

Abstract
Heart failure (HF) is associated with an increased propensity for atrial fibrillation (AF), causing higher mortality than AF or HF alone. It is hypothesized that HF-induced remodelling of atrial cellular and tissue properties promotes the genesis of atrial action potential (AP) alternans and conduction alternans that perpetuate AF. However, the mechanism underlying the increased susceptibility to atrial alternans in HF remains incompletely elucidated. In this study, we investigated the effects of how HF-induced atrial cellular electrophysiological (with prolonged AP duration) and tissue structural (reduced cell-to-cell coupling caused by atrial fibrosis) remodelling can have an effect on the generation of atrial AP alternans and their conduction at the cellular and one-dimensional (1D) tissue levels. Simulation results showed that HF-induced atrial electrical remodelling prolonged AP duration, which was accompanied by an increased sarcoplasmic reticulum (SR) Ca2+ content and Ca2+ transient amplitude. Further analysis demonstrated that HF-induced atrial electrical remodelling increased susceptibility to atrial alternans mainly due to the increased sarcoplasmic reticulum Ca2+-ATPase (SERCA) Ca2+ reuptake, modulated by increased phospholamban (PLB) phosphorylation, and the decreased transient outward K+ current (Ito). The underlying mechanism has been suggested that the increased SR Ca2+ content and prolonged AP did not fully recover to their previous levels at the end of diastole, resulting in a smaller SR Ca2+ release and AP in the next beat. These produced Ca2+ transient alternans and AP alternans, and further caused AP alternans and Ca2+ transient alternans through Ca2+→AP coupling and AP→Ca2+ coupling, respectively. Simulation of a 1D tissue model showed that the combined action of HF-induced ion channel remodelling and a decrease in cell-to-cell coupling due to fibrosis increased the heart tissue’s susceptibility to the formation of spatially discordant alternans, resulting in an increased functional AP propagation dispersion, which is pro-arrhythmic. These findings provide insights into how HF promotes atrial arrhythmia in association with atrial alternans.
Author summary
Atrial Fibrillation (AF) is the most common arrhythmia in adults, especially in the elderly, with the increased incidence of stroke being a major complication that increases morbidity and mortality. The occurrence of AF is often accompanied by heart failure (HF). AF and HF are also known to have the bidirectional relationship that AF worsens HF and HF promotes AF. HF can induce atrial remodelling, including electrical remodelling, atrial fibrosis, stretch and dilatation, and oxidative stress, in which many factors are associated with arrhythmogenic atrial alternans. HF-induced atrial remodelling varies during various stages and complications of HF, but possible mechanisms underlying their pro-susceptibility to alternans have not been completely elucidated. In this study, we investigated the effects of HF-induced atrial remodelling with prolonged action potential duration (APD) and decreased cell-to-cell coupling on susceptibility to atrial alternans. Simulation results showed that HF-induced an increase in sarcoplasmic reticulum Ca2+-ATPase (SERCA) Ca2+ reuptake caused by increased phospholamban phosphorylation and a decrease in transient outward K+ current played significant roles in the genesis of Ca2+ transient alternans and action potential alternans at the single-cell level. The HF-induced decline of cell-to-cell coupling and APD prolongation promoted the genesis of spatially discordant alternans in atrial tissue. This provides insights into how HF facilitates atrial arrhythmia in relation to atrial alternans.
Introduction
Atrial fibrillation (AF) and heart failure (HF) are common cardiovascular diseases that frequently coexist with each other, resulting in higher mortality than AF or HF alone [1]. For example, over one-third of patients with AF in the Framingham Heart Study had HF, and more than half of patients with HF also had AF [2]. This is because HF shares many similar risk factors (e.g. advanced age, coronary disease, hypertension, diabetes mellitus, sleep apnoea, etc.) and pathophysiology to AF such that HF and AF can induce and facilitate each other [3–6]. In particular, HF-induced atrial fibrosis, electrical remodelling, stretch and dilatation, and oxidative stress promote AF [3,7]. However, its exact mechanism remains to be completely elucidated.
HF may exhibit inconsistent pathological remodelling of the atria due to the various stages and complications of cardiac disease [8,9], as demonstrated by various techniques and experimental conditions [6]. For example, some studies reported shortened action potential duration (APD) [10,11] and reduced atrial cellular effective refractory periods (ERPs) [12] in HF, which had significant roles in promoting and maintaining AF due to the shortened wavelength [13–16], whereas the study by Molina et al. showed no significant difference in atrial APD between control and HF patients [17]. Even more confoundingly, other studies have reported prolonged APD [18–21] and increased atrial ERP [22] in HF.
When HF was present alongside prolonged atrial APD, experimental data obtained from patients and dogs indicated an increased propensity for AF [18,19,21,22]. This appears to be associated with atrial ion channel remodelling, Ca2+ handling abnormalities and structural remodelling. Previous studies have found that atrial action potential (AP) alternans (beat-to-beat alterations in APs) preceded AF episodes, indicating that atrial alternans plays an important role in promoting AF [23–25]. In the atria, many factors have been shown to be associated with arrhythmogenic atrial alternans, such as mechanical stretch, myocardial infarction, hypertension, and hypertrophy [26–29]. In addition, many aspects of HF-induced atrial remodelling have been identified experimentally to also be related to atrial alternans. These include a prolonged atrial APD, a reduced L-type Ca2+ current (ICa), an increased sarcoplasmic reticulum (SR) Ca2+ load, loss of t-tubules, decreased connexin expression and disrupted cell-to-cell coupling. Prolonged atrial APD caused by HF leads to a shortened diastolic interval (DI), resulting in an increased slope of the APD restitution curve. Some studies have demonstrated that APD alternans can be predicted by an APD restitution slope > 1 [30–33]. The reduced ICa may induce an increased susceptibility to Ca2+ alternans via a mechanism in which most ryanodine receptors (RyR2) are activated by Ca2+ waves that occur above a certain SR Ca2+ content threshold level; and an increased Ca2+ efflux with large Ca2+ transient causes a SR Ca2+ content to go below the threshold level, leading to a smaller Ca2+ transient in the next cycle [34]. In both experimental and simulation studies, SR Ca2+ load has been demonstrated to play a significant role in Ca2+ alternans [23,34–36]. In addition, the loss of t-tubules in atrial myocytes has been found in HF animal models [37], which may also contribute to the genesis of Ca2+ alternans [38]. Furthermore, decreased connexin expression and disrupted cell-to-cell coupling due to increased fibrosis increases tissue heterogeneity which may facilitate the development of spatially discordant alternans [39]. Though various atrial remodelling instances induced by HF have been identified and may vary at different stages, their pro-susceptibility to alternans are not completely understood yet.
This study focuses on HF with a prolonged APD and investigates the effects of atrial remodelling on the genesis of cardiac alternans based on short-term experimental data gathered from dogs who have had HF induced by no more than 5 weeks of ventricular tachypacing [18,19,40]. It aims to address how HF-induced atrial remodelling can increase susceptibility to alternans. We first modified the updated canine atrial cell model by Ramirez et al. [41] (the RNC model) to incorporate the effects of short-term HF-induced atrial remodelling. Then we sought to (i) illustrate the effects of this HF-induced atrial remodelling on atrial myocyte action potentials and Ca2+ transient; (ii) assess whether HF-induced atrial remodelling promotes susceptibility to atrial myocyte alternans and the mechanism by which this might occur; and (iii) investigate spatial alternans at the one-dimensional (1D) tissue level under the HF condition.
Methods
Single basal atrial cell model
This study was designed to assess the effects of HF-induced electrical remodelling on the genesis of atrial alternans, which was in association with a prolonged APD observed in canine atria with short-term HF [18,19]. Such effect was evaluated by using the canine atrial cell model developed by Ramirez et al. [41], with the incorporation of an updated Na/Ca exchanger (NCX) current (INCX) formulation developed by Weber et al. [42] to describe more detailed regulation of the current via intracellular Ca2+ concentration. The parameters of the Weber INCX model were derived from experimental data of mouse myocytes with overexpression of normal canine NCX [42]. As compared to the original RNC model, the modified model incorporating the Weber INCX produced similar Ca dynamics (S8 Fig). The modified model was validated by comparing the simulated action potential duration at 90% repolarization (APD90) under a quasi-steady state after pacing for 100 beats at 1 Hz to experimental data. The computed APD90 from the basal model was 185.8 ms, which was in concordance with previous experimental data [18].
Simulation of HF-induced atrial remodelling
HF-induced atrial remodelling has been identified in various ion channels and Ca2+ handling processes. For ion channel remodelling, we focused on HF-induced atrial remodelling associated with a prolonged APD, as observed in studies of dog HF models [18,19,40]. In the atria, it was shown that HF induced (i) a decrease in the transient outward K+ current (Ito), slow-delayed rectifier K+ current (IKs), and ICa; (ii) no change in the ultra-rapid-delayed rectifier K+ current (IKur), rapid-delayed rectifier K+ current (IKr),, and inward-rectifier K+ current (IK1); (iii) Ca2+ handling abnormalities; (iv) fibrosis populations. HF also induced an increase in the SR Ca2+ content [18], which was partly attributable to an increased sarcoplasmic reticulum Ca2+-ATPase (SERCA) Ca2+ reuptake modulated by increased fractional CaMKII phospholamban (PLB) phosphorylation [18]. Therefore, to simulate this increased PLB phosphorylation, the [Ca2+]i half-saturation constant for Ca2+ uptake into the network SR, Kup, was decreased as in [43]. In addition, Yeh et al. [18] showed a decrease in protein levels of SERCA2a and RyR2, a decrease in calsequestrin (Csqn, main SR Ca2+-binding protein), and no significant changes of fractional RyR2 phosphorylation.
A summary of HF-induced atrial ion channel and Ca2+ handling remodelling in association with prolonged APD at the single cell level is listed in Table 1. HF resulted in an increased APD90 and intracellular Ca2+ transient (CaT) amplitude matched to experimental observations, thus validating the need to decrease Kup in simulations [18].
Table 1. List of HF-induced atrial ion channels and Ca2+ handling remodelling based on data observed in HF dogs and model parameter alterations to simulate HF.
| Element | Experimental observation | Model parameters | Change from control |
|---|---|---|---|
| ICa | -≈30% [19], -31% (at +10mV) [40] | GCa | -30% |
| Ito | -≈50% [19], -54% (at +40mV) [40] | Gto | -50% |
| IKs | -≈30% [19], -46% (pulse to +50mV) [40] | GKs | -45% |
| IKur | not altered [19] | -- | not altered |
| IKr | not altered [19] | -- | not altered |
| IK1 | not altered [19,40] | -- | not altered |
| SERCA | Protein levels of SERCA2a -≈35% [18] | Jup(max) | -30% |
| Fractional CaMKII phosphorylation of PLB +≈120% [18] | Kup | -78% | |
| RyR2 | Protein levels -≈65% [18] | Jrel(max) | -60% |
| Fractional RyR2 phosphorylation not significant altered | -- | not altered | |
| Csqn | -≈15% [18] | [Csqn]max | -15% |
Abbreviations: GCa, maximal ICa conductance; Gto, maximal Ito conductance; GKs, maximal IKs conductance; NSR, network SR; Jup, Ca2+ uptake into the NSR; Kup, [Ca2+]i half-saturation constant for Jup; JSR, junctional SR; Jrel, Ca2+ release from the JSR; Jrel(max), maximal Ca2+ release rate for Jrel; [Csqn]max, total calsequestrin concentration in JSR.
Parameter sensitivity analysis
Seven parameters were changed from the control condition to simulate the HF-induced atrial remodelling at the single cell model. Multivariable regression with a population of 300 model variants was performed to assess the relative contributions of the different changes of these 7 parameters (Table 1) to the APD90, CaT amplitude and SR Ca2+ content [44–46]. In addition, each parameter was scaled at a time relative to control or HF conditions, and alternans behavior was assessed to identify the effects of the parameter changes on alternans genesis.
One-dimensional tissue model
Cell-to-cell electrical coupling in the tissue model can be described via a well-known reaction-diffusion equation [47]. For a 1D model (a cable within cardiac tissue), the mono-domain equation is
| (1) |
where V is the membrane potential, Iion is the total transmembrane ionic current, Cm is the cell capacitance, and D is the diffusion coefficient along the 1D fibre.
The 1D simulation was performed on a 50.1 mm strand that consisted of 300 cells with a spatial resolution of 0.167 mm, which was previously used in the canine atria tissue model [48,49]. Stimuli were applied to the left of four cells to evoke AP to propagate along the strand for control and the case when the diffusion coefficient was decreased to 0.028 mm2/ms. In the control simulation, the diffusion coefficient D was set to 0.167 mm2/ms to achieve a conduction velocity (CV) of approximately 0.60 m/s along the cable, which was in line with the CV previously used for tissue simulations [50–52]. The CV was calculated using the method in [53]. It has previously been reported that fibrosis may reduce the cell-to-cell coupling [39]. For HF simulations, the diffusion coefficient D was reduced from 0.167 mm2/ms to 0.084 mm2/ms and 0.028 mm2/ms in order to mimic the reduced CVs seen in HF (0.6, 0.4, and 0.2 m/s respectively).
Simulation protocols
At the single cell level, the dynamic pacing and standard S1-S2 protocols [53,54] were used to analyse the genesis of atrial alternans such as APD90 alternans and the intracellular CaT alternans (including the amplitude and decay time), as previously described in detail [55]. The rate dependent curves of APD90, CaT amplitude, and CaT decay time against various pacing cycle lengths (PCLs) were obtained by applying a dynamic pacing protocol. The APD90 restitution curves against various preceding DIs were obtained by applying the standard S1-S2 protocol.
At the 1D tissue level, the pacing protocol used by Narayan et al. [24] was used to induce the alternans. First, steady-state values of the single cell model under control and HF conditions at a PCL of 750 ms were used to initialize all cells in the 1D cable. Then, the 1D cable model was paced at 750 ms for 20 beats. This was followed by pacing for 50 beats at various PCLs.
Numerical implementation
All time-dependent variables were solved using the Forward Euler method with a time step of 0.02 ms. At the 1D tissue level, a space step was 0.167 mm and a no-flux boundary condition was implemented. Simulations were computed using an Intel Xeon E5-2637 v3 3.50GHz CPU rather than with GPU parallel computing due to increased performance when computing small amounts of cells.
Results
Effect of HF-induced electrical remodelling at the single cell level
The effects of HF-induced atrial ion channel and Ca2+ handling remodelling on the atrial cell APD90, CaT amplitude and SR Ca2+ content are shown in Fig 1, which were compared to those obtained in the control condition. These simulation results agreed with experimental data from canine heart failure animal model on observations of prolonged APD (Fig 1A) and increased CaT amplitude (Fig 1B) and SR Ca2+ content (Fig 1C) [18]. In the simulation, an APD90 increase of 61.4 ms was seen in HF cells relative to the control at 1 Hz which was similar to the experimental range of 50.8±8.2 ms in left atria (LA) and 51.6±8.0 ms in right atria (RA) observed in [18], and an APD90 increase of 29.6 ms relative to the control at 2 Hz which was within the experimental range of 36.9±8.4 ms in LA and 36.5±7.9 ms in RA observed in [18] (Fig 1A(ii)). HF resulted in an increased CaT amplitude of 185.4 nM and 138.8 nM relative to the control at 1 Hz and 2 Hz (Fig 1B(ii)). This was in agreement with experimental results at 1 Hz (RA: 180.2±40.2 nM) and 2 Hz (LA: 189.2±60.8 nM, RA: 183.6±56.6 nM) observed in [18]. HF produced an increased NSR Ca2+ content of 111.6% at 1 Hz, which was near the experimental results at 1 Hz (LA: 85%) observed in [18] (Fig 1C(ii)).
Fig 1. Effect of HF-induced electrical remodelling on the APD90, CaT amplitude and SR Ca2+ content.

(Ai), (Bi) and (Ci) Recordings of APs, intracellular Ca2+ concentration ([Ca2+]i) and Ca2+ concentration in NSR ([Ca2+]NSR) under control and HF conditions at 1Hz. (Aii) and (Bii) The increased APD90 (ΔAPD90) and increased CaT amplitude (ΔCaT amplitude) under HF conditions relative to control from experimental data [18] (grey and black) and simulations (while) at 1 Hz and 2Hz. (Cii) The increased SR Ca2+ content (ΔSR Ca2+ content) in HF relative to the control from experimental data [18] (grey and black) and simulations (while) at 1 Hz.
To identify the relative contributions of the 7 identified parameters (Table 1) to the APD90, CaT amplitude and SR Ca2+ content, parameter sensitivity analysis was performed using multivariable regression based on control model at 1 Hz. The results are shown in Fig 2. Sensitivity coefficients represented the quantitative contribution of each parameter. Increasing GCa and Jup(max), and decreasing Gto and Kup had significant contributions to APD90 (Fig 2A), CaT amplitude (Fig 2B) and SR Ca2+ content (Fig 2C), of which GCa had the largest contribution to APD90, and Jup(max) had the largest contribution to CaT amplitude and SR Ca2+ content. Decreasing Jrel(max) had a stronger contribution to SR Ca2+ content, and increasing Jrel(max) had only a limited effect on APD90 and CaT amplitude. There were minimal effects of GKs and [Csqn]max on APD90, CaT amplitude and SR Ca2+content. Furthermore, we varied each parameter by different degrees in the HF model one at a time to identify which parameters were the main determinants of HF-induced increases in APD90, CaT amplitude and SR Ca2+ content (S1 Table). It was shown that Kup remodelling played an important role in modulating electrical action potentials and Ca2+ handling characteristics in HF, as absence of which produced a decrease in APD90 and CaT amplitude, and only a small increase in SR Ca2+ content relative to control. As compared to the condition when all HF-remodelled factors were considered, absence of Jup(max) or GCa remodelling produced a larger increase in APD90, CaT amplitude and SR Ca2+ content; absence of Jrel(max) remodelling produced a smaller increase in SR Ca2+ content, and absence of Gto remodelling produced a smaller increase in APD90 relative to control. These results suggest that HF-induced changes on APD90, CaT amplitude and SR Ca2+ content are a consequence of all remodelling factors, though the individual role of each is different.
Fig 2. Relative contributions of 7 parameters to the APD90, CaT amplitude and SR Ca2+ content using parameter sensitivity analysis at 1 Hz.
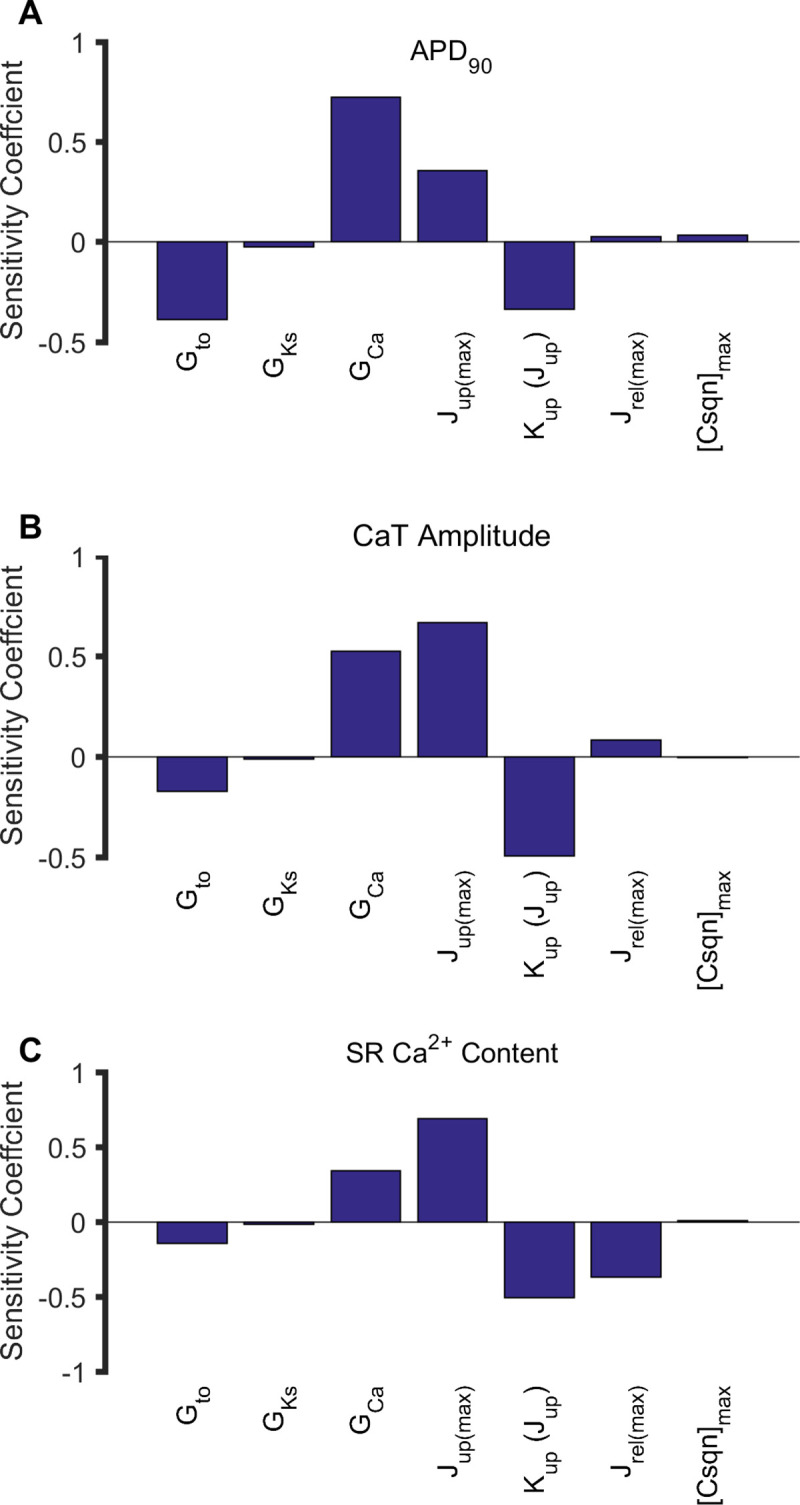
(A) APD90. (B) CaT amplitude. (C) SR Ca2+ content.
APD and CaT alternans in HF
Further simulations were conducted to investigate whether HF-induced atrial ion channel remodelling promotes the susceptibility to alternans. Results are shown in Fig 3 for the rate dependent curves of APD90 (Fig 3A), CaT amplitude (Fig 3B) and CaT decay time (Fig 3C) obtained from two consecutive beats using the dynamic pacing protocol. Alternans in APD, and CaT amplitude and decay time were observed under HF conditions, but not under the control condition. In HF, the three alternans were characterized as Eye-type alternans [56], indicating an on-setting PCL and a terminating PCL. In the three rate dependent curves, the computed on-setting PCL bifurcation point for alternans genesis was 153 ms under the HF condition (seen in orange in Fig 3A–3C). Under the control condition, the rate dependent curve of the CaT decay time was monotonic, by which the CaT decay time decreased as the PCL decreased (shown in blue in Fig 3C); however, the APD90 and CaT amplitude rate dependent curves were biphasic–they first decreased as the PCL decreased from 1000 ms to 180 ms, but then rose at a PCL during 147 ms–180 ms, and decreased again until the PCL reached 100 ms (shown in blue in Fig 3A and 3B). The increased APD90 upon decreasing the PCL from 180 ms was due to the supernormal premature action potential [57]. When the PCL was shorter than 180 ms, the stimulus was applied during the relative refractory period, resulting in a small peak action potential and small fast Na+ current (INa) and ICa that led to small Ito and IKur values which contributed to slowing repolarization to increase the APD90.
Fig 3. Effect of HF-induced electrical remodelling on rate dependent curves and the restitution curves.

(A) APD90 rate dependent curves, (B) CaT amplitude rate dependent curves, and (C) CaT decay time rate dependent curves from two consecutive beats in a quasi-steady state under control (shown in blue) and HF (shown in orange) conditions. (D) APD90 restitution curves at a quasi-steady state under control (blue) and HF (orange) conditions. (E) Slopes and (F) maximum slope of the restitution curves in (D). (ii) Enlarged view of (i) to more clearly reveal the PCL range with alternans.
It has been suggested that the slope of the APD restitution curve exceeding 1 may provide an indicator for the genesis of APD alternans [30–32], though this theory is limited due to short-term cardiac memory and calcium cycling dynamics [33]. In simulations, the standard S1-S2 protocol was also implemented to compute APD90 restitution curves from the dog atrial cell model under control and HF conditions, as well as their maximal slope. Results are shown in Fig 3D–3F. It was shown that the APD90 restitution curves under both control and HF conditions were non-monotonic (Fig 3D), as shown in APD90 dependent curves under the control condition (Fig 3A). The maximum APD90 restitution slope under the control condition was 0.84 (Fig 3E and 3F) which was less than 1; and the maximum slope of APD90 restitution in HF was 3.068 (Fig 3E and 3F), which exceeded 1.
The alternans of AP, Ca2+ transient, JSR Ca2+ content and ion channel currents are shown in Fig 4 at a PCL of 148 ms. Fig 4A–4C demonstrated alternating APs, Ca2+ transients and JSR Ca2+ content from two consecutive beats in the HF simulation whereas no alternans was observed in the control. It was shown that the AP alternans was in phase with the Ca2+ transient alternans and JSR Ca2+ content alternans, manifested by a larger AP amplitude with a longer APD being accompanied by larger CaT amplitude and JSR Ca2+ content; and a smaller AP amplitude with a shorter APD accompanied by smaller CaT amplitude and JSR Ca2+ content.
Fig 4. Traces of AP, Ca2+ transient, JSR Ca2+ concentration and ion channel currents during long-short alternans at a PCL of 148 ms.

(A) AP. (B) [Ca2+]i. (C) JSR Ca2+ concentration ([Ca2+]JSR). (D) INa. (E) ICa. (F) Jrel. (G) Ito. (H) INCX. (I) Jup.
Further sensitivity analysis of HF-induced remodelling was performed to identify the main determinants for alternans genesis in both control and HF models. Firstly, each HF-induced parameter change was incorporated into the control model one at a time (S2 Fig). In each case, no alternans was observed, suggesting that mechanism for HF-induced alternans was a multi-parameter action. Secondly, each HF-remodelled parameter was removed from the HF model one at a time. Fig 5A demonstrated that HF without Kup remodelling or Gto remodelling did not produce alternans, and HF without GCa remodelling produced alternans at longer PCLs. HF without the other 4 remodelled parameters had little effect on alternans (S3 Fig). These results suggested that decreased Kup and Gto were the main determinants underlying the occurrence of alternans, and decreased GCa reduced the susceptibility to alternans. Next, each parameter was scaled by different degrees relative to control to investigate how they influenced the occurrence of alternans (Kup, Gto and GCa in Fig 5B–5D, Jrel(max), GKs, Jup(max) and [Csqn]max in S3A–S3D Fig). For Kup, alternans occurred in HF until it was decreased by 78%, which may be related to increased SR Ca2+ content. As the degree of decreased Gto gradually increased, alternans occurred at longer PCLs, which may be caused by an increased APD. On the contrary, of the more we reduced GCa, the shorter PCLs were when alternans was observed, which may result from decreased APD and CaT amplitude. See Discussion for more details.
Fig 5. Sensitivity analysis of alternans to HF-induced remodelling in HF model.

(A) Each parameter remodelling was removed from the HF model one at a time compared with control and HF models. (B) Kup was reduced from 0% to 90% relative to the control in the HF model. (C) Gto was reduced from 0% to 90% relative to the control in the HF model. (D) GCa was reduced from 0% to 60% relative to the control in the HF model.
Discordant alternans in the 1D simulation
Conduction of the AP and its corresponding Ca2+ transient alternans were also investigated at the 1D tissue level. In simulations, both HF-induced ion channel remodelling and changes in intercellular coupling due to fibrosis (mimicked by a reduced diffusion coefficient, D) were considered. Pacing-induced spatial alternans were studied in the 1D simulation by increasing the pacing rate.
To illustrate the effects of PCLs and cell-to-cell coupling on the genesis of discordant alternans, various AP propagation patterns were investigated in a D-PCL parameter space under control and HF conditions, as shown in Fig 6A. There were three AP propagation patterns: normal conduction, discordant alternans, and 2:1 conduction block in a quasi-steady state. The 2:1 conduction block occurred when every other AP was not conducted along the cable. Fig 6B shows rate dependent curves of CV at the 1D tissue level by implementing three cell-to-cell couplings (D = 0.028 mm2/ms, 0.084 mm2/ms, and 0.167 mm2/ms) under control and HF conditions. The curves were flat at long PCLs and produced CVs of 0.60 m/s with D = 0.167 mm2/ms, 0.4 m/s with D = 0.084 mm2/ms, and 0.2 m/s with D = 0.028 mm2/ms. They decreased at short PCLs, producing reduced CVs and alternating large and small CVs under both HF and control conditions. The alternating CVs in Fig 6B corresponded to discordant alternans with the same PCLs as in Fig 6A. On the one hand, the PCLs in which discordant alternans occurred were longer under HF conditions than the control condition due to HF-induced APD prolongation. On the other hand, the genesis of discordant alternans was PCL-dependent. When the PCLs were decreased to within a particular range with alternating large and small CVs, discordant alternans occurred under both HF and control conditions. In addition, PCLs in which discordant alternans occurred vary over a wider range when D = 0.028 mm2/ms than that when D = 0.084 mm2/ms or 0.167 mm2/ms. These suggest that both HF-induced APD prolongation and decreased CVs caused by decreased PCLs and cell-to-cell coupling contribute to the genesis of discordant alternans at the tissue level.
Fig 6. 1D simulation results under control and HF conditions.

(A) Normal conduction, discordant alternans, and 2:1 conduction block in the D-PCL parameter space under control (i) and HF (ii) conditions. (B) The rate dependent curves of CV under control (i) and HF (ii) conditions, as measured by implementing three-member cell-to-cell coupling (D = 0.167 mm2/ms, 0.084 mm2/ms, and 0.028 mm2/ms). (Bii) is an enlarged view of (Bi) that more clearly reveals the 130 ms to 200 ms PCL range.
Discordant alternans was shown in Fig 7 with D = 0.028 mm2/ms at a PCL of 168 ms under HF conditions. The AP and Ca2+ transient propagation along the cable (vertically from the top 1st cell to the bottom 300th cell) as time increases (horizontally from the left to the right) were colour mapped in Fig 7A(i) and 7B(i), respectively. The APD node, a location between two consecutive beats with same APDs, is an important measurement related to discordant alternans [58]. In Fig 7A(ii), the APD node’s position was indicated by black dashed lines with an arrow, which moved towards the stimulus site (1st cell to 4th cell) as the beat number increased. Along the cable, the alternating areas were out of phase on both sides of the APD node, such as AP traces recorded from the 40th, 100th, and 170th cells in Fig 7A(iii) (marked by black lines in Fig 7A(i)). APs marked by black dashed lines with arrows exhibited long-short-long-short-long alternans at the 40th cell, short-long-short-long-short alternans at the 100th cell, and long-short-long-short-long alternans at the 170th cell, and these represented discordant alternans. Moreover, the AP and Ca2+ transient alternans were in phase (Fig 7A(i) and 7B(i)), with the longer APD90 accompanied by a bigger peak Ca2+ transient, and the shorter APD90 accompanied by a smaller peak Ca2+ transient (Fig 7A(ii) and 7B(ii)). This was consistent with observations at the single-cell level (Fig 4A and 4B). In Fig 7B(ii), the position of the peak Ca2+ transient node (with little variation and almost the same between two consecutive beats) marked by black dashed lines with an arrow was consistent with the APD nodes in Fig 7A(ii), and also moved towards the stimulus site. As with the AP alternans shown in Fig 7A(iii), the Ca2+ transient alternans (marked by black dashed lines with arrows in Fig 7B(iii)) exhibited discordant alternans at the 40th, 100th, and 170th cells. The larger AP activated the Ca2+ transient, but the smaller AP did not as the AP peak potential was insufficient to activate ICa and further activate the Ca2+ transient.
Fig 7. Representative 1D simulation of the discordant alternans with D = 0.028 mm2/ms at a PCL of 168 ms in HF.

(A(i)) Time-space plot of the membrane potential of a homogeneous, 1D cable. (A(ii)) The corresponding APD90 of each beat, in which black dashed lines with arrows indicate the APD node position as the beat number increases. (A(iii)) Time course traces of AP for the 40th, 100th, and 170th cells, which are marked with black lines in (A(i)). (B(i)) Time-space plot of the Ca2+ transient in a homogeneous, 1D cable. (B(ii)) The corresponding peak Ca2+ transient of each beat, in which black dashed lines with arrows indicate the peak Ca2+ transient node position as the beat number increases. (B(iii)) Time course traces of Ca2+ transient for the 40th, 100th, and 170th cells, which are marked with black lines in (B(i)).
Further analyses of the effects of cell-to-cell coupling on the pattern of discordant alternans are shown in Fig 7 with D = 0.028 mm2/ms and S6 Fig with D = 0.167 mm2/ms at a PCL = 168 ms under HF conditions. When D = 0.167 mm2/ms, there was one APD node positioned in a relatively stable manner near the stimulus site (marked with a black arrow in S6A(ii) Fig). However, the number of APD nodes increased when D was reduced to 0.028 mm2/ms, and the nodes moved towards the stimulus site with time (Fig 7B(ii)), producing a profound functional spatial heterogeneity. This suggests that cell-to-cell coupling plays an important role in determining the APD node position and number at the 1D tissue level.
Moreover, the pattern of discordant alternans in Fig 7 and S6 Fig can be explained by the evolution of spatial heterogeneity, which are illustrated via the spatial distributions of APD90 and DI in Fig 8 and the spatial distribution of the CV in S7 Fig in HF conditions. When D = 0.028 mm2/ms, the discordant alternans had two characteristics of nodes moving towards the stimulus site with more nodes forming with continued pacing. This could be interpreted as a larger APD and DI spatial dispersion caused by a slower CV. Reduced PCL and cell-to-cell coupling contributed to decreased CV. For the first two consecutive stimuli (beats 15 and 16 when D = 0.167 mm2/ms and beats 11 and 12 when D = 0.028 mm2/ms), the DI node occurred with no APD or CV node. This DI node then produced APD and CV nodes during the second and third stimuli. As the beat number increased, the spatial dispersions of DI and APD90 were enhanced for two consecutive beats. A shorter distance was required for the DI and APD90 distribution curves to intersect and produce DI and APD nodes. Thus, the nodes gradually moved towards the stimulus site with continued pacing. When D = 0.167 mm2/ms with a CV of ≈ 0.53 m/s at the CV nodes (S7A Fig), the APD90 and DI nodes finally docked at a nearby stimulus site (Fig 8A). When D = 0.028 mm2/ms with a CV of 0.185 m/s–0.195 m/s at the CV nodes (S7B Fig), the APD and DI dispersion were larger, causing the nodes to move faster (Fig 8B). Additionally, because of small drive force for AP propagation under conditions of weak cell-to-cell coupling, the stability of alternating tissue near the stimulus site could be destroyed, producing nodes disappearing at the stimulus site. Therefore, steeper DI and APD curve slopes promoted faster node movement when the beat number increased, resulting in more node formation.
Fig 8. Effect of cell-to-cell coupling on the node position and number with PCL = 168 ms under the HF condition.

(A) Results with D = 0.167 mm2/ms. (B) Results with D = 0.028 mm2/ms. (i) APD90 and (ii) DI spatial distribution for consecutive beats.
Discussion
In this study, the effects of HF-induced atrial remodelling with prolonged APD and decreased cell-to-cell coupling on atrial alternans were investigated in silico using the updated canine atrial cell model developed by Ramirez et al. [41] at the single cell and 1D tissue level. The simulation results at the single cell level illustrate the effects of HF-induced atrial electrical remodelling (decreased ICa, Ito, IKs, SRECA2a and RyR2 protein expression, total Csqn, and increased PLB phosphorylation) on the AP and Ca2+ transient alternans. Further simulation results at the 1D tissue level demonstrate the effects of HF-induced atrial electrical and structural remodelling (reduced cell-to-cell coupling caused by atrial fibrosis) on spatial alternans. Our major findings follow: (i) The increased SR Ca2+ content caused by increased Ca2+ uptake by SERCA and decreased RyR2 protein expression contributes to the increased Ca2+ transient amplitude, leading to an increase in inward INCX. This combined with the decreased Ito and IKs results in APD prolongation even though with reduced ICa in HF simulations. (ii) The AP and Ca2+ transient alternans can be induced in HF simulations, but could not in control simulations, suggesting that HF-induced atrial electrical remodelling can increase the atrial susceptibility to alternans. HF-induced increased PLB phosphorylation and decreased Ito are the main determinants underlying the occurrence of alternans through [Ca2+]i→AP coupling and AP→[Ca2+]i coupling, respectively. Failure of the increased SR Ca2+ content and prolonged AP recovery at the end of diastole underlies the genesis of AP and Ca2+ transient alternans in HF. (iii) Cell-to-cell coupling plays an important role in discordant alternans patterns. Reduced cell-to-cell coupling caused by HF-induced atrial fibrosis results in a decreased CV and can enhance spatial dispersion of AP propagation to produce discordant alternans. Together with HF-induced APD prolongation, the enhanced spatial dispersion of AP propagation increases susceptibility to spatial discordant alternans in atrial tissue.
Model development and relative contribution of parameters in HF simulations
In this study, the effects of HF-induced atrial electrical remodelling were incorporated into the canine atrial cell model. Experimental studies have reported reduced atrial ICa densities in sheep [11], rats [59], dogs [10, 19,40], and humans [17,60,61] with HF. In previous experimental studies, the observations of HF-induced atrial K+ current remodelling were not consistent, such as whether Ito was increased or reduced [10,12,19,40] and whether IKur and IK1 were unchanged or reduced [10,12,19,40]. In contrast to the inconsistency in Ito, IKur and IK1 remodelling, IKs has generally been found to decrease in HF [10,19,40] and IKr has been reported as unchanged [19]. The changes of INCX in HF atria were also inconsistent. Li et al. [19] and Cha et al. [40,62] demonstrated increased transient inward INCX in HF dogs. However, Yeh et al. [18] reported no significant changes in NCX1 in control and HF dogs. Given that the increased Ca2+ transient peak observed in HF atria can increase the transient inward INCX, INCX is likely to be unchanged in HF.
Atrial Ca2+ handling abnormalities were also reported in HF, with an increased atrial SR Ca2+ content being typically observed in the atria of larger animals and humans [11,17,18,63]. The increased SR Ca2+ load could be explained with increased SERCA Ca2+ reuptake modulated by increased PLB phosphorylation via CaMKII at Thr17 [11,18] or decreased sarcolipin [17,64] in atria with HF, even though the SERCA2a and PLB expressions were unaltered or decreased [11, 17,18,64]. In addition, RyR protein expression and RyR phosphorylation were found to be either unchanged or decreased in large animals and humans with HF [11,17,18,64].
As the computational model used was a canine model, this study focused on reported observations in HF dogs with prolonged APD. The ICa, Ito, IKs, SERCA2a and RyR2 protein expressions, total Csqn and Kup were decreased in HF simulations, producing a prolonged APD (S1A Fig) and an increased Ca2+ transient amplitude (S1B Fig) and SR Ca2+ content (S1D Fig), which are in agreement with experimental results [18]. These are attributable to the integral action of HF-induced remodelling. In spite of a decrease in SERCA2a protein expression, SERCA Ca2+ reuptake into the SR was increased (S1H Fig) due to the increased PLB phosphorylation (by decreasing Kup) by CaMKII [18]. This, combined with the decreased RyR2 protein expression and total Csqn, contributed to the increased SR Ca2+ content. Even though ICa and RyR2 protein expressions were decreased, the SR Ca2+ release (S1F Fig) and CaT amplitude was increased, caused by increased SR Ca2+ content. Although it was expected that a decrease in ICa in HF (S1G Fig) might lead to APD shortening, decreases in Ito and IKs (S1C and S1E Fig) may counteract such an effect. In addition, an increase in CaT amplitude augmented inward INCX and produced APD prolongation, which is consistent with the observations of increased transient inward INCX in previous experimental studies [19,40].
Mechanism of HF-induced alternans at the single cell level
Previous study in the ventricle has showed that the HF-enhanced genesis of cardiac alternans has been found to be closely related to impairment of Ca2+ cycling [65]. In the atria, HF-induced Ca2+ handling abnormalities may be related to atrial alternans, including a decrease in ICa and an increase in SR Ca2+ content [11,17,18,34–36,63]. In this study, AP, Ca2+ transient and JSR Ca2+ content alternans were observed in HF simulations, but no alternans were observed in control simulations (Figs 3 and 4), suggesting that HF-induced remodelling enhances susceptibility to cardiac alternans. While numerous clinical and animal experiments and simulations on ventricles have explored the underlying mechanism of alternans [31,34,66–68], there is still controversy whether abnormalities in AP or Ca2+ handling is the predominant factor [39]. The results in this study suggest that Kup and Gto remodelling are the main determinants underlying the occurrence of atrial alternans. This demonstrates that both abnormalities of AP and Ca2+ handling may play important role in alternans.
HF-induced an increase in PLB phosphorylation was simulated by decreasing Kup ([Ca2+]i half-saturation constant for Ca2+ uptake into the network SR), and the underlying mechanism of alternans could be supported by [Ca2+]i→AP coupling in HF. Decreasing Kup resulted in an increase in SERCA Ca2+ reuptake (S4A Fig), leading to increasing the SR Ca2+ content (S4B Fig). Increases in the SR Ca2+ content produced larger SR Ca2+ release (S4C Fig) and Ca2+ transients (S4D Fig). When the PCL was decreased to 148 ms, SR Ca2+ concentrations could not be restored to the level at the end of diastole that led to a smaller SR Ca2+ release and Ca2+ transient in the next beat. This caused beat-to-beat alternation in the intracellular Ca2+ transient (S4D Fig), and therefore induced INCX alternans (S4E Fig). Therefore, in two consecutive pacing cycles, a larger inward INCX contributed to slower repolarization in one beat while a smaller inward INCX contributed to faster repolarization in the following beat, forming the alternans of AP repolarization (APD alternans) as shown in S4F Fig. Beat-to-beat alternation in APD alternans caused beat-to-beat alternation in the INa (S4G Fig) and ICa (S4H Fig), as the AP could not be restored to basal level at the end of diastole when the APD was longer. Further, the alternans of ICa acted as a positive feedback on the alternans of the Ca2+ transient amplitude via Ca2+-induced Ca2+ release from SR.
Another AP→[Ca2+]i coupling can explain the underlying mechanism by which HF-induced decreases in Ito (simulated by decreasing Gto) increases the susceptibility to alternans in HF. Decreasing Gto (S5A Fig) resulted in an increase in the APD (S5B Fig). When the PCL was decreased to 148 ms, the AP could not be restored to the level at the end of diastole that led to a smaller AP in the next beat, forming the beat-to-beat alternation in AP and APD. AP alternans caused alternans of the INa (S5C Fig) and ICa (S5D Fig). ICa alternans was then responsible for alternans of both SR Ca2+ release (S5E Fig) and Ca2+ transients (S5F Fig). Further, the plateau phase of the AP was elevated and prolonged by reducing Gto, which led to increased transmembrane Ca2+ influx through ICa (S5D Fig) resulting in SR Ca2+ accumulation (S5G Fig), in agreement with experimental research [19]. The increased SR Ca2+ content also played a key role in alternans of SR Ca2+ concentrations and Ca2+ transients as described above. Moreover, the alternans of Ca2+ transient caused alternans of INCX (S5H Fig) which in turn affected APD alternans.
Mechanism of HF-induced discordant alternans at the cable tissue level
Spatially discordant alternans in atria underlies the development of atrial arrhythmia [25,39,69] and is characterized by opposite alternating phases at different areas of tissue. In this study, we demonstrated the effects of the PCL and cell-to-cell coupling on spatial discordant alternans in control and HF conditions. Only discordant alternans were observed in this study. When the PCLs was decreased to a certain range, discordant alternans occurred with different cell-to-cell coupling (D = 0.167 mm2/ms, 0.084 mm2/ms, and 0.028 mm2/ms) in both control and HF conditions (Fig 8). Compared with strong cell-to-cell coupling (D = 0.167 mm2/ms), additional APD nodes were produced under conditions with weak cell-to-cell coupling (D = 0.028 mm2/ms), and the range of PCLs at which discordant alternans occurred was longer especially in HF conditions (Figs 6 and 7, and S6 Fig).
Patterns of discordant alternans vary with the strength of cell-to-cell coupling. (i) The DI and APD nodes moved towards the stimulus site as pacing continued (Fig 8), which is consistent with observations by Watanabe et al. [58]. With strong cell-to-cell coupling, the DI and APD nodes finally docked at a site near the stimulus site (Fig 8A). However, these nodes moved much faster before disappearing at stimulus sites with weak cell-to-cell coupling (Fig 8B). Alternans occurred at the stimulus site and propagated along the cable. With strong cell-to-cell coupling, the driving force for AP propagation was larger and created relatively stable in-phase alternans near the stimulation site (Fig 8A). Even if the DI and APD nodes move towards the stimulation site, they are unable to destroy the stability near the stimulation site. In contrast, if the cell-to-cell coupling is weak and the driving force is small, stability near the stimulation site can be destroyed where DI and APD nodes move to the stimulus site and will disappear (Fig 8B). (ii) The number of DI and APD nodes increased when cell-to-cell coupling was weak (Fig 8B). This can be interpreted as reduced intercellular coupling enhancing the dispersion of AP distribution between adjacent myocytes [39], producing steeper DI and APD distribution curves. This promotes faster node movement as the beat number increases, resulting in more node formation. Furthermore, experimental results from rabbit heats demonstrated that the gap junction modifier rotigaptide could reduce the repolarization heterogeneity and suppress the discordant alternans [70]. This adds evidence of the significant role of cell-to-cell coupling in the genesis of discordant alternans, which contributes to increased susceptibility to arrhythmias.
Limitations
HF-induced atrial electrical remodelling varies with various stages and complications of cardiac disease [8,9] and various experimental techniques and conditions [6]. Simulation in this study focused on HF with prolonged APD, which was observed in dog atria. Further research should be undertaken to investigate whether HF with unaltered or shortened APD also makes a contribution to atrial arrhythmia. The canine atrial cell model developed by Ramirez et al. [41] did not incorporate the CaMKII regulatory pathway. For this reason, Kup was decreased to simulate the increased Ca2+ reuptake by SECRA which is modulated by increased PLB phosphorylation due to CaMKII as in [43]. Though some preliminary studies have been done here (S1 File), but detailed studies warrant to be conducted in the future to update the canine atrial cell model with more detailed calcium handling and CaMKII regulatory pathway based on atrial experimental data.
For the 1D tissue model, we only considered the homogeneous tissue and did not consider heterogeneous tissue. Cell-to-cell coupling disrupted by HF-induced fibrosis was also only included in the 1D tissue simulation. This warrants further studies by incorporating the fibroblast model coupled with atrial myocytes and the arrangement of fibroblasts in atrial tissue. Furthermore, the 1D tissue model should be extended to 2D or 3D tissue model to explore the transition from discordant alternans to re-entry in HF.
Despite these limitations, the canine HF model we developed in the present study can simulate experimental observations in cellular electrical activities in HF dogs, such as the prolonged APD. The developed model may be incorporated into a computational platform in future for further investigations of pro-arrhythmic effects of HF in the atria, particularly with respect to atrial alternans, results of which may provide new insights into the clinical treatment of AF with HF.
Supporting information
(A) AP. (B) [Ca2+]i. (C) Ito. (D) Junctional SR Ca2+ concentration ([Ca2+]JSR). (E) IKs. (F) Jrel. (G) ICa. (H) Jup. (I) INCX.
(TIF)
Each parameter remodelling induced by HF was incorporated into the control model at a time.
(TIF)
(A) Jrel(max), (B) GKs, (C) Jup(max), and (D) [Csqn]max were reduced from 0% to 90% relative to the control in the HF model.
(TIF)
(A) Jup. (B) [Ca2+]JSR. (C) Jrel. (D) [Ca2+]i. (E) INCX. (F) AP. (G) INa. (H) ICa.
(TIF)
(A) Ito. (B) AP. (C) INa. (D) ICa. (E) Jrel. (F) [Ca2+]i. (G) [Ca2+]JSR. (H) INCX.
(TIF)
(A(i)) Time-space plot of the membrane potential of a homogeneous, 1D cable. (A(ii)) The corresponding APD90 of each beat, in which black dashed lines with arrows indicate the APD node position as the beat number increases. (B(i)) Time-space plot of the Ca2+ transient in a homogeneous, 1D cable. (B(ii)) The corresponding peak Ca2+ transient of each beat, in which black dashed lines with arrows indicate the peak Ca2+ transient node position as the beat number increases.
(TIF)
(A) Results with D = 0.167 mm2/ms. (B) Results with D = 0.028 mm2/ms.
(TIF)
(A) AP. (B) [Ca2+]i. (C) INCX. (D) [Ca2+]JSR. (E) Jrel. (F) Jup.
(TIF)
The values of APD90, CaT amplitude, and SR Ca2+ content were computed relative to control, and the bold font demonstrated the HF model.
(PDF)
(DOCX)
Data Availability
All relevant data are within the manuscript and its Supporting Information files.
Funding Statement
The work was supported by the National Natural Science Foundation of China (NSFC) under Grant Nos. 61572152 (to HZ), 61601143 (to QL), 61571165 (to KW), and 81770328 (to QL), and the Heilongjiang and China Postdoctoral Science Foundation under Grant No. 2015M581448 (to QL). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Verma A, Kalman JM, Callans DJ. Treatment of Patients With Atrial Fibrillation and Heart Failure With Reduced Ejection Fraction. Circulation. 2017;135(16):1547–63. 10.1161/CIRCULATIONAHA.116.026054 [DOI] [PubMed] [Google Scholar]
- 2.Santhanakrishnan R, Wang N, Larson MG, Magnani JW, McManus DD, Lubitz SA, et al. Atrial Fibrillation Begets Heart Failure and Vice Versa: Temporal Associations and Differences in Preserved Versus Reduced Ejection Fraction. Circulation. 2016;133(5):484–92. 10.1161/CIRCULATIONAHA.115.018614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ling LH, Kistler PM, Kalman JM, Schilling RJ, Hunter RJ. Comorbidity of atrial fibrillation and heart failure. Nat Rev Cardiol. 2016;13(3):131–47. 10.1038/nrcardio.2015.191 [DOI] [PubMed] [Google Scholar]
- 4.Kotecha D, Lam CS, Van Veldhuisen DJ, Van Gelder IC, Voors AA, Rienstra M. Heart Failure With Preserved Ejection Fraction and Atrial Fibrillation: Vicious Twins. J Am Coll Cardiol. 2016;68(20):2217–28. 10.1016/j.jacc.2016.08.048 [DOI] [PubMed] [Google Scholar]
- 5.Patel RB, Vaduganathan M, Shah SJ, Butler J. Atrial fibrillation in heart failure with preserved ejection fraction: Insights into mechanisms and therapeutics. Pharmacol Ther. 2017;176:32–9. 10.1016/j.pharmthera.2016.10.019 [DOI] [PubMed] [Google Scholar]
- 6.Denham NC, Pearman CM, Caldwell JL, Madders GWP, Eisner DA, Trafford AW, et al. Calcium in the Pathophysiology of Atrial Fibrillation and Heart Failure. Front Physiol. 2018;9:1380 10.3389/fphys.2018.01380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kotecha D, Piccini JP. Atrial fibrillation in heart failure: what should we do? Eur Heart J. 2015;36(46):3250–7. 10.1093/eurheartj/ehv513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation. 2019;139(10):e56–e528. 10.1161/CIR.0000000000000659 [DOI] [PubMed] [Google Scholar]
- 9.Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr., Colvin MM, et al. 2017 ACC/AHA/HFSA Focused Update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Circulation. 2017;136(6):e137–e61. 10.1161/CIR.0000000000000509 [DOI] [PubMed] [Google Scholar]
- 10.Sridhar A, Nishijima Y, Terentyev D, Khan M, Terentyeva R, Hamlin RL, et al. Chronic heart failure and the substrate for atrial fibrillation. Cardiovasc Res. 2009;84(2):227–36. 10.1093/cvr/cvp216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clarke JD, Caldwell JL, Horn MA, Bode EF, Richards MA, Hall MC, et al. Perturbed atrial calcium handling in an ovine model of heart failure: potential roles for reductions in the L-type calcium current. J Mol Cell Cardiol. 2015;79:169–79. 10.1016/j.yjmcc.2014.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Workman AJ, Pau D, Redpath CJ, Marshall GE, Russell JA, Norrie J, et al. Atrial cellular electrophysiological changes in patients with ventricular dysfunction may predispose to AF. Heart Rhythm. 2009;6(4):445–51. 10.1016/j.hrthm.2008.12.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iwasaki YK, Nishida K, Kato T, Nattel S. Atrial fibrillation pathophysiology: implications for management. Circulation. 2011;124(20):2264–74. 10.1161/CIRCULATIONAHA.111.019893 [DOI] [PubMed] [Google Scholar]
- 14.Dobrev D, Carlsson L, Nattel S. Novel molecular targets for atrial fibrillation therapy. Nat Rev Drug Discov. 2012;11(4):275–91. 10.1038/nrd3682 [DOI] [PubMed] [Google Scholar]
- 15.Nattel S, Harada M. Atrial remodeling and atrial fibrillation: recent advances and translational perspectives. J Am Coll Cardiol. 2014;63(22):2335–45. 10.1016/j.jacc.2014.02.555 [DOI] [PubMed] [Google Scholar]
- 16.Nattel S, Xiong F, Aguilar M. Demystifying rotors and their place in clinical translation of atrial fibrillation mechanisms. Nat Rev Cardiol. 2017;14(9):509–20. 10.1038/nrcardio.2017.37 [DOI] [PubMed] [Google Scholar]
- 17.Molina CE, Abu-Taha IH, Wang Q, Rosello-Diez E, Kamler M, Nattel S, et al. Profibrotic, Electrical, and Calcium-Handling Remodeling of the Atria in Heart Failure Patients With and Without Atrial Fibrillation. Front Physiol. 2018;9:1383 10.3389/fphys.2018.01383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yeh YH, Wakili R, Qi XY, Chartier D, Boknik P, Kaab S, et al. Calcium-handling abnormalities underlying atrial arrhythmogenesis and contractile dysfunction in dogs with congestive heart failure. Circ Arrhythm Electrophysiol. 2008;1(2):93–102. 10.1161/CIRCEP.107.754788 [DOI] [PubMed] [Google Scholar]
- 19.Li D, Melnyk P, Feng J, Wang Z, Petrecca K, Shrier A, et al. Effects of experimental heart failure on atrial cellular and ionic electrophysiology. Circulation. 2000;101(22):2631–8. 10.1161/01.cir.101.22.2631 [DOI] [PubMed] [Google Scholar]
- 20.Koumi S, Arentzen CE, Backer CL, Wasserstrom JA. Alterations in muscarinic K+ channel response to acetylcholine and to G protein-mediated activation in atrial myocytes isolated from failing human hearts. Circulation. 1994;90(5):2213–24. 10.1161/01.cir.90.5.2213 [DOI] [PubMed] [Google Scholar]
- 21.Fedorov VV, Glukhov AV, Ambrosi CM, Kostecki G, Chang R, Janks D, et al. Effects of KATP channel openers diazoxide and pinacidil in coronary-perfused atria and ventricles from failing and non-failing human hearts. J Mol Cell Cardiol. 2011;51(2):215–25. 10.1016/j.yjmcc.2011.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanders P, Morton JB, Davidson NC, Spence SJ, Vohra JK, Sparks PB, et al. Electrical remodeling of the atria in congestive heart failure: electrophysiological and electroanatomic mapping in humans. Circulation. 2003;108(12):1461–8. 10.1161/01.CIR.0000090688.49283.67 [DOI] [PubMed] [Google Scholar]
- 23.Kanaporis G, Blatter LA. Membrane potential determines calcium alternans through modulation of SR Ca(2+) load and L-type Ca(2+) current. J Mol Cell Cardiol. 2017;105:49–58. 10.1016/j.yjmcc.2017.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Narayan SM, Franz MR, Clopton P, Pruvot EJ, Krummen DE. Repolarization alternans reveals vulnerability to human atrial fibrillation. Circulation. 2011;123(25):2922–30. 10.1161/CIRCULATIONAHA.110.977827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hiromoto K, Shimizu H, Furukawa Y, Kanemori T, Mine T, Masuyama T, et al. Discordant repolarization alternans-induced atrial fibrillation is suppressed by verapamil. Circ J. 2005;69(11):1368–73. 10.1253/circj.69.1368 [DOI] [PubMed] [Google Scholar]
- 26.Tsai CT, Chiang FT, Tseng CD, Yu CC, Wang YC, Lai LP, et al. Mechanical stretch of atrial myocyte monolayer decreases sarcoplasmic reticulum calcium adenosine triphosphatase expression and increases susceptibility to repolarization alternans. J Am Coll Cardiol. 2011;58(20):2106–15. 10.1016/j.jacc.2011.07.039 [DOI] [PubMed] [Google Scholar]
- 27.Kettlewell S, Burton FL, Smith GL, Workman AJ. Chronic myocardial infarction promotes atrial action potential alternans, afterdepolarizations, and fibrillation. Cardiovasc Res. 2013;99(1):215–24. 10.1093/cvr/cvt087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pluteanu F, Hess J, Plackic J, Nikonova Y, Preisenberger J, Bukowska A, et al. Early subcellular Ca2+ remodelling and increased propensity for Ca2+ alternans in left atrial myocytes from hypertensive rats. Cardiovasc Res. 2015;106(1):87–97. 10.1093/cvr/cvv045 [DOI] [PubMed] [Google Scholar]
- 29.Pluteanu F, Nikonova Y, Holzapfel A, Herzog B, Scherer A, Preisenberger J, et al. Progressive impairment of atrial myocyte function during left ventricular hypertrophy and heart failure. J Mol Cell Cardiol. 2018;114:253–63. 10.1016/j.yjmcc.2017.11.020 [DOI] [PubMed] [Google Scholar]
- 30.Qu Z, Garfinkel A, Chen PS, Weiss JN. Mechanisms of discordant alternans and induction of reentry in simulated cardiac tissue. Circulation. 2000;102(14):1664–70. 10.1161/01.cir.102.14.1664 [DOI] [PubMed] [Google Scholar]
- 31.Goldhaber JI, Xie LH, Duong T, Motter C, Khuu K, Weiss JN. Action potential duration restitution and alternans in rabbit ventricular myocytes: the key role of intracellular calcium cycling. Circ Res. 2005;96(4):459–66. 10.1161/01.RES.0000156891.66893.83 [DOI] [PubMed] [Google Scholar]
- 32.Koller ML, Maier SK, Gelzer AR, Bauer WR, Meesmann M, Gilmour RF Jr., Altered dynamics of action potential restitution and alternans in humans with structural heart disease. Circulation. 2005;112(11):1542–8. 10.1161/CIRCULATIONAHA.104.502831 [DOI] [PubMed] [Google Scholar]
- 33.Weiss JN, Karma A, Shiferaw Y, Chen PS, Garfinkel A, Qu Z. From pulsus to pulseless: the saga of cardiac alternans. Circ Res. 2006;98(10):1244–53. 10.1161/01.RES.0000224540.97431.f0 [DOI] [PubMed] [Google Scholar]
- 34.Diaz ME, O'Neill SC, Eisner DA. Sarcoplasmic reticulum calcium content fluctuation is the key to cardiac alternans. Circ Res. 2004;94(5):650–6. 10.1161/01.RES.0000119923.64774.72 [DOI] [PubMed] [Google Scholar]
- 35.Nivala M, Qu Z. Calcium alternans in a couplon network model of ventricular myocytes: role of sarcoplasmic reticulum load. Am J Physiol Heart Circ Physiol. 2012;303(3):H341–52. E 10.1152/ajpheart.00302.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rovetti R, Cui X, Garfinkel A, Weiss JN, Qu Z. Spark-induced sparks as a mechanism of intracellular calcium alternans in cardiac myocytes. Circ Res. 2010;106(10):1582–91. 10.1161/CIRCRESAHA.109.213975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dibb KM, Clarke JD, Horn MA, Richards MA, Graham HK, Eisner DA, et al. Characterization of an extensive transverse tubular network in sheep atrial myocytes and its depletion in heart failure. Circ Heart Fail. 2009;2(5):482–9. Epub 2009/10/08. 10.1161/CIRCHEARTFAILURE.109.852228 [DOI] [PubMed] [Google Scholar]
- 38.Li Q, O'Neill SC, Tao T, Li Y, Eisner D, Zhang H. Mechanisms by which cytoplasmic calcium wave propagation and alternans are generated in cardiac atrial myocytes lacking T-tubules-insights from a simulation study. Biophys J. 2012;102(7):1471–82. 10.1016/j.bpj.2012.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kanaporis G, Blatter LA. Alternans in atria: Mechanisms and clinical relevance. Medicina (Kaunas). 2017;53(3):139–49. 10.1016/j.medici.2017.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cha TJ, Ehrlich JR, Zhang L, Nattel S. Atrial ionic remodeling induced by atrial tachycardia in the presence of congestive heart failure. Circulation. 2004;110(12):1520–6. 10.1161/01.CIR.0000142052.03565.87 [DOI] [PubMed] [Google Scholar]
- 41.Ramirez RJ, Nattel S, Courtemanche M. Mathematical analysis of canine atrial action potentials: rate, regional factors, and electrical remodeling. Am J Physiol Heart Circ Physiol. 2000;279(4):H1767–85. 10.1152/ajpheart.2000.279.4.H1767 [DOI] [PubMed] [Google Scholar]
- 42.Weber CR, Ginsburg KS, Philipson KD, Shannon TR, Bers DM. Allosteric regulation of Na/Ca exchange current by cytosolic Ca in intact cardiac myocytes. J Gen Physiol. 2001;117(2):119–31. 10.1085/jgp.117.2.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Voigt N, Heijman J, Wang Q, Chiang DY, Li N, Karck M, et al. Cellular and molecular mechanisms of atrial arrhythmogenesis in patients with paroxysmal atrial fibrillation. Circulation. 2014;129(2):145–56. 10.1161/CIRCULATIONAHA.113.006641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sobie EA. Parameter sensitivity analysis in electrophysiological models using multivariable regression. Biophys J. 2009;96(4):1264–74. 10.1016/j.bpj.2008.10.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Devenyi RA, Sobie EA. There and back again: Iterating between population-based modeling and experiments reveals surprising regulation of calcium transients in rat cardiac myocytes. J Mol Cell Cardiol. 2016; 96:38–48. 10.1016/j.yjmcc.2015.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Onal B, Gratz D, Hund TJ. Ca2+/calmodulin-dependent kinase II-dependent regulation of atrial myocyte late Na+ current, Ca2+ cycling, and excitability: a mathematical modeling study. Am J Physiol Heart Circ Physiol. 2017;313(6):H1227–H1239. 10.1152/ajpheart.00185.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Clayton RH, Bernus O, Cherry EM, Dierckx H, Fenton FH, Mirabella L, et al. Models of cardiac tissue electrophysiology: progress, challenges and open questions. Prog Biophys Mol Biol. 2011;104(1–3):22–48. 10.1016/j.pbiomolbio.2010.05.008 [DOI] [PubMed] [Google Scholar]
- 48.Kneller J, Zou R, Vigmond EJ, Wang Z, Leon LJ, Nattel S. Cholinergic atrial fibrillation in a computer model of a two-dimensional sheet of canine atrial cells with realistic ionic properties. Circ Res. 2002;90(9):E73–87. 10.1161/01.res.0000019783.88094.ba [DOI] [PubMed] [Google Scholar]
- 49.Zou R, Kneller J, Leon LJ, Nattel S. Development of a computer algorithm for the detection of phase singularities and initial application to analyze simulations of atrial fibrillation. Chaos. 2002;12(3):764–78. 10.1063/1.1497505 [DOI] [PubMed] [Google Scholar]
- 50.Aguilar-Shardonofsky M, Vigmond EJ, Nattel S, Comtois P. In silico optimization of atrial fibrillation-selective sodium channel blocker pharmacodynamics. Biophys J. 2012;102(5):951–60. 10.1016/j.bpj.2012.01.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matene E, Vinet A, Jacquemet V. Dynamics of atrial arrhythmias modulated by time-dependent acetylcholine concentration: a simulation study. Europace. 2014;16 Suppl 4:iv11–iv20. 10.1093/europace/euu255 [DOI] [PubMed] [Google Scholar]
- 52.Jacquemet V, Henriquez CS. Genesis of complex fractionated atrial electrograms in zones of slow conduction: a computer model of microfibrosis. Heart Rhythm. 2009;6(6):803–10. 10.1016/j.hrthm.2009.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang W, Zhang S, Ni H, Garratt CJ, Boyett MR, Hancox JC, et al. Mechanistic insight into spontaneous transition from cellular alternans to arrhythmia-A simulation study. PLoS Comput Biol. 2018;14(11):e1006594 10.1371/journal.pcbi.1006594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Koller ML, Riccio ML, Gilmour RF Jr. Dynamic restitution of action potential duration during electrical alternans and ventricular fibrillation. Am J Physiol. 1998;275(5):H1635–42. Epub 1998/11/14. 10.1152/ajpheart.1998.275.5.H1635 [DOI] [PubMed] [Google Scholar]
- 55.Zhao N, Li Q, Wang K, He R, Yuan Y, Zhang H. Effect of Blocking IKur on the Genesis of Action Potential Alternans in Canine Atrium. 2019. Computing in Cardiology (CinC). 10.23919/CinC49843.2019.9005821 [DOI] [Google Scholar]
- 56.Zhou X, Bueno-Orovio A, Orini M, Hanson B, Hayward M, Taggart P, et al. In Vivo and In Silico Investigation Into Mechanisms of Frequency Dependence of Repolarization Alternans in Human Ventricular Cardiomyocytes. Circ Res. 2016;118(2):266–78. 10.1161/CIRCRESAHA.115.307836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Franz MR, Schaefer J, Schottler M, Seed WA, Noble MI. Electrical and mechanical restitution of the human heart at different rates of stimulation. Circ Res. 1983;53(6):815–22. 10.1161/01.res.53.6.815 [DOI] [PubMed] [Google Scholar]
- 58.Watanabe MA, Fenton FH, Evans SJ, Hastings HM, Karma A. Mechanisms for discordant alternans. J Cardiovasc Electrophysiol. 2001;12(2):196–206. 10.1046/j.1540-8167.2001.00196.x [DOI] [PubMed] [Google Scholar]
- 59.Boixel C, Gonzalez W, Louedec L, Hatem SN. Mechanisms of L-type Ca(2+) current downregulation in rat atrial myocytes during heart failure. Circ Res. 2001;89(7):607–13. 10.1161/hh1901.096702 [DOI] [PubMed] [Google Scholar]
- 60.Dinanian S, Boixel C, Juin C, Hulot JS, Coulombe A, Rucker-Martin C, et al. Downregulation of the calcium current in human right atrial myocytes from patients in sinus rhythm but with a high risk of atrial fibrillation. Eur Heart J. 2008;29(9):1190–7. 10.1093/eurheartj/ehn140 [DOI] [PubMed] [Google Scholar]
- 61.Ouadid H, Albat B, Nargeot J. Calcium currents in diseased human cardiac cells. J Cardiovasc Pharmacol. 1995;25(2):282–91. 10.1097/00005344-199502000-00014 [DOI] [PubMed] [Google Scholar]
- 62.Cha TJ, Ehrlich JR, Zhang L, Shi YF, Tardif JC, Leung TK, et al. Dissociation between ionic remodeling and ability to sustain atrial fibrillation during recovery from experimental congestive heart failure. Circulation. 2004;109(3):412–8. 10.1161/01.CIR.0000109501.47603.0C [DOI] [PubMed] [Google Scholar]
- 63.Aistrup GL, Arora R, Grubb S, Yoo S, Toren B, Kumar M, et al. Triggered intracellular calcium waves in dog and human left atrial myocytes from normal and failing hearts. Cardiovasc Res. 2017;113(13):1688–99. 10.1093/cvr/cvx167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shanmugam M, Molina CE, Gao S, Severac-Bastide R, Fischmeister R, Babu GJ. Decreased sarcolipin protein expression and enhanced sarco(endo)plasmic reticulum Ca2+ uptake in human atrial fibrillation. Biochem Biophys Res Commun. 2011;410(1):97–101. 10.1016/j.bbrc.2011.05.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wilson LD, Jeyaraj D, Wan X, Hoeker GS, Said TH, Gittinger M, et al. Heart failure enhances susceptibility to arrhythmogenic cardiac alternans. Heart Rhythm. 2009;6(2):251–9. 10.1016/j.hrthm.2008.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jordan PN, Christini D. Characterizing the contribution of voltage- and calcium-dependent coupling to action potential stability: implications for repolarization alternans. Am J Physiol Heart Circ Physiol. 2007. 293(4):H2109–18. 10.1152/ajpheart.00609.2007 [DOI] [PubMed] [Google Scholar]
- 67.Jordan PN, Christini DJ. Action potential morphology influences intracellular calcium handling stability and the occurrence of alternans. Biophys J 2006;90:672–80. 10.1529/biophysj.105.071340 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shkryl VM, Maxwell JT, Domeier TL, Blatter LA. Refractoriness of sarcoplasmic reticulum Ca2+ release determines Ca2+ alternans in atrial myocytes. Am J Physiol Heart Circ Physiol 2012;302:H2310–2. 10.1152/ajpheart.00079.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gong Y, Xie F, Stein KM, Garfinkel A, Culianu CA, Lerman BB, et al. Mechanism underlying initiation of paroxysmal atrial flutter/atrial fibrillation by ectopic foci: a simulation study. Circulation. 2007;115(16):2094–102. Epub 2007/04/11. 10.1161/CIRCULATIONAHA.106.656504 [DOI] [PubMed] [Google Scholar]
- 70.Hsieh YC, Lin JC, Hung CY, Li CH, Lin SF, Yeh HI, et al. Gap junction modifier rotigaptide decreases the susceptibility to ventricular arrhythmia by enhancing conduction velocity and suppressing discordant alternans during therapeutic hypothermia in isolated rabbit hearts. Heart Rhythm. 2016;13(1):251–61. 10.1016/j.hrthm.2015.07.023 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) AP. (B) [Ca2+]i. (C) Ito. (D) Junctional SR Ca2+ concentration ([Ca2+]JSR). (E) IKs. (F) Jrel. (G) ICa. (H) Jup. (I) INCX.
(TIF)
Each parameter remodelling induced by HF was incorporated into the control model at a time.
(TIF)
(A) Jrel(max), (B) GKs, (C) Jup(max), and (D) [Csqn]max were reduced from 0% to 90% relative to the control in the HF model.
(TIF)
(A) Jup. (B) [Ca2+]JSR. (C) Jrel. (D) [Ca2+]i. (E) INCX. (F) AP. (G) INa. (H) ICa.
(TIF)
(A) Ito. (B) AP. (C) INa. (D) ICa. (E) Jrel. (F) [Ca2+]i. (G) [Ca2+]JSR. (H) INCX.
(TIF)
(A(i)) Time-space plot of the membrane potential of a homogeneous, 1D cable. (A(ii)) The corresponding APD90 of each beat, in which black dashed lines with arrows indicate the APD node position as the beat number increases. (B(i)) Time-space plot of the Ca2+ transient in a homogeneous, 1D cable. (B(ii)) The corresponding peak Ca2+ transient of each beat, in which black dashed lines with arrows indicate the peak Ca2+ transient node position as the beat number increases.
(TIF)
(A) Results with D = 0.167 mm2/ms. (B) Results with D = 0.028 mm2/ms.
(TIF)
(A) AP. (B) [Ca2+]i. (C) INCX. (D) [Ca2+]JSR. (E) Jrel. (F) Jup.
(TIF)
The values of APD90, CaT amplitude, and SR Ca2+ content were computed relative to control, and the bold font demonstrated the HF model.
(PDF)
(DOCX)
Data Availability Statement
All relevant data are within the manuscript and its Supporting Information files.