

Abstract
Identifying structural variation (SV) is essential for genome interpretation but has been historically difficult to resolve. Detection methods using ensemble algorithms and emerging sequencing technologies that mitigate short-read limitations have enabled discovery of thousands of SVs, uncovering information about their ubiquity, relationship to disease, and possible effects on biological mechanisms. Given the variability in SV type and size, along with unique detection biases of emerging genomic platforms, multiplatform discovery is necessary to resolve the full spectrum of variation. Here, we review modern approaches for investigating SVs and proffer that, moving forward, studies integrating biological information with detection will be necessary to comprehensively understand the impact of SV in the human genome.
Introduction
Widespread application of whole-genome high throughput sequencing (HTS) for the detection of genetic variants has shown that differences between individuals are typically present as single nucleotide variants (SNVs), small insertions and deletions (indels; < 50bp), and structural variations (SVs)1. SVs are extremely diverse in type and size, ranging anywhere from ~50 bp to well over megabases of sequence, affecting more of the genome per nucleotide changes than any other class of sequence variant2–6. They comprise a myriad of subclasses consisting of unbalanced copy number variants (CNVs) including deletions, duplications and insertions of genetic material, as well as balanced rearrangements such as inversions and inter and intrachromosomal translocations. Additionally, SVs include mobile element insertions, multi-allelic CNVs of highly variable copy number, segmental duplications, and complex arrangements which consist of multiple combinations of these described events. SVs are present in every human genome, affecting molecular and cellular processes, regulatory functions, 3D structure, and transcriptional machinery5,7,8. Thus, increasing our knowledge of SV structure and prevalence is necessary to discern the genomics of physiological and pathophysiological processes.
Many of the prevalent tools and algorithms to detect SVs use short-read signatures to infer the presence of SVs compared to a reference genome9. While short-read approaches are highly effective at resolving SNVs, SV detection is unable to completely overcome the limited sequence and insert sizes of standard short-read HTS10. There are still considerable limitations on what can be achieved in SV analysis owing to technical difficulties in resolving exact structures of SVs given their substantial diversity and proximity to repetitive regions5,9,11–13. SNVs detected by short-reads can be sequence-resolved during the discovery stage owing to their smaller size whereas most SVs would require computational inference post hoc. Because of this, the degree to which contemporary genomics has studied SNVs compared to SVs is significantly skewed. Specifically, standardized best practices, robust detection platforms, high-quality reference sets, and extensive functional data from genome-wide association studies are available for SNV research14–20. Comparatively, progress in SV analysis is significantly behind, as detection is suboptimal and reference sets are lacking in diversity, sample size, and depth.
A considerable increase in the development and availability of novel sequencing technologies that leverage specialized flow cells, advanced microfluidics, and protein pores, among others, has led to platforms that produce reads several orders of magnitude longer than those generated from short-read HTS, enabling direct detection of many SVs21. In this Review, we discuss methods for resolving SVs in human genomes that bypass the limitations of individual short-read approaches through algorithmic ensembles and by leveraging new technologies. In particular, we discuss the findings of applying new technologies to genome assembly and population-scale variant mapping as they relate to germline SVs (for recent reviews on somatic SVs, see REFS22,23). Along with integrating short-read SV callers, we consider integrating data generated from multiple genomic platforms as a way to comprehensively detect the broad range of SVs. As each approach has different strengths, we highlight the individual strategies, their applications, and recent findings. We discuss future directions and consider incorporating multimodal biological information as a way to interpret the impact of SVs in their molecular contexts.
Ensemble Algorithms
Sequencing-based SV detection primarily leverages signatures that result from mapping discordance between a sample read and the reference genome: read-pair (RP) assesses the orientation and distance of paired-ends; read-depth (RD) detects deletions or duplications based on divergences in mapping depth; split-read (SR) approaches leverage alignments that map over breakpoints; and alternatively de novo or local assembly (AS) reassembles contigs before pairwise comparison to a reference24–26. Many early SV callers like PEMer, Breakdancer, and CNVnator specialized in leveraging only one of four approaches which inherently limits detection (reviewed in Alkan et al.)27–29. Hybrid-signature algorithms such as Genome STRiP, Delly, Manta, and LUMPY, among others, mitigate the limited scope of single-approach algorithms, improving sensitivity by integrating two or more disparate signatures to call putative SVs based on combined supporting evidence30–36. However, even with signal integration, no individual caller has been shown to be capable of identifying the complete range of SV owing to the large diversity in viable detection approaches and the variability in SV subtype and size37–39. One strategy to attenuate this issue involves detecting SVs using multiple discrete algorithms on the same sequence data and integrating calls to generate a unified callset (FIG. 1). Combining multiple algorithms improves detection by leveraging the different heuristic approaches of each individual caller and has been shown to increase the concordance of SV calls when compared to reference datasets developed by large consortium projects40–42. From here, we refer to “ensemble algorithm” (EA) as the combination and integration of multiple independent SV detection algorithms.
Figure 1 |. Overview of ensemble algorithms.

This flowchart outlines the major steps in an ensemble algorithm. Step 1, discordantly mapped reads result in signatures that are used to infer SVs. Step 2, multiple independent algorithms detect SVs in parallel. Step 3, filters and heuristics based on the project aims are applied to remove false-positives and merge calls (see BOX 2 for details). Step 4, final decisions are made to designate and preserve high-confidence calls and they are output as a consolidated list of putative variants.
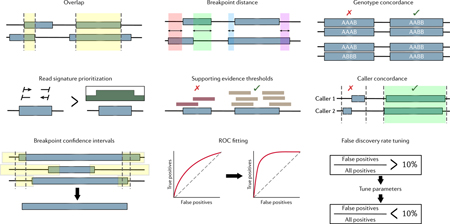
Most EA methods are “in-house,” meaning the algorithm ensemble and heuristic filters are unique to individual projects. Thus, the combination of algorithms employed are non-standardized but typically consist of one or several algorithms to cover each signature type, e.g. combining CNVnator with BreakDancer and Pindel to cover RD, RP and SR, respectively, though recent approaches use the hybrid-signature callers discussed above. Following multi-algorithm detection, the resultant calls are merged, combining potentially duplicate SVs while delineating SVs called uniquely by each algorithm. The methods to integrate, combine, and score calls varies significantly between studies and thus far have used breakpoint confidence interval overlap, breakpoint distances, false-discovery rate (FDR) cutoffs, read-signature prioritization (SR > RP > RD), caller concordance, and supporting signatures thresholds (BOX 2) 4,5,43–46. A fifth factor, coordinate overlap, is considered by all EA methods to varying degrees. Depending on the level of sensitivity a project aims to achieve, applications will either intersect calls or take a union, decreasing and increasing sensitivity while decreasing and increasing the FDR, respectively.
Box 2 |. Factors in integrating structural variant calls.
As detection methods vary significantly in their resolution and approach, a large variety of heuristics have been applied to merge calls derived from different algorithms. (1) Almost all integration methods consider the immediate intuitive option, overlap, with a common requirement of 50% reciprocity. Overlap analysis can require a minimum or maximum length difference between the called SVs to improve stringency. Alternative to coordinate overlap, one can use sequence similarity as employed by the Genome in a Bottle consortium102. (2) Computing the distance between breakpoints as opposed to overlap is useful for higher-resolution methods like split-read analysis. (3) Algorithms may require that calls to be merged have consistent genotypes for additional accuracy. (4) Read signatures are often prioritized such that if two calls overlap, the call supported with a higher-resolution read signature is chosen. (5) Calls may be required to have support from a minimum number of reads containing a given signature before merging. (6) Intersection, or caller concordance, requires that calls are detected by a minimum number of multiple algorithms, most often two. This opposes taking the union of calls which requires no caller overlap. (7) Breakpoint confidence intervals were estimated by local reassembly in the 1KGP phase 1 and by comparisons to high-quality long-read SVs in Chaisson et al.4,39. In both studies calls were merged if their breakpoint confidence intervals overlapped. (8) Parameters of individual callers can be adjusted to better fit a receiver operating characteristic curve by benchmarking against a truth set of choice, though high-confidence calls within a given callset have also been used as a benchmark43. (9) Projects with orthogonal data can adjust caller parameters to keep FDR at a certain threshold (typically < 10%) before merging calls5. These factors and teschniques have been primarily considered for short-read integration but they carry over to multiplatform approaches as well.
There are standalone tools for EAs that help standardize these integrative pipelines. SpeedSeq employs LUMPY and CNVnator to cover SR, PE, and RD detection before validating calls with a Bayesian likelihood genotyper (SVTyper), an approach implemented in the population-scale specific svtools47,48. HugeSeq, SVMerge, iSVP, and Parliament2 are all EA callers that primarily intersect by coordinate overlap whereas MetaSV takes the union40,41,49–51. SVMerge and MetaSV both validate their consensus calls with local reassembly but MetaSV prioritizes SV signatures with higher resolution (e.g. SR over RP). Parliament2 allows users to decide on a combination of six short-read algorithms, merges calls with SURVIVOR, and genotypes with SVTyper as well47,52. EA callers are beginning to implement meta-level heuristics to improve precision beyond using simple overlap: (1) Parliament2 scores each SV call with a caller concordance metric trained on HG002; (2) FusorSV implements a data-mining method to learn how well different SV algorithms perform compared to a truth set to promote the most complementary and highest performing ensemble; (3) CN-Learn, an algorithm for whole-exome data, extracts features from a truth set and uses these features to train a Random Forest classifier that differentiates CNV calls as true or false50,53,54.
Population-scale SV detection.
EA approaches have been widely used in studies characterizing SVs across populations. The 1000 Genomes Project (1KGP) initially integrated nineteen algorithms to detect SVs in Yoruban, Japanese, Han, and European individuals to create a sequencing-based SV reference map4. This early work provided one of the first frameworks for using ensemble approaches to detect SVs at population-scale and revealed 51 SV hotspots in the genome, 80% of which were dominated by a single formation mechanism, non-allelic homologous recombination, some at loci associated with known genetic conditions. At the completion of phase 3, the 1KGP sequenced 2,504 individuals across 26 populations and investigated all major SV classes in contrast to the deletion focus of the phase 1 marker paper5. The authors generated one of the most comprehensive and diverse reference sets of human SVs estimating that typical human genomes contain between 2,100–2,500 SVs affecting ~20 million nucleotides, finding that SVs are enriched up to 50X more for expression quantitative trait loci compared to SNVs. While the 1KGP was an enormous effort that set the stage for large-scale SV detection by sequencing, the relatively low ~6–7x coverage per sample limited power to detect rare variants55.
SV projects with larger and deeper datasets have emerged to improve upon the 1KGP reference set. Abel et al. applied svtools to ~18,000 human genomes, detecting 118,973 and 241,426 SVs from datasets aligned to GRCh37 and GRCh38, respectively44. Abel and colleagues estimated a mean of 4,442 high-confidence SVs per human genome and notably find: (1) ~4/4,442 directly alter exons, (2) ~19/4,442 are rare non-coding deletions that, using predictive functional annotation, (3) were up to 800 times more likely to be strongly deleterious than rare SNVs and exhibited levels of purifying selection comparable to small loss-of-function variants. To improve rare SV detection, The Genome Aggregation Database (gnomAD) systematically processed data from fewer individuals (~15,000) but at increased mean coverage (~32X vs 20X) relative to Abel et al.43. The authors detected 498,257 SVs from an ensemble of four algorithms finding an average of 8,202 SVs per human genome nonuniformly distributed through the genome by SV subclass. Collins et al. revealed 253/8,202 SVs in the average genome are intragenic and 8/8,202 are rare SVs that likely alter gene function. Strikingly, they found 57% of the human reference genome “hg19” is covered by at least 1 CNV. The 1KGP and subsequent population-scale SV analyses show the potential for SVs to impact gene expression and reveal the prodigious ubiquity of SVs far beyond the ~12 CNVs per human genome estimated in 200456.
In contrast to global approaches, some projects focus on detecting SVs from populations deriving from a recent common ancestry. SVs were twice analyzed in ~750 genomes derived from 250 Dutch families, once for de novo SVs and another for phased SVs (note that SVs were defined as variants >20 bp in this project), revealing Dutch-specific SVs and SV hotspots undetected by the 1KGP45,57. Similar work by Nagasaki et al. used an EA to detect SVs in 1,070 Japanese individuals to develop a Japanese-specific reference panel58. An increase in similar population-specific SV detection projects will be necessary to shift the diversity gap in genetics research and help identify rare SVs specific to ancestral backgrounds59. Indeed, some groups are still extremely underrepresented: Hispanic and Latin American individuals make up only 7.8% and 16% of the gnomAD-SV and Abel et al. datasets, respectively43,44.
Limitations.
EA studies are confounded by highly variable coverage, which has ranged from 3X to 90X in different projects, leading to the application of ad hoc heuristics and filtering which appreciably influence sensitivity and detection outcomes. Projects employ anywhere from three to nineteen distinct algorithms – variation in sensitivity and precision between algorithm choices will directly affect the consensus callset as the accuracy of ensembles are highly influenced by algorithm combinations38. The truth-sets used to benchmark calls and the filters applied for stringency are also highly variable, leading to parameterizations that may sacrifice precision for recall, or vice versa. Additionally, standalone EA tools are largely immature and mostly rely on simple overlap. Larger projects optimize EAs with truth sets generated from validation data, implementing FDR cutoffs and ROC tuning, but standalone methods do not possess such specifically generated benchmarks, making it difficult to implement these methods. The development of standardized variant benchmarks is an active area of research that may help formalize development of EAs by providing high-quality reference datasets that are thoroughly validated computationally and experimentally42,60. Further, EAs focused on integrating only short-read data do not overcome the limitations of short-insert sizes: they poorly detect small insertions and continue to suffer in repetitive regions39,61,62.
Emerging genomic technologies
A plethora of emerging technologies seek to expand beyond the capabilities of short-reads. Connected-molecule strategies, such as 10x Genomics Linked-Reads (LR), Strand-seq, and Hi-C, expand upon short-reads by inferring long connections between distally mapped short-read pairs. These strategies are similar to long-insert short-read libraries (reviewed elsewhere)63, which trade lowered sequence coverage for high physical coverage, improving and decreasing power to detect large and small variants, respectively. Alternatively, single-molecule strategies generate contiguous reads tens to hundreds of kilobases long, thus enabling direct detection of many SVs and improving alignment of unique reads in repetitive regions. Single-molecule strategies exist in two dominant forms: (1) long-read sequencing by Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT), and (2) optical mapping (OM) by Bionano. Comparatively, connected-molecule strategies have high specificity for defined size ranges and SV subtypes, whereas single-molecule strategies have higher overall sensitivity. Many of the above technologies are thoroughly reviewed in Goodwin et al21.
Connected-molecule strategies
10x Genomics Linked-Reads.
A number of methods, such as pooled-clone sequencing and Illumina Synthetic Long Reads, represent “synthetic long reads” which use specific library preparations to infer long range information from existing short-read sequencers64,65. Linked-Reads (LRs) from 10x Genomics are currently the most commonly used synthetic long-read platform, which partitions and barcodes diluted high-molecular weight DNA using a microfluidic device prior to short-read sequencing such that the origin of the short-read fragments can be determined from their respective barcodes and long-range information is reconstructed in silico66. Additionally, LRs retain their underlying short-read information and have greatly increased physical coverage resulting from coverage of the constructed molecule combined with coverage of each underlying short-fragment. The physical coverage makes LRs well suited for SV detection whereas the low error rate and long-molecule length (up to 100 kb) makes it useful for haplotype phasing67. Detection methods such as Long Ranger and GROC-SVs leverage read clouds which are clusters of short-reads implied to derive from the same underlying molecule due to identical barcodes. Read cloud methods look at two criteria: (1) density of overlapping barcodes where sudden increases or drops in barcode “coverage” determine SV breakpoints; (2) distant genomic loci that share more barcode overlap than would occur by chance (FIG. 2)66,68,69. GROC-SVs additionally performs local reassembly to detect complex SVs 10 kb −100 kb in length. A second approach analyzes split alignments within “molecules” which are the reconstructed long-reads from shared barcodes, analogous to split-reads. NAIBR, LinkedSV, and VALOR2 are SV callers that use split molecule approaches to detect SVs while ZoomX considers discrepancies in molecule coverage.70–74.
Figure 2 |. Structural variation signatures in single-molecule and connected-molecule strategies.

Emerging technologies vary in how they detect SVs. 10x Genomics linked-reads detect SVs based on barcode overlap between genomic loci. Split-molecule approaches infer SVs from splitting of linked-reads, examples of which are displayed below each barcode matrix (each color represents a shared barcode and linked-molecules are separated by haplotype; only homozygous variants are shown for simplicity). Strand-seq determines SVs based on read-depth or sudden changes in mapping orientation. For deletions and duplications, only two of four possible daughter cell configurations are shown for simplicity (Watson-Watson and Watson-Crick, Crick-Crick not shown). For inversions, only a homozygous inversion in Watson-Watson and Crick-Crick daughter cells are shown as Watson-Crick daughter cells mask homozygous inversions (homozygous for simplicity; for more detail on inversion detection see REF81. Hi-C detects SVs by looking for unusually high-frequency contacts between genomic loci. Underneath each interaction matrix is a schematic of the expected chromosomal contacts resulting from each SV. Single-molecule sequencing methods infer SVs based on discordant mapping signatures that can involve one (intra) or many (inter) reads. SVs derive from intra-read signatures, which result from reads that span an entire SV, or inter-read signatures, which require multiple reads to cover the event. Insertions differ from deletions by an increase in the expected distance between the two split pairs marked by the white soft-clip between the reads and inversions involve reads that map best to the complimentary strand. Optical maps detect SVs based on increased presence, absence or change in the orientation of restriction enzyme sites compared to a reference (blue: sample; green: reference). Resolution is dependent on the distribution of restriction enzyme sites.
LR approaches have various strengths owing to their barcoding, a key feature being the ability to determine if fragments mapping to distant genomic loci derive from the same molecule, making the visual interpretation of translocations and large SVs exceptionally effective66. LRs are able to detect comparable amounts of deletions compared to single-molecule approaches but there is a discrepancy in detectable insertions68. While assembly-based LR studies have found megabases of novel insertional sequence across different populations75,76, single-molecule approaches will typically detect more insertion events77. This may result from the fact that LRs have a coverage drawback compared to single-molecule reads: no molecule within a read cloud has complete coverage of the DNA fragment such that there are substantial gaps between the read-pairs underlying each molecule, decreasing mappability in repetitive regions. The decrease in insertion detection may also result from the higher algorithmic difficulty of calling insertions through mapping versus assembly approaches which use simple pairwise comparisons78. Indeed, one of the most widely used algorithms, Long Ranger, cannot currently call insertions. However, recent efforts to develop algorithms that augment the mapping of LRs to repetitive regions are improving the ability of LRs to detect novel sequence insertions77,79.
Strand-seq.
Strand-seq independently sequences template DNA strands by incorporating bromodeoxyuridine into the non-template strand during replication followed by UV-induced photolysis at bromodeoxyuridine sites to selectively ablates the nascent strand80. As libraries only contain independent parental strands, Strand-seq is especially suited for haplotype phasing. The inherent directionality enables highly efficient detection of inversions which manifest as segments of opposing strand orientation (FIG. 2)39,81. Indeed, Strand-seq has been used to identify polymorphic inversions, showing that they are enriched for certain chromosomes over others, and revealing that the reference genome carries the minor allele or is misoriented at many inverted loci81. Deletions and duplications can be detected by read-depth while translocations are detected as changes in template state, as implemented in BAIT82. However, Strand-seq requires many enzymatic cleanup steps that end up reducing sequence coverage to an average of .01-.05X per library making it inappropriate to detect smaller sized SVs until improvements in single-cell library preparation are made83. Additionally, as inversions and translocations in Strand-seq look similar to sister chromatid exchanges, events must be consistent across multiple libraries for identification, thus SV detection with Strand-seq requires preparation of many individual single-cells.
Hi-C.
Hi-C involves sequencing crosslinked chromatin to provide information about DNA sequences that may be far in the linear genome but proximal in 3D space84. Hi-C read pairs can span megabases making it useful for detecting large SVs, especially translocations. However, as Hi-C relies on the presence of digestion sites kilobases apart in the linear genome, its resolution is limited. Hi-C also relies on underlying read-pairs and suffers from low sequence coverage as LRs and Strand-seq. Chromosomal interactions derived from Hi-C are represented in a contact frequency heat map across all possible pairs of genomic loci. Interactions between proximal loci are shown in the diagonal and contacts off of the diagonal are indicative of long-range interactions. Unusually elevated contact frequencies between distal loci represent possible deletions, inversions, and translocations, while elevated contact frequencies at proximal loci are indicative of duplications (FIG. 2)85. While Hi-C has mostly been used to detect translocations within cancer cells, methods to detect other SVs, such as HiCNV which uses read coverage to detect CNVs, are starting to emerge85–89. Delineating potential SVs from regular fluctuations in 3D structure remains a significant challenge. Recent work shows that large CNVs can affect chromatin organization across the chromosome, further confounding the ability to differentiate between variation in chromatin interaction and putative rearrangements90. To address this, Hi-C Breakfinder uses a probabilistic model that incorporates information about expected spatial features when determining aberrant contact frequencies91. However, most of the intrachromosomal SVs detected by this method are > 2 Mb as distinction from local interactions is still difficult. Additionally, Hi-C currently requires cell culture of millions of cells, though there are recent developments that aim to decrease this limitation92. A deeper understanding of 3D architecture will be necessary before Hi-C can reliably call SVs independent of orthogonal support.
Single-molecule strategies
PacBio.
PacBio single-molecule real-time (SMRT) sequencing leverages a stationary polymerase attached to the bottom of a nanosized well and passages single DNA strands through the enzyme to produce long-reads that significantly improve unambiguous mappability across the genome93. Algorithms detect SVs from SMRT data by leveraging intra and inter-read signatures (FIG. 2). Intra-read signatures enable direct detection of SVs and are derived from reads spanning entire SV events, resulting in missing sequence (deletion) or a soft-clip (insertion) within properly aligned flanking sequences. Inter-read signatures involve multiple reads and detect SVs from inconsistencies in orientation, location, and size during mapping, analogous to SR signatures. After signature detection, callers typically cluster and merge similar signatures from multiple reads, delineate proximal but different signatures, and choose the highest quality reads that support the putative SV. PBHoney, pbsv, SMRT-SV, Sniffles, CORGi, and SVIM detect SVs through combinations of intra-and-inter-read signatures but differ in their discovery heuristics61,94–98. Sniffles filters SVs by evaluating similarities between breakpoint position and size, and additionally clusters SVs supported by the same set of reads to detect nested SVs. SVIM evaluates how signature clusters overlap each other or nearby breakpoints to differentiate between interspersed duplications, tandem duplications, and novel sequence insertions. Some methods, such as SMRT-SV and CORGi, locally reassemble loci with SV signatures and call SVs based on consensus sequences derived from these assemblies. NextSV integrates Sniffles and PBHoney analogous to EA approaches discussed above99.
Single-molecule sequencing studies have so far been used to investigate fewer genomes due to higher operational costs, a large input DNA requirement, and lower sample throughput. Thus, while many short-read studies sequence across numerous genomes, long-reads have been mostly applied to single genome assemblies. While the base-calling error rate for Pacbio sequencing is higher than for short-reads, one can overcome this by increasing coverage or utilizing circular concensus sequencing100. It is pertitent to note that higher SMRT coverage results in more accurate consensus sequences but at a tradeoff for shorter median read lengths due to enzyme degradation – researchers must “sweet spot” coverage according to project aims101. Nonetheless, these single-molecule applications are challenging the SV detection landscape and its reliance on short-read technology. Sequencing of the CHM1 human hydatidiform mole genome served as proof of concept for using long-reads to resolve SVs, detecting > 20,000 SVs in this haploid genome compared to ~2,500 SVs per diploid genome in the 1KGP5,61. A recent analysis found that PacBio long-reads were approximately three times more sensitive than a short-read ensemble maximized for sensitivity, implying that a large subset of SVs, many 50 – 2000 bp in length, are unresolvable without long-reads39. Approximately half of the novel variants detectable by long-reads are insertions ~ 500 bp in length embedded within mobile elements and tandem repeats. SMRT assembly or SV detection in 19 other human genomes all find comparably large magnitudes of SVs and exhibit the corresponding insertional bias39,95,96,102–108. As it is impossible to tell the difference between a novel insertion or missing sequence in the reference, the magnitude of SVs that have been detected questions the completeness of the human reference genome. To investigate, Audano et al. performed SV discovery in 15 individuals long-read sequenced to an average ~57X and found 86,761 SVs absent from the 1KGP and the Genomes of the Netherlands project datasets109. A significant amount of the SVs shared between these 15 genomes are not present in the GRCh38 version of the human reference sequence implying it may contain errors or minor alleles at many SV loci. Remarkably, ~50% of the detected SVs intersect genes or regulatory elements. Overall, long-read technology enables detection of previously unresolveable SVs and may be pivotal in deciding how the field of genomics evolves from using a single human reference genome.
Oxford Nanopore Technologies.
Algorithms to detect SVs from nanopore sequencing are still emerging but have gradually become available, primarily through studies utilizing ONT. ONT threads single-stranded DNA through a protein pore and discriminates sequences based on current110,111. As nanopore is a variation of single-molecule sequencing, the signatures to detect SVs are similar to those used in PacBio data (FIG. 2). Callers that detect SVs from nanopore data include NanoSV, SVIM, Picky, and Sniffles; the latter three also detect SVs from PacBio data. Both NanoSV and Picky leverage split-reads to detect SVs and apply heuristics that consider coordinates, orientation, and breakpoint sites. NanoSV iteratively clusters all reads that support a breakpoint junction whereas Picky stitches together split-reads with surrounding reads and calls SVs from the best alignments. Studies that use ONT find similar numbers of SVs as PacBio detection but show many nanopore-specific small deletions112,113. However, Sedlazeck et al. found the overwhelming majority of ~10,000 unique ONT SVs were small deletions located within repeat regions and likely derived from base-calling errors, compared to ~800 unique PacBio SVs of which ~40% overlapped repeats, and De Coster et al. found that ONT SV algorithms detect small SVs poorly96,114. ONT provides improved read-lengths, an exceptionally small footprint, lower adaption costs, high throughput, and is effective at detecting many SVs, but lower specificity stemming from higher error rates make ONT less suitable for smaller SVs (< 100–200 bp), though recent improvements in base-calling error may mitigate this issue. Overall, the single-molecule approaches provided by PacBio and ONT enable highly sensitive SV detection and are the most powerful methods to detect novel sequence insertions.
Optical mapping.
OM, an alternative to sequencing-based technologies, linearizes single DNA strands in nanochannels and intermittently marks them with a nicking endonuclease to create physical maps known as genome maps115–117. OM-based methods call structural variation by comparing divergences in the nicks of DNA strands against an in silico digested reference: missing or extra labels and the spacing between labels are used to determine deletions or insertions; repeated labels indicate repeats and copy number changes; the presence of unique nicks on non-reference loci indicate translocations; and reversed nicking patterns indicate inversions (FIG. 2). The generated DNA fragments are up to 1 Mb long making OM well suited to detect large genomic rearrangements, particularly insertions, and is effective at identifying SVs within repetitive regions75,118–120. OM excels at deconvoluting zygosity as long as there is sufficent coverage such that molecules spanning each haplotype can be directly observed119. Due to reliance on restriction enzyme sites, OM does not produce sequence and therefore lacks base-pair resolution, instead providing breakpoint estimations based on the most proximal nicks. As a result, OM detects significantly fewer SVs than long-read methods and is typically limited to sizes ~ 6 kb and larger, though newer applications improve resolution by utilizing more than one restriction enzyme21‘75,107,119–121. Thus, most OM applications detect large SVs through de novo assembly of genome maps but use short-read sequencing to detect smaller variants104,105. New detection algorithms such as OSMV and Bionano Solve call SVs without de novo assembly by using alignment-based strategies121,122. It is important to note that OM suffers from a high error rate where errors manifest as missing or extra labels from incomplete and uneven stretching of individual molecules in their nanochannels. Resolution and error rate withholding, OM is amplification-free and significantly cheaper than HTS even at 60X coverage, making it an economical choice to investigate large cohorts119. Recent work by Levy-Sakin et al. used OM on 154 genomes from the 1KGP to find ~60 Mb of sequence not present in the reference genome as well as 55 loci in the genome that are both structurally complex and harbored by complex SVs120.
Multiplatform Discovery
Currently, no single method or technology has been shown to be comprehensive enough to detect all SV within a genome. Multiplatform approaches have emerged as a result, which combine strengths of various genomic platforms to enhance detection of SVs across all types and sizes. The platforms discussed can be employed combinatorially to complement strengths and mitigate weaknesses102. Due to their high base-calling accuracy, bioinformatic maturity, and affordability, short-reads are regularly used to correct errors in long-reads (‘polishing’, reviewed and evaluated elsewhere)78,123–125, whereas newer technologies are used for exhaustive variant detection and resolution of complex structures. A practical example includes combining short-read sequencing at higher coverage (> 30X) with lower coverage single-molecule sequencing (~10X) to optimize economy and sensitivity. The use of individual technologies will depend on logistical variables such as cost, required resolution, and project scope. Technical variables including sensitivity, variant size, repetitive nature of the target region, and haplotype information must be considered as well. A review of the advantages and disadvantages of each technology is provided in (TABLE 1).
Table 1 |.
Algorithms to detect genome-wide SVs from ensemble, single-molecule, and connected-molecule approaches
| Platform | Strengths | Limitations | Selected methods | Approach | Detection | URL | Refs |
|---|---|---|---|---|---|---|---|
| Ensemble Algorithms | Affordability; accessible, as infrastructure is widely available; high base-calling accuracy; detection of well characterized SVs; low cost makes read-depth methods more effective; deletion detection; high throughput | Amplification bias; insert sizes are inherently limiting; ambiguous mapping to repetitive regions; low phasing power; lack of standardized merging and ensemble choice; poor insertion detection | SVMerge | PE, SR, and RD signals with integration of two specialized insertion callers. Calls are merged on overlap with coordinate thresholds and validated by local reassembly | DEL, INS, INV, CNG, CPX | http://svmerge.sourceforge.net | 40 |
| Huge-Seq | PE, SR, and RD signals, along with breakpoint junction mapping. Calls are merged by 50% reciprocal coordinate overlap | DEL, DUP, INS, INV | https://github.com/StanfordBioinformatics/HugeSeq | 41 | |||
| iSVP | PE, SR, and RD signals. Additional calls are made with GATK HaplotypeCaller, which uses local reassembly. Calls are merged by overlap | DEL | http://nagasakilab.csml.org/en/isvp | 51 | |||
| MetaSV | PE, SR, and RD signals, along with breakpoint junction mapping, Calls are merged by overlap that prioritizes read signatures by their respective resolution and are refined with local reassembly | DEL, DUP, INS, INV, TRX | https://github.com/bioinform/metasv | 49 | |||
| SpeedSeq | PE and SR signals, along with a Bayesian likelihood genotyper. Uses a RD caller to annotate copy number at each variant locus | DEL, DUP, INS, INV, TRX, CNG | https://github.com/hall-lab/speedseq | 47 | |||
| Parliament2 | User choice of six individual callers. Calls are merged based on coordinate overlap and scored with a precision metric based trained on HG002 | DEL, DUP, INS, INV, TRX | https://github.com/dnanexus/parliament2 | 50 | |||
| FusorSV | Fits a model that determines which combinations of eight individual callers performs best according to a user-input truth set | Dependent on input truth set | https://github.com/timothyjamesbecker/FusorSV | 152 | |||
| PacBio | Short insertions and deletions 500 bp – 2000 bp; high sensitivity over a wide range of SVs; resolving SVs in repetitive regions; detecting complex SVs and mobile element insertions; amplification free | High base-calling error rate (stochastic); high input DNA requirement; high operating costs; poor detection of long inversions; low throughput | PBHoney | Unmapped split-read tails (PBHoney-Tails) and intra-read discordance (PBHoney-Spots) | DEL, INS, INV, TRX | https://sourceforge.net/projects/pb-jelly/ | 94 |
| pbsv | Split-read and intra-read signatures | DEL, DUP, INS, INV, TRX | https://github.com/PacificBiosciences/pbsv | n/a | |||
| SMRT-SV | Local assembly at loci with intra-or-inter read signatures; SVs subsequently called from consensus sequences derived from each assembly | DEL, DUP, INS, INV | https://github.com/EichlerLab/smrtsv2 | 61,95 | |||
| Snifflesa | Split-read and intra-read signatures | DEL, DUP, INS, INV, CPX, TRX | https://github.com/fritzsedlazeck/Sniffles | 96 | |||
| NextSV | Combines calls from PBHoney and Sniffles by union (sensitive callset) or intersect (stringent callset) | DEL, DUP, INS, INV, CPX, TRX | https://github.com/Nextomics/nextsv | 99 | |||
| CORGi | Chooses the highest scoring putative SV from a collection of possible SVs generated by realigning loci with split-read and intra-read signatures multiple times | DEL, DUP (tandem and dispersed), INS, INV, CPX, CNG | https://github.com/zstephens/CORGi | 98 | |||
| SVIMa | Split-read and intra-read signatures | DEL, DUP (tandem and dispersed), INS, INV | https://github.com/eldariont/svim | 97 | |||
| Oxford Nanopore | High sensitivity over a wide range of SVs; small footprint, extremely useful for field work; fast turnaround and high throughput; detection of SVs > 200 bp; amplification free; low operating cost | High base-calling error rate; high input DNA requirement; deletion artifacts impede detection of small SVs; poor detection of long inversions | NanoSV | Split-read signatures and evidence from reads that map to putative breakpoint junctions | DEL, DUP, INS, INV, TRX | https://github.com/mroosmalen/nanosv | 113 |
| Pickyb | Spit-read signatures from long-read alignments that are linked together to improve coverage | DEL, DUP, INS, INV, TRX | https://github.com/TheJacksonLaboratory/Picky | 112 | |||
| Optical Mapping | Large SVs > 5 kb; insertions are easily visualized; long single-molecule strands suitable for haplotype phasing; detecting SVs in repetitive regions; amplification free; cheaper than HTS platforms | High labeling error rate; low resolution; dependent on restriction enzyme sites; detects significantly fewer SVs overall | OMSV | Discordance in the number of and distances between restriction label sites | DEL, DUP, INS, INV, TRX | http://yiplab.cse.cuhk.edu.hk/omsv/ | 121 |
| Bionano Solve | Discordance in the number of and distances between restriction label sites | DEL, DUP, INS, INV, TRX | https://bionanogenomics.com/support/softwaredownloads/ | n/a | |||
| 10x Genomics Linked-Reads | Haplotype phasing due to long length of reconstructed molecules (~100kb); large SVs > 30 kb; translocations and gene fusions are easily visualized and quantified with barcodes; high base-calling accuracy; low adoptability cost and footprint; high physical coverage; low input DNA requirement | Low sequence coverage of each molecule fragment; poor detection of insertions; low sequence coverage; poor detection of small variants | Long Ranger | Read pair barcode overlap between distant loci and changes in barcode density | DEL, DUP, INV, TRX | https://support.10xgenomics.com/genomeexome/software/pipelines/latest/what-is-longranger | 66 |
| GROC-SVs | Read pair barcode overlap between distant loci and changes in barcode density. SVs are reconstructed with local reassembly | Reports reconstructed breakends that can derive from any SV type | https://github.com/grocsvs/grocsvs | 69 | |||
| LinkedSV | Molecule barcode overlap between distant loci and barcodes from two distance loci mapped to adjacent positions | DEL, DUP, INV, TRX | https://github.com/WGLab/LinkedSV | 99 | |||
| VALOR2 | Split-read signatures from linked molecules, read-pair signatures, and molecule depth for filtering | DEL, DUP, INV, TLC, INV-DUP, INV-TRX | https://github.com/BilkentCompGen/valor | 71,72 | |||
| Novel-X | Assembly of unmapped read with other reads of associated barcodes to obtain anchors in unique sequence followed by mapping of these long, reassembled insertions | INS | https://github.com/1dayac/novel_insertions | 77 | |||
| NAIBR | Combines split-read signatures from linked molecules with the PE signatures from the underlying short-reads into a probabilistic model | DEL, DUP, INS, INV, TRX | https://github.com/raphael-group/NAIBR | 70 | |||
| ZoomX | Changes in linked molecule coverage | DEL, DUP, INV, TRX | https://bitbucket.org/charade/zoomx | 74 | |||
| Strand-Seq | Highly accurate large inversion detection; haplotype phasing due to innate directionality of libraries; low input DNA requirement; high physical coverage | Low sequence coverage; poor detection of small variants; poor detection of translocations and homozygous inversions; requires multiple libraries to differentiate SVs from sister chromatid exchanges | BAIT | Changes in the ratio of reads mapped in opposing directionality and sudden changes in template state that are consistent across loci | DEL, DUP, INV, TRX | https://sourceforge.net/p/bait/wiki/Home/ | 82 |
| Invert.R | Changes in the ratio of reads mapped to opposing directionalities | INV | https://sourceforge.net/projects/strandseq-invertr/ | 81 | |||
| Hi-C | Translocations are easily visualized as high frequency interchromosomal contacts; very large SVs (> 2 Mb); high physical coverage | Low sequence coverage; dependent on sparse short-read pairs; poor detection of insertions; poor detection of small variants; difficult to delineate between chromosome interactions due to 3D structure vs. rearrangements; large input requirement | HiCNV + HiCtrans | Read depth of restriction enzyme fragments and high frequency interchromosomal contacts | DEL, DUP, TLC |
https://github.com/ay-lab/HiCnv https://github.com/ay-lab/HiCtrans |
89 |
| Hi-C Breakfinder | Clusters of interaction frequencies that deviate from expected | DEL, DUP, INV, TRX | https://github.com/dixonlab/hic_breakfinder | 91 | |||
| Multiplatform | Comprehensive, allowing detection across the entire SV spectrum; provides orthogonal validation; highest sensitivity | Costly; batch effects must be controlled for; methods to integrate interplatform calls are ad hoc | MultiBreak-SV | Clusters all possible short-and-long read alignments that support a putative SV into a combined probabilistic model | DEL, INV, TRX | https://github.com/raphael-group/multibreak-sv | 212 |
| HySA | Clusters short-reads with PE and SR signals with long-reads. SVs are called from contigs assembled by the reads in each cluster | DEL, INS, CPX | https://bitbucket.org/xianfan/hybridassemblysv | 132 |
also able to detect SVs from ONT data
also able to detect SVs from PacBio data
DEL, deletions; DUP, duplications; INS, insertions; INV, inversions; TLC, translocations; CNG, copy-number gain; CPX, complex rearrangement.
Multiplatform discovery is often employed to investigate SVs in cancer. Two studies on leukemia and prostate cancer genomes integrated short-reads with OM and found that many SVs detected uniquely by OM have breakpoints within low mappability regions, whereas SVs detected uniquely by short-reads are typically smaller and below the resolution of OM126,127. Analysis combining an EA, LRs, and long-insert libraries to detect and phase SVs in the K562 and HepG2 cancer genomes finds thousands of calls unique to each platform128,129. Similarly, combining OM, short-reads, and Hi-C to detect SVs in eight different cancer genomes found only 20% of interchromosomal translocations were detected by two or more platforms, demonstrating the necessity of multiplatform discovery to detect all variants91. In another case, short-reads were not used to improve sensitivity across the detection size spectrum but were used to resolve ambiguity in unique, unaligned OM fragments from a liposarcoma genome. While OM was necessary to reveal large fragments, the short-read signatures provided the necessary resolution to reveal ~6 SV breakpoints within the unaligned maps, suggesting that the fragments consisted of complex SVs130.
Genome assemblies typically integrate platforms when detecting SVs to increase sensitivity and produce orthogonal validation. In one example, assembly of genome NA12878 merged PacBio contigs with OM genome maps to create highly contiguous scaffolds with an N50 of 28.8 Mb103. As 55% of inversions called from these scaffolds were enriched for arrangement complexity and colocation with other SVs, they would be difficult to detect without the improved contiguity from integration. A similar approach was used by Ameur et al.106 In another example, English et al. generated short and long-read sequences in genome HS1011 and detected SVs by combining an EA, PacBio, and hybrid local reassembly131. While the authors found many SVs overlapping from the three approaches, they revealed bona fide SVs that were unique to their respective detection method. Additionally, hybrid reassembly detection performed with FDR < 10% whereas popular short-read callers (CNVnator, BreakDancer, Delly, Pindel) exhibited FDRs between 31–80%, showing greatly improved detection with integration. A recent comprehensive multiplatform discovery of SVs integrated nine platforms across three family trios discovering ~27,622 SVs per genome39. Chaisson et al. combined an EA, PacBio, OM, Strand-seq, and long-insert libraries to detect deletions, insertions, and inversions, with additional technologies applied for phasing, assembly, and orthogonal validation. While PacBio contributed the highest number of unique deletions and insertions, Strand-seq contributed the highest number of inversions, and each platform identified high-confidence unique calls. Each of these studies illustrate that combining platforms is necessary for comprehensive detection across the full range of SVs.
Integration of SV calls from differing technologies is analogous to EA approaches: most methods are “in-house” and consider coordinate overlap, breakpoint proximity, mapping orientation, read support, putative SV type, and resolution of the underlying technology. There are few standalone multiplatform detection tools; most combine short and long-reads, such as MultiBreak-SV and HySA95,132. MultiBreak-SV considers all possible short and long-read alignments that support a putative SV in a combined probabilistic model, whereas HySA clusters short-reads with PE and SR signals with the long-reads that support them before calling SVs from contigs assembled with the long-reads in each cluster. New “platform ensemble” tools are expected to develop as the cost of sequencing continues to drop and access to new technologies improves.
Perspectives and future directions
Tremendous improvements in variant calling have made the ubiquity, complexity, and pertinence of SVs in human genomes clearer than ever. Many advancements contributed to an explosion in detection, including the application of ensemble algorithms, which have been essential in characterizing SVs across populations4,5,43–45,58, and single-molecule and connected-molecule strategies, which enable detection of thousands of previously undiscoverable variants61,66,80,85,113,115. Indeed, we now estimate that each human genome contains >20,000 SVs, many of which are located in regions where short-reads are unmappable61,95,104,105,109. Each emerging platform possess unique strengths, but they also exhibit inherent biases. A philosophical ideal would involve sequencers that read entire genomes, without bias, as a contiguous whole. Until this is possible, the integration of multiple platforms will be necessary to resolve all SVs within a given human genome. Though there are no human genomes where all classes of structural variants have been completely resolved, multiplatform discovery approaches are dramatically closing this gap39,102.
In spite of these improvements, we are still unable to interpret the functional consequences of the vast majority of variants. Strategies to ascertain functional impact are more necessary than ever given the expansive increase in detectable and novel SVs. Moving forward, integrating SV detection across layers of biological information shows promise for elucidating the biological impact of variants. Studies using short-reads have shown the potential of integrative frameworks in interpreting SV function133–140 and now a subset of studies employing the emerging detection methods discussed are starting to integrate SVs with layered biological data such as expression, epigenetics, and 3D structure, to understand the effects of SVs holistically (Weischefelt, mcpherson 2012, mcpherson 2011, yorukoglu 2012, spielmann, franke, gheldof, fudenberg, quigley). Building on seminal work by Stranger et al.141, Chiang and colleagues detected SVs with an EA before mapping SV-expression quantitative trait loci, finding that SVs had a larger median effect and were up to 53 times more likely to affect gene expression compared to SNVs or indels142. Indeed, other studies integrating emerging detection methods with expression data, long-read transcriptome sequencing, and transcriptome assembly have revealed the high potential for rearrangements to affect genes, demonstrating differential expression, alternatively spliced transcripts, and complex gene fusions resulting from novel SVs75,105,127,143–149. While the transcriptome is often integrated given its immediacy to the genome, more efforts to integrate the methylome are emerging and so far have revealed inconsistent methylation patterns around SVs, suggesting complex regulatory consequences128,129,150,151. Another datatype that should be considered with SVs are small variants and their effects. For example, ONT analysis identified a heterozygous point mutation and an exon disrupting deletion in a disease individual where the disease genotype involves bi-allelic point mutations144. Additionally, a study investigating non-reccurent SVs with arrays, short-reads, and long-reads found enrichment of de novo SNVs and indels near SV breakpoints, the majority of which are intragenic152. These studies imply and show the potential for multimodal integration to provide insight into the biological mechanisms affect by SVs.
Ideally, the field moves toward integration across multiple layers, which can reveal relationships that reconstruct molecular contexts (for a strategy that can be generalized to functionally interpret SVs within multiple molecular contexts, see FIG. 6 in REF8). LRs found that AR was co-amplified with upstream tandem duplications in cancer cells153. DNase hypersensitivity peaks and increased nucleosome spacing predicted an enhancer within the duplicated region, Hi-C data revealed the duplications and AR lie within the same topologically associating domain, and paired RNA-seq revealed increased expression of AR in samples with the upstream SV, implicating that duplication of a distal enhancer element results in upregulation of the oncogene (FIG. 3). In another example, Dixon et al. combined short-reads, OM, and Hi-C to detect large and complex SVs in cancer cells, which can possibly disrupt TAD structure8,91,154. RNA-seq analysis of cancer genes within disrupted-TADs revealed that TADs containing an SV show greater allelic-bias and altered gene expression in cis, suggesting that the SVs create neo-TADs that rewire regulatory environments. In a final example, OM and short-reads detected a 3.4 kb deletion in a copy-number amplified region, H3K27ac ChlP-seq peaks predicted that part of the removed sequence acted as an enhancer, Hi-C linked the deleted enhancer to upstream GNB4, and RNA-seq revealed decreased expression of GNB4 but increased expression of all other proximal genes91. These relationships, discovered by integrating multimodal data, paint a clearer picture of the role of this variant in perturbing biological mechanisms (FIG 3). These studies show immense potential and provide frameworks to interpret the effects of SVs, but largely rely on manual curation.
Figure 3 |. Resolving the molecular context behind structural variants by integrating multimodal information.

a | Layers of biological data that can be integrated with SV calls to interpret a possible mechanistic chain of events. Each layer possesses quantifiable readouts that can be tested for association with genomic variants. Studies have focused less on the integration with more distal layers, such as the proteome, metabolome and microbiome (later two not shown), but future efforts focused here should have just as much potential to be informative. b | Linked-reads detect tandem duplications upstream of AR153. Previous studies showed that this region contains an enhancer (green boxes) for AR which are consistent with DNase hypersensitivity peaks. Hi-C analysis shows that both the enhancer and gene body are located within the same topologically associating domain, further suggesting their interaction. Paired expression data from multiple samples shows that duplication of the enhancer leads to increased AR expression when compared to cases without the duplication. Integration of layered data suggests that tandem duplications cause gain of an enhancer element that drives AR expression in castration-resistant prostate cancer. c | A 3.4 kb deletion was detected by OM and read-depth from short-read HTS91. The authors use H3K27ac ChIP-seq data to determine that the deletion overlapped an enhancer element (green) and Hi-C data to determine that the enhancer interacts with an upstream promoter (yellow oval) to regulate GNB4. Comparisons of expression data against HMEC reveals that nearby genes show increased expression but GNB4 expression is notably decreased. This information taken together illustrates that decreased expression of GNB4 may result from deletion of a downstream enhancer in spite of amplification of the gene body.
Detection is essential to characterizing individual genomes, but detection alone is not enough. Indeed, the technologies and methods discussed have resulted in an aggressive influx of detectable variants but there is little ability to assign impact. Lists of thousands of newly detected SVs will be more useful for the field if we are able to interpret their functional effects. Thus, we believe that the field should consider concurrent detection and integration. We anticipate that moving from manual curation to the development of multivariate models generalizable to projects with layered data bears great potential to provide insight into the complex genomic architecture affected by SV. Ultimately, detecting SVs is a piece of the larger puzzle that is understanding the genome, its disparate parts, and all of its connections. Improvements in, and applications of, new emerging genomic technologies, and the integration of variants with disparate layers of biological information, will pave the way for a future where we understand the possible function and effects of every nucleotide in the human genome.
Table box 3 |.
Structural variation reference sets
| Selected reference datasets | Reference type, platform, coverage | Raw data publicly available | Sample number | SVs detected | Description; orthogonal validation if applicable | URLs and accessions | Ref |
|---|---|---|---|---|---|---|---|
| 1000 Genomes Project phase 3 | Population-scale Illumina short-read, 7.4 | Y | 2,504 | 68,818 | Individuals across 26 populations; PCR, orthogonal short-read platforms, genome.org/data PacBio, and microarrays | http://www.international | 5 |
| 1000 Genomes Project – High coverage | Population-scale Illumina short-read, ~30 | N/A | 2,504 | n/a | High coverage sequencing of the individuals from phase 3 of the 1KGP | https://www.ebi.ac.uk/ena/data/view/PRJEB3l736 | N/A |
| Genome of the Netherlands release 6.1 | Population-scale Illumina short-read, 12 | N | 769 | 59,358* | 769 individuals from 250 Dutch families; PCR amplification of breakpoint junctions followed by Sanger or short-read sequencing | http://www.nlgenome.nl | 45 |
| Tohoku Medical Megabank Organization, 1KJPN | Population-scale Illumina short-read, 32.4 | N | 1,070 | 56,697* (> 100 bp) |
Individuals of Japanese ancestry; digital droplet PCR | https://ijgvd.megabank.tohoku.ac.jp/download_lkjpn/ | 58 |
| GTEx | Population-scale Illumina short-read, 49.9 | N | 147 | 23,602 | SVs detected across 13 different human tissues; microarray data | https://gtexportal.org/home/datasets | 142 |
| Abel et al. | Population-scale Illumina short-read, >= 20 | N | 17,795 | 118,973 / GRCh37 241,426 / GRCh38 | African American, Latino, Finnish European, non-Finnish European, East Asian, Pacific Islander, and South Asian ancestry | https://www.biorxiv.org/content/10.1101/508515v1.supplementary-material | 44 |
| Sherman et al. | Population-scale Illumina short-read, 30–40 | Y | 910 | 125,715 | Novel insertion detection in individuals of African ancestry | https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001123.v1.p1 | 175 |
| gnomAD-SV | Population-scale Illumina short-read, 32 | N/A | 14,216 | 498,257 | Individuals of African, East Asian, European, Latino, and admixed ancestry | https://gnomad.broadinstitute.org/downloads | 43 |
| Venter/HuRef | Highly curated Sanger reads, 7.5 10× Genomics LR, 42 Illumina short-read, 92, 36 Illumina 2 kb mate-pair, 7 Illumina 5 kb mate-pair, 6 IIlumina 12 kb mate-pair, 3 |
Y | 1 | 808,346* | De novo assembly of a European-American adult male; Sanger sequencing-based assembly, a wide suite of microarray data, and BAC and fosmid libraries | NCBI:SRR7G97858, SRR7097859, SRR6951312, SRR6951313, SRR6951310, SRR6951311 GenBank: AADDGGGGGGGG, ABBAG1GGGGGG GEO:GSE20290 |
170,176,177 |
| CHM1 | Highly curated PacBio, ~40 PacBio, 62.4 |
Y | 1 | 20,602 | De novo assembly of a haploid human hydatidiform mole; short-reads and Sanger capillary-based sequencing; target sequencing of BAC clones, de novo PacBio assemblies, Sanger sequencing, and targeted PCR |
https://eichlerlab.gs.washington.edu/publications/chml-structural-variation/ https://www.ncbi.nlm.nih.gov/dbvar/studies/nstd137/ NCBI:PRJNA246220 |
61,95 |
| CHM13 | Highly curated PacBio, 66.3 ONT, 32 10X Genomics LR, 50 Bionano OM, 430 Hi-C, 40 Illumina short-read, ~30 |
Y | 1 | 20,470 | Haploid human hydatidiform mole; target sequencing of BAC clones, de novo PacBio assemblies, Sanger sequencing, and targeted PCR |
https://www.ncbi.nlm.nih.gov/dbvar/studies/nstd137/ NCBI: PRJNA269593 https://github.com/nanopore-wgs-consortium/CHMl3 |
95,178 |
| HX1 | Highly curated PacBio, 103 Bionano OM, 101 Illumina short-read, 143 |
Y | 1 | 20,175 | De novo assembly of a Chinese adult male |
http://hx1.wglab.org NCBI:PRJNA301527 |
104 |
| AK1 | Highly curated PacBio, 101 Bionano OM, 97 & 108 10x Genomics LR, 30 Illumina short-read, 72 |
Y | 1 | 18,210 | De novo assembly of a Korean adult male; BAC clone assembly | NCBI:PRJNA298944 | 105 |
| Audano et al. | Population-scale PacBio, ~57 |
Y | 15 | 99,604 | African, Asian, European, American, and South Asian ancestry; BAC and fosmid libraries |
https://www.ncbi.nlm.nih.gov/dbvar/studies/nstd162/ HG00514, NCBI:PRJNA300843; HG00733, NCBI:PRJNA300840 ;NA19240, NCBI:PRJNA288807; HG02818, NCBI:PRJNA339722; NA19434, NCBI:PRJNA385272; HG01352, NCBI:PRJNA339719; HG02059, NCBI:PRJNA339726; NA12878, NCBI:PRJNA323611; HG04217, NCBI:PRJNA481794; HG02106, NCBI:PRJNA480858; HG00268, NCBI:PRJNA480712 |
109 |
| Swe1 & Swe2 | Highly-curated PacBio, 78.8 (Swe1) PacBio, 77.8 (Swe2) Bionano OM, >100 |
N | 2 | 17,936 / Swe1 17,687 / Swe2 |
One male and one female Swedish individual | https://www.mdpi.com/2073-4425/9/10/486/s1 | 106 |
| Levy-Sakin et al. | Population-scale Bionano OM, 79 10x Genomics LR, 60 |
Y | 156 | 15,601 | 156 samples from the 1KGP; concordance with 10x-Genomics LRs |
https://www.ncbi.nlm.nih.gov/dbvar/studies/nstd168/ NCBI:PRJNA418343 |
120 |
| Pendleton et al. & Jain et al., NA12878 | Highly curated PacBio, 22 and 24 Bionano OM, 80 ONT, 26* |
Y | 1 | 34,237 | Two separate de novo assemblies of a Caucasian adult female; PCR |
https://github.com/nanopore-wgs-consortium/NA12878/blob/master/nanopore-human-genome/NA12878.hq.sv.vcf NCBI:PRJNA253696; ENA:PRJEB23027 |
103,172 |
| Genome in a Bottle, NA12878 | Highly curated PacBio, ~44 |
Y | 1 | 10,594 | One Caucasian adult female |
ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/data/NA12878/N A12878_PacBio_MtSinai |
N/A |
| Wong et al. | Population-scale 10x Genomics LR, 60 |
Y | 17 | 1,842 | De novo assembly and non-reference insertion detection in individuals of African, American, East Asian, European, and South Asian ancestry; insertions > 2kb were validated with OM | NCBI:MH533022-MH534863, PRJNA418343, PRJNA435626 |
75 |
| Genome in a Bottle, HG005, HG003, HG004 (son/father/mother) | Highly-curated Illumina short-read, 300 (son), 100 (parent) Complete Genomics, 98 Ion Proton, 1036 Bionano OM, 57 PacBio, 60 (son), 30 (parents) |
Y | 3 | 59,973 | A preliminary callset containing deletions and insertions from a Han Chinese family trio | ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/data/ChineseTrio/analysis/ | 179,180 |
| Genome in a Bottle, HG002, HG003, HG004 (son/father/mother) | Highly curated Illumina short-read, ~300, ~14.5, ~25, ~208.5, ~101, ~100 10x Genomics LR, 47 (mother), 36 (father), 86 (son) Complete Genomics, ~101, 100 Ion Proton, 1020 Bionano OM, 92 (mother), 87 (father), 112 (son) PacBio, ~31 (parent), 69 (son) ONT, 0.017 (son) |
Y | 3 | 12,745 | Contains high-confidence deletions and insertions from an Ashkenazi family trio; concordance across multiple trios |
ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/data/AshkenazimTrio/analysis/NIST_SVs_Integration_v0.6 NCBI: PRJNA200694 |
102 |
| Human Genome Structural Variation Consortium | Highly curated PacBio. ~40X ONT, 18.9X Illumina short-read, 74.5 Illumina 3 kb mate-pair, 3 Illumina 7 kb mate-pair, 1.1 10x Genomics LR, 82.4 Bionano, N/A Tru-Seq SLR, 3.47 Strand-seq, N/A Hi-C, 19.49 |
Y | 3 (data available fo 9) | 103,985 | Three family trios of Han Chinese, Puerto Rican, and Yoruban Nigerian ancestry; concordance across multiple genomic platforms |
https://www.ncbi.nlm.nih.gov/dbvar/studies/nstd152/ http://www.internationalgenome.org/data-portal/data-collection/structural-variation |
39 |
non-standard definition of SVs
Box 5 Table |.
Callers specialized in resolving complexity
Box 1 |. From microarrays to short-read sequencing and beyond.
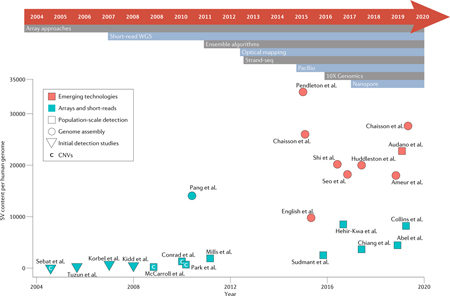
The prevalence of SVs in human genomes has historically been determined by the resolution of available technologies. Molecular cytogenetics techniques, particularly chromosome-banding and fluorescence in situ hybridization, powered seminal work involving the detection of microscopic chromosomal aberrations but were unable to identify submicroscopic variants (for brief historical perspectives on cytogenetic-based SV detection, see REFS22,155). Microarrays then became the primary technology to identify CNVs in the 2000s due to improved resolution over karyotype-based analysis. Array-comparative genomic hybridization enabled the first reports of global structural variation, identifying ~300 copy-number variable loci and informing the wide presence of SVs in phenotypically normal human genomes56,156. One of the first sequence mapping approaches performed with a single fosmid library reported a similar number of SVs, ~300 variants11. These numbers were highly preliminary as SNP arrays would soon detect 1,447 and 1,320 CNVs across 270 individuals157,158. At this time, sequencing-based approaches were dropping in cost; their proof-of-principle studies exhibited similar sensitivity compared to arrays but with significantly fewer samples: Korbel et al. employed paired-end 454 pyrosequencing in two human genomes while Kidd et al. used a fosmid-clone based mapping approach in nine human genomes to detect ~1,700 and ~1,300 SVs, respectively24,159. Large, population-scale detection efforts then started to emerge. In 2010, high-density microarrays employing millions of probes ascertained 11,700 CNVs across 450 individuals2. A sequencing based-approach proved to be more comprehensive when in 2011 Mills et al. applied an ensemble approach (reviewed below) to ~4X short-read HTS of 185 individuals to detect a three-fold increase of SVs in comparison4. Throughout these studies, two main advantages made short-read HTS superior for exhaustive SV detection: (1) detection of balanced variants and sequences not in the reference (novel insertions), which are missed by arrays; (3) higher overall resolution. Thus, short-read HTS has been the major driver of progress in SV detection over the last decade given its improved sensitivity over array platforms, though arrays are still regularly utilized for their low cost and high throughput160. Improvements in short-read technology have enabled detection of millions of variants, improving the number of detectable SVs from ~2,100 to ~8,000 SVs per human genome5,43. The emerging sequencing technologies discussed in this Review push these estimates further, to >25,000 SVs per individual. Below are selected studies that either estimate the extent of SV content or provide estimates of detectable SVs according to technology within phenotypically healthy human genomes, showing the relationship between detectable SVs and available technologies.
For a more comprehensive overview of the methods and algorithms used to detect SVs before adoption of the technologies discussed in this Review, we suggest the following references: molecular cytogenetics techniques, REF162; the application of molecular cytogenetics to understand clinical disorders, REF163; array and clone-based approaches to detect SVs, REF155; a comprehensive survey of the first SV detection studies, REF161; short-read discovery and genotyping, REFS9,164,165; detecting complex SVs, REF166; clinically relevant CNVs and SV detection from whole-exome sequencing, REFS167–169. (additional citations for the figure in this box)2,4,5,11,24,39,43–45,61,95,103–106,109,131,142,156,158,159,170,171
Box 3 |. Structural variation reference sets.
Reference datasets are essential for the development of SV discovery methods. Many algorithms validate detection ability by benchmarking against or training with datasets released by population-scale sequencing, de novo genome assemblies, or projects that perform comprehensive discovery with multiple orthogonal platforms5,39,43–45,58,61,75,95,102,104–106,108,109,120,142,172. The type of chosen reference sets should be appropriate for each application, e.g. highly curated discovery sets are appropriate for benchmarking detection methods whereas population-scale sets are useful for determining callset novelty or rarity. These datasets differ in sample size, ancestry, depth, platform, merging methodology, sensitivity, and specificity, all of which should be considered before deciding which set is right to utilize, as biases influenced by these choices are inherently passed to the applications that employ them. Reference sets also vary widely when it comes to orthogonal validation where some reference sets employ multiple orthogonal platforms while others perform none, opting to maximize quality metrics instead. Given this large variation, projects often use more than one reference set to maximize inclusivity and avoid overfitting. Reference sets undergo an iterative process where newer datasets are typically more sensitive and exhaustive due to technological improvements, thus, developing algorithms should focus their benchmarks on more recent resources to avoid confounding issues stemming from technological limitations in legacy data. Indeed, a recent study finds numerous batch effects within the 1KGP release set173. Selected sequencing-driven reference datasets representing phenotypically “normal” individuals are listed below. We choose datasets that include called SVs, focus on collections with available raw data, and list orthogonal data from multiple sources for some reference sets. Additional resources can be found at dbVar174.
Box 4 |. Detecting structural variation in disease.
SVs are associated with diverse diseases and are a notable hallmark of cancer genomes181. Long-reads, linked-reads, Hi-C, and optical mapping resolve structures that short-reads struggle to detect in the majority of cancers such as inter-and-intra chromosomal translocations, complex rearrangements, chromoplexy, chromothripsis, chained fusions, and extremely large (> 30 kb) SVs69,74,87,130,128,129,145,182–186. PacBio reads were used to analyze the breast cancer cell line SKBR3, detecting > 17,000 SVs including SVs that overlap COSMIC genes148. The single-molecule approach detected 76% more SVs than an ensemble of 3 short-read callers (with 2 caller concordance), most of which derive from repetitive regions. The long-reads enabled identification of clustered, complex translocations and inverted duplications that amplified the oncogene ERBB2 to > 32 copies, later confirmed in a separate long-read analysis by Sedlazeck et al., providing insight into a possible breast-cancer specific mechanism96,148. LRs have been used to detect and phase translocations and gene fusions in cancer genomes finding loci where heterozygous SVs impact allele-specific expression128,129. Another LR study resolved an extremely complex haplotype-specific SV in a lung cancer cell line where one haplotype harbors an EML4-ALK gene fusion and the other an ALK-PTPN3 fusion66. Viswanathan et al. also used LRs to study the genomic architecture of the AR oncogene in castration-resistant prostate cancer and found that SVs were likely to inactivate tumor-suppressor genes in complex patterns where each haplotype could harbor a different type of inactivating SV153. Each of these findings are examples of complex genomic architectures now resolvable through the improved resolution of emerging technologies.
CNVs and de novo mutations play pertinent roles in the etiology of several neuropsychiatric diseases such as intellectual disability, schizophrenia and particularly Autism Spectrum Disorder (ASD)187–189. Application of EAs in ASD family genomes has revealed CNVs that disrupt known neurodevelopmental genes, clustering of de novo SNVs proximal to de novo CNV regions, an abundance of complex duplication-associated SVs, and elevated numbers of de novo CNVs compared to unaffected individuals190–193. However, it is pertinent to note the challenges and disagreement in extrapolating association between rare noncoding variants and ASD risk: risk: a dearth of both rigorous analytical approaches and replicated associations between studies significantly encumbers the interpretation of noncoding SVs in these diseases46,194,195.
Emerging methods have additionally been applied to mendelian disorders, clinical phenotypes, and structural haplotypes to identify SVs that are traditionally difficult to characterize. For example, OM is effective at detecting the D4Z4 repeats in facioscapulohumeral muscular dystrophy which are challenging to resolve with classical techniques due to their size150,183. In individuals where short-reads were uninformative PacBio sequencing was able to detect disease-causal SVs, such as a de novo ~2.1 kb SV overlapping PRKAR1A in Carney complex and a 4.6 kb repeat expansion and 12.4 kb deletion in benign adult familial myoclonic epilepsy located in GA and GC-rich regions, respectively143,196,197. Similarly, in a glycogen storage disease type 1a patient where whole-exome and Sanger sequencing failed to determine a genetic cause, nanopore sequencing detected a compound heterozygous structure containing a point mutation and a 7.1 kb deletion in G6PC on separate alleles144. New detection methods have also identified complex SVs that are insufficiently resolved with short reads in patients with congenital abnormalities and severe quality-of-life disorders: they contain numerous breakpoints, cluster closely with other SVs, affect considerable nucleotides, and are flanked by repetitive sequences113,146,193,198–200. In a final example, OM was used to construct and determine the frequency of segmental duplication haplotypes LCR22A and LCR22D, which are involved in 22q11 deletion syndrome and escape short-read resolution. The large fragment sizes of OM enabled the authors to find extensive copy number variation differing up to 1.75 Mb between individuals and reveal that the reference genome does not represent the major allele at this locus201.
Box 5 |. Confounding Complexity.
The detection studies discussed have revealed SVs consisting of complex arrangements are more prevalent than previously perceived in both phenotypically “normal” and disease individuals5,43,45,61,103,120,128,129,132,146,148,152,153. Additionally, new technologies reveal significant amounts of SVs in areas that are difficult to resolve with short-reads: these loci are either extremely low in complexity such as tandem repeats, telomeres, and mobile element insertions, or high in complexity such as segmental duplications, centromeres, the major histocompatability complex, and other areas of high polymorphism5,39,61,95,104–106,109,115,118–120,120,126,201. Indeed, mechanisms behind SV formation such as non-allelic homologous recombination and replication-based mechanisms are dependent on local repeat structures which leads to breakpoints within repetitive regions (reviewed in Carvalho and Lupski)202. “Complexity” confounds detection in two senses: (1) in terms of complex SV events and (2) in terms of the variable complexity at genomic loci. It is essential to consider specialized methods that can leverage new technologies to detect SVs in complex regions, detect SVs of complex arrangements, and methods that reassemble complex regions to decrease unambiguous mapping. Indeed, specific tools such as SDA which resolves segmental duplications, CORGI which resolves complex events, and rMETL which detects mobile element insertions, and other tools taking a specificity-first approach will help in resolving difficult-to-detect SVs that cannot be ascertained from generalized whole-genome approaches due complicated genomic loci or irregular compounded structure69,96,98,132,203–211. Eventually, generalized SV detection methods should either implement the strategies used from specialized callers or be utilized concurrently for a more comprehensive assessment of genome-wide SV.
Acknowledgements
We thank Y. Wang, W. Zhou, A. Weber, and B. Zhou for their valuable comments and help with proofreading the manuscript. S.S.H. was supported through the Michigan Predoctoral Training in Genetics grant (T32 GM007544). A.E.U. acknowledges funding by the NIH, by the Simons Foundation, and is a Tashia and John Morgridge Faculty Scholar of the Stanford Child Health Research Institute.
Glossary
- STRUCTURAL VARIATION
(SV) Operationally defined as sequence variants > 50 bp in size. The most recognized forms of structural variation include deletions, duplications, inversions, insertions, and translocations
- COMPLEX STRUCTURAL VARIATION
A structural variant that consists of multiple combinations of structural variant types nested or clustered with one another
- SHORT-READS
Standard sequencing libraries fragmented to ~ 600–800 bp in length. Two ends are sequenced ~ 100–250 bp with an unsequenced insert size of ~100–600 bp
- REFERENCE SET
High-resolution SV datasets typically deriving from de novo genome assemblies, population-scale sequencing, or projects employing multiple orthogonal detection methods. Reference sets are used to benchmark detection algorithms and determine the novelty and rarity of SV calls
- CALL
Each putative SV detected by a program is an individual ‘call’. ‘Call’ derives from computer science, meaning to invoke a particular task: detected SVs are the result of each performed ‘task
- CALLSET
The set of all putative SVs detected by a or a combination of SV detection programs.
- READ SIGNATURES
Specific marks that result from reads that map discordantly to the reference genome.
- SENSITIVITY
The ability to detect known variants correctly. Low sensitivity implies low ability to detect bona fide variants.
- SPECIFICITY
The ability to detect the absence of variants correctly. Low specificity implies many false positives.
- FALSE POSITIVE
Designating a false call, often from noise or sequencing error, as true. An important metric when evaluating the detection abilities of calling algorithms.
- FALSE-DISCOVERY RATE
The expected number of calls that should be false but are marked as true within the final callset.
- RECEIVER OPERATING CHARACTERISTIC CURVE
(ROC) Plots the true positive rate against the false positive rate showing the relationship between sensitivity and specificity.
- ENSEMBLE ALGORITHM
A detection method that combines the resulting callsets from multiple independent algorithms.
- SINGLE MOLECULE STRATEGIES
Genomic methods that read the entirety of long strands of DNA.
- CONNECTED MOLECULE STRATEGIES
Genomic methods that connect shorter reads of a DNA molecule together to provide long range information.
- SEQUENCE COVERAGE
The average number of times a given locus is covered by a sequenced read.
- PHYSICAL COVERAGE
The average number of times a given locus is covered by the cumulative length of the reads, including unsequenced inserts.
- INTER-READ SIGNATURES
Discordant signatures obtained from multiple reads that do not individually overlap the entire SV, analogous to SR signals
- INTRA-READ SIGNATURES
Discordant signatures obtained from reads that overlap the entire SV.
- BASE-CALLING ERROR
Errors in determining the respective nucleotide from raw signals during sequencing.
- HYBRID ASSEMBLIES
Genome assemblies that leverage sequencing data from multiple platforms to reconstruct the original sequence, using the orthogonal data to extend the contig lengths or to branch contigs to one another.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.The 1000 Genomes Project Consortium et al. A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.The Wellcome Trust Case Control Consortium et al. Origins and functional impact of copy number variation in the human genome. Nature 464, 704–712 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sudmant PH et al. Diversity of Human Copy Number Variation and Multicopy Genes. Science 330, 641–646 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mills RE et al. Mapping copy number variation by population-scale genome sequencing. Nature 470, 59–65 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.The 1000 Genomes Project Consortium et al. An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sudmant PH et al. Global diversity, population stratification, and selection of human copy-number variation. Science 349, aab3761 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weischenfeldt J, Symmons O, Spitz F & Korbel JO Phenotypic impact of genomic structural variation: insights from and for human disease. Nat. Rev. Genet 14, 125–138 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Spielmann M, Lupianez DG & Mundlos S Structural variation in the 3D genome. Nat. Rev. Genet 19, 453–467 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Alkan C, Coe BP & Eichler EE Genome structural variation discovery and genotyping. Nat. Rev. Genet 12, 363–376 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lappalainen T, Scott AJ, Brandt M & Hall IM Genomic Analysis in the Age of Human Genome Sequencing. Cell 177, 70–84 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tuzun E et al. Fine-scale structural variation of the human genome. Nat. Genet 37, 727–732 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Sharp AJ et al. Segmental Duplications and Copy-Number Variation in the Human Genome. Am. J. Hum. Genet 77, 78–88 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hastings PJ, Lupski JR, Rosenberg SM & Ira G Mechanisms of change in gene copy number. Nat. Rev. Genet 10, 551–564 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sherry ST dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29, 308–311 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.†The International HapMap Consortium. The International HapMap Project. Nature 426, 789–796 (2003). [DOI] [PubMed] [Google Scholar]
- 16.DePristo MA et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet 43, 491–498 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.The Geuvadis Consortium et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 501, 506–511 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.The UK10K Consortium. The UK10K project identifies rare variants in health and disease. Nature 526, 82–90 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zook JM et al. Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat. Biotechnol 32, 246–251 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Exome Aggregation Consortium et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goodwin S, McPherson JD & McCombie WR Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet 17, 333–351 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Macintyre G, Ylstra B & Brenton JD Sequencing Structural Variants in Cancer for Precision Therapeutics. Trends Genet. 32, 530–542 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Yi K & Ju YS Patterns and mechanisms of structural variations in human cancer. Exp. Mol. Med 50, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Korbel JO et al. Paired-End Mapping Reveals Extensive Structural Variation in the Human Genome. Science 318, 420–426 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoon S, Xuan Z, Makarov V, Ye K & Sebat J Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res. 19, 1586–1592 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hajirasouliha I et al. Detection and characterization of novel sequence insertions using paired-end next-generation sequencing. Bioinformatics 26, 1277–1283 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Korbel JO et al. PEMer: a computational framework with simulation-based error models for inferring genomic structural variants from massive paired-end sequencing data. Genome Biol. 10, R23 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen K et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat. Methods 6, 677–681 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abyzov A, Urban AE, Snyder M & Gerstein M CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 21, 974–984 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Handsaker RE, Korn JM, Nemesh J & McCarroll SA Discovery and genotyping of genome structural polymorphism by sequencing on a population scale. Nat. Genet 43, 269–276 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Layer RM, Chiang C, Quinlan AR & Hall IM LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 15, R84 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rausch T et al. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28, i333–i339 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sindi SS, Önal S, Peng LC, Wu H-T & Raphael BJ An integrative probabilistic model for identification of structural variation in sequencing data. Genome Biol. 13, R22 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao X, Emery SB, Myers B, Kidd JM & Mills RE Resolving complex structural genomic rearrangements using a randomized approach. Genome Biol. 17, 126 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen X et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32, 1220–1222 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Michaelson JJ & Sebat J forestSV: structural variant discovery through statistical learning. Nat. Methods 9, 819–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Telenti A et al. Deep sequencing of 10,000 human genomes. Proc. Natl. Acad. Sci 113, 11901–11906 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kosugi S et al. Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biol. 20, 117 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chaisson MJP et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun 10, 1784 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wong K, Keane TM, Stalker J & Adams DJ Enhanced structural variant and breakpoint detection using SVMerge by integration of multiple detection methods and local assembly. Genome Biol. 11, R128 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lam HYK et al. Detecting and annotating genetic variations using the HugeSeq pipeline. Nat. Biotechnol 30, 226–229 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Parikh H et al. svclassify: a method to establish benchmark structural variant calls. BMC Genomics 17, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Collins RL et al. An open resource of structural variation for medical and population genetics. bioRxiv (2019). doi: 10.1101/578674 [DOI] [Google Scholar]
- 44.Abel HJ et al. Mapping and characterization of structural variation in 17,795 deeply sequenced human genomes. bioRxiv (2018). doi: 10.1101/508515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.The Genome of the Netherlands Consortium et al. A high-quality human reference panel reveals the complexity and distribution of genomic structural variants. Nat. Commun 7, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Werling DM et al. An analytical framework for whole-genome sequence association studies and its implications for autism spectrum disorder. Nat. Genet 50, 727–736 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chiang C et al. SpeedSeq: ultra-fast personal genome analysis and interpretation. Nat. Methods 12, 966–968 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Larson DE et al. svtools: population-scale analysis of structural variation. Bioinformatics btz 492 (2019). doi: 10.1093/bioinformatics/btz492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mohiyuddin M et al. MetaSV: an accurate and integrative structural-variant caller for next generation sequencing. Bioinformatics 31, 2741–2744 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zarate S et al. Parliament2: Fast Structural Variant Calling Using Optimized Combinations of Callers. bioRxiv (2018). doi: 10.1101/424267 [DOI] [Google Scholar]
- 51.Mimori T et al. iSVP: an integrated structural variant calling pipeline from high-throughput sequencing data. BMC Syst. Biol 7, S8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jeffares DC et al. Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat. Commun 8, 14061 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Becker T et al. FusorSV: an algorithm for optimally combining data from multiple structural variation detection methods. Genome Biol. 19, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pounraja VK, Jayakar G, Jensen M, Kelkar N & Girirajan S A machine-learning approach for accurate detection of copy number variants from exome sequencing. Genome Res. 29, 1134–1143 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huddleston J & Eichler EE An Incomplete Understanding of Human Genetic Variation. Genetics 202, 1251–1254 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Iafrate AJ et al. Detection of large-scale variation in the human genome. Nat. Genet 36, 949–951 (2004). [DOI] [PubMed] [Google Scholar]
- 57.Kloosterman WP et al. Characteristics of de novo structural changes in the human genome. Genome Res. 25, 792–801 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.ToMMo Japanese Reference Panel Project et al. Rare variant discovery by deep whole-genome sequencing of 1,070 Japanese individuals. Nat. Commun 6, 8018 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morales J et al. A standardized framework for representation of ancestry data in genomics studies, with application to the NHGRI-EBI GWAS Catalog. Genome Biol. 19, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zook JM et al. A robust benchmark for germline structural variant detection. (bioRxiv, 2019). doi: 10.1101/664623 [DOI] [Google Scholar]
- 61.Chaisson MJP et al. Resolving the complexity of the human genome using single-molecule sequencing. Nature 517, 608–611 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Treangen TJ & Salzberg SL Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat. Rev. Genet 13, 36–46 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Medvedev P, Stanciu M & Brudno M Computational methods for discovering structural variation with next-generation sequencing. Nat. Methods 6, S13–S20 (2009). [DOI] [PubMed] [Google Scholar]
- 64.Kitzman JO et al. Haplotype-resolved genome sequencing of a Gujarati Indian individual. Nat. Biotechnol 29, 59–63 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McCoy RC et al. Illumina TruSeq Synthetic Long-Reads Empower De Novo Assembly and Resolve Complex, Highly-Repetitive Transposable Elements. PLOS ONE 9, 13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zheng GXY et al. Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nat. Biotechnol 34, 303–311 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bishara A et al. Read clouds uncover variation in complex regions of the human genome. Genome Res. 25, 1570–1580 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Marks P et al. Resolving the full spectrum of human genome variation using Linked-Reads. Genome Res. (2019). doi: 10.1101/gr.234443.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Spies N et al. Genome-wide reconstruction of complex structural variants using read clouds. Nat. Methods 14, 915–920 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Elyanow R, Wu H-T & Raphael BJ Identifying structural variants using linked-read sequencing data. Bioinformatics 34, 353–360 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Eslami Rasekh M et al. Discovery of large genomic inversions using long range information. BMC Genomics 18, 65 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Karaoglanoglu F et al. Characterization of segmental duplications and large inversions using Linked-Reads. (Bioinformatics, 2018). doi: 10.1101/394528 [DOI] [Google Scholar]
- 73.Fang L et al. LinkedSV: Detection of mosaic structural variants from linked-read exome and genome sequencing data. bioRxiv (2018). doi: 10.1101/409789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xia LC et al. Identification of large rearrangements in cancer genomes with barcode linked reads. Nucleic Acids Res. 46, e19–e19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wong KHY, Levy-Sakin M & Kwok P-Y De novo human genome assemblies reveal spectrum of alternative haplotypes in diverse populations. Nat. Commun 9, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Weisenfeld NI, Kumar V, Shah P, Church DM & Jaffe DB Direct determination of diploid genome sequences. Genome Res. 27, 757–767 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Meleshko D, Marks P, Williams S & Hajirasouliha I Detection and assembly of novel sequence insertions using Linked-Read technology. (2019). doi: 10.1101/551028 [DOI] [Google Scholar]
- 78.Sedlazeck FJ, Lee H, Darby CA & Schatz MC Piercing the dark matter: bioinformatics of long-range sequencing and mapping. Nat. Rev. Genet 19, 329–346 (2018). [DOI] [PubMed] [Google Scholar]
- 79.Shajii A, Numanagić I, Whelan C & Berger B Statistical Binning for Barcoded Reads Improves Downstream Analyses. Cell Syst. 7, 219–226.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Falconer E et al. DNA template strand sequencing of single-cells maps genomic rearrangements at high resolution. Nat. Methods 9, 1107–1112 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sanders AD et al. Characterizing polymorphic inversions in human genomes by single-cell sequencing. Genome Res. 26, 1575–1587 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hills M, O’Neill K, Falconer E, Brinkman R & Lansdorp PM BAIT: Organizing genomes and mapping rearrangements in single cells. Genome Med. 5, 82 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sanders AD, Falconer E, Hills M, Spierings DCJ & Lansdorp PM Single-cell template strand sequencing by Strand-seq enables the characterization of individual homologs. Nat. Protoc 12, 1151–1176 (2017). [DOI] [PubMed] [Google Scholar]
- 84.Lieberman-Aiden E et al. Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science 326, 289–293 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Harewood L et al. Hi-C as a tool for precise detection and characterisation of chromosomal rearrangements and copy number variation in human tumours. Genome Biol. 18, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Burton JN et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol 31, 1119–1125 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Steininger A et al. Genome-Wide Analysis of Interchromosomal Interaction Probabilities Reveals Chained Translocations and Overrepresentation of Translocation Breakpoints in Genes in a Cutaneous T-Cell Lymphoma Cell Line. Front. Oncol 8, 183 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Seaman L et al. Nucleome Analysis Reveals Structure-Function Relationships for Colon Cancer. Mol. Cancer Res 15, 821–830 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chakraborty A & Ay F Identification of copy number variations and translocations in cancer cells from Hi-C data. Bioinformatics 34, 338–345 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang X et al. Local and global chromatin interactions are altered by large genomic deletions associated with human brain development. Nat. Commun 9, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dixon JR et al. Integrative detection and analysis of structural variation in cancer genomes. Nat. Genet 50, 1388–1398 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Diaz N et al. Chromatin conformation analysis of primary patient tissue using a low input Hi-C method. Nat. Commun 9, 4938 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lee H & Schatz MC Genomic dark matter: the reliability of short read mapping illustrated by the genome mappability score. Bioinformatics 28, 2097–2105 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.English AC, Salerno WJ & Reid JG PBHoney: identifying genomic variants via long-read discordance and interrupted mapping. BMC Bioinformatics 15, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Huddleston J et al. Discovery and genotyping of structural variation from long-read haploid genome sequence data. Genome Res. 27, 677–685 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sedlazeck FJ et al. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 15, 461–468 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Heller D & Vingron M SVIM: structural variant identification using mapped long reads. 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Stephens Z, Wang C, Iyer RK & Kocher J-P Detection and visualization of complex structural variants from long reads. BMC Bioinformatics 19, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fang L, Hu J, Wang D & Wang K NextSV: a meta-caller for structural variants from low-coverage long-read sequencing data. BMC Bioinformatics 19, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wenger AM et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol (2019). doi: 10.1038/s41587-019-0217-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rhoads A & Au KF PacBio Sequencing and Its Applications. Genomics Proteomics Bioinformatics 13, 278–289 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zook JM et al. A robust benchmark for germline structural variant detection. [Google Scholar]
- 103.Pendleton M et al. Assembly and diploid architecture of an individual human genome via single-molecule technologies. Nat. Methods 12, 780–786 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shi L et al. Long-read sequencing and de novo assembly of a Chinese genome. Nat. Commun 7, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Seo J-S et al. De novo assembly and phasing of a Korean human genome. Nature 538, 243–247 (2016). [DOI] [PubMed] [Google Scholar]
- 106.Ameur A et al. De Novo Assembly of Two Swedish Genomes Reveals Missing Segments from the Human GRCh38 Reference and Improves Variant Calling of Population-Scale Sequencing Data. Genes 9, 486 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kronenberg ZN et al. High-resolution comparative analysis of great ape genomes. Science 360, eaar6343 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nagasaki M Construction of JRG (Japanese reference genome) with single-molecule real-time sequencing. 18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Audano PA et al. Characterizing the Major Structural Variant Alleles of the Human Genome. Cell 176, 663–675.e19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Clarke J et al. Continuous base identification for single-molecule nanopore DNA sequencing. Nat. Nanotechnol 4, 265–270 (2009). [DOI] [PubMed] [Google Scholar]
- 111.Eid J et al. Real-Time DNA Sequencing from Single Polymerase Molecules. 323, 7 (2009). [DOI] [PubMed] [Google Scholar]
- 112.Gong L et al. Picky comprehensively detects high-resolution structural variants in nanopore long reads. Nat. Methods 15, 455–460 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cretu Stancu M et al. Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nat. Commun 8, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.De Coster W et al. Structural variants identified by Oxford Nanopore PromethlON sequencing of the human genome. Genome Res. 29, 1178–1187 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lam ET et al. Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nat. Biotechnol 30, 771–776 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schwartz D et al. Ordered restriction maps of Saccharomyces cerevisiae chromosomes constructed by optical mapping. Science 262, 110–114 (1993). [DOI] [PubMed] [Google Scholar]
- 117.Teague B et al. High-resolution human genome structure by single-molecule analysis. Proc. Natl. Acad. Sci 107, 10848–10853 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cao H et al. Rapid detection of structural variation in a human genome using nanochannel-based genome mapping technology. GigaScience 3, 34 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mak ACY et al. Genome-Wide Structural Variation Detection by Genome Mapping on Nanochannel Arrays. Genetics 202, 351–362 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Levy-Sakin M et al. Genome maps across 26 human populations reveal population-specific patterns of structural variation. Nat. Commun 10, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Li L et al. OMSV enables accurate and comprehensive identification of large structural variations from nanochannel-based single-molecule optical maps. Genome Biol. 18, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hastie AR et al. Rapid Automated Large Structural Variation Detection in a Diploid Genome by NanoChannel Based Next-Generation Mapping. bioRxiv (2017). doi: 10.1101/102764 [DOI] [Google Scholar]
- 123.Lima L et al. Comparative assessment of long-read error correction software applied to Nanopore RNA-sequencing data. Brief. Bioinform bbz058 (2019). doi: 10.1093/bib/bbz058 [DOI] [PubMed] [Google Scholar]
- 124.Fu S, Wang A & Au KF A comparative evaluation of hybrid error correction methods for error-prone long reads. Genome Biol. 20, 26 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhang H, Jain C & Aluru S A comprehensive evaluation of long read error correction methods. (Bioinformatics, 2019). doi: 10.1101/519330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Jaratlerdsiri W et al. Next generation mapping reveals novel large genomic rearrangements in prostate cancer. Oncotarget 8, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Xu J et al. An Integrated Framework for Genome Analysis Reveals Numerous Previously Unrecognizable Structural Variants in Leukemia Patients’ Samples. bioRxiv (2019). doi: 10.1101/563270 [DOI] [Google Scholar]
- 128.Zhou B et al. Comprehensive, integrated, and phased whole-genome analysis of the primary ENCODE cell line K562. Genome Res. 29, 472–484 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhou B et al. Haplotype-resolved and integrated genome analysis of the cancer cell line HepG2. Nucleic Acids Res. (2019). doi: 10.1093/nar/gkz169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chan EKF et al. Optical mapping reveals a higher level of genomic architecture of chained fusions in cancer. Genome Res. 28, 726–738 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.English AC et al. Assessing structural variation in a personal genome—towards a human reference diploid genome. BMC Genomics 16, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Fan X, Chaisson M, Nakhleh L & Chen K HySA: a Hybrid Structural variant Assembly approach using next-generation and single-molecule sequencing technologies. Genome Res. 27, 793–800 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Weischenfeldt J et al. Pan-cancer analysis of somatic copy-number alterations implicates IRS4 and IGF2 in enhancer hijacking. Nat. Genet 49, 65–74 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.McPherson A et al. deFuse: An Algorithm for Gene Fusion Discovery in Tumor RNA-Seq Data. PLoS Comput. Biol 7, e1001138 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.McPherson A et al. nFuse: Discovery of complex genomic rearrangements in cancer using high-throughput sequencing. Genome Res. 22, 2250–2261 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yorukoglu D et al. Dissect: detection and characterization of novel structural alterations in transcribed sequences. Bioinformatics 28, i179–i187 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Franke M et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 538, 265–269 (2016). [DOI] [PubMed] [Google Scholar]
- 138.Gheldof N et al. Structural Variation-Associated Expression Changes Are Paralleled by Chromatin Architecture Modifications. PLoS ONE 8, e79973 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Fudenberg G & Pollard KS Chromatin features constrain structural variation across evolutionary timescales. Proc. Natl. Acad. Sci 116, 2175–2180 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Quigley DA et al. Genomic Hallmarks and Structural Variation in Metastatic Prostate Cancer. Cell 174, 758–769.e9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Stranger BE et al. Relative Impact of Nucleotide and Copy Number Variation on Gene Expression Phenotypes. Science 315, 848–853 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Chiang C et al. The impact of structural variation on human gene expression. Nat. Genet 49, 692–699 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Merker JD et al. Long-read genome sequencing identifies causal structural variation in a Mendelian disease. Genet. Med 20, 159–163 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Miao H et al. Long-read sequencing identified a causal structural variant in an exome-negative case and enabled preimplantation genetic diagnosis. Hereditas 155, 32 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Roberts DS et al. Linked-read Sequencing Analysis Reveals Tumor-specific Genome Variation Landscapes in Neurofibromatosis Type 2 (NF2) Patients: Otol. Neurotol. 40, e150–e159 (2019). [DOI] [PubMed] [Google Scholar]
- 146.Sanchis-Juan A et al. Complex structural variants in Mendelian disorders: identification and breakpoint resolution using short- and long-read genome sequencing. Genome Med. 10, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Cantsilieris S et al. Recurrent structural variation, clustered sites of selection, and disease risk for the complement factor H (CFH) gene family. Proc. Natl Acad. Sci 115, E4433–E4442 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Nattestad M et al. Complex rearrangements and oncogene amplifications revealed by long-read DNA and RNA sequencing of a breast cancer cell line. Genome Res. 28, 1126–1135 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Aneichyk T et al. Dissecting the Causal Mechanism of X-Linked Dystonia-Parkinsonism by Integrating Genome and Transcriptome Assembly. Cell 172, 897–909.e21 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sharim H et al. Long-read single-molecule maps of the functional methylome. Genome Res. 29, 646–656 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lee I et al. Simultaneous profiling of chromatin accessibility and methylation on human cell lines with nanopore sequencing: Supplemental Materials. (Genomics, 2018). doi: 10.1101/504993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Beck CR et al. Megabase Length Hypermutation Accompanies Human Structural Variation at 17p 11.2. Cell 176, 1310–1324.e10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Viswanathan SR et al. Structural Alterations Driving Castration-Resistant Prostate Cancer Revealed by Linked-Read Genome Sequencing. Cell 174, 433–447.e19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Huynh L & Hormozdiari F TAD fusion score: discovery and ranking the contribution of deletions to genome structure. Genome Biol. 20, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Feuk L, Carson AR & Scherer SW Structural variation in the human genome. Nat. Rev. Genet 7, 85–97 (2006). [DOI] [PubMed] [Google Scholar]
- 156.Sebat J Large-Scale Copy Number Polymorphism in the Human Genome. Science 305, 525–528 (2004). [DOI] [PubMed] [Google Scholar]
- 157.Redon R et al. Global variation in copy number in the human genome. Nature 444, 444–454 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.McCarroll SA et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat. Genet 40, 1166–1174 (2008). [DOI] [PubMed] [Google Scholar]
- 159.Kidd JM et al. Mapping and sequencing of structural variation from eight human genomes. Nature 453, 56–64 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zhou B et al. Whole-genome sequencing analysis of CNV using low-coverage and paired-end strategies is efficient and outperforms array-based CNV analysis. J. Med. Genet 55, 735–743 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Scherer SW et al. Challenges and standards in integrating surveys of structural variation. Nat. Genet 39, S7–S15 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Speicher MR & Carter NP The new cytogenetics: blurring the boundaries with molecular biology. Nat. Rev. Genet 6, 782–792 (2005). [DOI] [PubMed] [Google Scholar]
- 163.Lee C, Iafrate AJ & Brothman AR Copy number variations and clinical cytogenetic diagnosis of constitutional disorders. Nat. Genet 39, S48–S54 (2007). [DOI] [PubMed] [Google Scholar]
- 164.Tattini L, D’Aurizio R & Magi A Detection of Genomic Structural Variants from Next-Generation Sequencing Data. Front. Bioeng. Biotechnol 3, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Guan P & Sung W-K Structural variation detection using next-generation sequencing data. Methods 102, 36–49 (2016). [DOI] [PubMed] [Google Scholar]
- 166.Quinlan AR & Hall IM Characterizing complex structural variation in germline and somatic genomes. Trends Genet. 28, 43–53 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Tan R et al. An Evaluation of Copy Number Variation Detection Tools from Whole-Exome Sequencing Data. Hum. Mutat 35, 899–907 (2014). [DOI] [PubMed] [Google Scholar]
- 168.Hehir-Kwa JY, Tops BBJ & Kemmeren P The clinical implementation of copy number detection in the age of next-generation sequencing. Expert Rev. Mol. Diagn 18, 907–915 (2018). [DOI] [PubMed] [Google Scholar]
- 169.Hehir-Kwa JY, Pfundt R & Veltman JA Exome sequencing and whole genome sequencing for the detection of copy number variation. Expert Rev. Mol. Diagn 15, 1023–1032 (2015). [DOI] [PubMed] [Google Scholar]
- 170.Pang AW et al. Towards a comprehensive structural variation map of an individual human genome. 14 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Park H et al. Discovery of common Asian copy number variants using integrated high-resolution array CGH and massively parallel DNA sequencing. Nat. Genet 42, 400–405 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Jain M et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol 36, 338–345 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Anderson-Trocme L et al. Legacy Data Confounds Genomics Studies. (2019). doi: 10.1101/624908 [DOI] [PubMed] [Google Scholar]
- 174.Lappalainen I et al. dbVar and DGVa: public archives for genomic structural variation. Nucleic Acids Res. 41, D936–D941 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Sherman RM et al. Assembly of a pan-genome from deep sequencing of 910 humans of African descent. Nat. Genet 51, 30–35 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Zhou B et al. Extensive and deep sequencing of the Venter/HuRef genome for developing and benchmarking genome analysis tools. Sci. Data 5, 180261 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Levy S et al. The Diploid Genome Sequence of an Individual Human. PLoS Biol. 5, e254 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Miga KH et al. Telomere-to-telomere assembly of a complete human X chromosome. bioRxiv 735928 (2019). doi: 10.1101/735928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Wang Y-C et al. High-coverage, long-read sequencing of Han Chinese trio reference samples. (Genomics, 2019). doi: 10.1101/562611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Zook JM et al. Extensive sequencing of seven human genomes to characterize benchmark reference materials. Sci. Data 3, 160025 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Mitelman F, Johansson B & Mertens F The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer 7, 233–245 (2007). [DOI] [PubMed] [Google Scholar]
- 182.Greer SU & Ji HP Structural variant analysis for linked-read sequencing data with gemtools. Bioinformatics (2019). doi: 10.1093/bioinformatics/btz239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zhang Q et al. Clinical application of single-molecule optical mapping to a multigeneration FSHD1 pedigree. Mol. Genet. Genomic Med 7, e565 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Norris AL, Workman RE, Fan Y, Eshleman JR & Timp W Nanopore sequencing detects structural variants in cancer. Cancer Biol. Ther 17, 246–253 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Euskirchen P et al. Same-day genomic and epigenomic diagnosis of brain tumors using real-time nanopore sequencing. Acta Neuropathol. (Berl.) 134, 691–703 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Jacobson EC et al. Hi-C detects novel structural variants in HL-60 and HL-60/S4 cell lines. Genomics S0888754318306700 (2019). doi: 10.1016/j.ygeno.2019.05.009 [DOI] [PubMed] [Google Scholar]
- 187.Sebat J et al. Strong Association of De Novo Copy Number Mutations with Autism. Science 316, 445–449 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Marshall CR et al. Structural Variation of Chromosomes in Autism Spectrum Disorder. Am. J. Hum. Genet 82, 477–488 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Sullivan PF & Geschwind DH Defining the Genetic, Genomic, Cellular, and Diagnostic Architectures of Psychiatric Disorders. Cell 177, 162–183 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Turner TN et al. Genome Sequencing of Autism-Affected Families Reveals Disruption of Putative Noncoding Regulatory DNA. Am. J. Hum. Genet 98, 58–74 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Yuen RK et al. Genome-wide characteristics of de novo mutations in autism. Npj Genomic Med. 1, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Brand H et al. Paired-Duplication Signatures Mark Cryptic Inversions and Other Complex Structural Variation. Am. J. Hum. Genet 97, 170–176 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Collins RL et al. Defining the diverse spectrum of inversions, complex structural variation, and chromothripsis in the morbid human genome. Genome Biol. 18, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Brandler WM et al. Paternally inherited cis-regulatory structural variants are associated with autism. Science 360, 327–331 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Turner TN et al. Genomic Patterns of De Novo Mutation in Simplex Autism. Cell 171, 710–722.e12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Mizuguchi T et al. Detecting a long insertion variant in SAMD12 by SMRT sequencing: implications of long-read whole-genome sequencing for repeat expansion diseases. J. Hum. Genet (2018). doi: 10.1038/s10038-018-0551-7 [DOI] [PubMed] [Google Scholar]
- 197.Mizuguchi T et al. A 12-kb structural variation in progressive myoclonic epilepsy was newly identified by long-read whole-genome sequencing. J. Hum. Genet 64, 359–368 (2019). [DOI] [PubMed] [Google Scholar]
- 198.Barseghyan H et al. Next-generation mapping: a novel approach for detection of pathogenic structural variants with a potential utility in clinical diagnosis. Genome Med. 9, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Eisfeldt J et al. Comprehensive structural variation genome map of individuals carrying complex chromosomal rearrangements. PLOS Genet. 15, e1007858 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Dutta UR et al. Breakpoint mapping of a novel de novo translocation t(X;20)(q11.1;p13) by positional cloning and long read sequencing. Genomics S0888754318302994 (2018). doi: 10.1016/j.ygeno.2018.07.005 [DOI] [PubMed] [Google Scholar]
- 201.Demaerel W et al. The 22q11 low copy repeats are characterized by unprecedented size and structure variability. bioRxiv (2018). doi: 10.1101/403873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Carvalho CMB & Lupski JR Mechanisms underlying structural variant formation in genomic disorders. Nat. Rev. Genet 17, 224–238 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Vollger MR et al. Long-read sequence and assembly of segmental duplications. Nat. Methods 16, 88–94 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Shao H et al. nplnv: accurate detection and genotyping of inversions using long read sub-alignment. BMC Bioinformatics 19, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Bakhtiari M, Shleizer-Burko S, Gymrek M, Bansal V & Bafna V Targeted genotyping of variable number tandem repeats with adVNTR. Genome Res. 28, 1709–1719 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Ummat A & Bashir A Resolving complex tandem repeats with long reads. Bioinformatics 30, 3491–3498 (2014). [DOI] [PubMed] [Google Scholar]
- 207.Liu Q, Zhang P, Wang D, Gu W & Wang K Interrogating the “unsequenceable” genomic trinucleotide repeat disorders by long-read sequencing. Genome Med. 9, 65 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Mitsuhashi S Tandem-genotypes: robust detection of tandem repeat expansions from long DNA reads. 17 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Frith MC & Khan S A survey of localized sequence rearrangements in human DNA. Nucleic Acids Res. 46, 1661–1673 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Jiang T, Liu B, Li J & Wang Y rMETL: sensitive mobile element insertion detection with long read realignment. Bioinformatics (2019). doi: 10.1093/bioinformatics/btz106 [DOI] [PubMed] [Google Scholar]
- 211.Meng G et al. TSD: A computational tool to study the complex structural variants using PacBio targeted sequencing data. (Bioinformatics, 2018). doi: 10.1101/474445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Ritz A et al. Characterization of structural variants with single molecule and hybrid sequencing approaches. Bioinformatics 30, 3458–3466 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]