

Abstract
The overexpression of the ATP-binding cassette (ABC) transporter ABCG2 has been linked to clinical multidrug resistance in solid tumors and blood cancers, which remains a significant obstacle to successful cancer chemotherapy. For years, the potential modulatory effect of bioactive compounds derived from natural sources on ABCG2-mediated multidrug resistance has been investigated, as they are inherently well tolerated and offer a broad range of chemical scaffolds. Licochalcone A (LCA), a natural chalcone isolated from the root of Glycyrrhiza inflata, is known to possess a broad spectrum of biological and pharmacological activities, including pro-apoptotic and anti-proliferative effects in various cancer cell lines. In this study, the chemosensitization effect of LCA was examined in ABCG2-overexpressing multidrug-resistant cancer cells. Experimental data demonstrated that LCA inhibited the drug transport function of ABCG2 and reverses ABCG2-mediated multidrug resistance in human multidrug-resistant cancer cell lines in a concentration-dependent manner. Results of LCA-stimulated ABCG2 ATPase activity and the in silico docking analysis of LCA to the inward-open conformation of human ABCG2 suggest that LCA binds ABCG2 in the transmembrane substrate-binding pocket. This study provides evidence that LCA should be further evaluated as a modulator of ABCG2 in drug combination therapy trials against ABCG2-expressing drug-resistant tumors.
The ATP-Binding Cassette (ABC) transporters are membrane proteins that can utilize energy derived from ATP hydrolysis to translocate a wide range of chemicals, including a large proportion of therapeutic drugs, away from their targets.1–3 Consequently, the overexpression of one or more ABC transporters in cancer cells can significantly reduce the efficacy of chemotherapeutic drugs that are functionally and structurally unrelated, resulting in the development of multidrug resistance (MDR) phenotype in cancer cells.4 Although many ABC transporters were reported to be linked to MDR, ABCB1 (P-glycoprotein/MDR1) and ABCG2 (BCRP; MXR) are currently the two ABC transporters with evidence to support their role in clinical drug resistance.4 In contrast, the involvement of ABCC1 (also known as multidrug resistance-associated protein 1 or MRP1) in clinical drug resistance remains ambiguous.5 Studies have provided evidence that ABCB1 and ABCG2 are contributing, at least in part, to the development of MDR phenotype in blood tumors such as multiple myeloma (MM),6–9 chronic lymphocytic leukemia (CLL),10 acute lymphocytic leukemia (ALL), acute myelogenous leukemia (AML),11–13 as well as in solid tumors such as metastatic breast cancer14 and advanced non-small cell lung cancer.15 Collectively, ABCB1 and ABCG2 can confer resistance to some of the most widely used conventional anticancer agents, such as anthracyclines, methotrexate, topotecan, SN-38, vinca alkaloids, as well as numerous protein kinase inhibitors.16–19 Moreover, high expression of ABCB1 and ABCG2 in cells forming the blood-tissue barrier sites can have a significant impact on the oral bioavailability, distribution, metabolism, and elimination of most drugs in patients.2, 17, 20
At this time, treatment failure associated with the development of MDR remains a major challenge for drug developers and clinicians.3, 4 Since directly attenuating the drug efflux function of the ABC drug transporters is one of the most effective approaches to overcome MDR, tremendous efforts have been made to develop potent and specific inhibitors of ABCB1 and ABCG2 in an attempt to improve therapeutic outcomes in patients.21 In addition to developing novel synthetic inhibitors of ABC drug transporters, the use of bioactive compounds originated from natural sources, which are inherently well tolerated and offer the widest variety of novel chemical scaffolds, for the resensitization of multidrug-resistant cancer cells has also been the focus of many research groups.22, 23 Novel antiproliferative compounds and kinase inhibitors derived from natural products have been tested in clinical trials, with some approved by the U.S. Food and Drug Administration (FDA), for cancer prevention and treatment.24–26 Consequently, many more flavonoids and marine natural products are now being tested for the reversal of multidrug resistance in cancer.27, 28 However, it is worth noting that due to the lack of selectivity and/or high intrinsic toxicity, not all natural product modulators of ABCG2 will be suitable for clinical applications.22, 23 For instance, despite being one of the most potent inhibitors of ABCG2,29 the neurotoxic nature of the diketopiperazine fungal mycotoxin fumitremorgin C (FTC) limits its use in clinical practice.30
Licochalcone A (LCA) is a natural chalcone (chemical structure given in Figure 8) and the primary active compound that can be isolated from the root of Glycyrrhiza inflata (licorice).31, 32 LCA is known to exhibit a wide range of pharmacological effects,33 including antiprotozoal,34, 35 anti-viral,36–38 antifungal,39 antimicrobial,37, 40 and anti-inflammatory activities.41 Moreover, the antiproliferative action of LCA has been shown in a variety of cancer cell lines and in vivo models.42–47 Interestingly, LCA was also reported to inhibit the proliferation of human breast cancer cells and oral squamous cancer cells through suppression of specificity protein 1 (Sp1),43, 44 a novel therapeutic target for cancers such as colon cancer,48 cervical cancer49 and oral squamous cell carcinoma (OSCC).50 Chalcones were originally discovered able to inhibit the activity of both ABCB1 and ABCG2,51 but later studies reported that synthetic chalcones can be chemically modified into selective modulators of ABCG2.52–54 Juvale et al. demonstrated that chalcones with dimethoxy substituents at ring B are highly effective in reversing ABCG2-mediated SN-38 resistance in ABCG2-overexpressing MCF7-MX cells and ABCG2-transfected MDCK-BCRP cells, with the maximum inhibitory activity of approximately 3-fold less than Ko143,53 a benchmark inhibitor of ABCG2. Moreover, by studying a series of 44 chalcones and analogues, Valdameri et al. described the importance of methoxy substituents on the toxicity of chalcones and their activity against ABCG2.54 A more recent study by Winter et al. demonstrated that amongst the analogs tested, chalcones with quinoxaline substituent were the most potent inhibitors of ABCG2.55 It is worth noting that although many chalcones were found effective against the activity of ABCG2, the chemosensitization effect of these chalcones in ABCG2-overexpressing cancer cells was not evaluated.54, 55
Figure 8.

A schematic illustration of licochalcone A reversing ABCG2-mediated drug resistance by competing with the binding of ABCG2 substrate drugs (triangles) at the substrate-binding pocket within the transmembrane domains of ABCG2, which increases the intracellular accumulation and the efficacy of ABCG2 substrate chemotherapeutic drugs.
In the present study, the chemosensitization effect of LCA was examined in multidrug-resistant cancer cells. By modulating the drug transport function of ABCG2, LCA significantly enhanced topotecan-induced apoptosis and restored the chemosensitivity of ABCG2-overexpressing multidrug-resistant cancer cells to anticancer drugs. Moreover, LCA is selective for ABCG2 relative to ABCB1 and ABCC1, two other well-characterized MDR-linked ABC drug transporters. Further evidence of the interaction between LCA and ABCG2 was provided by ABCG2 ATPase assays and an in silico docking analysis of LCA to the drug-binding pocket of human ABCG2. Overall, this study demonstrates the potential use of LCA for the combination therapy of multidrug-resistant tumors.
RESULTS AND DISCUSSION
Licochalcone A Reverses Multidrug Resistance Mediated by ABCG2.
Knowing that ABCB1, ABCC1, and ABCG2 can contribute significantly to the development of resistance to multiple chemotherapeutic agents in solid tumors and blood cancers,4, 12, 15, 56, 57 the reversal effect of LCA on multidrug resistance mediated by these three major MDR-linked ABC drug transporters was examined. Previously, Nabekura et al. reported that LCA could increase the accumulation of daunorubicin and rhodamine 123 in ABCB1-overexpressing KB/MDR1 cells.58 More recently, while examining the interaction between flavonoids with ABCG2, Fan et al. discovered that many flavonoids, including LCA, increased the uptake of mitoxantrone into the BCRP-MDCKII cells and enhanced the cytotoxicity of doxorubicin and temozolomide in ABCG2-expressing cells.59 Unfortunately, because of the high concentration of LCA (50 μM) that was used in these studies, the true potential of LCA on multidrug resistance mediated by ABC drug transporters remains to be clarified. The human ABCB1 transfected HEK293 cells (MDR19-HEK293), human ABCC1 transfected HEK293 cells (MRP1-HEK293) and ABCG2 transfected HEK293 cells (R482-HEK293) were treated with an increasing concentration of respective substrate drugs in the absence or presence of nontoxic concentrations of LCA, or a reference inhibitor as a positive control. LCA had no significant effect on ABCB1-mediated colchicine resistance in MDR19-HEK293 cells (Figure 1A) or ABCC1-mediated etoposide resistance in MRP1-HEK293 cells (Figure 1B). However, LCA significantly restored the chemosensitivity of R482-HEK293 cells to known ABCG2 substrates mitoxantrone (Figure 1C) and topotecan (Table 1), two well-known substrates of ABCG2,60–62 in a concentration-dependent manner. Consequently, the effect of LCA was further examined in ABCG2-overexpressing multidrug-resistant S1-M1–80 human colon cancer and H460-MX20 human non-small cell lung cancer (NSCLC) cell lines, and in their respective drug-sensitive S1 and H460 parental cancer cell lines. As summarized in Table 2, LCA significantly reversed ABCG2-mediated mitoxantrone and topotecan resistance in both S1-M1–80 (Figure 1D and 1E) and H460-MX20 (Figure 1F and 1G) cancer cell lines in the same manner as in R482-HEK293 cells. Of note, the fold-reversal (FR) value63 here represents the extent of chemosensitization by LCA in a particular cell line, which is calculated by dividing the 50% inhibitory concentration (IC50) value of cells treated with a substrate drug in the absence of LCA or a reference inhibitor by the IC50 value of cells treated with the same substrate drug in the presence of an inhibitor. Moreover, LCA had no significant effect on the proliferation of drug-sensitive parental cell lines, and tariquidar, MK-571, and Ko143 were used as reference inhibitors for ABCB1, ABCC1, and ABCG2, respectively (Table 1). Notably, an antiproliferative effect of LCA on BT-20 breast cancer cells was reported by Komoto et al.,64 therefore, we examined the cytotoxic effect of LCA in our cell lines. As shown in Figure 2, LCA is cytotoxic to R482-HEK293, S1-M1–80, H460-MX20 cells and the corresponding drug-sensitive parental cells, with IC50 values ranging from 10 to 50 μM, which is comparable to the IC50 value of LCA determined by Komoto et al.64 It is worth noting that in addition to cancerous cell lines65–67, the cytotoxicity of LCA is also low in non-cancerous cell lines.65, 68–71. For instance, Liou et al. discovered that LCA was not cytotoxic up to 12 μM in human hepatocellular carcinoma HepG2 cells, whereas Wu et al. reported an IC50 value of LCA higher than 180 μM in normal gastric epithelium GES-1 cells.65 Moreover, the ABCG2-overexpressing cells and the corresponding drug-sensitive parental cells are equally sensitive to LCA, suggesting that LCA is not actively effluxed out of cancer cells by ABCG2.
Figure 1.

Licochalcone A reverses drug resistance mediated by ABCG2. Parental pcDNA-HEK293 cells (A - C, left panel), S1 human colon cancer cells (D and E, left panel), and H460 human lung cancer cells (F and G, left panel), as well as ABCB1-transfected MDR19-HEK293 cells (A, right panel), ABCC1-transfected MRP1-HEK293 cells (B, right panel), ABCG2-transfected R482-HEK293 cells (C, right panel), and ABCG2-overexpressing multidrug-resistant S1-M1–80 human colon cancer cells (D and E, right panel) and H460-MX20 human lung cancer cells (F and G, right panel) were treated with increasing concentrations of colchicine (A), etoposide (B), mitoxantrone (C, D and F) or topotecan (E and G) in the presence of DMSO (open circles) or licochalcone A (LCA) at 0.5 μM (open squares), 1.0 μM (filled squares), 2.0 μM (open triangles), 3.0 μM (filled triangles), or ABCB1 reference inhibitor tariquidar at 1.0 μM (A, filled circles), ABCC1 reference inhibitor MK-571 at 25 μM (B, filled circles) or ABCG2 reference inhibitor Ko143 at 1.0 μM (C – G, filled circles) for 72 h before processed as described in the Experimental section under Cytotoxicity Study. Points, mean values from at least three independent experiments; bars; SEM.
Table 1.
The Effect of Licochalcone A on Drug Resistance Mediated by ABCB1, ABCC1 or ABCG2
| Treatment | Concentration (μM) | Mean IC50a ± SD and (FRb) | |
|---|---|---|---|
| pcDNA-HEK293 [nM] | MDR19-HEK293 [nM] |
||
| Colchicine (ABCB1 substrate) | - | 8.42 ± 3.13 (1.0) | 97.58 ± 17.62 (1.0) |
| + licochalcone A | 0.5 | 8.67 ± 2.87 (1.0) | 94.92 ± 13.25 (1.0) |
| + licochalcone A | 1.0 | 7.99 ± 2.31 (1.1) | 114.81 ± 15.32 (0.8) |
| + licochalcone A | 2.0 | 8.90 ± 2.90 (0.9) | 121.20 ± 11.48 (0.8) |
| + licochalcone A | 3.0 | 9.03 ± 3.02 (0.9) | 105.41 ± 9.50 (0.9) |
| + tariquidar | 1.0 | 7.62 ± 2.36 (1.1) | 7.83 ± 1.37*** (12.5) |
| pcDNA-HEK293 [μM] |
MRP1-HEK293 [μM] |
||
| Etoposide (ABCC1 substrate) | - | 0.21 ± 0.04 (1.0) | 63.34 ± 7.92 (1.0) |
| + licochalcone A | 0.5 | 0.31 ± 0.07 (0.7) | 53.57 ± 4.65 (1.2) |
| + licochalcone A | 1.0 | 0.30 ± 0.07 (0.7) | 49.26 ± 4.31 (1.3) |
| + licochalcone A | 2.0 | 0.30 ± 0.06 (0.7) | 66.65 ± 12.38 (1.0) |
| + licochalcone A | 3.0 | 0.28 ± 0.06 (0.8) | 56.47 ± 7.41 (1.1) |
| + MK-571 | 25 | 0.16 ± 0.03 (1.0) | 11.87 ± 1.65*** (5.4) |
| pcDNA-HEK293 [nM] |
R482-HEK293 [nM] |
||
| Mitoxantrone (ABCG2 substrate) | - | 4.70 ± 0.99 (1.0) | 50.66 ± 6.88 (1.0) |
| + licochalcone A | 0.5 | 5.11 ± 1.01 (0.9) | 18.75 ± 2.69** (2.7) |
| + licochalcone A | 1.0 | 4.60 ± 0.91 (1.0) | 7.09 ± 0.82*** (7.1) |
| + licochalcone A | 2.0 | 4.74 ± 0.90 (1.0) | 7.83 ± 1.45*** (6.5) |
| + licochalcone A | 3.0 | 4.78 ± 0.93 (1.0) | 6.22 ± 0.85*** (8.1) |
| + Ko143 | 1.0 | 4.57 ± 0.91 (1.0) | 4.62 ± 0.52*** (10.9) |
| [nM] | [nM] | ||
| Topotecan (ABCG2 substrate) | - | 24.60 ± 5.98 (1.0) | 201.31 ± 25.89 (1.0) |
| + licochalcone A | 0.5 | 26.83 ± 6.50 (0.9) | 90.05 ± 13.54** (2.2) |
| + licochalcone A | 1.0 | 24.72 ± 5.57 (1.0) | 51.47 ± 10.61*** (3.9) |
| + licochalcone A | 2.0 | 25.89 ± 5.53 (1.0) | 44.11 ± 8.51*** (4.6) |
| + licochalcone A | 3.0 | 25.55 ± 5.49 (1.0) | 42.41 ± 8.71*** (4.7) |
| + Ko143 | 1.0 | 25.15 ± 5.75 (1.0) | 17.88 ± 2.31*** (11.3) |
IC50 values are mean ± SD calculated from dose-response curves obtained from three independent experiments.
FR,. fold-reversal values were obtained by dividing the IC50 values of cells treated with a particular drug in the absence of a modulator by the IC50 values of cells treated with the same drug in the presence of a modulator.
p < 0.05;
p < 0.01;
p < 0.001.
Table 2.
The Effect of Licochalcone A on Drug Resistance Mediated by ABCG2 in Human Cancer Cell Lines
| Treatment | Concentration (μM) |
Mean IC50a ± SD and (FRb) | |
|---|---|---|---|
| S1 [nM] | S1-M1–80 [μM] | ||
| Mitoxantrone | - | 4.83 ± 0.83 (1.0) | 73.62 ± 7.20 (1.0) |
| + licochalcone A | 0.5 | 4.44 ± 0.71 (1.1) | 39.47 ± 10.55** (1.9) |
| + licochalcone A | 1.0 | 4.53 ± 0.85 (1.1) | 6.80 ± 1.77*** (10.8) |
| + licochalcone A | 2.0 | 4.55 ± 0.71 (1.1) | 2.63 ± 0.57*** (28.0) |
| + licochalcone A | 3.0 | 4.73 ± 0.85 (1.0) | 1.53 ± 0.38*** (48.1) |
| + Ko143 | 1.0 | 4.65 ± 0.83 (1.0) | 0.58 ± 0.08*** (126.9) |
| [nM] | [μM] | ||
| Topotecan | - | 9.21 ± 1.45 (1.0) | 10.14 ± 2.82 (1.0) |
| + licochalcone A | 0.5 | 9.73 ± 1.55 (0.9) | 1.03 ± 0.32** (9.8) |
| + licochalcone A | 1.0 | 11.01 ± 2.19 (0.8) | 0.61 ± 0.17** (16.6) |
| + licochalcone A | 2.0 | 10.70 ± 1.98 (0.9) | 0.41 ± 0.10** (24.7) |
| + licochalcone A | 3.0 | 11.74 ± 2.39 (0.9) | 0.31 ± 0.06** (32.7) |
| + Ko143 | 1.0 | 10.22 ± 1.75 (0.9) | 0.17 ± 0.05** (59.6) |
| H460 [nM] | H460-MX20 [nM] | ||
| Mitoxantrone | - | 21.61 ± 5.85 (1.0) | 1160.60 ± 365.72 (1.0) |
| + licochalcone A | 0.5 | 25.91 ± 5.01 (0.8) | 441.96 ± 144.66* (2.6) |
| + licochalcone A | 1.0 | 19.84 ± 4.56 (1.1) | 86.46 ± 35.83** (13.4) |
| + licochalcone A | 2.0 | 20.10 ± 4.47 (1.1) | 74.77 ± 28.33** (15.5) |
| + licochalcone A | 3.0 | 20.34 ± 4.56 (1.1) | 69.86 ± 28.94** (16.6) |
| + Ko143 | 1.0 | 20.07 ± 4.51 (1.1) | 87.81 ± 27.51** (13.2) |
| [nM] | [nM] | ||
| Topotecan | - | 37.42 ± 5.45 (1.0) | 1319.20 ± 356.14 (1.0) |
| + licochalcone A | 0.5 | 25.54 ± 4.88* (1.5) | 366.91 ± 124.29** (3.6) |
| + licochalcone A | 1.0 | 22.41 ± 5.11* (1.7) | 143.89 ± 49.12** (9.2) |
| + licochalcone A | 2.0 | 21.69 ± 4.97* (1.7) | 147.56 ± 49.33** (8.9) |
| + licochalcone A | 3.0 | 22.52 ± 5.25* (1.7) | 130.38 ± 44.27** (10.1) |
| + Ko143 | 1.0 | 21.89 ± 4.30* (1.7) | 47.47 ± 16.66** (27.8) |
IC50 values are mean ± SD calculated from dose-response curves obtained from three independent experiments.
FR,. fold-reversal values were obtained by dividing the IC50 values of cells treated with a particular drug in the absence of a modulator by the IC50 values of cells treated with the same drug in the presence of a modulator.
p < 0.05;
p < 0.01;
p < 0.001.
Figure 2.
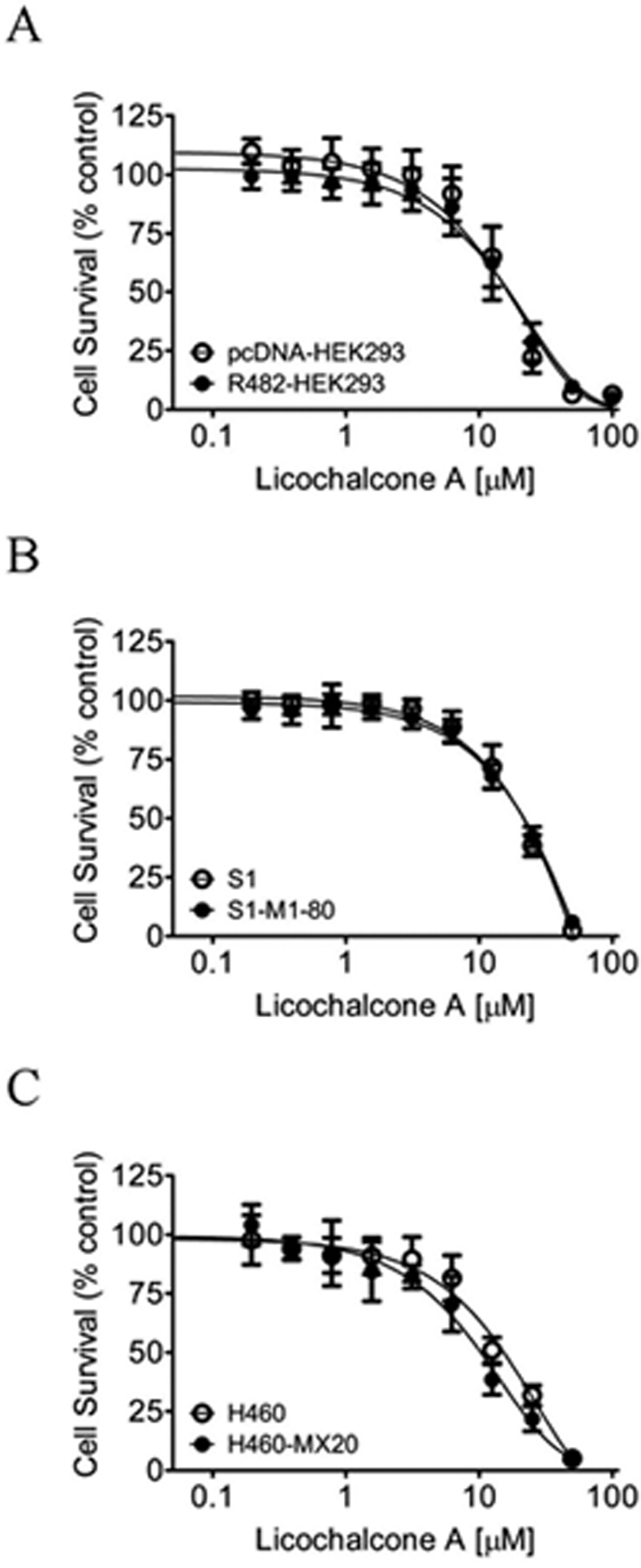
Licochalcone A is equally cytotoxic to drug-sensitive and ABCG2-overexpressing multidrug-resistant cells. The cytotoxicity of licochalcone A was determined in (A) HEK293 cells transfected with human ABCG2 (R482-HEK293, filled circles) and the parental pcDNA-HEK293 cells (open circles), (B) ABCG2-overexpressing multidrug-resistant S1-M1–80 human colon cancer cells (filled circles) and the corresponding drug-sensitive parental S1 cells (open circles), and (C) ABCG2-overexpressing multidrug-resistant H460-MX20 human lung cancer cells (filled circles) and the corresponding drug-sensitive H460 cells (open circles).
Licochalcone A Attenuates the Function of ABCG2.
Next, the effect of LCA on the drug transport function of ABCB1, ABCC1 and ABCG2 was examined in MDR19-HEK293 cells, MRP1-HEK293 cells, and ABCG2-HEK293 cells, respectively. MDR19-HEK293 and MRP1-HEK293 cells were incubated with calcein-AM for 10 min, whereas R482-HEK293 cells were incubated with PhA for 1 h in the presence or absence of licochalcone A, tariquidar, MK-571 or Ko143 as described in the Experimental Section. It is worth noting that in contrast to a 72h end-point experiment such as cytotoxicity assay, higher concentrations of LCA were used here in a shorter time steady-state fluorescent drug accumulation assay to demonstrate the effect of LCA on ABCG2-mediated transport to the fullest extent. Consistent with the results of the MDR reversal assays (Figure 1), 20 μM of LCA did not substantially increase the intracellular accumulation of fluorescent substrate drug calcein72 in either MDR19-HEK293 cells (Figure 3A, right panel) or MRP1-HEK293 cells (Figure 3B, right panel). In contrast, LCA at the same concentration significantly increased the intracellular accumulation of PhA, an ABCG2-specific fluorescent substrate73 in R482-HEK293 cells (Figure 3C, right panel), and in ABCG2-overexpressing S1-M1–80 (Figure 3D, right panel) and H460-MX20 cancer cells (Figure 3E, right panel). Of note, LCA had no significant effect on the accumulation of any fluorescent probes in drug-sensitive parental cell lines (Figure 3, left panels). Moreover, LCA inhibited ABCG2-mediated PhA efflux in R482-HEK293 cells, S1-M1–80 and H460-MX20 cancer cells (Figure 3F). Experimental results here demonstrated that LCA inhibited the function of ABCG2 in a similar manner as the synthetic chalcones,53–55 and LCA is selective for ABCG2 as compared to ABCB1 and ABCC1. In addition to direct inhibition of ABCG2-mediated drug efflux, drug-induced transient downregulation of ABCG2 protein is another common mechanism in which ABCG2-overexpressing multidrug-resistant cancer cells can become resensitized to drug treatment.74–76 To this end, the protein expression of ABCG2 was examined in S1-M1–80 and H460-MX20 cancer cells after treating cells with increasing concentrations of LCA (0.5 – 3 μM) for 72 h followed by immunoblotting as described in the Experimental Section. No significant change in ABCG2 expression was detected in either S1-M1–80 (Figure 4A) or H460-MX20 cancer cell lines (Figure 4B), suggesting that LCA reverses MDR in ABCG2-overexpressing cells by attenuating the drug transport function of ABCG2. It is worth noting that several previous studies have investigated the effect of LCA on the expression of ABCB1 and ABCG2. He et al. reported that treating LS-180 human colorectal adenocarcinoma cells with 1 μM of LCA could upregulate the basal mRNA expression of ABCB1 and ABCG2 by approximately 2–6 fold.77 However, the upregulation effect of LCA on ABC transporters may be cell line-specific as Komoto et al. reported an opposite observation that LCA could reduce the protein expression of ABCB1 in BT-20 breast cancer cells.64 Nevertheless, although He et al. reported that LCA could upregulate the mRNA level of ABCG2, LCA had no significant effect on ABCG2 protein expression in LS-180 cells after an induction period of 7 days.77
Figure 3.

Licochalcone A inhibits the drug efflux function of ABCG2. The accumulation of fluorescent calcein in human HEK293 cells (A and B, left panel), MDR19-HEK293 cells (A, right panel) and MRP1-HEK293 cells (B, right panel) for 10 min, or fluorescent pheophorbide A (PhA) in human HEK293 cells (C, left panel) and R482-HEK293 (C, right panel), as well as in human S1 colon cancer cells (D, left panel) and the ABCG2-overexpressing variant S1-M1–80 cancer cells (D, right panel), human H460 lung cancer cells (E, left panel) and the ABCG2-overexpressing variant H460-MX20 cancer cells (E, right panel) for 1 h, was determined in the absence (solid lines) or presence of licochalcone A (LCA) at 20 μM (shaded, solid lines), or ABCB1 reference inhibitor tariquidar at 3 μM (A, dotted lines) or ABCC1 reference inhibitor MK-571 at 25 μM (B, dotted lines) or ABCG2 reference inhibitor Ko143 at 3 μM (C - E, dotted lines). (F) Concentration-dependent inhibition of ABCG2-mediated efflux of PhA by LCA in R482-HEK293 cells (open circles), S1-M1–80 cancer cells (open triangles) and H460-MX20 cancer cells (open squares). Experimental data were analyzed immediately by flow cytometry as described previously.94 Representative histograms of at least three independent experiments are shown. Values are presented as mean ± SD calculated from three independent experiments.
Figure 4.

Immunoblot detection and quantification of ABCG2 protein in (A) ABCG2-overexpressing S1-M1–80 human colon cancer cells and (B) ABCG2-overexpressing H460-MX20 human lung cancer cells treated with DMSO (vehicle control) or increasing concentrations (0.5 – 3.0 μM) of licochalcone (LCA) for 72 h according to the method described previously.79 α-Tubulin was used as an internal loading control. Values are presented as mean ± SEM calculated from three independent experiments.
Licochalcone A Potentiates Topotecan-Induced Apoptosis in ABCG2-Overexpressing Cancer Cells.
To confirm that LCA resensitizes ABCG2-overexpressing multidrug-resistant cancer cells to cytotoxic ABCG2 substrate drugs (Table 2), the effect of LCA on topotecan-induced apoptosis was examined in S1-M1–80 cancer cells to substantiate cytotoxicity and growth retardation induced by a substrate drug of ABCG2. The drug-sensitive parental S1 cancer cells and ABCG2-overexpressing S1-M1–80 cancer cells were treated with DMSO (control), 10 μM of LCA, 5 μM of topotecan or a combination of 5 μM of topotecan and 10 μM of LCA for 48 h, and analyzed by flow cytometry according to the method described previously.78, 79 As shown in Figure 5, treatment with topotecan induced substantial early and late apoptosis in S1 cells (from approximately 6% basal level to 54% total apoptosis), but not in S1-M1–80 cells (from approximately 6% basal level to 9% total apoptosis). Interestingly, treatment with LCA alone did not induce apoptosis in either S1 or S1-M1–80 cells, but topotecan-induced apoptosis in S1-M1–80 cells was significantly potentiated by LCA (from approximately 9% total apoptosis to 32% total apoptosis). These results confirmed that by modulating the drug efflux function of ABCG2, LCA improves drug-induced apoptosis in ABCG2-overexpressing cancer cells and consequently resensitizes these multidrug-resistant cancer cells to chemotherapeutic agents.
Figure 5.
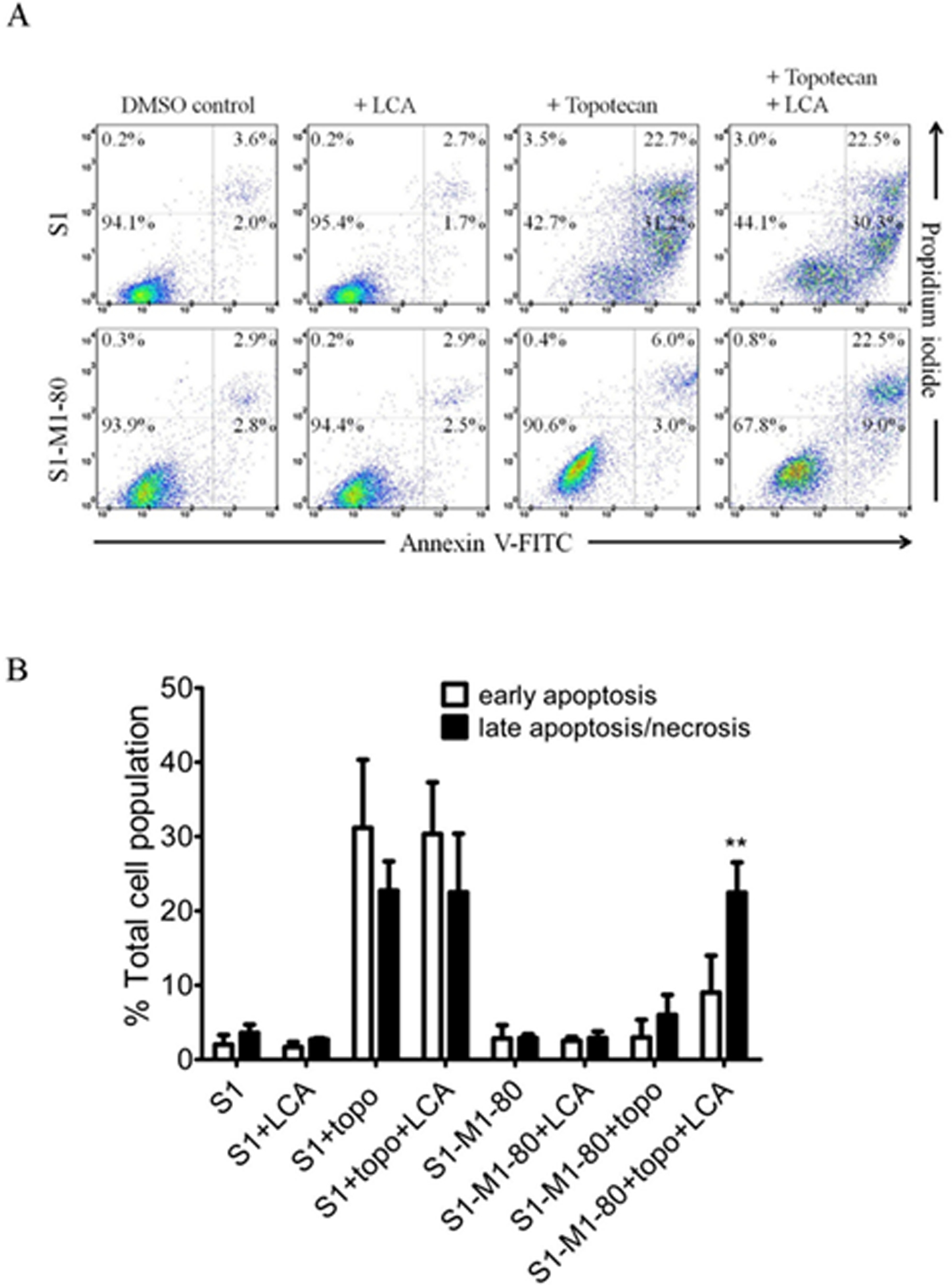
Licochalcone A potentiates apoptosis induced by ABCG2 substrate drug topotecan in ABCG2-overexpressing S1-M1–80 cancer cells. (A) Dot plots and (B) quantification of annexin V-FITC and PI double staining for apoptosis in drug-sensitive S1 human colon cancer cells (upper dot plot panels) and ABCG2-overexpressing MDR variant S1-M1–80 cancer cells (lower dot plot panels) treated with DMSO (control), 10 μM of licochalcone A (+ LCA), 5 μM of topotecan (+ Topotecan) or a combination of 5 μM of topotecan and 10 μM of licochalcone A (+ Topotecan + LCA) for 48 h. Cells were isolated and analyzed by flow cytometry according to the method described previously.95 Representative dot plots and the mean ± S.D. calculated from three independent experiments are shown. **p < 0.01, versus the same treatment in the absence of licochalcone A.
Licochalcone A Stimulates the ATPase Activity of ABCG2.
Since the inhibition of ABCG2 function by LCA appears to play a major role in the resensitization of ABCG2-overexpressing cells to chemotherapeutic agents, the binding interactions between LCA and the substrate-binding site of ABCG2 were investigated. In order to gain further insight into the interaction between LCA and the substrate-binding site of ABCG2, the effect of LCA on the vanadate (Vi)-sensitive ATPase activity of ABCG2 and the docking of LCA to the substrate-binding pocket of ABCG2 were examined. As shown in Figure 6, LCA stimulated Vi-sensitive ABCG2 ATP hydrolysis in a concentration-dependent manner, producing a maximal stimulation of approximately 150% of basal level (139.22 ± 9.34 nmole Pi/min/mg protein) and a half-maximal effective concentration (EC50) value of 14 μM. Moreover, the lowest energy docking pose analysis of LCA with the inward-open structure of human ABCG2 (PDBID:5NJ3)80, which is the conformation of ABCG2 with the binding of substrate to the ABCG2 transmembrane region, revealed numerous hydrophobic and aromatic interactions between LCA and the predicted hydrophobic residues within the transmembrane domain (TMD) of ABCG2 (Figure 7). Knowing that the stimulation of ATP hydrolysis is coupled to the transport activity of ABCG2,81, 82 the results here indicate the interaction between LCA and the substrate-binding pocket within the transmembrane region of ABCG2. Our results are in agreement with previous studies reporting that ABCG2-modulating agents stimulated the ATPase activity of ABCG2 by binding to its drug-binding pocket.63, 79, 83). If any information on toxicity is available about in vivo models such as xenograft or mice—add here. Otherwise state that at present there is no data available on in vivo toxicity under physiological conditions and this information will be useful to determine therapeutic index in a clinical setting.
Figure 6.

Licochalcone A stimulates the vanadate-sensitive ATPase activity of ABCG2. The effect of licochalcone A at indicated concentrations ranging from 0 to 390 μM on vanadate (Vi)-sensitive ABCG2 ATP hydrolysis was determined by endpoint Pi liberation assays as described in the Experimental section as described previously.91, 96 Data are presented as mean ± S.D. from three independent experiments.
Figure 7.
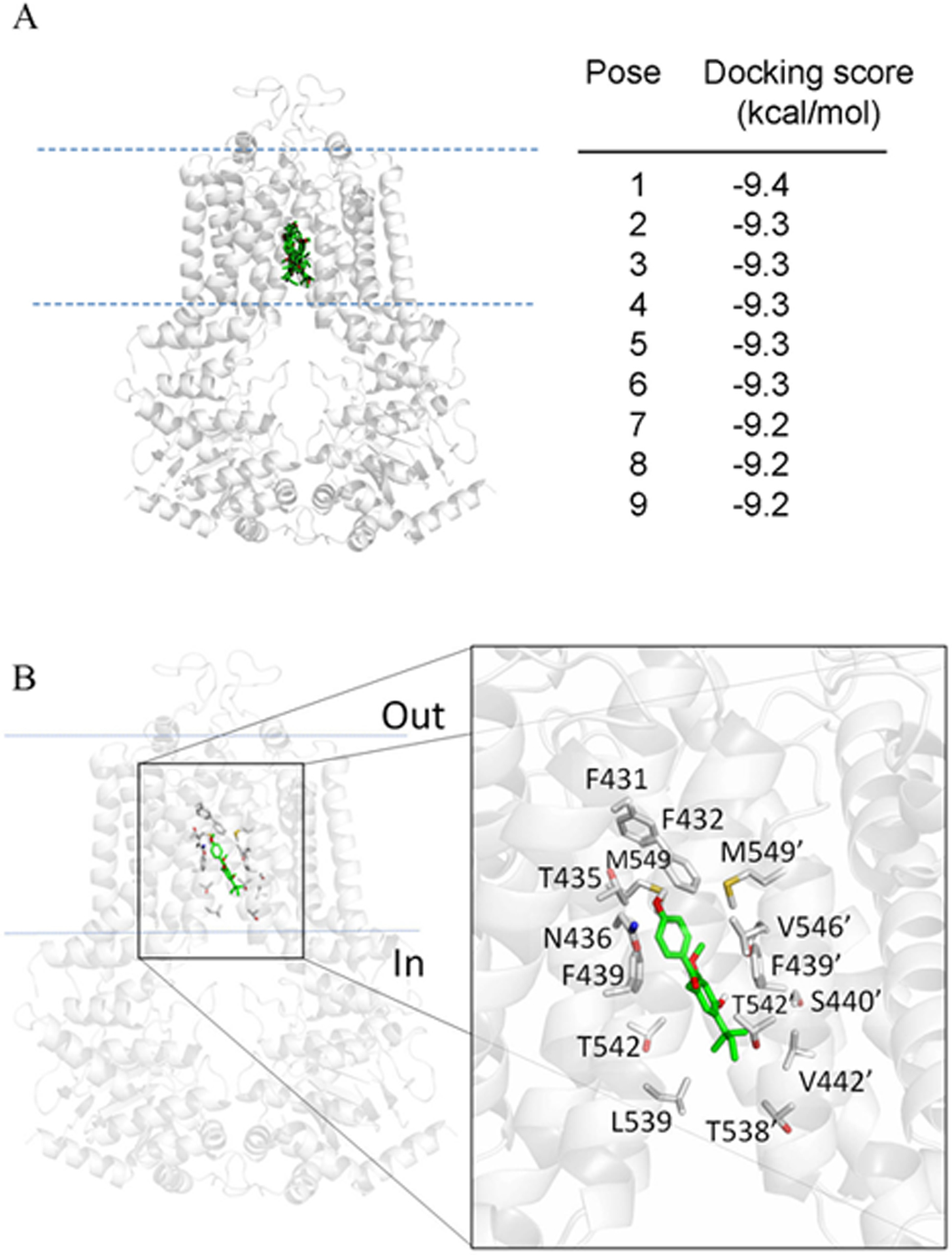
Docking of licochalcone A with cryo-electron microscopy structure of ABCG2 (PDB: 5NJ3) obtained after exhaustive docking using AutoDock Vina software,93 as described in the Experimental section. The lowest energy pose for licochalcone A with the atoms colored as carbon-green and oxygen-red, in the transmembrane region of ABCG2 is shown to illustrate the residues that are within 4 Å of the ligand (shown in gray). Figures were prepared using the PYMOL molecular graphics system, Version 1.7 Shrödinger, LLC.
Although one cannot exclude other possible mechanisms of resistance that may also be involved, the experimental results here support the notion that LCA reverses ABCG2-mediated MDR by competing directly with the binding of co-administered chemotherapeutic drugs at the same site, and the down-regulation of ABCG2 is unlikely to play a significant role (Figure 8). In summary, although combination therapy of conventional anticancer agents and natural bioactive compounds may sometimes lead to unforeseen drug-drug interactions, the experimental data support the inclusion of natural chalcone LCA as a therapeutic strategy against cancer multidrug resistance associated with the overexpression of ABCG2, deserving further investigation in preclinical studies.
EXPERIMENTAL SECTION
Chemicals and Cell Culture.
Licochalcone A (purity ≥96.0% by HPLC), tariquidar, MK-571, Ko143, and all other chemicals were obtained from Sigma-Aldrich (St. Louis, MO, USA) unless stated otherwise. Annexin V: FITC Apoptosis Detection Kit was purchased from BD Pharmingen (San Diego, CA, USA). Tools Cell Counting Kit-8 was purchased from Biotools (Biotools Co., Ltd, Taipei, Taiwan). Penicillin-Streptomycin, fetal calf serum (FCS), Phosphate-buffered saline (PBS), Dulbecco’s Modified Eagle’s Medium (DMEM), Iscove’s Modified Dulbecco’s Medium (IMDM) and RPMI medium were purchased from Gibco, Invitrogen (Gibco Invitrogen, CA, USA). The pcDNA3.1-HEK293, HEK293 cells transfected with human ABCB1 (MDR19-HEK293)84, HEK293 cells transfected with ABCC1 (MRP1-HEK293)85 and HEK293 cells transfected with ABCG2 (R482-HEK293),84 as well as the H460 human non-small cell lung cancer cells and the mitoxantrone-selected, ABCG2-overexpressing variant H460-MX20, were cultured in DMEM. The S1 human colon cancer cell line and the mitoxantrone-selected, ABCG2-overexpressing variant S1-M1–80 were cultured in RPMI-1640. HEK293 cells and HEK293 transfected cells were maintained in medium containing 2 mg/mL G418,83 whereas the S1-M1–80 cells and H460-MX20 cells were maintained in medium containing 80 μM mitoxantrone83 or 20 nM mitoxantrone,86 respectively. All cell lines were cultured in medium supplemented with 10% FCS, 2 mM L-glutamine and 100 units of penicillin/streptomycin/mL, maintained at 37 °C in 5% CO2 humidified air and placed in drug-free medium 7 days prior to assay.
Cytotoxicity Study.
Cytotoxicity assays were performed based on the method described by Ishiyama et al.87 Briefly, cells were plated in each well of 96-well plates at a density of 5000 cells per well in 100 μL of culture medium and maintained at 37 °C. After 24 h, an additional 100 μL of a tested drug at various concentrations was added to each well and incubated for an additional 72 h. Drug solutions were prepared in DMSO (up to 0.5% v/v). HEK293 cells, MDR19-HEK293, MRP1-HEK293 and R482-HEK293 cells were developed using Tools Cell Counting Kit-8 (Biotools Co., Ltd, Taipei, Taiwan), whereas attached human cancer cell lines were developed using MTT reagent as described previously.78 The half-maximal inhibitory concentration (IC50) value for each treatment was calculated from a fitted dose-response curve acquired from at least three independent experiments. For the reversal assay, a nontoxic concentration of LCA or tariquidar (a reference inhibitor of ABCB1), MK-571 (a reference inhibitor of ABCC1) or Ko143 (a reference inhibitor of ABCG2), was added to the respective cytotoxicity assays to calculate the respective fold-reversal (FR) value, which represents the extent of reversal by the tested drug.63
Apoptosis Assay.
The fraction of apoptotic cells in the total cell population induced by the indicated regimens was determined based on the conventional Annexin V–FITC and propidium iodide (PI) staining method,88 as described previously.78 Briefly, cells were treated with DMSO (control), 10 μM of LCA, 5 μM of topotecan or a combination of topotecan and LCA as indicated for 48 h before harvested, centrifuged and resuspended in FACS buffer containing 1.25 μg/mL Annexin V–FITC and 0.1 mg/mL PI and incubated for 15 min at room temperature. The labeled cells (10000 per sample) were analyzed by FACScan using CellQuest software (BD Biosciences) according to the method described by Anderson et al.89
Fluorescent Drug Accumulation Assay.
The intracellular accumulation of fluorescent probe calcein (excitation and emission wavelengths of 485 and 535 nm) was used to monitor the function of ABCB1 and ABCC1, whereas pheophorbide A (PhA) (excitation and emission wavelengths of 395 and 670 nm) was used to monitor the function of ABCG2. Briefly, 3 × 105 cells were harvested and resuspended in 4 mL of IMDM supplemented with 5% FCS before respective fluorescent substrate was added to the cell suspension in the presence or absence of licochalcone A, tariquidar, MK-571 or Ko143 for 10 min (for ABCB1- and ABCC1-mediated efflux) or 1 h (for ABCG2-mediated efflux) as described previously.73 The fluorescence signal was recorded using a FACScan flow cytometer (BD Biosciences) and analyzed using CellQuest software (BD Bioscience) according to the method described by Gribar et al.90
Immunoblotting.
Cells were treated with different concentrations of LCA for 72 h, harvested and then subjected to SDS-polyacrylamide electrophoresis. Primary antibodies BXP-21 (1:15000) and α-tubulin (1:100000) were used in Western blot immunoassay to detect ABCG2 and positive control tubulin, respectively. The horseradish peroxidase-conjugated goat anti-mouse IgG (1:100000) was used as the secondary antibody. Signals were detected as described previously.83
ATPase Assay.
The effect of LCA on vanadate (Vi)-sensitive ABCG2 ATPase activity was determined using a colorimetric method based on the endpoint inorganic phosphate (Pi) assay that measures the amount of Pi released during the reaction as described previously.91 Briefly, membrane vesicles of ABCG2-expressing High-Five cells (Thermo Fisher Scientific, Waltman, MA, USA) were incubated with LCA in the absence or presence of 0.3 mM sodium orthovanadate in ATPase buffer (50 mM MES-Tris pH 6.8, 1 mM ouabain, 2 mM DTT, 5 mM NaN3, 1 mM EGTA, 50 mM KCl), and the ATPase activity of ABCG2 was monitored at 37 °C. The reaction was stopped after 20 min by the addition of 50 μL of Pi reagent containing 1% ammonium molybdate in 2.5 N H2SO4 and 0.014% antimony potassium tartrate. The liberated inorganic phosphate was quantified by the addition of a 150 μL of 0.33% sodium L-ascorbate and measured (absorbance at 880 nm) using the Spectramax iD3 microplate reader (Molecular Devices, San Jose, CA, USA). The Vi-sensitive activity was calculated as the ATPase activity in the absence of vanadate minus the ATPase activity in the presence of vanadate, as described previously.91
Docking Analysis of Licochalcone A with ABCG2.
The structure of LCA and the inward-open structure of ABCG2 (PDB: 5NJ3)80 were prepared using MGLtools software package (The Scripps Research Institute).92 The docking of LCA to the ABCG2 template was conducted using AutoDock Vina.93 The following residues within the drug-binding pocket of ABCG2 were set as flexible: N393, A397, N398, V401, L405, I409, T413, N424, F431, F432, T435, N436, F439, S440, V442, S443, Y538, L539, T542, I543, V546, F547, M549, I550, L554, L555. The receptor grid was centered at x = 125, y = 125 and z = 130, and a box with inner box dimensions 34 Å × 30 Å × 50 Å. Due to the large box size and the number of flexible residues, the exhaustiveness level was set at 100 for ABCG2 protein to ensure that the global minimum of the scoring function would be found. Analysis of the docked poses was performed using PYMOL molecular graphics system, Version 1.7 (Shrödinger, LLC).
Quantification and Statistical Analysis.
The experimental data and IC50 values are presented as mean ± standard deviation (SD) calculated from at least three independent experiments unless stated otherwise. Curve fitting and statistical analysis were performed with GraphPad Prism (GraphPad Software, La Jolla, CA, USA) and KaleidaGraph (Synergy Software, Reading, PA, USA) software. The improvement in fit was analyzed by two-sided Student’s t-test and labeled “statistically significant” if the probability, p, was less than 0.05.
ACKNOWLEDGMENTS
This work was supported by the Ministry of Science and Technology of Taiwan (MOST-108-2320-B-182-038-MY3 and MOST-106-2320-B-182-017), Chang Gung Medical Research Program (BMRPC17 and CMRPD1J0281) and the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research to SL and SVA.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Gottesman MM Annu Rev Med 2002, 53, 615–27. [DOI] [PubMed] [Google Scholar]
- (2).Szakacs G; Paterson JK; Ludwig JA; Booth-Genthe C; Gottesman MM Nat Rev Drug Discov 2006, 5, (3), 219–34. [DOI] [PubMed] [Google Scholar]
- (3).Gillet JP; Gottesman MM Methods in molecular biology 2010, 596, 47–76. [DOI] [PubMed] [Google Scholar]
- (4).Robey RW; Pluchino KM; Hall MD; Fojo AT; Bates SE; Gottesman MM Nat Rev Cancer 2018, 18, (7), 452–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Larkin A; O’Driscoll L; Kennedy S; Purcell R; Moran E; Crown J; Parkinson M; Clynes M Int J Cancer 2004, 112, (2), 286–94. [DOI] [PubMed] [Google Scholar]
- (6).Pilarski LM; Szczepek AJ; Belch AR Blood 1997, 90, (9), 3751–9. [PubMed] [Google Scholar]
- (7).Schwarzenbach H Med Oncol 2002, 19, (2), 87–104. [DOI] [PubMed] [Google Scholar]
- (8).Turner JG; Gump JL; Zhang C; Cook JM; Marchion D; Hazlehurst L; Munster P; Schell MJ; Dalton WS; Sullivan DM Blood 2006, 108, (12), 3881–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Hofmeister CC; Yang X; Pichiorri F; Chen P; Rozewski DM; Johnson AJ; Lee S; Liu Z; Garr CL; Hade EM; Ji J; Schaaf LJ; Benson DM Jr.; Kraut EH; Hicks WJ; Chan KK; Chen CS; Farag SS; Grever MR; Byrd JC; Phelps MA J Clin Oncol 2011, 29, (25), 3427–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Matthews C; Catherwood MA; Larkin AM; Clynes M; Morris TC; Alexander HD Leuk Lymphoma 2006, 47, (11), 2308–13. [DOI] [PubMed] [Google Scholar]
- (11).Ross DD; Karp JE; Chen TT; Doyle LA Blood 2000, 96, (1), 365–8. [PubMed] [Google Scholar]
- (12).Steinbach D; Sell W; Voigt A; Hermann J; Zintl F; Sauerbrey A Leukemia 2002, 16, (8), 1443–7. [DOI] [PubMed] [Google Scholar]
- (13).Uggla B; Stahl E; Wagsater D; Paul C; Karlsson MG; Sirsjo A; Tidefelt U Leuk Res 2005, 29, (2), 141–6. [DOI] [PubMed] [Google Scholar]
- (14).Kovalev AA; Tsvetaeva DA; Grudinskaja TV Exp Oncol 2013, 35, (4), 287–90. [PubMed] [Google Scholar]
- (15).Yoh K; Ishii G; Yokose T; Minegishi Y; Tsuta K; Goto K; Nishiwaki Y; Kodama T; Suga M; Ochiai A Clinical cancer research : an official journal of the American Association for Cancer Research 2004, 10, (5), 1691–7. [DOI] [PubMed] [Google Scholar]
- (16).Wu C-P; Hsieh C-H; Wu Y-S Molecular Pharmaceutics 2011, 8, (6), 1996–2011. [DOI] [PubMed] [Google Scholar]
- (17).Gottesman MM; Fojo T; Bates SE Nat Rev Cancer 2002, 2, (1), 48–58. [DOI] [PubMed] [Google Scholar]
- (18).Hegedus C; Ozvegy-Laczka C; Szakacs G; Sarkadi B Curr Cancer Drug Targets 2009, 9, (3), 252–272. [DOI] [PubMed] [Google Scholar]
- (19).Noguchi K; Katayama K; Sugimoto Y Pharmgenomics Pers Med 2014, 7, 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Bodo A; Bakos E; Szeri F; Varadi A; Sarkadi B Toxicol Lett 2003, 140–141, 133–43. [DOI] [PubMed] [Google Scholar]
- (21).Wu CP; Calcagno AM; Ambudkar SV Curr Mol Pharmacol 2008, 1, (2), 93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Shukla S; Wu CP; Ambudkar SV Expert Opin Drug Metab Toxicol 2008, 4, (2), 205–223. [DOI] [PubMed] [Google Scholar]
- (23).Wu CP; Ohnuma S; Ambudkar SV Curr Pharm Biotechnol 2011, 12, (4), 609–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Sanders K; Moran Z; Shi Z; Paul R; Greenlee H Semin Oncol Nurs 2016, 32, (3), 215–40. [DOI] [PubMed] [Google Scholar]
- (25).Li T; Wang N; Zhang T; Zhang B; Sajeevan TP; Joseph V; Armstrong L; He S; Yan X; Naman CB Mar Drugs 2019, 17, (9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Khalifa SAM; Elias N; Farag MA; Chen L; Saeed A; Hegazy MF; Moustafa MS; Abd El-Wahed A; Al-Mousawi SM; Musharraf SG; Chang FR; Iwasaki A; Suenaga K; Alajlani M; Goransson U; El-Seedi HR Mar Drugs 2019, 17, (9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Ye Q; Liu K; Shen Q; Li Q; Hao J; Han F; Jiang RW Front Oncol 2019, 9, 487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kumar A; Jaitak V Eur J Med Chem 2019, 176, 268–291. [DOI] [PubMed] [Google Scholar]
- (29).Rabindran SK; Ross DD; Doyle LA; Yang W; Greenberger LM Cancer research 2000, 60, (1), 47–50. [PubMed] [Google Scholar]
- (30).Allen JD; Schinkel AH Mol Cancer Ther 2002, 1, (6), 427–34. [PubMed] [Google Scholar]
- (31).Hatano T; Yasuhara T; Fukuda T; Noro T; Okuda T Chem Pharm Bull (Tokyo) 1989, 37, (11), 3005–9. [DOI] [PubMed] [Google Scholar]
- (32).Hajirahimkhan A; Simmler C; Dong H; Lantvit DD; Li G; Chen SN; Nikolic D; Pauli GF; van Breemen RB; Dietz BM; Bolton JL Chem Res Toxicol 2015, 28, (11), 2130–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Wang ZF; Liu J; Yang YA; Zhu HL Curr Med Chem 2018. [DOI] [PubMed] [Google Scholar]
- (34).Chen M; Theander TG; Christensen SB; Hviid L; Zhai L; Kharazmi A Antimicrob Agents Chemother 1994, 38, (7), 1470–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Si H; Xu C; Zhang J; Zhang X; Li B; Zhou X; Zhang J Int J Parasitol Drugs Drug Resist 2018, 8, (2), 238–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Adianti M; Aoki C; Komoto M; Deng L; Shoji I; Wahyuni TS; Lusida MI; Soetjipto Fuchino, H.; Kawahara N; Hotta H Microbiol Immunol 2014, 58, (3), 180–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Wang L; Yang R; Yuan B; Liu Y; Liu C Acta Pharm Sin B 2015, 5, (4), 310–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Fu X; Wang Z; Li L; Dong S; Li Z; Jiang Z; Wang Y; Shui W Sci Rep 2016, 6, 29680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Messier C; Grenier D Mycoses 2011, 54, (6), e801–6. [DOI] [PubMed] [Google Scholar]
- (40).Friis-Moller A; Chen M; Fuursted K; Christensen SB; Kharazmi A Planta Med 2002, 68, (5), 416–9. [DOI] [PubMed] [Google Scholar]
- (41).Kwon HS; Park JH; Kim DH; Kim YH; Park JH; Shin HK; Kim JK J Mol Med (Berl) 2008, 86, (11), 1287–95. [DOI] [PubMed] [Google Scholar]
- (42).Fu Y; Hsieh TC; Guo J; Kunicki J; Lee MY; Darzynkiewicz Z; Wu JM Biochem Biophys Res Commun 2004, 322, (1), 263–70. [DOI] [PubMed] [Google Scholar]
- (43).Cho JJ; Chae JI; Yoon G; Kim KH; Cho JH; Cho SS; Cho YS; Shim JH Int J Oncol 2014, 45, (2), 667–74. [DOI] [PubMed] [Google Scholar]
- (44).Kang TH; Seo JH; Oh H; Yoon G; Chae JI; Shim JH J Cell Biochem 2017, 118, (12), 4652–4663. [DOI] [PubMed] [Google Scholar]
- (45).Qiu C; Zhang T; Zhang W; Zhou L; Yu B; Wang W; Yang Z; Liu Z; Zou P; Liang G Int J Mol Sci 2017, 18, (8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Tsai JP; Lee CH; Ying TH; Lin CL; Lin CL; Hsueh JT; Hsieh YH Oncotarget 2015, 6, (30), 28851–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Hao W; Yuan X; Yu L; Gao C; Sun X; Wang D; Zheng Q Sci Rep 2015, 5, 10336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Zhao Y; Zhang W; Guo Z; Ma F; Wu Y; Bai Y; Gong W; Chen Y; Cheng T; Zhi F; Zhang Y; Wang J; Jiang B Oncol Rep 2013, 30, (4), 1782–92. [DOI] [PubMed] [Google Scholar]
- (49).Choi ES; Nam JS; Jung JY; Cho NP; Cho SD Sci Rep 2014, 4, 7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Chae JI; Lee R; Cho J; Hong J; Shim JH J Biomed Sci 2014, 21, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Liu XL; Tee HW; Go ML Bioorg Med Chem 2008, 16, (1), 171–80. [DOI] [PubMed] [Google Scholar]
- (52).Han Y; Riwanto M; Go ML; Ee PL Eur J Pharm Sci 2008, 35, (1–2), 30–41. [DOI] [PubMed] [Google Scholar]
- (53).Juvale K; Pape VF; Wiese M Bioorg Med Chem 2012, 20, (1), 346–55. [DOI] [PubMed] [Google Scholar]
- (54).Valdameri G; Gauthier C; Terreux R; Kachadourian R; Day BJ; Winnischofer SM; Rocha ME; Frachet V; Ronot X; Di Pietro A; Boumendjel A J Med Chem 2012, 55, (7), 3193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Winter E; Gozzi GJ; Chiaradia-Delatorre LD; Daflon-Yunes N; Terreux R; Gauthier C; Mascarello A; Leal PC; Cadena SM; Yunes RA; Nunes RJ; Creczynski-Pasa TB; Di Pietro A Drug Des Devel Ther 2014, 8, 609–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Wu CP; Hsieh CH; Wu YS Mol Pharm 2011, 8, (6), 1996–2011. [DOI] [PubMed] [Google Scholar]
- (57).Tang C; Schafranek L; Watkins DB; Parker WT; Moore S; Prime JA; White DL; Hughes TP Leukemia & lymphoma 2011, 52, (11), 2139–47. [DOI] [PubMed] [Google Scholar]
- (58).Nabekura T; Hiroi T; Kawasaki T; Uwai Y Biomed Pharmacother 2015, 70, 140–5. [DOI] [PubMed] [Google Scholar]
- (59).Fan X; Bai J; Zhao S; Hu M; Sun Y; Wang B; Ji M; Jin J; Wang X; Hu J; Li Y Toxicol In Vitro 2019, 61, 104642. [DOI] [PubMed] [Google Scholar]
- (60).Doyle LA; Yang W; Abruzzo LV; Krogmann T; Gao Y; Rishi AK; Ross DD Proceedings of the National Academy of Sciences of the United States of America 1998, 95, (26), 15665–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Brangi M; Litman T; Ciotti M; Nishiyama K; Kohlhagen G; Takimoto C; Robey R; Pommier Y; Fojo T; Bates SE Cancer Res. 1999, 59, (23), 5938–46. [PubMed] [Google Scholar]
- (62).Honjo Y; Hrycyna CA; Yan QW; Medina-Perez WY; Robey RW; van de Laar A; Litman T; Dean M; Bates SE Cancer Res 2001, 61, (18), 6635–6639. [PubMed] [Google Scholar]
- (63).Dai CL; Tiwari AK; Wu CP; Su XD; Wang SR; Liu DG; Ashby CR Jr.; Huang Y; Robey RW; Liang YJ; Chen LM; Shi CJ; Ambudkar SV; Chen ZS; Fu LW Cancer Res 2008, 68, (19), 7905–7914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Komoto TT; Bernardes TM; Mesquita TB; Bortolotto LFB; Silva G; Bitencourt TA; Baek SJ; Marins M; Fachin AL Molecules 2018, 23, (8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Wu J; Zhang X; Wang Y; Sun Q; Chen M; Liu S; Zou X Oncol Rep 2018, 39, (3), 1181–1190. [DOI] [PubMed] [Google Scholar]
- (66).Liou CJ; Lee YK; Ting NC; Chen YL; Shen SC; Wu SJ; Huang WC Cells 2019, 8, (5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Lin RC; Yang SF; Chiou HL; Hsieh SC; Wen SH; Lu KH; Hsieh YH Cells 2019, 8, (11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Liu X; Ma Y; Wei X; Fan T J Cell Biochem 2018, 119, (4), 3210–3219. [DOI] [PubMed] [Google Scholar]
- (69).Guo W; Liu B; Yin Y; Kan X; Gong Q; Li Y; Cao Y; Wang J; Xu D; Ma H; Fu S; Liu J Front Immunol 2019, 10, 287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Huang WC; Liu CY; Shen SC; Chen LC; Yeh KW; Liu SH; Liou CJ Cells 2019, 8, (6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Mann T; Eggers K; Rippke F; Tesch M; Buerger A; Darvin ME; Schanzer S; Meinke MC; Lademann J; Kolbe L Photodermatol Photoimmunol Photomed 2020, 36, (2), 135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Hollo Z; Homolya L; Davis CW; Sarkadi B Biochim Biophys Acta 1994, 1191, (2), 384–8. [DOI] [PubMed] [Google Scholar]
- (73).Robey RW; Steadman K; Polgar O; Morisaki K; Blayney M; Mistry P; Bates SE Cancer Res 2004, 64, (4), 1242–1246. [DOI] [PubMed] [Google Scholar]
- (74).Cuestas ML; Castillo AI; Sosnik A; Mathet VL Bioorganic & medicinal chemistry letters 2012, 22, (21), 6577–9. [DOI] [PubMed] [Google Scholar]
- (75).Bhullar J; Natarajan K; Shukla S; Mathias TJ; Sadowska M; Ambudkar SV; Baer MR PLoS One 2013, 8, (8), e71266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Natarajan K; Bhullar J; Shukla S; Burcu M; Chen ZS; Ambudkar SV; Baer MR Biochemical pharmacology 2013, 85, (4), 514–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).He Y; Ci X; Xie Y; Yi X; Zeng Y; Li Y; Liu C Phytomedicine 2019, 56, 175–182. [DOI] [PubMed] [Google Scholar]
- (78).Hsiao SH; Lu YJ; Li YQ; Huang YH; Hsieh CH; Wu CP Mol Pharm 2016. [DOI] [PubMed] [Google Scholar]
- (79).Wu CP; Hsiao SH; Murakami M; Lu MJ; Li YQ; Hsieh CH; Ambudkar SV; Wu YS Cancer Lett 2017, 409, 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Taylor NMI; Manolaridis I; Jackson SM; Kowal J; Stahlberg H; Locher KP Nature 2017, 546, (7659), 504–509. [DOI] [PubMed] [Google Scholar]
- (81).Ambudkar SV; Dey S; Hrycyna CA; Ramachandra M; Pastan I; Gottesman MM Annu Rev Pharmacol Toxicol 1999, 39, 361–98. [DOI] [PubMed] [Google Scholar]
- (82).Ambudkar SV; Kimchi-Sarfaty C; Sauna ZE; Gottesman MM Oncogene 2003, 22, (47), 7468–85. [DOI] [PubMed] [Google Scholar]
- (83).Wu CP; Shukla S; Calcagno AM; Hall MD; Gottesman MM; Ambudkar SV Mol Cancer Ther 2007, 6, (12 Pt 1), 3287–3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Robey RW; Honjo Y; Morisaki K; Nadjem TA; Runge S; Risbood M; Poruchynsky MS; Bates SE Br J Cancer 2003, 89, (10), 1971–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Muller M; Yong M; Peng XH; Petre B; Arora S; Ambudkar SV Biochemistry 2002, 41, (31), 10123–32. [DOI] [PubMed] [Google Scholar]
- (86).Henrich CJ; Bokesch HR; Dean M; Bates SE; Robey RW; Goncharova EI; Wilson JA; McMahon JB J Biomol Screen 2006, 11, (2), 176–83. [DOI] [PubMed] [Google Scholar]
- (87).Ishiyama M; Tominaga H; Shiga M; Sasamoto K; Ohkura Y; Ueno K Biol Pharm Bull 1996, 19, (11), 1518–20. [DOI] [PubMed] [Google Scholar]
- (88).Vermes I; Haanen C; Steffens-Nakken H; Reutelingsperger C J Immunol Methods 1995, 184, (1), 39–51. [DOI] [PubMed] [Google Scholar]
- (89).Anderson HA; Maylock CA; Williams JA; Paweletz CP; Shu H; Shacter E Nat Immunol 2003, 4, (1), 87–91. [DOI] [PubMed] [Google Scholar]
- (90).Gribar JJ; Ramachandra M; Hrycyna CA; Dey S; Ambudkar SV The Journal of membrane biology 2000, 173, (3), 203–14. [DOI] [PubMed] [Google Scholar]
- (91).Ambudkar SV Methods Enzymol 1998, 292, 504–514. [DOI] [PubMed] [Google Scholar]
- (92).Sanner MF J Mol Graph Model 1999, 17, (1), 57–61. [PubMed] [Google Scholar]
- (93).Trott O; Olson AJ J Comput Chem 2010, 31, (2), 455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Wu CP; Sim HM; Huang YH; Liu YC; Hsiao SH; Cheng HW; Li YQ; Ambudkar SV; Hsu SC Biochemical pharmacology 2013, 85, (3), 325–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Wu CP; Hsiao SH; Su CY; Luo SY; Li YQ; Huang YH; Hsieh CH; Huang CW Biochem Pharmacol 2014, 92, (4), 567–76. [DOI] [PubMed] [Google Scholar]
- (96).Wu CP; Hsieh CH; Hsiao SH; Luo SY; Su CY; Li YQ; Huang YH; Huang CW; Hsu SC Mol Pharm 2015, 12, (11), 3885–95. [DOI] [PubMed] [Google Scholar]