

Abstract
Intracranial aneurysm (IA) is a kind of cerebrovascular disorder, which may result in the subarachnoid hemorrhage with high lethality and disability. The purpose of this study was to reveal the pathogenesis and identify novel biomarkers in IA.
We processed the raw microRNA (miRNA) expression profile data of IA obtained from Gene Expression Omnibus. Then weighted correlation network analysis was performed to identify the hub miRNAs in IA. Target genes of hub miRNAs were predicted using multiR package. In addition, a protein-protein network as well as miRNA-mRNA network was constructed and functional and pathway enrichment analyses were done. Finally, the prediction value of hub miRNAs in IA was tested in validation set.
Two modules that had relation with IA were identified and 10 hub miRNAs in each module with higher gene-module association were selected. The protein-protein network and miRNA-mRNA network contained 243 nodes and 1496 edges. Functional and pathway enrichment analyses showed that they were mainly enriched in cell cycle, cell proliferation, and PI3K/Akt signaling pathways. Besides, hsa-miR-191-3p, hsa-miR-423-5p, hsa-miR-424-5p, hsa-miR-425-3p were proven to be valuable in prediction IA occurrence.
In a word, this study reveals hub miRNAs, target genes and pathways potentially participating in formation and development of IA and screens out some candidate biomarkers. Our findings provide some new perspectives for research and treatment of IA.
Keywords: bioinformatics analysis, biomarkers, intracranial aneurysm, microRNA, weighted correlation network analysis
1. Introduction
Subarachnoid hemorrhage is a devastating subtype of stroke affecting younger patients with high mortality and disability. Nearly 85% of cases are due to the rupture of intracranial aneurysm (IA) which results in the increase of intracranial pressure, decrease of cerebral blood flow as well as decrease of cerebral perfusion pressure. It is reported that about 2% to 3% of the general population are affected by IA.[1,2] The diagnosis of unruptured IA relies on the radiological technologies which may cost lots of money and time. However, there is little biomarkers of IA based on peripheral blood test. Furthermore, the specific mechanisms of IA formation and rupture are still largely unknown. MicroRNAs (miRNAs) are the best characterized non-coding RNA, consisting of 18 to 25 nucleotides. These short RNAs are able to inhibit gene expression by binding to the mRNAs. The binding sites of miRNAs are multiple, so that miRNAs can regulate many pathophysiological processes.[3] Previous studies have demonstrated that the miRNAs can be detected in circulation including plasma and serum, and these circulating miRNAs can avoid enzymatic degradation via various mechanisms.[4] The expression level and type of circulating miRNAs are changeable under different disease conditions suggesting that they play a significant part in the generation and development of diseases and may be served as potent diagnosis biomarkers. Lots of studies have proven that miRNAs are important in modulating the cell proliferation, apoptosis, inflammation so that they can influence many diseases such as myocardial infarction, hypertension, cancer even aneurysms.[5] However, we know little about the function of circulating miRNAs in aneurysms especially in IA. Compared with the miRNAs localized in tissue, circulating miRNAs are more convenient for detecting and have a wider range of influence.
The invention of novel algorithm including weighted correlation network analysis (WGCNA),[6] gene set enrichment analysis[7] and establishment of database such as gene ontology (GO) knowledgebase,[8,9] Kyoto encyclopedia of genes and genomes (KEGG) database[10] make it possible to explore the pathophysiology mechanisms of diseases in a more comprehensive and profounder way. Therefore, in this study, we will use the bioinformatics methods combined with public databases to comprehensively explore the potent function of circulating miRNAs in IA and screen out the hub miRNAs that may be considered as potential biomarkers.
2. Materials and methods
2.1. Microarray data processing
The raw data of a human IA miRNAs microarray dataset (GSE50867) were downloaded from GEO.[11] The R package Limma was used to process the raw data.[12] Probe IDs were converted into miRNA names according to the proper GPL file downloaded from GEO. To facilitate the further analyses, we transformed the miRNA names into latest version according to the miRBase (v22.0).[13] Expression clusters and box plots were done to check whether there was a batch effect or v outliers.
2.2. Construction of co-expression networks
The WGCNA R package was used to contrast the co-expressed gene networks. Modules were generated using the dynamic tree-cutting function with following parameters: TOMType = “signed,” deepSplit = 2, pamStage = TRUE, cutHeight = 0.99, and minClusterSize = 30-3∗deepSplit. (The module preservation function (nPermutations = 200) of the WGCNA package (Langfelder et al, 2011) was utilized. Modules showing 75% similarity are merged together. Module eigengene were correlated with clinical traits and the correlations were calculated by linear regression model. The higher correlation means the more closely connection between modules and clinical traits. Hub miRNAs are those that show higher gene-module association KME values.
2.3. Gene set variation analysis (GSVA) score
To verify the high correlation between hub miRNAs and disease state, we used R package GSVA to calculate the score of each control sample or IA sample.[14]
2.4. Target mRNA identification and network construction
Since miRNAs play a biological role by modulating the mRNA, we firstly identified target mRNA of hub miRNAs using R package multiMiR.[15] This package consist of multi miRNAs databases and all data can be divided into 2 parts according that the targets of miRNAs are validated or predicted. The validated data can be obtained from mirecords, mirtarbase and tarbase, and the predicted data can be acquired from diana_microt, elmmo, microcosm, Miranda, mirdb, pictar, pita as well as targetscan. The string database was used to explore the protein-protein interaction (PPI) of target, and cytoscape was used to construct and show the miRNAs-mRNAs network.[16]
2.5. GO and KEGG pathway analysis of gene expression networks
The validated and predicted target gene gained via multiR were processed utilizing the R package clusterprofile.[17] We tried to annotate they biological function via GO and KEGG databases. The output combined scores and adjusted P-values were used to determine significance.
2.6. Validating the prediction performance of hub miRNAs
To verify the prediction performance of hub miRNAs, another independent expression set downloaded from GEO (GSE66239) was used as validation set.[18] Receiver operating characteristic (ROC) curves were constructed using R package pROC.[19]
3. Results
3.1. Raw data processing and quality control
The raw data of GSE50867 were downloaded from GEO and handled with the Limma package. The box plot of processed data suggested that there is no obvious bias among each group, and the expression clustering results showed no outlier samples. Principal component analysis shows significant segregation of different group samples. All these results are shown in Figure 1.
Figure 1.
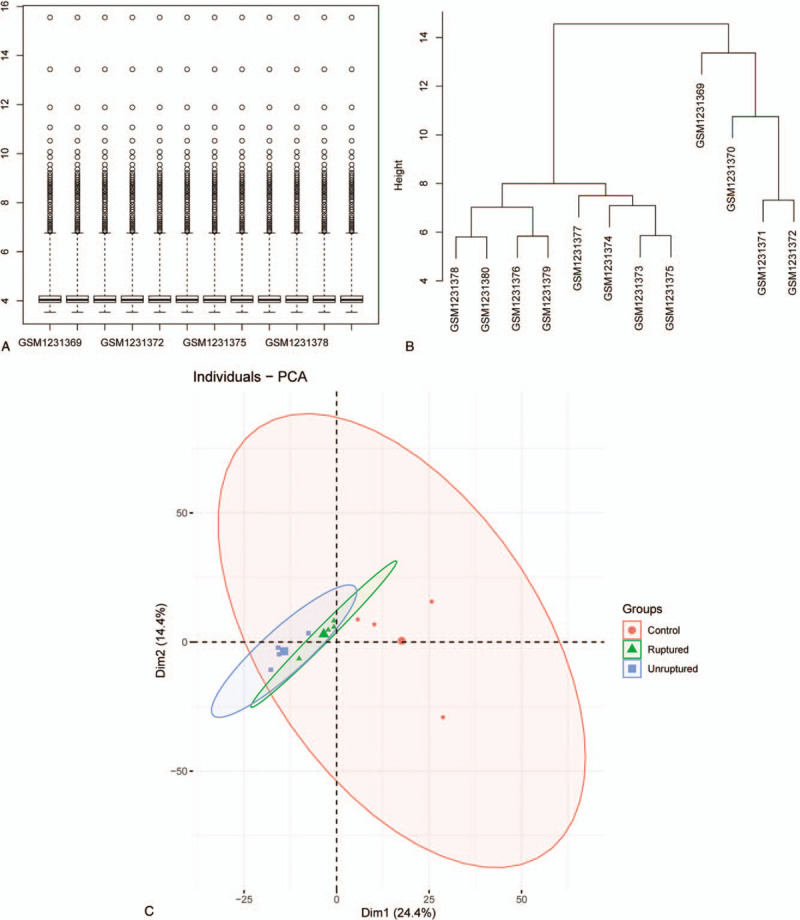
Quality control of processed data. (A) Boxplot of processed data. (B) Expression clusters of processed data. (C) Principal component analysis of processed data.
Figure 2.
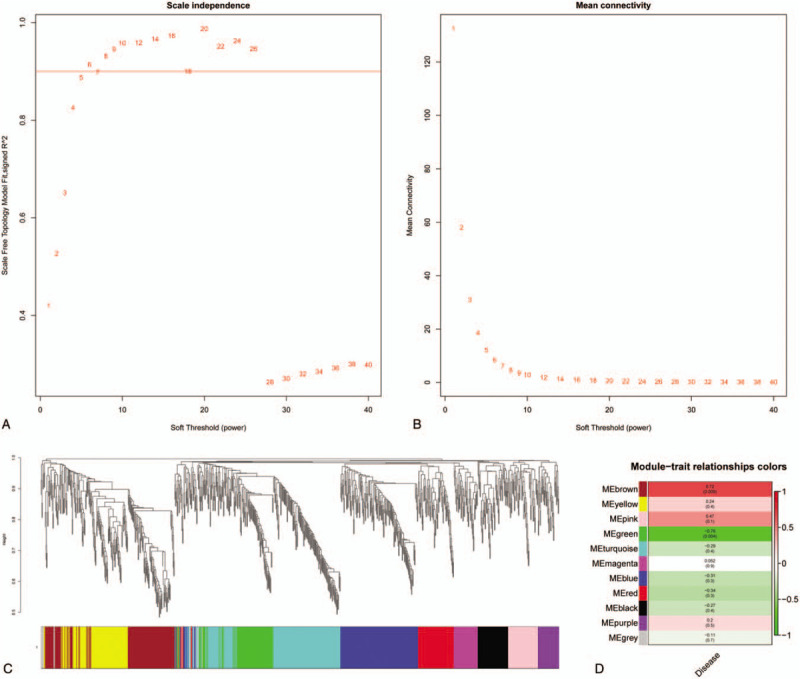
Weighted correlation network analysis (WGCNA) of processed data. (A, B) Scale independence and mean connectivity analysis. (C) Cluster dendrogram among modules. (D) Module-trait relationships. The darker the module color, the more significant their relationship.
3.2. WGCNA and hub gene identification
To analyze relationships between miRNAs and IA, the entire data were employed to constructed a weighted gene co-expression network which clusters miRNAs based on their expression in all investigated samples into co-expression modules. According to the Figure 1A, a scale-free network distribution with higher connectivity was formed when the soft-threshold power β was set to 5. As we can learn from Figure 1C, brown module showed the highest positive correlation with IA (the P-value was .0088), meanwhile, the green module had the most significant negative correlation (the P-value was .0039). Thus, we further screened the top 10 hub miRNAs in 2 modules based on the module membership (kME) values. hsa-miR-363-3p, hsa-miR-192-5p, hsa-miR-425-5p, hsa-miR-25-3p, hsa-miR-423-5p, hsa-miR-20b-5p, hsa-miR-103a-3p, hsa-miR-424-5p, hsa-miR-140-5p, hsa-miR-93-5p are 10 hub miRNAs in brown module and hsa-miR-1281, hsa-miR-1825, hsa-miR-498-5p, hsa-miR-1280, hsa-miR-1234-3p, hsa-miR-720, hsa-miR-1228-3p, hsa-miR-425-3p, hsa-miR-191-3p, hsa-miR-1225-3p are 10 hub miRNAs in green module. All these miRNAs may have major impact on the generation and development of IA, and may regarded as the potential biomarkers. To verify the reliability of these hub miRNAs, we referred them to 2 different gene sets according their affiliation and used GSVA to calculate the scores. As shown in Figure 3A, the score of brown module in control was significant lower than that in other 2 groups (scores: control group, −0.78 ± 0.10; unruptured group, 0.65 ± 0.11; ruptured group, 0.44 ± 0.11, P < .05, 1 way ANNOVA). Meanwhile, Figure 3B showed that compared with control group, the scores of green in IA groups were decreased. (scores: control group, 0.75 ± 0.05; unruptured group, −0.55 ± 0.17; ruptured group, −0.52 ± 0.13, P < .001, 1 way ANNOVA). A conclusion drawn from these 2 distinct algorithms was that hub miRNAs identified above were highly related to IA and may play an extremely important role in the pathophysiology of this disease. What's more, considering their great correlation with IA, they may be served as forecast indicators.
Figure 3.
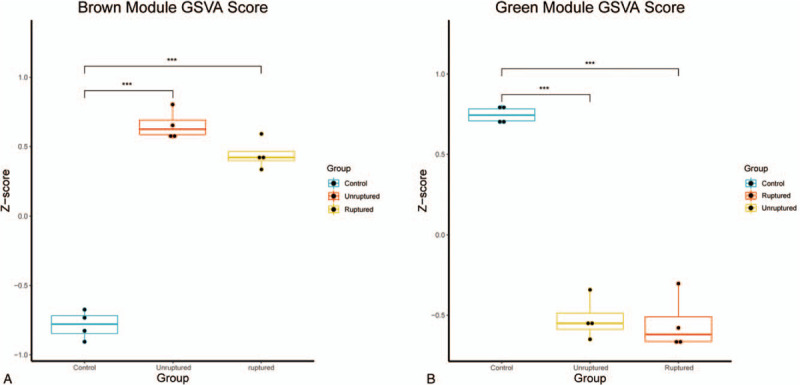
Gene set variation analysis (GSVA) scores. (A) GSVA score of brown module hub miRNAs in GSE50867. (B) GSVA score of green module hub miRNAs in GSE50867. miRNAs = microRNAs.
3.3. Target mRNA identification and enrichment analysis
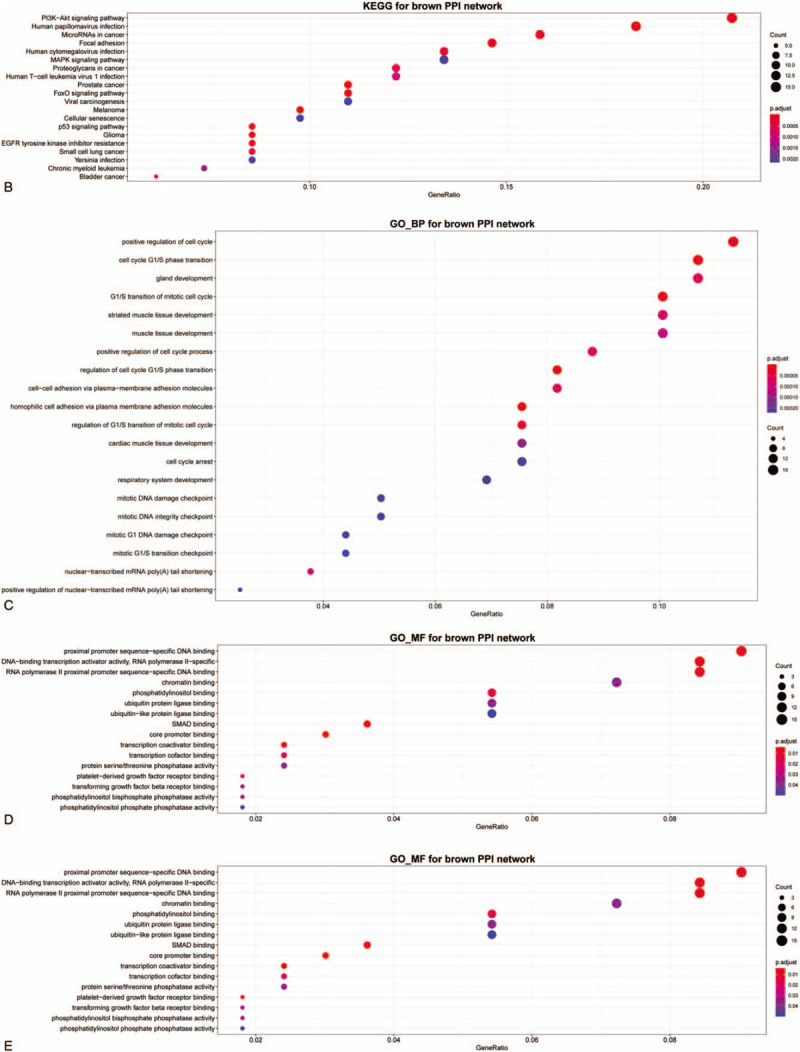
Since miRNAs work through regulating mRNAs, to further understand the function of hub miRNAs in IA, the multiMiR was employed to identify the target mRNAs. MultiMiR package provides 2 kinds of databases including validated and predicted data. To filter out the mRNAs that may have huge influence on IA, we included targets which are modulated by more than 3 disparate miRNAs, then enrichment analysis was done to reveal their function. When it comes to the target genes of green module hub miRNAs, there is no terms can be enriched through GO or KEGG analysis, consequently, we paid attention to the brown module hub miRNAs in the following. As shown in Figure 4A, when it referred to genes controlled by brown module hub miRNAs, GO biological process terms were mainly associated with tissue development, cell cycle, cell proliferation, cell division, cell adhesion. In the molecular function group, GO terms mainly involved promoter binding, transcription activator or coactivator binding (Fig. 4B). There are no items enriched in cellular components (CC) and KEGG pathway.
Figure 4.
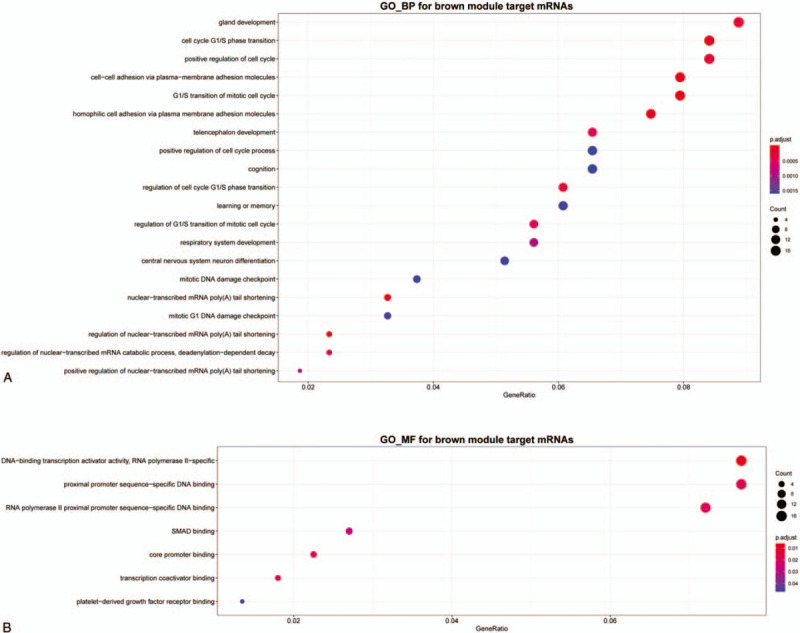
Gene Ontology (GO) enrichment analysis of brown module hub miRNAs target mRNAs. (A) Biological process (BP). (B) Molecular function (MF). miRNAs = microRNAs.
3.4. PPI and miRNA-mRNA network
With the help of STRING online database (https://string-db.org), a PPI network was constructed to study the relationship among each mRNA and cytoscape was utilized to build and visualized the miRNA-mRNA network.[20] The network containing 233 different genes and 10 hub miRNAs, connected with 1496 edges (Fig. 5 A). Among the 243 nodes, the most significant 10 node degree genes were PTEN, VEGFA, CCND1, MDM2, CREB1, AGO2, BCL2L11, TNRC6B, SMAD7, and KIF23, and the hub miRNA that connected with others most extensively is hsa-miR-93-5p. To further understand the purposes of this network, annotation was performed. As we can conclude from Figure 5 B, PI3K-Akt signaling pathway, FoxO signaling pathway, p53 signaling pathway as well as other cancer related pathways were significantly enriched, suggesting the involvement of cell proliferation, cell adhesion. Besides, another important pathway named (mitogen-activated protein kinase) MAPK was also enriched, indicating that miRNAs may extensively participate in IA generation and development. CC analysis showed that this PPI network is strongly correlated with cell adhesion, different junction, and extrinsic component of membrane (Fig. 5 E).
Figure 5.
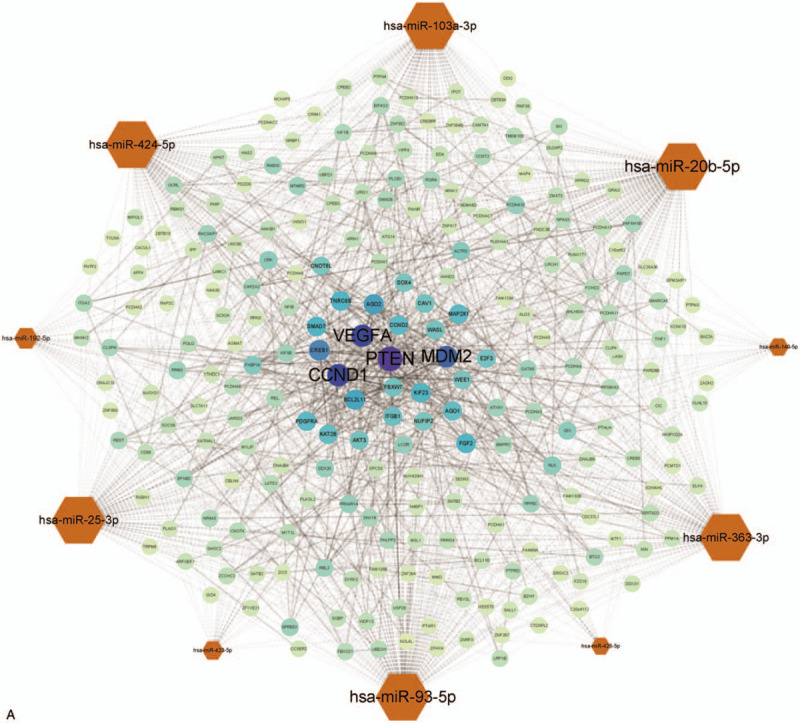
Protein-protein interaction (PPI) and miRNA-mRNA network and enrichment analysis. (A) The bigger of label size, the more significant of node in network. The hexagon represents the miRNAs and the circle represents the target mRNAs. (B) Signaling pathways enrichment of PPI network. (C) BP. (D) MF. (E) Cellular component (CC). BP = biological process, MF = molecular function, miRNAs = microRNAs.
3.5. Expression level and prediction performance of hub miRNAs
To validate the prediction performance of hub miRNAs, another independent dataset was utilized. All identified hub miRNAs belonging to green or brown module were used respectively to draw the ROC curve with the assistance of R package (pROC). We chose the hub miRNAs whose area under curve (AUC) value is higher than 0.75. As shown in Figure 6, the hsa-miR-191-3p performs better than others with the AUC being 100%. Meanwhile, hsa-miR-423-5p (AUC 75.9%), hsa-miR-424-5p (AUC 83.3%), hsa-miR-425-3p (AUC 75.9%) also had good prediction performance. In order to clarify their potential biomarker roles more clearly, we show the expression level of hub miRNAs in Figure 6E.
Figure 5 (Continued).
Protein-protein interaction (PPI) and miRNA-mRNA network and enrichment analysis. (A) The bigger of label size, the more significant of node in network. The hexagon represents the miRNAs and the circle represents the target mRNAs. (B) Signaling pathways enrichment of PPI network. (C) BP. (D) MF. (E) Cellular component (CC). BP = biological process, MF = molecular function, miRNAs = microRNAs.
Figure 6.
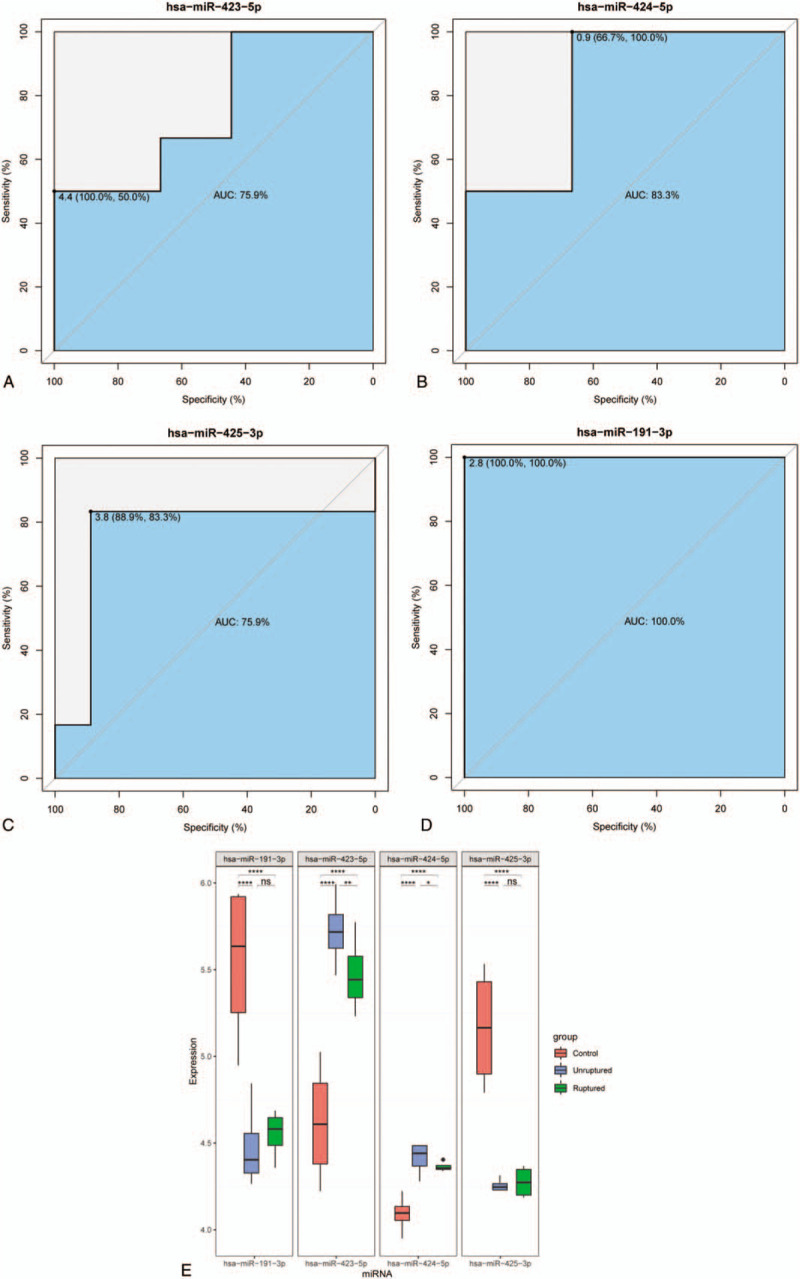
Receiver operating characteristic (ROC) curves for hsa-miR-423-5p (AUC 75.9%), hsa-miR-424-5p (AUC 83.3%), hsa-miR-425-3p (AUC 75.9%), hsa-miR-191-3p (AUC 100.0%). AUC indicates area under the curve. (E) The expression level of hub miRNAs. miRNAs = microRNAs.
4. Discussion
IA is a kind of cerebrovascular disorder which mainly performs the abnormal dilatation or ballooning in the blood vessel, localized in the thickness part of the vascular wall. The specific mechanism of its generation and development is still unclear and little potent biomarkers are identified. Recently, circulating miRNAs are viewed as potentially valuable predictors for their stability, sensitivity as well as broadly influence.[4] Furthermore, previous studies have demonstrated that miRNAs are significant in regulating the IA.[11] Thus, in this studies, with the assistance of WGCNA, we obtained the modules highly associated with IA and screened twenty key miRNAs. Then, the GO terms and pathway enrichment analysis of target mRNAs or PPI network were enriched in biology associated with cellular characteristics, especially for the cell cycle, cell proliferation, cell adhesion, and promoter binding. Many former investigations have demonstrated that the apoptosis and proliferation of endothelial cell and smooth muscle cell, immune cells infiltration are 2 major causes of IA.[21–24] Our results further prove that these 2 main pathophysiological processes can be adjusted by miRNAs and indicates that miRNAs play an important role in IA.
According to our results, PI3K/Akt signaling pathway is important in IA which is consistent with previous studies. Li et al have proven that PI3K/Akt signaling pathway has effect on the production of IA in rat model.[25] The involvement of this pathway in other kinds of aneurysm such as abdominal aortic aneurysm and thoracic aortic aneurysm is also detected by different groups.[26,27] Based on existing knowledge, we speculate that PI3K/Akt may affect IA via following ways. First, it may regulate the tight junction of endothelial cells, resulting in the immune cells infiltration and intimal structure changes.[28] Second PI3K/Akt may modulate the proliferation and migration of vascular smooth muscle cell (VSMC).[29] The balance between apoptosis and proliferation of VSMCs as well as the correction arrangement are crucial in IA growth. Thirdly, PI3K/Akt pathway can control the activation of NF-κB via IκB, published data support that NF-κB essential in regulating the inflammation.[30] Dysregulated inflammation exacerbates the IA via various ways, for example, degrading the extracellular matrix through matrix metalloproteinase and recruiting the immune cells.[21,22] What's more, FOXO1, one of the downstream transcription factor of PI3K/Akt pathway, has been proven to be capable of binding with the promoter region of CCND1. This process can modulate the proliferation of VSMCs and vascular remodeling, which influence the development of IA.[31]
Besides the PTEN which is vital molecule in above mentioned PI3K/Akt pathway, VEGFA also takes up a core position in the PPI network. Li et al have verified that when binding with its receptors, VEGFA contribute to abdominal aortic aneurysm formation by multiple ways, for instance, suppressing mural angiogenesis, producing matrix metalloproteinase and inducing myeloid cell as well as monocytes chemotaxis.[32] The similar functions of VEGFA in IA have also been found in IA by a different group.[33]
Serving circulating miRNAs as biomarkers have attracting considerable interest and pioneering studies have revealed the changes in plasma miRNAs in response to IA.[11] In this study, we used WGCNA and identified 4 miRNAs which might be potential biomarkers in IA, with AUC value higher than 75%. In addition to concerning the sensitivity and specificity, the expression levels of latent biomarkers are also crucial, because of the possibility of miRNAs degradation. All these 4 miRNAs are abundant in samples. Consequently, our results suggest that they may be biological markers that are useful in evaluation the probability of IA occurrence.
However, there are still many limitations in our research. First of all, we only used the data obtained from GEO, but we do not check this result in our specimens. Second, the total number of sample is 12 (4 per group), which might be too little. Third, the functions of hub miRNAs predicted through databases are needed to be further verified using cell experimentation and clinical samples. At last, the validation set and training set come from different organizations. Therefore, the results, especially the predictive performance of hub miRNAs could be influence by samples derived from different sources.
In conclusion, we identified the key miRNAs, their target mRNAs as well as crucial pathways. What's more the possibility of using hub miRNAs as biomarkers are verified. These findings are beneficial to comprehensive understanding of potential mechanism in IA and provide some novel perspectives on the IA therapeutic targets.
Author contributions
Data curation: Ming Zhao, Longbiao Xu.
Formal analysis: Ming Zhao.
Funding acquisition: Ming Zhao, Hui Qian.
Investigation: Hui Qian.
Project administration: Ming Zhao.
Software: Ming Zhao, Longbiao Xu.
Visualization: Ming Zhao, Longbiao Xu.
Writing – original draft: Ming Zhao, Longbiao Xu.
Writing – review and editing: Ming Zhao, Hui Qian.
Footnotes
Abbreviations: AUC = area under curve, CC = cellular components, GEO = gene expression omnibus, GO = gene ontology, GSVA = gene set variation analysis, IA = intracranial aneurysm, KEGG = Kyoto encyclopedia of genes and genomes, miRNA = microRNA, PPI = protein-protein interaction, ROC = receiver operating characteristic, VSMC = vascular smooth muscle cell, WGCNA = weighted correlation network analysis.
How to cite this article: Zhao M, Xu L, Qian H. Bioinformatics analysis of microRNA profiles and identification of microRNA-mRNA network and biological markers in intracranial aneurysm. Medicine. 2020;99:31(e21186).
The data in this article are obtained from GEO database.
The datasets generated during and/or analyzed during the current study are publicly available.
This study was funded by the Clinical research fund of Zhejiang Medical Association (2019ZYC-A52), Natural Science Foundation of Zhejiang (LQ16H090002), Medical and health technology project of Zhejiang Province (2020RY081).
The authors have no conflicts of interest to disclose.
References
- [1].Macdonald RL, Schweizer TA. Spontaneous subarachnoid haemorrhage. Lancet 2017;389:655–66. [DOI] [PubMed] [Google Scholar]
- [2].Molyneux AJ, Birks J, Clarke A, et al. The durability of endovascular coiling versus neurosurgical clipping of ruptured cerebral aneurysms: 18 year follow-up of the UK cohort of the International Subarachnoid Aneurysm Trial (ISAT). Lancet 2015;385:691–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004;116:281–97. [DOI] [PubMed] [Google Scholar]
- [4].Creemers EE, Tijsen AJ, Pinto YM. Circulating microRNAs: novel biomarkers and extracellular communicators in cardiovascular disease? Circ Res 2012;110:483–95. [DOI] [PubMed] [Google Scholar]
- [5].van Rooij E, Olson EN. MicroRNA therapeutics for cardiovascular disease: opportunities and obstacles. Nat Rev Drug Discov 2012;11:860–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008;9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Subramanian A1, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ashburner M1, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000;25:25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].The Gene Ontology Consortium. The Gene Ontology Resource: 20 years and still going strong. Nucleic Acids Res 2019;47:D330–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 2000;28:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Li P, Zhang Q, Wu X, et al. Circulating microRNAs serve as novel biological markers for intracranial aneurysms. J Am Heart Assoc 2014;3:e000972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ritchie ME, Phipson B, Wu D. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kozomara A, Birgaoanu M, Griffiths-Jones S. miRBase: from microRNA sequences to function. Nucleic Acids Res 2019;47:D155–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hänzelmann S1, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 2013;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ru Y, Kechris KJ, Tabakoff B. The multiMiR R package and database: integration of microRNA-target interactions along with their disease and drug associations. Nucleic Acids Res 2014;42:e133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Shannon P, Markiel A, Ozier O. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003;13:2498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Yu G, Wang LG, Han Y, et al. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 2012;16:284–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bekelis K, Kerley-Hamilton JS, Teegarden A, et al. MicroRNA and gene expression changes in unruptured human cerebral aneurysms. J Neurosurg 2016;125:1390–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Robin X, Turck N, Hainard A. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 2011;12:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 2019;47:D607–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Turjman AS, Turjman F, Edelman ER. Role of fluid dynamics and inflammation in intracranial aneurysm formation. Circulation 2014;129:373–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hosaka K, Hoh BL. Inflammation and cerebral aneurysms. Transl Stroke Res 2014;5:190–8. [DOI] [PubMed] [Google Scholar]
- [23].Chalouhi N, Hoh BL, Hasan D. Review of cerebral aneurysm formation, growth, and rupture. Stroke 2013;44:3613–22. [DOI] [PubMed] [Google Scholar]
- [24].Diagbouga MR, Morel S, Bijlenga P. Role of hemodynamics in initiation/growth of intracranial aneurysms. Eur J Clin Invest 2018;48:e12992. [DOI] [PubMed] [Google Scholar]
- [25].Li XG, Wang YB. SRPK1 gene silencing promotes vascular smooth muscle cell proliferation and vascular remodeling via inhibition of the PI3K/Akt signaling pathway in a rat model of intracranial aneurysms. CNS Neurosci Ther 2019;25:233–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Escudero P, Navarro A, Ferrando C. Combined treatment with bexarotene and rosuvastatin reduces angiotensin-II-induced abdominal aortic aneurysm in apoE(-/-) mice and angiogenesis. Br J Pharmacol 2015;172:2946–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chen S, Chen H, Yu C. Long noncoding RNA myocardial infarction associated transcript promotes the development of thoracic aortic by targeting microRNA-145 via the PI3K/Akt signaling pathway. J Cell Biochem 2019;120:14405–13. [DOI] [PubMed] [Google Scholar]
- [28].Cong X, Kong W. Endothelial tight junctions and their regulatory signaling pathways in vascular homeostasis and disease. Cell Signal 2020;66:109485. [DOI] [PubMed] [Google Scholar]
- [29].Muller DW. The role of proto-oncogenes in coronary restenosis. Prog Cardiovasc Dis 1997;40:117–28. [DOI] [PubMed] [Google Scholar]
- [30].Hoesel B, Schmid JA. The complexity of NF-(B signaling in inflammation and cancer. Mol Cancer 2013;12:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Cao W, Chang T, Li XQ, et al. Dual effects of fructose on ChREBP and FoxO1/3α are responsible for AldoB up-regulation and vascular remodelling. Clin Sci (Lond) 2017;131:309–25. [DOI] [PubMed] [Google Scholar]
- [32].Xu B, Iida Y, Glover KJ, et al. Inhibition of VEGF (Vascular endothelial growth factor)-A or its receptor activity suppresses experimental aneurysm progression in the aortic elastase infusion model. Arterioscler Thromb Vasc Biol 2019;39:1652–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Liu P, Shi Y, Fan Z, et al. Inflammatory smooth muscle cells induce endothelial cell alterations to influence cerebral aneurysm progression via regulation of integrin and VEGF expression. Cell Transplant 2019;28:713–22. [DOI] [PMC free article] [PubMed] [Google Scholar]