

Abstract
Background
Functional movement disorders (FMDs), part of the wide spectrum of functional neurological disorders (conversion disorders), are common and often associated with a poor prognosis. Nevertheless, little is known about their neurobiological underpinnings, particularly with regard to the contribution of genetic factors. Because FMD and stress-related disorders share a common core of biobehavioural manifestations, we investigated whether variants in stress-related genes also contributed, directly and interactively with childhood trauma, to the clinical and circuit-level phenotypes of FMD.
Methods
Sixty-nine patients with a ‘clinically defined’ diagnosis of FMD were genotyped for 18 single-nucleotide polymorphisms (SNPs) from 14 candidate genes. FMD clinical characteristics, psychiatric comorbidity and symptomatology, and childhood trauma exposure were assessed. Resting-state functional connectivity data were obtained in a subgroup of 38 patients with FMD and 38 age-matched and sex-matched healthy controls. Amygdala–frontal connectivity was analysed using a whole-brain seed-based approach.
Results
Among the SNPs analysed, a tryptophan hydroxylase 2 (TPH2) gene polymorphism—G703T—significantly predicted clinical and neurocircuitry manifestations of FMD. Relative to GG homozygotes, T carriers were characterised by earlier FMD age of onset and decreased connectivity between the right amygdala and the middle frontal gyrus. Furthermore, the TPH2 genotype showed a significant interaction with childhood trauma in predicting worse symptom severity.
Conclusions
This is, to our knowledge, the first study showing that the TPH2 genotype may modulate FMD both directly and interactively with childhood trauma. Because both this polymorphism and early-life stress alter serotonin levels, our findings support a potential molecular mechanism modulating FMD phenotype.
Introduction
Functional movement disorders (FMDs) represent one of the more common disorders referred to the modern neurology clinic,1 yet they pose unresolved etiological and diagnostic challenges. In the last two decades, important advances in neuroscience have begun to unravel the neurobiological underpinnings of FMD, although several relevant aspects still need to be investigated. In particular, little is known about the contribution of genetic factors to the pathophysiology of FMD.
Several studies have indicated that positive family history for FMD is associated with increased morbidity risk among family members.2 3 However, large-scale genetic epidemiology studies (eg, twin-based, family-based, adoption-based and other population-based studies), which have provided a necessary first step in establishing heritability and exploring genetic interactions in several neuropsychiatric disorders, have not yet been carried out in patients with FMD.
This dearth of data may be related to several factors, including difficulties in case definition and ascertainment, absence of a clinical phenotype of FMD rooted in their neurobiology, as well as the lack of adequate animal models. Furthermore, the heterogeneous phenotype characterising patients with FMD suggests that the prevalence and severity of this disorder unlikely stems from a single dysfunctional gene but could be modulated by multiple genes of small effect, in interaction with each other and in conjunction with environmental events.
Since numerous genetic variants can be implicated in the pathophysiology of FMD, very large samples are needed for gene discovery. Alternatively, shifting the focus from the entire FMD phenotype to common domains of symptoms and behaviours shared across FMD and other disorders may represent a first, important step to identify genetic contributors. In line with this perspective, increasing evidence has indicated that stress response dysfunctions are commonly observed in individuals with FMD, as well as in other functional neurological disorders.4 Patients with FMD exhibit hyperarousal, increased stress reactivity,5 6 alterations in threat responses and traumatic memory recall, and dysregulation in neurocircuitry implicated in emotion regulation.7 8 These manifestations are also commonly observed in stress-related disorders (eg, mood and anxiety disorders), which are often comorbid with FMD, suggesting they may share overlapping biological substrates.4 In stress-related disorders, neurocircuitry and behavioural alterations associated with impaired stress response mechanisms mainly result from interaction between genes, particularly stress-related genes (ie, serotonin (5-HT)-related genes and hypothalamic pituitary adrenal (HPA) axis-related genes), and adverse life experiences, particularly early in life (eg, childhood trauma).9
Taken together, these findings lend support to an evolving concept of a core phenotype shared across FMD and stress-related disorders, which is linked to dysregulation of neurobiological systems implicated in stress response, and may be modulated by a common set of genes and gene×environment interactions. To begin addressing this set of questions, we investigated the relationship between single-nucleotide polymorphism (SNPs) previously implicated with stress-related disorders and/or associated endophenotypes, childhood trauma, and the behavioural and circuit-level phenotypes of FMD.
Materials and methods
Subjects
Sixty-nine patients with clinically definite FMD, as diagnosed by at least two movement disorder specialists using Fahn and Williams criteria,10 were recruited from the Human Motor Control Clinic at the NIH between October 2011 and December 2018. Participants partially overlap with those reported in a previous article.11 In brief, all patients were enrolled in a 3-day inpatient study aimed at investigating the clinical and neurobiological correlates of FMD. Exclusion criteria included comorbid neurological disease; psychosis, bipolar disorder or current substance abuse; a history of traumatic brain injury; active autoimmune disorder; current suicidality; disease severity requiring hospitalisation; contraindication for MRI; abnormal clinical MRI brain; and pregnancy.
Assessments and main outcomes measures
Functional motor symptoms were assessed using a modified version of the Psychogenic Movement Disorders Rating Scale.12 We specifically assessed for age of onset and disease duration, type of symptoms (eg, rest tremor, action tremor, dystonia and myoclonus), and the body regions and/or functions (gait/speech) affected, and patients rated the level of disability and severity associated (scoring is based on 0–4 ordinal scales). Scores are added together to obtain total disability and severity scores, respectively. During the study, all participants were screened for psychiatric diagnoses using the Structured Clinical Interview for the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-IV-TR), Patient Edition.13 Anxious and depressive symptomatology were assessed using the Hamilton Anxiety Rating Scale (HAM-A)14 and the Hamilton Rating Scale for Depression (HAM-D).15 These clinician-rated scales are considered among the most common and useful tools to evaluate the severity of psychiatric symptoms across different functional neurological disorders.16 Participants also completed the Childhood Trauma Questionnaire (CTQ),17 which is one of the most widely used self-reports of early adversity used in research settings. This questionnaire measures five types of maltreatment: emotional, physical, and sexual abuse, and emotional and physical neglect, and has been shown to be a reliable, internally consistent and valid instrument.17
Gene and SNP selection
Genes implicated in the pathophysiology of stress-related disorders, directly and interactively with childhood trauma, were selected. Specifically, we included in our analysis 5-HT-related genes as serotonergic modulation of the acute response to stress and the adaptation to chronic stress is mediated by numerous molecules controlling 5-HT synthesis (tryptophan hydroxylase 1 (TPH1) and 2 isozymes and tryptophan hydroxylase 2 (TPH2)), signal transduction (5-HTR2A, 5-HTR1A and 5-HTR1B) and reuptake (5-HT transporter, SLC6A4).18 Furthermore, previous studies in patients with somatoform disorders seem to indicate a role for serotonergic pathway genes also in FMD.19 20
In addition to these genes, we also selected genes from the HPA axis and endocannabinoid system (CNR1, FAAH, FKBP5, CRHR1 and NR3C2), given that these systems play a critical role in regulating stress response21 and that several of these genes have been indicated as potential candidate genetic factors for FMD.22 Both HPA axis and endocannabinoid-related genetic variations interact with early-life stress in predicting mood and anxiety symptoms, as well as threat-related amygdala function.23 An important exception is represented by a common variant on the FAAH gene (C385A, rs324420), which has been associated with reduced anxiety levels in patients with stress-related disorders.24 Finally, polymorphisms on catechol-O-methyltransferase (COMT), brain-derived neurotrophic factor (BDNF) and glutamate solute carrier family 1 member 3 (SLC1A3) genes were also included in our analysis, given the large body of literature implicating polymorphisms in these genes with impairment in stress response.25–27
Eighteen SNPs from the 14 selected genes (HTR1A, HTR2A, HTR1B, TPH1, TPH2, SLC6A4, CNR1, FAAH, FKBP5, CRHR1, NR3C1, COMT, BDNF and SLC1A3) were included in our analysis (table 1). Priority was given to functional SNPs or SNPs within regulatory regions using online genetic database ensemble (http://www.ensembl.org/index.html) and DbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/).
Table 1.
Candidate genes and SNPs and genotype frequencies
| Gene symbol | Rs number | Wild type>variant allele | Genotype frequency | Hardy-Weinberg equilibrium (P value) |
| HTR1A | rs6295 | C/G | CC=0.23, GC=0.48, GG=0.29 | 0.73 |
| HTR1B | rs6296 | C/G | CC=0.64, GC=0.29, GG=0.07 | 0.48 |
| HTR2A | rs6313 | G/A | GG=0.42, AG=0.46, AA=0.12 | 0.85 |
| rs6311 | C/T | CC=0.82, TC=0.16, TT=0.01 | 0.58 | |
| TPH1 | rs1800532 | A/C | AA=0.30, CA=0.42, AA=0.27 | 0.18 |
| TPH2 | rs4570625 | G/T | GG=0.53, TG=0.42, TT=0.04 | 0.36 |
| SLC6A4* | rs25531 | A/G | S=0.44, LA=0.52, LG=0.04 | 0.64 |
| CRHR1 | rs110402 | G/A | GG=22, AG=31, AA=16 | 0.43 |
| rs1876831 | C/T | CC=50, TC=17, TT=2 | 0.70 | |
| CNR1 | rs1049353 | G/A | GG=48, AG=18, AA=3 | 0.44 |
| FAAH | rs324420 | A/C | CC=0.63; AC=0.30 AA=0.07 | 0.48 |
| FKBP5 | rs9470080 | A/G | AA=11, GA=34, GG=24 | 0.85 |
| rs3800373 | C/A | CC=9, AC=28, AA=32 | 0.47 | |
| rs1360780 | T/C | TT=9, CT=32, CC=28 | 0.97 | |
| NR3C1 | rs10482672 | G/A | GG=54, AG=14, AA=1 | 0.93 |
| BDNF | rs6265 | G/A | GG=46, AG=20, AA=3 | 0.66 |
| COMT | rs4680 | G/A | GG=19, AG=39, AA=11 | 0.22 |
| SLC1A3 | rs2269272 | C/T | CC=48, TC=20, TT=1 | 0.49 |
*SLC6A4 rs25531 was genotyped as part of the triallelic 5-HTTLPR locus (high activity LA allele; low transcriptional activity S allele, low transcriptional activity LG allele). Alleles were grouped as low activity (SS, S LG, LGLG) (0.23), intermediate activity (S LA, LALG) (0.49) and high activity (LALA) (0.28) variants.
†
BDNF, brain-derived neurotrophic factor; CNR1, cannabinoid receptor type 1; COMT, catechol-O-methyltransferase; CRHR1, corticotropin-releasing hormone receptor 1; FAAH, fatty acid amide hydrolase; FKBP5, FK506 binding protein 5; HTR1A, 5-Hydroxytryptamine Receptor 1A; HTR2A, 5-Hydroxytryptamine Receptor 2A; HTR1B, 5-Hydroxytryptamine Receptor 1B; NR3C1, nuclear receptor subfamily 3 group C member 1; TPH1, tryptophan hydroxylase 1; TPH2, tryptophan hydroxylase 2.
Genotyping
Genomic DNA was extracted from whole blood using standard protocols. DNA samples were genotyped using the Illumina OmniExpress BeadChip array (Illumina, San Diego, California, USA), including more than 700 000 SNPs. For the triallelic 5-HTTLPR locus, genotyping was performed in two stages using size discrimination for the S (103 bp) and L (146 bp) alleles and for the rs25531 (LA (146 bp) and LG (61 bp) alleles. Further details are provided in the online supplement section.
jnnp-2019-322636supp001.pdf (78.1KB, pdf)
Assessment of population stratification was performed using ancestry informative markers (AIMs). AIMs (n=2500) were extracted from the Illumina array to calculate ancestral proportions for all study participants. Using methods described previously for an AIM panel including 186 markers,28 which were not available for the current data set, the ancestry assessment identified six ethnic factors (Africa, Europe, Asia, Far East Asia, Oceania and Americas).
MRI data acquisition and processing
Structural and functional images were available for a subgroup of study participants (n=38). Images were acquired with a 3T Siemens (Munich, Germany) Skyra scanner using a 32-channel head coil. The MEMPRAGE images were averaged across echoes and used for registration. Functional magnetic resonance imaging (fMRI) data processing and analysis were performed using the Analysis of Functional Neuroimages (AFNI) software.29 Further details are provided in the online supplement section.
Data analysis
Genotype and behavioural data
For each SNP, allele frequencies and Hardy-Weinberg equilibrium were calculated (table 1). Analyses were conducted using three genotype groups (homozygous for the wild-type allele, heterozygous for the variant allele and homozygous for the variant allele). For several SNPs, given the small number of subjects homozygous for the variant allele, analyses were conducted using two genotype groups (homozygous for the wild-type allele versus homozygous and heterozygous for the variant allele). Demographic and clinical data were tested for normal distribution using the Shapiro-Wilk test. Prior to each analysis, we tested the differences between genotype groups using Fisher’s exact test for binary variables or Student’s t-test and the Mann-Whitney U test for continuous variables. Variables analysed included age, gender, disease duration, HAM-A and HAM-D scores, the most common comorbid axis I psychiatric disorder in our sample (depressive and anxiety disorders), and the CTQ total score. Subgroup analyses were performed if there were differences between genotype groups.
Our primary analyses were aimed at establishing the contribution of individual genetic variants and SNP×CTQ interaction to the prediction of FMD age of onset and severity score. To this extent, linear regression analyses were conducted with (1) FMD age of onset and (2) FMD symptom severity total continuous score as the dependent variables. The selected SNPs and the CTQ total score were included as independent variables, together with the SNP×CTQ interaction term. Secondary analyses included linear regression analyses with HAM-A and HAM-D total continuous scores as dependent variables, and logistic regression analysis for the dichotomous axis I psychiatric diagnosis (depressive disorders) score.
All models included as covariates age, gender and the ancestry scores determined from AIMs; additional covariates were evaluated for inclusion on a model-by-model basis, such that covariates that significantly predicted the outcome measure were retained in the model. Covariates that were evaluated included HAM-A and HAM-D scores, diagnosis of depressive and anxiety disorders, and CTQ score (unless they were examined as outcomes in the model). The level of statistical significance was set at p<0.05 for all tests. Bonferroni correction was applied to account for testing for multiple SNPs. All statistical analyses were performed using R V.3.0.2.
Resting-state functional connectivity (rsFC) data
We investigate whether identified genotypic and gene×CTQ effects on FMD clinical phenotype were also associated with effects on brain connectivity, with a focus on amygdala–frontal circuitry. Alterations in this circuitry have been associated with hyperarousal and impaired emotion regulation.30 Furthermore, prior studies have indicated that both genetic variants at the level of stress-related genes and childhood trauma exposure exert effects on amygdala–frontal connectivity.31 32 To investigate whether similar effects were also observed in patients with FMD, we conducted whole-brain seed-based analysis in AFNI29 to identify the cluster in amygdala–frontal connectivity showing significant group difference, as well as to comprehensively evaluate amygdala connectivity. Details on rsFC analysis are provided in the online supplement section.
rsFC analyses were restricted to SNPs showing an effect on the behavioural phenotype of FMD.
Results
Clinical and psychological features of study population
Study subjects were predominantly female (73%) and Caucasian (89%), with a mean age of 46.7 years±8.3 (21–60). The average disease duration was 5.9±3.7 years, and abnormal movements reported by patients included tremor (75%), jerking movements (65%), abnormal gait/balance (71%), abnormal speech (50%), abnormal posturing/dystonia (35%) and paresis (35%). Fifty-three per cent of the subjects reported exposure to childhood trauma (as measured by the CTQ). Depressive symptom severity was in the mild range (9.9±6.1), while anxiety symptom severity was in the mild to moderate range (13.6±7.7). The most commonly comorbid psychiatric diagnosis was depressive disorder (42% of the study subjects had a lifetime diagnosis). The characteristics of our study population are comparable to those reported in two recent studies analysing the demographic and clinical features of patients with FMD33 34 and are also similar to those observed in a large cohort of patients with dissociative seizures.35
SNPs and SNPs×CTQ effects on FMD clinical phenotype
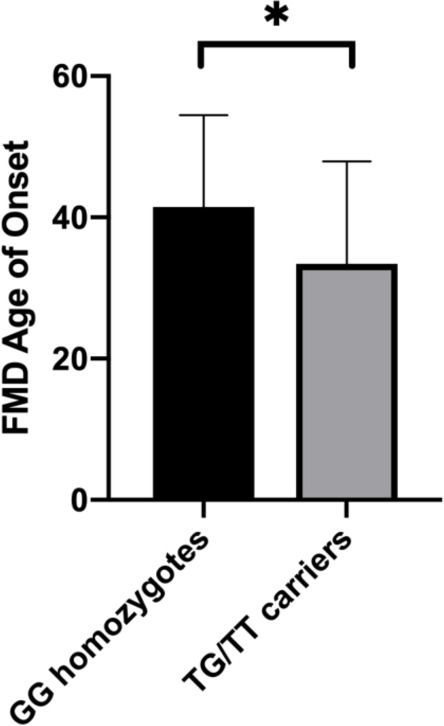
As shown in table 1, none of the 25 genetic loci considered in our analysis significantly deviated from the Hardy-Weinberg equilibrium. Results of linear regression analysis indicated that, among the SNPs analysed, the G-703T polymorphism (rs4570625) in the tryptophan hydroxylase-2 (TPH2) gene was a significant predictor of FMD age of onset (β=−12.83, SE=4.69, p=0.011), after controlling for possible confounders such as age, gender, HAM-A and HAM-D scores, psychiatric comorbidity, CTQ score and AIM score (table 2). Specifically, in individuals carrying the T allele, the mean FMD age of onset was significantly lower relative to non-carriers (mean age of onset: 33.4 years in T carriers, 41.4 in GG homozygotes; figure 1). TPH2 polymorphism was not a significant predictor of FMD symptom severity (β=−1.34, SE=1.51, p=0.37); however, we found a significant interaction between TPH2 genotype (T-allele carriers) and childhood trauma on FMD symptom severity (β=0.08, SE=0.04, p=0.03), after controlling for age, gender, severity of depressive and anxiety symptoms, and comorbid psychiatric diagnosis. In T-allele carriers, increasing levels of childhood trauma were associated with greater FMD symptom severity, while in GG homozygotes, there was no relationship between CTQ total score and FMD symptom severity (figure 2).
Table 2.
Demographic and clinical characteristics of TPH2 GG homozygotes and T carriers
| GG (n=37) | TT/TG (n=32) | P value | |
| Age, mean (SD) | 47.1 (11.6) | 40.5 (12.1) | 0.03 |
| Female, n (%) | 31 (84) | 23 (72) | 0.26 |
| Disease duration, mean (SD) | 5.4 (7.3) | 7.4 (8.6) | 0.32 |
| HAM-A, mean (SD) | 13.1 (7.4) | 14.2 (8.5) | 0.58 |
| HAM-D, mean (SD) | 9.4 (5.9) | 10.8 (7.2) | 0.37 |
| CTQ, mean (SD) | 38.4 (18.6) | 43.6 (14.5) | 0.21 |
| Depression Dx (SCID), n (%) | 3 (8) | 4 (13) | 0.70 |
CTQ, Childhood Trauma Questionnaire; Dx, diagnosis; HAM-A, Hamilton Anxiety Rating Scale; HAM-D, Hamilton Rating Scale for Depression; SCID, Structured Clinical Interview for DSM-IV-TR, Patient Edition.
Figure 1.
Effect of TPH2 G703T polymorphism on FMD age of onset. In individuals carrying the T allele, the mean FMD age of onset was significantly higher relative to GG homozygotes (mean age of onset: 33.4 years in T carriers, 41.4 in GG homozygotes). Sample size: 69 patients with FMD. FMD, functional movement disorder. *P ≤ 0.05
Figure 2.

TPH2 703T moderates the relationship between childhood trauma and FMD symptom severity. In T carriers, increasing levels of childhood trauma were associated with worsening FMD symptom severity, while no relationship was observed in GG homozygotes (β=0.08, SE=0.04, p=0.03). CTQ, Childhood Trauma Questionnaire; FMD, functional movement disorder.
Within the linear regression model for HAM-A and HAM-D, childhood trauma had a marginally significant effect on the HAM-A (β=0.15, SE=0.07, p=0.04) and HAM-D scores (β=0.12, SE=0.06, p=0.04), while no significant TPH2 703T main effect or interaction with childhood trauma was observed. Logistic regression analysis for the dichotomous psychiatric disorder score showed a significant effect of childhood trauma (β=0.10, SE=0.04, p=0.02) and a trend effect for TPH2 703T (β=−3.46, SE=1.89, p=0.07), but no significant interaction (data not shown).
Resting-state functional connectivity
We next explored the neurobiological underpinnings of the identified relationship between the TPH2 polymorphism, childhood trauma exposure and FMD clinical phenotype by investigating whether TPH2 703T and CTQ were associated with alterations in amygdala–frontal connectivity. Subjects included in this analysis were 38 patients with FMD (28 women, mean age of 46.7 years, mean CTQ total score=39.5, GG=22, TG/TT=16) and 38 age-matched and gender-matched healthy volunteers (mean CTQ=26), for whom genetic data were not available.
Using the whole-brain seed-based approach, we first examined whether right amygdala–frontal connectivity differed between genotype groups, after controlling for age, gender, AIM score, CTQ total score, psychometric scores and comorbid psychiatric diagnosis. T carriers exhibited decreased connectivity between the right amygdala and the right middle frontal gyrus (MFG), a node of the default mode network, whereas rsFC between these regions was increased in GG homozygotes (voxel p=0.01; cluster size: 37 voxels; xyz: −37.5, –33.5, 32.5; figure 3). The difference in right amygdala–MFG connectivity between genotype groups was not moderated by CTQ total score, as also indicated by linear regression analysis (p=0.4). As a quality control, we verified that the spontaneous neural activity in the right amygdala did not vary between genotype groups by comparing the amplitude of low frequency fluctuations (ALFF) between the two groups (data not shown, p>0.05).
Figure 3.

Decreased resting-state functional connectivity between the right amygdala and the MFG in TPH2 T carriers. Maps demonstrate group differences in right amygdala–MFG resting state FC between GG homozygotes and T carriers. Images show decreased FC in T carriers between the right amygdala (seed) and the right MFG, compared with GG homozygotes. The threshold for display was set at voxel p=0.01, cluster size >30 contiguous voxels. Sample size: 38 right-handed patients with FMD. FMD, functional movement disorder; MFG, middle frontal gyrus.
We then tested whether the group difference in right amygdala–MFG rsFC was distinctly associated with allele status by comparing the mean cluster z-scores between T carriers, GG homozygotes and healthy controls (HC). Post hoc two-sample t-tests indicated that T carriers exhibited reduced right amygdala functional connectivity with right MFG when compared with GG homozygotes (t=3.61, df=39, p=0.0009) and HC (t=3.803, df=48, p=0.0004), who both exhibited increased connectivity between these regions (figure 4). No differences were observed between GG homozygotes and HC (t=0.612, df=53, p>0.5)
Figure 4.
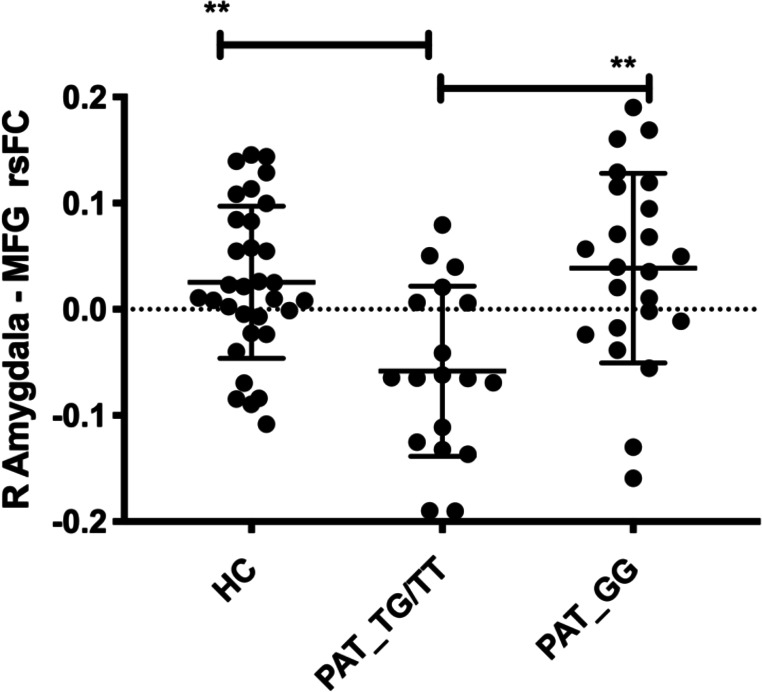
Group differences in right amygdala–MFG connectivity between T carriers, GG homozygotes and HCs. Mean z-scores were extracted for the right amygdala–MFG rsFC cluster. Post hoc two-sample t-tests indicated that patients with FMD carrying the T allele (PAT_TX) exhibited reduced right amygdala–right MFG functional connectivity when compared with GG carriers (PAT_GG) (t=3.61, df=39, p=0.0009) and HCs (t=3.803, df=48, p=0.0004), who both exhibited increased connectivity between these regions. Sample size: 38 patients with FMD and 38 age-matched and gender-matched HCs. FMD, functional movement disorder; HC, healthy control; MFG, middle frontal gyrus; rsFC, resting-state functional connectivity. **P ≤ 0.01
We also explored whether right amygdala–MFG connectivity was correlated with FMD phenotype (age of onset and symptom severity). Across genotype groups, decreased right amygdala–MFG rsFC was associated with greater symptom severity (r=–0.47, p=0.001; figure 5), whereas no correlation was observed with FMD age of onset (r=–0.06, p=0.68).
Figure 5.
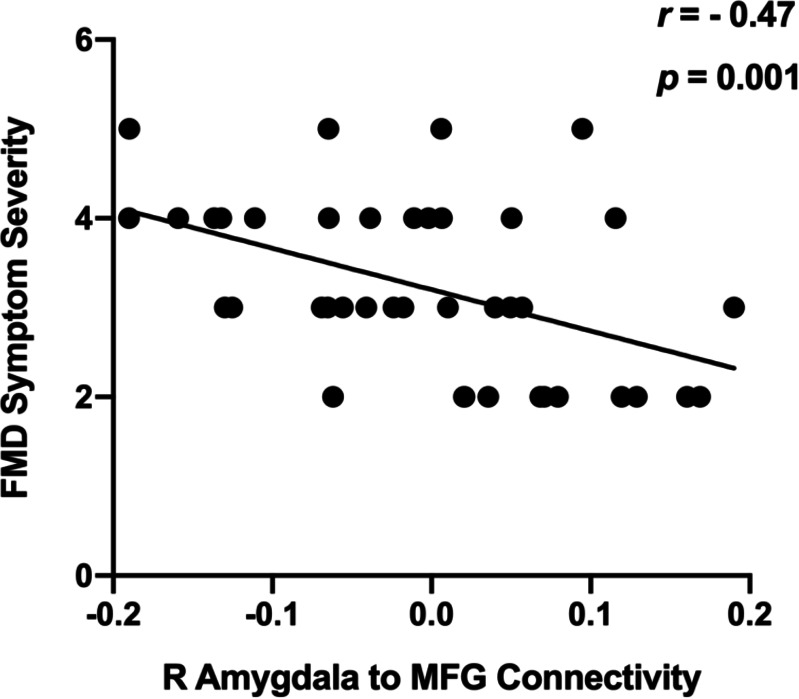
Relationship between right amygdala–MFG rsFC. Across genotype groups, decreased right amygdala–MFG rsFC was associated with greater symptom severity (r=–0.47, p=0.001). Sample size: 38 right-handed patients with FMD. MFG, middle frontal gyrus; rsFC, resting state functional connectivity.
Although our focus was on group differences in amygdala–frontal connectivity, we also observed that connectivity between right amygdala and the left lingual gyrus was reduced in T carriers compared with GG homozygotes, after controlling for age, gender, AIM score, CTQ total score, psychometric scores and comorbid psychiatric diagnosis (uncorrected p=0.01; cluster size: 54 voxels; xyz: 1.5, 62.5, 2.5).
Discussion
To our knowledge, this is the first study investigating the contribution of stress-related genetic polymorphisms to the clinical and circuit-level phenotype of FMD. Using a candidate gene approach, we found that the variant T allele of the TPH2 −703 G/T polymorphism was associated with earlier FMD age of onset and significantly interacted with childhood trauma in predicting worse symptom severity. Furthermore, TPH2 703T had an impact on amygdala–frontal connectivity, with T carriers exhibiting decreased right amygdala–MFG rsFC, compared with both GG homozygotes and HCs.
These findings are consistent with results from animal and human studies indicating that serotonergic dysfunctions, due to genetic factors and/or early-life stress, are critically associated with neuropsychiatric disorders characterised by impairment in stress response and emotion regulation, and by alterations in the neurocircuits underlying these behaviours.36 Importantly, beyond the identification of a particular allele, our results indicate for the first time that a serotonergic mechanism not only underlies psychological manifestations but may also modulate functional motor symptoms.
Serotonergic pathway genes have been previously investigated in relation to somatoform disorders, with inconsistent results.19 20 More recently, Khoury and colleagues37 found that a polymorphism in the DDC gene was associated with a greater frequency of somatoform symptoms in patients with temporomandibular disorders, which belong to the spectrum of functional disorders. Interestingly, this gene encodes the amino acid decarboxylase enzyme, which contributes to the biosynthesis of both 5-HT and dopamine.37 Although the functional meaning of the specific variant identified in this study needs to be further characterised, the authors reported a negative correlation between 5-HT plasma levels and the frequency of somatoform symptoms, thus suggesting that variations in 5-HT levels could represent a mechanism underlying these symptoms.
Our results provide support to this hypothesis. The human TPH2 gene, located on chromosome 12, encodes the neuron-specific enzyme isoform catalysing the rate-limiting step in 5-HT synthesis.38 Animal studies have corroborated that the TPH2 enzyme is a key player in the modulation of 5-HT neurotransmission. For example, TPH2 knockout mice exhibited a >90% reduction in brain 5-HT levels in various brain regions, including hippocampus and frontal cortex.39 Furthermore, TPH2 gene deletion in rat brain is associated with altered serotonergic neuronal circuitry formation and prodepressive and anxiogenic behaviours.39
A growing body of evidence indicates that TPH2 gene expression is highly inducible and is under the control of genetic and environmental factors, including stressors (internal or external). Specifically, chronic stress has been shown to affect TPH2 mRNA expression, with studies reporting elevated TPH2 mRNA levels in animals exposed to early-life stress (for a review, see Chen and Miller39). In addition to stressful events, functional genetic polymorphisms also alter TPH2 gene expression/function both directly and interactively with early-life stress, leading to variations in 5-HT neurotransmission, which critically modulate the stress response as well as the development of phenotypical traits in late life.40 These findings indicate that there is a reciprocal and complex interaction between 5-HT and stress response, as consistently demonstrated in both rodents and non-human primates.39
Similarly, polymorphisms in the human TPH2 gene have also been associated with altered serotonergic neurotransmission. In particular, the G-703T variant, which leads to a T substitution for G in the 5′ regulatory region of the gene, appears to exert a significant effect on TPH2 gene expression in vitro41 (but see also Scheuch and colleagues 42). This polymorphism has been linked to differences in promoter activity and 5-HT levels, with TPH2 T carriers belonging to a high 5-HT-level group.43 Support to this hypothesis has been provided by a positron emission tomography study indicating that in individuals with social anxiety disorder (SAD), the T variant predicted higher 5-HT synthesis rates in frontal–limbic circuits.44
Early-life exposure to increased 5-HT levels, as a consequence of TPH2 genetic mutations and/or early-life stress, has been identified as one of the key mechanisms affecting the development and function of limbic–prefrontal circuits, and impacting on adult emotional and cognitive behaviours.41 In line with these findings, our results indicated that childhood trauma significantly predicted depressive and anxious symptomatology in our sample and was associated with comorbid psychiatric diagnosis, an effect also observed for the T variant, although at a trend level.
Indeed, the TPH2 G-703T polymorphism has been linked to altered emotional and cognitive processing, depressive symptoms after exposure to stressful life events, and treatment response to both antidepressants and cognitive–behavioural treatment of SAD (for a review, see Chen and Miller39 40). The clinical findings related to TPH2 G-703T variation converge, with evidence from the neuroimaging literature indicating that the T variant is associated with increased amygdala responsiveness during negative emotional stimulation, reduced amygdala and hippocampal volumes and altered associations between trait anxiety and amygdala–hippocampal connectivity.45
In our study, right amygdala–MFG functional connectivity was reduced in T carriers, compared with GG homozygotes and HCs. Previous studies have indicated that FC between these regions is associated with cognitive reappraisal of emotion and downregulation of amygdala activity.46 Individuals with FMD exhibited increased right amygdala reactivity and impairment in habituation in response to emotional salient stimuli.7 8 Dysfunctions in amygdala activity and amygdala–medial prefrontal cortex (PFC) connectivity have been consistently associated with genetically driven perturbations of the serotoninergic system.36 Importantly, these brain features have been considered critical endophenotypes of stress-related disorders. Our results suggest that amygdala–medial PFC connectivity may also modulate functional motor manifestations, as indicated by the negative correlation between amygdala–MFG connectivity and FMD symptom severity observed in our sample.
Furthermore, we observed that T-allele carrier status was also associated with decreased connectivity between amygdala and lingual gyrus. The lingual gyrus has been related to visual form and word processing, but not typically with emotion processing. However, right amygdala–lingual gyrus connectivity has been shown to mediate negative mood/depression.47
Our study should be interpreted in light of some limitations. First, we used a candidate gene and not a genome‐wide approach to search for genetic association with the FMD phenotype. Although a candidate gene approach limits the possibility of discovery, a well‐selected gene panel can look for pathological processes in modestly sized cohorts by focusing on a limited number of pathways with higher neurobiological relevance. Pathways investigated in our study are relevant to stress response, emotion regulation and cognitive processing, which are all domains related to FMD, as well as to other functional neurological disorders. Importantly, the SNPs in our analysis were selected based on potential functionality and previous evidence for associations with stress-related disorders and related endophenotypes.
Second, while our results indicate an effect of TPH2 703T and TPH2×childhood trauma interaction on FMD phenotype, we could not assess whether this SNP directly or interactively with childhood trauma also represented a risk factor for FMD, given the lack of genetic data for the HC group. Further studies are needed to examine the contribution of TPH2 703T, as well gene–environment and gene–gene interactions to the susceptibility for FMD.
Third, we investigated TPH2 703T effects on amygdala–frontal connectivity under uncorrected condition. While this choice was motivated by our modest sample size, our results are consistent with previous studies indicating that genetic variations at the level of 5-HT pathway genes are associated with impaired limbic–frontal connectivity.36
Finally, we cannot exclude that other polymorphisms on 5-HT-related genes, as well as on other stress-related genes, including those investigated in the current study, modulate the vulnerability and severity of FMD. Leading models for neuropsychological health and disease suggest that FMD, similarly to stress-related disorders, would be expected to manifest only when the net effect of multiple gene variants causes a system-level failure to maintain homeostasis in the face of persistent environmental challenge.4 However, the identification of genetic variants implicated in complex and polygenic disorders (ie, the genetic component includes hundreds or thousands of individual rare and common variants) can be extremely difficult, as indicated by genetic studies in stress-related disorders. Only few specific genetic risk variants have been consistently linked to these disorders, despite substantial efforts in this area.9 Collaborative, consortium-based research is filling this gap, thus indicating that large sample sizes are crucial for gene discovery. With more cohorts being collected relevant to functional neurological disorders, we hope to see further progress also in the field of FMD.
In summary, our findings indicate, for the first time, that alterations in serotonergic neurotransmission, due to TPH2 variations and environmental stressors, may represent a neurobiological mechanism modulating functional motor symptoms. The effects of TPH2 genotype and childhood trauma were identified both at the clinical and neurocircuitry levels in our sample and are in line with a recent study implicating serotoninergic dysfunctions in somatoform symptoms.37 We believe our findings provide an important first step for future studies aimed at better understanding the role of the 5-HT system in FMD, via pathway analyses of functionally related genes, the development of animal assays of FMD and by assessing the relationship between 5-HT levels and FMD symptoms severity and frequency in clinical populations. These results may open the door to treatment options that target the 5-HT system in individuals with more severe FMD symptoms, particularly those carrying the TPH2 703T allele. To this extent, it will be crucial to replicate our findings on TPH2 in large-scale studies, which may also facilitate the identification of other genetic contributors to FMD.
Footnotes
Contributors: PAS and MH conceived the idea for this study; PAS wrote the manuscript. PAS and SH performed the resting-state functional connectivity (rsFC) analysis. GN performed the statistical analysis; GD and CH performed the genotyping of DNA samples; CM collected behavioural and imaging data and preprocessed rsFC data. All authors provided critical feedback and helped shape the research, analysis and manuscript.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Patient consent for publication: Not required.
Ethics approval: All participants provided written informed consent. The NIH Institutional Review Board approved the study.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data availability statement: Data are available upon reasonable request. Deidentified participant data can be shared upon reasonable request.
References
- 1. Stone J, Carson A, Duncan R, et al. Who is referred to neurology clinics?--the diagnoses made in 3781 new patients. Clin Neurol Neurosurg 2010;112:747–51. 10.1016/j.clineuro.2010.05.011 [DOI] [PubMed] [Google Scholar]
- 2. Ljungberg L. Hysteria: a clinical. Prognostic and Genetic Study: Munksgaard, 1957. [PubMed] [Google Scholar]
- 3. Stamelou M, Cossu G, Edwards MJ, et al. Familial psychogenic movement disorders. Mov Disord 2013;28:1295–8. 10.1002/mds.25463 [DOI] [PubMed] [Google Scholar]
- 4. Keynejad RC, Frodl T, Kanaan R, et al. Stress and functional neurological disorders: mechanistic insights. J Neurol Neurosurg Psychiatry 2019;90:813–21. 10.1136/jnnp-2018-318297 [DOI] [PubMed] [Google Scholar]
- 5. Seignourel PJ, Miller K, Kellison I, et al. Abnormal affective startle modulation in individuals with psychogenic [corrected] movement disorder. Mov Disord 2007;22:1265–71. 10.1002/mds.21451 [DOI] [PubMed] [Google Scholar]
- 6. Apazoglou K, Mazzola V, Wegrzyk J, et al. Biological and perceived stress in motor functional neurological disorders. Psychoneuroendocrinology 2017;85:142–50. 10.1016/j.psyneuen.2017.08.023 [DOI] [PubMed] [Google Scholar]
- 7. Voon V, Brezing C, Gallea C, et al. Emotional stimuli and motor conversion disorder. Brain 2010;133:1526–36. 10.1093/brain/awq054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Aybek S, Nicholson TR, O'Daly O, et al. Emotion-motion interactions in conversion disorder: an fMRI study. PLoS One 2015;10:e0123273. 10.1371/journal.pone.0123273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Smoller JW. The genetics of stress-related disorders: PTSD, depression, and anxiety disorders. Neuropsychopharmacology 2016;41:297–319. 10.1038/npp.2015.266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Williams DT, Ford B, Fahn S. Phenomenology and psychopathology related to psychogenic movement disorders. Adv Neurol 1995;65:231–57. [PubMed] [Google Scholar]
- 11. Maurer CW, LaFaver K, Ameli R, et al. Impaired self-agency in functional movement disorders: a resting-state fMRI study. Neurology 2016;87:564–70. 10.1212/WNL.0000000000002940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hinson VK, Cubo E, Comella CL, et al. Rating scale for psychogenic movement disorders: scale development and Clinimetric testing. Mov Disord 2005;20:1592–7. 10.1002/mds.20650 [DOI] [PubMed] [Google Scholar]
- 13. First MB SR, Gibbon M, Williams JBW. Structured clinical interview for DSM-IV patient edition: biometric research 1995.
- 14. Hamilton M. The assessment of anxiety states by rating. Br J Med Psychol 1959;32:50–5. 10.1111/j.2044-8341.1959.tb00467.x [DOI] [PubMed] [Google Scholar]
- 15. Hamilton M. A RATING SCALE FOR DEPRESSION. Journal of Neurology, Neurosurgery &. Psychiatry 1960;23:56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pick S, Anderson DG, Asadi-Pooya AA, et al. Outcome measurement in functional neurological disorder: a systematic review and recommendations. J Neurol Neurosurg Psychiatry 2020;91:638–49. 10.1136/jnnp-2019-322180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bernstein DP, Fink L, Handelsman L, et al. Initial reliability and validity of a new retrospective measure of child abuse and neglect. Am J Psychiatry 1994;151:1132–6. 10.1176/ajp.151.8.1132 [DOI] [PubMed] [Google Scholar]
- 18. Graeff FG, Guimarães FS, De Andrade TG, et al. Role of 5-HT in stress, anxiety, and depression. Pharmacol Biochem Behav 1996;54:129–41. 10.1016/0091-3057(95)02135-3 [DOI] [PubMed] [Google Scholar]
- 19. Hennings A, Zill P, Rief W. Serotonin transporter gene promoter polymorphism and somatoform symptoms. J Clin Psychiatry 2009;70:1536–9. 10.4088/JCP.08m04613 [DOI] [PubMed] [Google Scholar]
- 20. Koh KB, Choi EH, Lee Y-J, et al. Serotonin-Related gene pathways associated with undifferentiated somatoform disorder. Psychiatry Res 2011;189:246–50. 10.1016/j.psychres.2011.04.002 [DOI] [PubMed] [Google Scholar]
- 21. Micale V, Drago F. Endocannabinoid system, stress and HPA axis. Eur J Pharmacol 2018;834:230–9. 10.1016/j.ejphar.2018.07.039 [DOI] [PubMed] [Google Scholar]
- 22. Ellenstein A, Kranick SM, Hallett M. An update on psychogenic movement disorders. Curr Neurol Neurosci Rep 2011;11:396–403. 10.1007/s11910-011-0205-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Di Iorio CR, Carey CE, Michalski LJ, et al. Hypothalamic-Pituitary-Adrenal axis genetic variation and early stress moderates amygdala function. Psychoneuroendocrinology 2017;80:170–8. 10.1016/j.psyneuen.2017.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Spagnolo PA, Ramchandani VA, Schwandt ML, et al. Faah gene variation moderates stress response and symptom severity in patients with posttraumatic stress disorder and comorbid alcohol dependence. Alcohol Clin Exp Res 2016;40:2426–34. 10.1111/acer.13210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bernard R, Kerman IA, Thompson RC, et al. Altered expression of glutamate signaling, growth factor, and glia genes in the locus coeruleus of patients with major depression. Mol Psychiatry 2011;16:634–46. 10.1038/mp.2010.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jabbi M, Kema IP, van der Pompe G, et al. Catechol-O-Methyltransferase polymorphism and susceptibility to major depressive disorder modulates psychological stress response. Psychiatr Genet 2007;17:183–93. 10.1097/YPG.0b013e32808374df [DOI] [PubMed] [Google Scholar]
- 27. Jabbi M, Korf J, Kema IP, et al. Convergent genetic modulation of the endocrine stress response involves polymorphic variations of 5-HTT, COMT and MAOA. Mol Psychiatry 2007;12:483–90. 10.1038/sj.mp.4001975 [DOI] [PubMed] [Google Scholar]
- 28. Hodgkinson CA, Yuan Q, Xu K, et al. Addictions biology: haplotype-based analysis for 130 candidate genes on a single array. Alcohol Alcohol 2008;43:505–15. 10.1093/alcalc/agn032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cox RW. AFNI: software for analysis and visualization of functional magnetic resonance NeuroImages. Comput Biomed Res 1996;29:162–73. 10.1006/cbmr.1996.0014 [DOI] [PubMed] [Google Scholar]
- 30. Banks SJ, Eddy KT, Angstadt M, et al. Amygdala-frontal connectivity during emotion regulation. Soc Cogn Affect Neurosci 2007;2:303–12. 10.1093/scan/nsm029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pagliaccio D, Luby JL, Bogdan R, et al. Stress-system genes and life stress predict cortisol levels and amygdala and hippocampal volumes in children. Neuropsychopharmacology 2014;39:1245–53. 10.1038/npp.2013.327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. van der Werff SJA, Pannekoek JN, Veer IM, et al. Resting-State functional connectivity in adults with childhood emotional maltreatment. Psychol Med 2013;43:1825–36. 10.1017/S0033291712002942 [DOI] [PubMed] [Google Scholar]
- 33. Baizabal-Carvallo JF, Jankovic J. Gender differences in functional movement disorders. Mov Disord Clin Pract 2020;7:182–7. 10.1002/mdc3.12864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. O'Connell N, Nicholson TR, Wessely S, et al. Characteristics of patients with motor functional neurological disorder in a large UK mental health service: a case-control study. Psychol Med 2020;50:446–55. 10.1017/S0033291719000266 [DOI] [PubMed] [Google Scholar]
- 35. Goldstein LH, Robinson EJ, Reuber M, et al. Characteristics of 698 patients with dissociative seizures: a UK multicenter study. Epilepsia 2019;60:2182–93. 10.1111/epi.16350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Holmes A. Genetic variation in cortico-amygdala serotonin function and risk for stress-related disease. Neurosci Biobehav Rev 2008;32:1293–314. 10.1016/j.neubiorev.2008.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Khoury S, Piltonen MH, Ton A-T, et al. A functional substitution in the L-aromatic amino acid decarboxylase enzyme worsens somatic symptoms via a serotonergic pathway. Ann Neurol 2019;86:168–80. 10.1002/ana.25521 [DOI] [PubMed] [Google Scholar]
- 38. Patel PD, Pontrello C, Burke S. Robust and tissue-specific expression of TPH2 versus TPH1 in rat raphe and pineal gland. Biol Psychiatry 2004;55:428–33. 10.1016/j.biopsych.2003.09.002 [DOI] [PubMed] [Google Scholar]
- 39. Chen G-L, Miller GM. Advances in tryptophan hydroxylase-2 gene expression regulation: new insights into serotonin-stress interaction and clinical implications. Am J Med Genet B Neuropsychiatr Genet 2012;159B:152–71. 10.1002/ajmg.b.32023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen G-L, Miller GM. Tryptophan hydroxylase-2: an emerging therapeutic target for stress disorders. Biochem Pharmacol 2013;85:1227–33. 10.1016/j.bcp.2013.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lin Y-MJ, Chao S-C, Chen T-M, et al. Association of functional polymorphisms of the human tryptophan hydroxylase 2 gene with risk for bipolar disorder in Han Chinese. Arch Gen Psychiatry 2007;64:1015–24. 10.1001/archpsyc.64.9.1015 [DOI] [PubMed] [Google Scholar]
- 42. Scheuch K, Lautenschlager M, Grohmann M, et al. Characterization of a functional promoter polymorphism of the human tryptophan hydroxylase 2 gene in serotonergic raphe neurons. Biol Psychiatry 2007;62:1288–94. 10.1016/j.biopsych.2007.01.015 [DOI] [PubMed] [Google Scholar]
- 43. Hahn T, Heinzel S, Notebaert K, et al. The tricks of the trait: neural implementation of personality varies with genotype-dependent serotonin levels. Neuroimage 2013;81:393–9. 10.1016/j.neuroimage.2013.05.037 [DOI] [PubMed] [Google Scholar]
- 44. Furmark T, Henningsson S, Appel L, et al. Genotype over-diagnosis in amygdala responsiveness: affective processing in social anxiety disorder. J Psychiatry Neurosci 2009;34:30–40. [PMC free article] [PubMed] [Google Scholar]
- 45. Gutknecht L, Jacob C, Strobel A, et al. Tryptophan hydroxylase-2 gene variation influences personality traits and disorders related to emotional dysregulation. Int J Neuropsychopharmacol 2007;10:309–20. 10.1017/S1461145706007437 [DOI] [PubMed] [Google Scholar]
- 46. Buhle JT, Silvers JA, Wager TD, et al. Cognitive reappraisal of emotion: a meta-analysis of human neuroimaging studies. Cereb Cortex 2014;24:2981–90. 10.1093/cercor/bht154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Barch D, Pagliaccio D, Belden A, et al. Effect of hippocampal and amygdala connectivity on the relationship between preschool poverty and school-age depression. Am J Psychiatry 2016;173:625–34. 10.1176/appi.ajp.2015.15081014 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jnnp-2019-322636supp001.pdf (78.1KB, pdf)