invest Ophthalmol Vis Sci. 2006; 47: 2080–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ooto S, Hangai M, Takayama K, Ueda-Arakawa N, Hanebuchi M, Yoshimura N. Photoreceptor damage and foveal sensitivity in surgically closed macular holes: an adaptive optics scanning laser ophthalmoscopy study. Am J Ophthalmol. 2012; 154: 174–186.e2. [DOI] [PubMed] [Google Scholar]
- 47. Rangaswamy NV, Patel HM, Locke KG, Hood DC, Birch DG. A comparison of visual field sensitivity to photoreceptor thickness in retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2010; 51: 4213–4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Supriya D, Shwetha M, Kiran Anupama K, et al.. Structural and function correlation of cone packing utilizing adaptive optics and microperimetry. Biomed Res Int. 2015; 2015: 968672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sung CH, Davenport CM, Nathans J. Rhodopsin mutations responsible for autosomal dominant retinitis pigmentosa. Clustering of functional classes along the polypeptide chain. J Biol Chem. 1993; 268: 26645–26649. [PubMed] [Google Scholar]
- 50. Charng J, Cideciyan AV, Jacobson SG, et al.. Variegated yet non-random rod and cone photoreceptor disease patterns in RPGR-ORF15-associated retinal degeneration. Hum Mol Genet. 2016; 25: 5444–5459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Khanna H. More than meets the eye: current understanding of RPGR function. Adv Exp Med Biol. 2018; 1074: 521–538. [DOI] [PubMed] [Google Scholar]
- 52. Wang DY, Chan WM, Tam PO, et al.. Gene mutations in retinitis pigmentosa and their clinical implications. Clin Chim Acta. 2005; 351: 5–16. [DOI] [PubMed] [Google Scholar]
- 53. Gao B, Wenzel A, Grimm C, et al.. Localization of organic anion transport protein 2 in the apical region of rat retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2002; 43: 510–514. [PubMed] [Google Scholar]
- 54. Garafalo AV, Cideciyan AV, Heon E, et al.. Progress in treating inherited retinal diseases: early subretinal gene therapy clinical trials and candidates for future initiatives. Prog Retin Eye Res. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Aleman TS, LaVail MM, Montemayor R, et al.. Augmented rod bipolar cell function in partial receptor loss: an ERG study in P23H rhodopsin transgenic and aging normal rats. Vision Res. 2001; 41: 2779–2797. [DOI] [PubMed] [Google Scholar]
- 56. Tuten WS, Vergilio GK, Young GJ, et al.. Visual function at the atrophic border in choroideremia assessed with adaptive optics microperimetry. Ophthalmol Retina. 2019; 3: 888–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Aleman TS, Cideciyan AV, Sumaroka A, et al.. Retinal laminar architecture in human retinitis pigmentosa caused by Rhodopsin gene mutations. Invest Ophthalmol Vis Sci. 2008; 49: 1580–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chrysostomou V, Stone J, Valter K. Life history of cones in the rhodopsin-mutant P23H-3 rat: evidence of long-term survival. Invest Ophthalmol Vis Sci. 2009; 50: 2407–2416. [DOI] [PubMed] [Google Scholar]