

INTRODUCTION
Intratumoral (IT) immunotherapy is approved for stage IIIB to IV melanoma1-5 and under evaluation in other malignancies with novel immune-stimulatory products.6-18 Standardized efficacy evaluation is essential for drug development. Current oncology response criteria, such as Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST 1.1) and guidelines for immunotherapeutic trials (iRECIST), were designed only to assess response to systemic therapy.19-25
RECIST is an evolving standardized framework for evaluating changes in tumor size, that is used in clinical trials to define treatment responses and disease progression.19 RECIST 1.1 and iRECIST are unsuitable for IT immunotherapy trials for several reasons. Because they were designed for systemic therapy, focal intervention renders treated lesions nonevaluable. RECIST 1.1 does not allow separate response assessment in injected and noninjected lesions, which is critical for IT immunotherapy trials. Moreover, there is no consensus on injected lesion assessment when lesions chosen for injection may change during treatment because of regression, loss of accessibility, or growth of other lesions. iRECIST has limited usefulness because the purpose of assessment after initial progression is solely to exclude pseudoprogression; it does not consider that the lesions selected for injection may change at progression.
Nevertheless, the experience of developing iRECIST by revising RECIST 1.1 for immunotherapy25 provides valuable guidance. Before the consensus effort of the RECIST Working Group, stakeholders devised divergent approaches to RECIST modification for immunotherapy, resulting in confusion and incomparability among trials.25 Without standardization, these issues may recur for IT therapy.
The goal of IT RECIST (itRECIST) is to create guidelines for capturing data and assessing response in IT immunotherapy trials. As with iRECIST, the standardized data collection and initial suggestions for response assessment of itRECIST will be refined based on collected data. We anticipate itRECIST will initially be used for exploratory analyses, with primary and secondary end points based on RECIST 1.1, until evidence indicates that itRECIST improves efficacy assessment.
itRECIST
itRECIST is designed to address the unique needs of IT immunotherapy trials but, where possible, aligns with RECIST 1.1 and iRECIST. It does not dictate which lesions to inject at each visit, but rather provides guidelines for assessing responses as treatment evolves. The key questions, and the approaches to answering them, are as follows:
What is the overall response? Overall response is determined as per RECIST 1.1 (or per iRECIST, after initial progression).
What is the maximal effect of IT therapy (with or without systemic therapy) on noninjected lesions? The smallest (nadir) total size of predesignated noninjected lesions is compared with pretreatment size.
What is the effect of therapy on injected lesions? During treatment, an iterative assessment accounts for changes in lesions selected for injection. After treatment, a combined response compares the smallest size achieved by each injected target lesion with its size before injection.
It is important to define a lexicon of precise and simple terms for these criteria; novel, nonintuitive terminology hinders understanding and adoption. Therefore, lesions are classified as injected or noninjected, and the terms injected response and noninjected response describe response in injected and noninjected lesions, respectively. The choice not to use the term abscopal effect was deliberate, because this implies causality: injecting lesion A causes a response in lesion B. Many IT immunotherapies are administered with systemic immunotherapies; hence, noninjected lesions may be affected by systemic therapy alone.
LESION MEASUREMENT
Lesion measurements should be performed per RECIST 1.1, with one exception.24 Briefly, either computed tomography (CT) or magnetic resonance imaging should be used to measure target lesions.24 For skin lesions, RECIST 1.1 recommends color photography documentation, including a size standard or caliper for scale.24
RECIST 1.1 does not allow ultrasound for lesion measurement because of operator dependence and difficulty with standardization.24 However, in practice, ultrasound may be the only practical choice for some subcutaneous lesions. Therefore, itRECIST permits ultrasound measurement if no other lesions are available for quantitative assessment (Data Supplement). When feasible, the same operator should perform the ultrasound at all visits using the same equipment and acquisition parameters, capturing lesion images in a similar orientation, with anatomic landmarks to align with preceding scans. Standard RECIST 1.1 thresholds apply to consider a lesion measurable (≥ 10-mm longest diameter for extranodal lesions, ≥ 15-mm short axis for lymph nodes).24
Most importantly, investigators should use the same imaging technique for a given target lesion at each assessment to evaluate changes over time. For instance, if a patient underwent CT at baseline and ultrasound-guided IT immunotherapy for liver metastasis, response assessments should be based on repeat CT. Although pre-IT injection ultrasound assessments might yield information about the kinetics of response, ultrasound should not be used in itRECIST calculations in this specific example.
The intent with itRECIST is to capture both systemic and local effects of IT therapy. Thus, unlike in RECIST 1.1, injected lesions remain evaluable for overall response assessment even after local procedures, such as electroporation or low-dose irradiation, as long as these are integral to the IT regimen to support or enhance the injection effect. Although intralesional administration techniques and intrinsic tumor factors add variability to changes resulting from injection, no obvious adjustment to measurement methods would improve response assessment. Tumor biopsies are often performed as part of a clinical trial. Excisional biopsy renders a lesion nonevaluable in itRECIST. Although core needle biopsy would not automatically make a lesion nonevaluable, its use is discouraged for target lesions. When feasible, biopsies should be restricted to nontarget lesions.
BASELINE DOCUMENTATION OF TUMOR BURDEN
At baseline, lesions are classified as measurable (eligible for selection as target lesions) or nonmeasurable per RECIST 1.1 guidelines on size and reproducibility.24 Baseline lesions are categorized as target injected (T-I), target noninjected (T-NI), nontarget injected (NT-I), and nontarget noninjected (NT-NI) according to an algorithm (Fig 1A). As in RECIST 1.1, target refers strictly to lesions that are selected for measurement; it has no relationship to lesions selected for injection. One to five measurable lesions are designated as T-I and are used to evaluate the injected lesion response. One to five measurable lesions are designated as T-NI and remain noninjected for as long as possible to allow assessment of the maximal noninjected lesion response, as discussed in a later section. A sum of diameters (SOD; longest diameters for extranodal lesions and short axis for lymph nodes) is calculated for all target lesions combined, and separately for T-I and T-NI lesions (Fig 1A).
FIG 1.
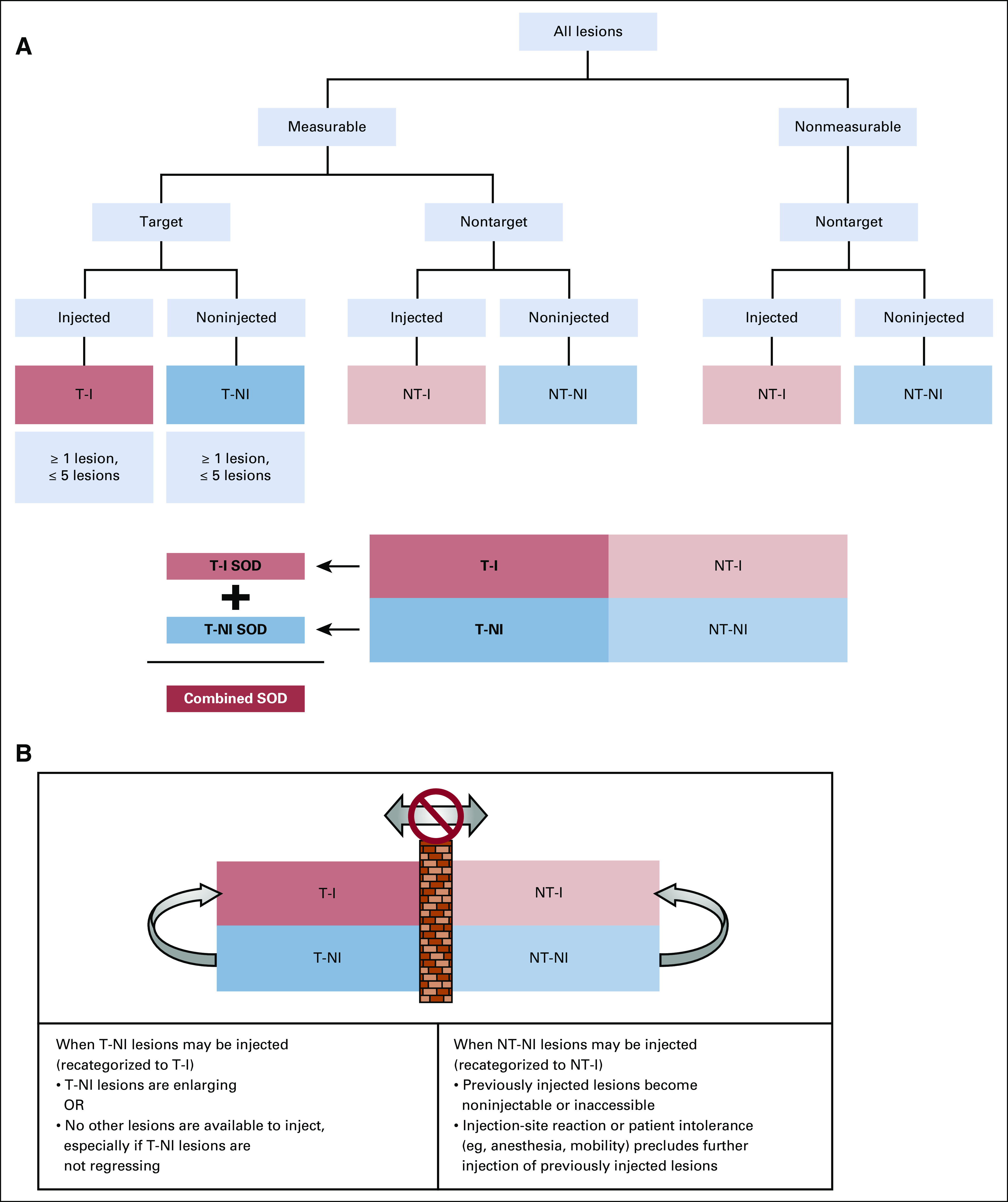
Algorithm for classification of lesions into 4 categories at baseline and recategorization after baseline. (A) Classification of lesions at baseline. Lesions are classified first as measurable or nonmeasurable using the standard RECIST 1.1 rules for measurability. Measurable lesions (those eligible for selection as target lesions) are then classified as target (selected to be observed quantitatively) or nontarget (selected to be observed qualitatively), and the decisions about which lesions are to be injected are made based on the prioritization rules discussed. Lesions selected for injection may be either target or nontarget in RECIST 1.1 terms. Between 1 and 5 lesions should be classified as target injected (T-I), and between 1 and 5 should be classified as target noninjected (T-NI), for a maximum of 10 target lesions. All lesions not chosen as target are observed qualitatively as nontarget, and some of these may be selected for injection at baseline. T-I lesions and T-NI lesions each have their own distinct sum of diameters (SOD; longest diameters for extranodal lesions, short axis for lymph nodes). A combined SOD also includes all target lesions, injected and noninjected. Nontarget injected (NT-I) and nontarget noninjected (NT-NI) lesions are observed qualitatively, exactly as in RECIST 1.1, classified in aggregate as showing complete response (CR), unequivocal progressive disease (PD), or neither (called non-CR/non-PD in RECIST 1.1). (B) T-NI or NT-NI lesions can be recategorized as injected lesions if the decision is made to inject them after baseline assessment. NT-NI lesions may be injected if previously injected nontarget lesions regress completely or become inaccessible, or if a patient factor such as injection-site reaction or patient intolerance precludes further injection. Lesions initially selected as T-NI should remain noninjected for as long as possible so that the maximal noninjected effect can be evaluated, but they may be injected if they are enlarging or if no other lesions are available for injection, especially if the lesions initially designated as T-NI are not regressing. The barrier between target and nontarget categories means that all lesions remain target and nontarget in accordance with the initial designation, regardless of whether they are subsequently injected.
If only 1 lesion is measurable, although others are accessible for injection but not suitable for reproducible quantification, the measurable lesion should be designated as T-NI, because it may be more important to detect objective responses in noninjected lesions than in injected lesions as a means of assessing treatment efficacy. This suggestion must, of course, be considered in light of other clinically significant factors, such as whether the measurable lesion should be injected to palliate symptoms and whether the other injectable lesions offer sufficiently attractive injection targets to achieve the overall treatment goals.
RECLASSIFICATION OF LESIONS AFTER BASELINE
Injected lesions may change if those initially injected regress or become inaccessible, or if others enlarge. Nevertheless, target lesions always remain target, and nontarget lesions remain nontarget, regardless of whether they receive injections (Fig 1B). If initially noninjected lesions enlarge, the treating physician may decide the enlarging T-NI lesions can be controlled by injection (especially if injected lesions are regressing). Once injected, these lesions are recategorized as T-I lesions. T-NI lesions can also be injected when previously injected lesions regress or become noninjectable, particularly when initially selected T-NI lesions are not regressing (maximal noninjected effect has been achieved). NT-NI lesions may be recategorized as NT-I and injected when the original NT-I lesions can no longer be injected because of regression, inaccessibility, injection-site reaction, patient intolerance, or need for more aggressive anesthesia.
Guidelines for Prioritization of Lesion Injection for IT Therapy
Selection and prioritization of lesions for IT injection is a complex set of decisions made at each treatment visit and is ultimately based on clinical judgment. A complete description of the process is beyond the scope of this guidance, which is focused on response assessment, but a set of guiding principles follows.
The first priority is patient safety. Lesions are selected to minimize the potential for procedural complications and operational complexity. One important safety concern is vascularity within and adjacent to a lesion. To minimize systemic administration, injection into tumor vasculature should be avoided. To minimize bleeding risk, vessels adjacent to a tumor should not be traversed, and areas of vascular encasement should be avoided in high-risk locations (eg, inferior vena cava encasement for liver lesions, great vessel encasement for head and neck tumors).
The next priority is accessibility. Preference is given to visible cutaneous lesions, and superficial subcutaneous lesions and lymph nodes which are easily palpable. Deeper lesions, including nonpalpable lymph nodes and extranodal lesions in viscera or body cavities, are more difficult to access and typically require imaging guidance, increasing procedural complexity. Deciding to inject a lesion based on accessibility must be balanced against potential clinical benefits such as symptom relief.
At initiation of therapy, other factors guiding lesion prioritization include size and amount of viable tumor tissue. Other factors being equal, larger lesions are preferred because of the greater amount of tissue and because the likely older age of the lesion may indicate the potential to release a wider breadth of tumor-specific antigens to stimulate a broader repertoire of antigen-specific T cells. Very large lesions should be approached cautiously because of possible central necrosis, increased bleeding risk, and difficulty dispersing immunotherapeutics. Radiographically visible necrosis should be avoided, with IT therapy directed at viable portions of lesions. A larger lesion that is predominantly necrotic may have lower priority than a smaller lesion with little or no radiographic necrosis. Lesions with radiographic evidence of aggressiveness (eg, local invasiveness) should have higher priority.
If additional lesions are injected after therapy begins, new or enlarging lesions should be given priority over lesions selected based on size or imaging features, but safety and accessibility are still paramount. These lesions contain actively dividing cells and therefore may be more responsive to injection. In addition, new or enlarging lesions may contain cancer cells that represent the vanguard of the disease as it attempts to evolve under the selective pressure of immunotherapy. These lesions could harbor new tumor antigens not strongly represented in previously injected lesions. Although some lesion types or anatomic locations may be better for stimulating systemic immune responses, evidence is insufficient to use such information for lesion prioritization. Nonetheless, data related to lesion response by disease site will inform such choices in the future.
Response Assessment Before Radiographic Progression
Overall response.
The principle that target lesions remain target and that nontarget lesions remain nontarget regardless of injection status allows an overall assessment for each imaging visit similar to that for RECIST 1.1 (different only in allowing more target lesions, in injected lesions not becoming nonevaluable, and allowing ultrasound). Target lesion response, nontarget lesion response, and new lesion appearance are defined as they are for RECIST 1.1 and combined similarly to determine overall response for each visit (Fig 2). The overall response should include all lesions classified as target at baseline (SOD of T-I and T-NI combined v SOD at baseline and at nadir) and all nontarget lesions (NT-I and NT-NI) combined (classified as absent, present, or collectively showing unequivocal progression). Of note, the rare instances of seeding along a needle track should not be reported as new lesions unless they show growth on subsequent imaging.
FIG 2.

Overall response until disease progression per RECIST 1.1. The injected response at each visit is based only on the changes in the sum of diameters (SOD) of the lesions designated as target injected (T-I). The noninjected response at each visit is based only on the changes in the SOD of the target noninjected (T-NI) lesions. The overall response is based on the changes in the SOD of all target lesions together, the qualitative assessment of all nontarget lesions together, and the evaluation for possible new lesions and uses the same response categories and logical combination of these that RECIST 1.1 uses. NT-I, nontarget injected; NT-NI, nontarget noninjected.
The role of fluorodeoxyglucose (FDG)–positron emission tomography and biopsy in assessing response must be further evaluated. Because radiographic assessment might not correlate with tissue response and loss of FDG uptake in injected lesions may represent necrosis, biopsy may provide additional information in case of doubt.26,27
In the neoadjuvant setting, IT immunotherapy may yield pathologic complete response (pCR) rates surpassing clinical response rates (which include radiographic objective response and clinical assessment). For example, after 12 weeks of neoadjuvant talimogene laherparepvec in resectable stage IIIB to IVM1a melanoma, 3 patients achieved clinical CR and all achieved pCR. Additionally, 1 of 7 patients with clinical partial response (PR) achieved pCR, 6 of 21 patients with clinical stable disease (SD) achieved pCR, and even 2 of 35 patients with clinical progressive disease (PD) achieved pCR.27 Subanalysis of noninjected response may not apply in the neoadjuvant setting if only a single lesion is present initially.
Noninjected response.
Noninjected response is based entirely on T-NI lesions. The SOD for these lesions at each time point is compared with those at baseline and nadir, similar to target lesion response assessments in RECIST 1.1 (Table 1). Lesions designated T-NI at baseline should remain noninjected for as long as possible to allow assessment of maximal systemic response to IT therapy in noninjected lesions. The treating physician may choose to inject T-NI lesions when they enlarge (systemic therapy alone is not restraining their growth) or when previously injected lesions have become noninjectable, especially if the T-NI lesions are not regressing. Once any T-NI lesion is injected, the noninjected response becomes nonevaluable. Overall response, however, remains evaluable because it is based on all target lesions together. As discussed below in the section on end points, the best noninjected response and maximal tumor shrinkage are determined based on assessments before injection of any T-NI lesion.
TABLE 1.
Response by Lesion Category
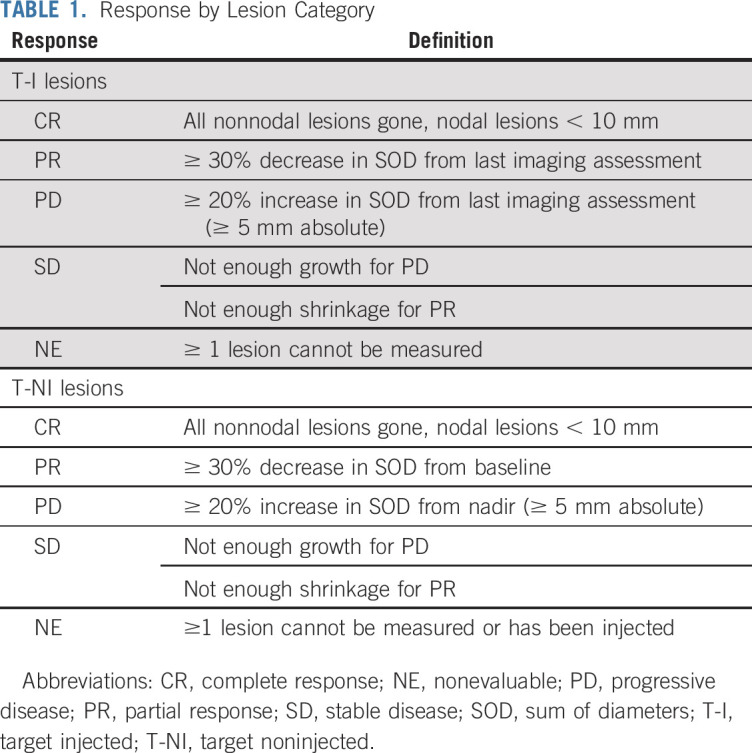
Injected response.
Lesions selected for injection may change at each treatment visit, so there is no stable baseline for comparison. Therefore, during treatment, the response assessment for injected lesions is iterative. At each assessment, the current SOD for all target lesions injected during the preceding treatment visit (whether originally classified or reclassified as T-I) should be compared with their SOD at the preceding assessment (Fig 3). The injected response is based on SOD change from the previous assessment (Table 1). The decision about which lesions to inject should be made at this time, based on the guidelines for lesion prioritization outlined here. The new T-I SOD should be calculated and used as the comparator for the next assessment. After treatment discontinuation or during an interim analysis, the best response for injected lesions is determined by comparing the size of each injected lesion at its smallest with its size before first injection, as discussed in the section on end points.
FIG 3.
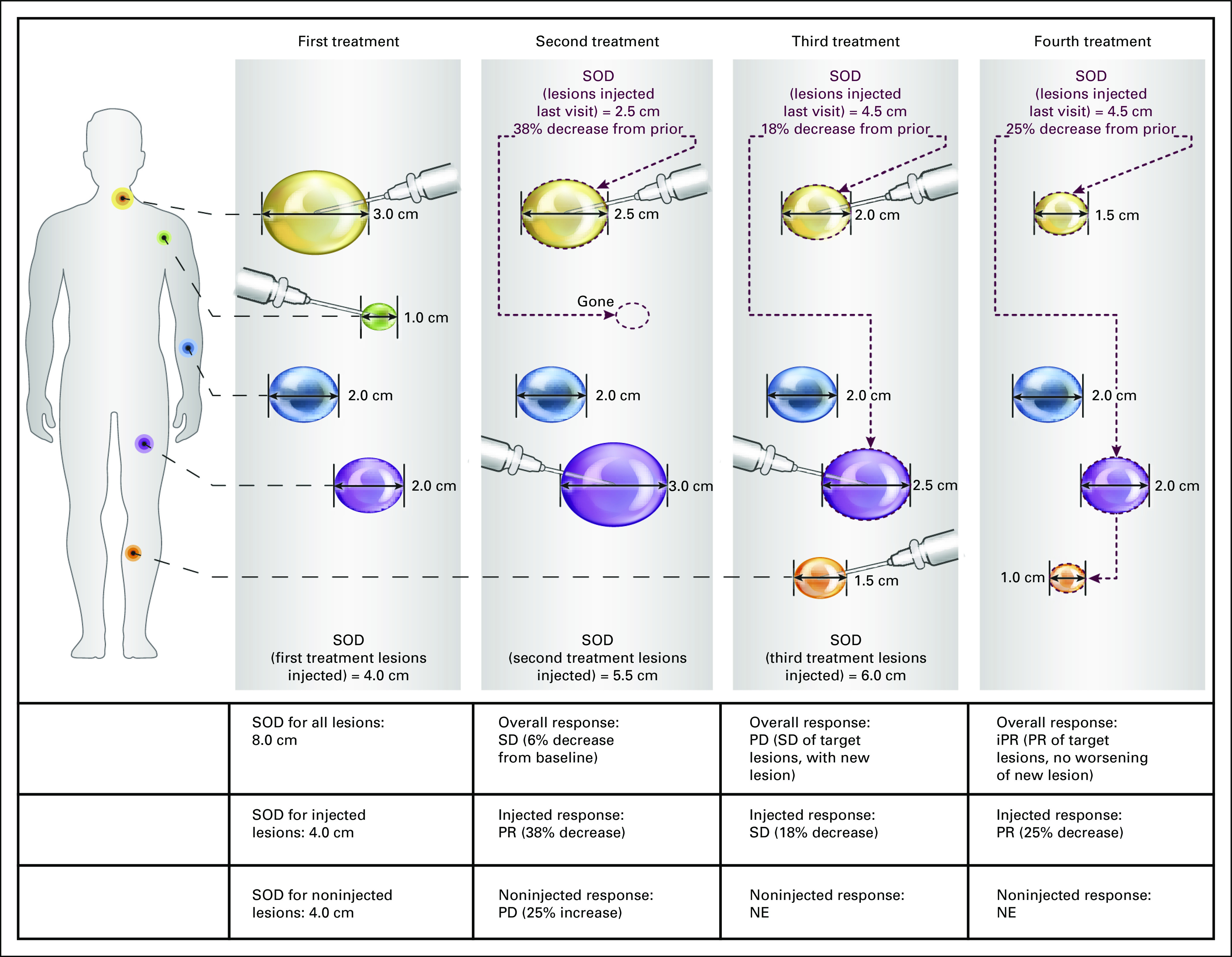
Example of iterative assessment of injected lesion response during treatment. This is an illustration of overall, injected, and noninjected response assessment, with a particular focus on the iterative assessment of injected lesions. All lesions from a single patient are displayed in simple schematic form and are not meant to be anatomically adjacent. For purposes of this illustration, the yellow and green lesions were selected at baseline as target injected (T-I), and the purple and blue lesions were selected as target noninjected (T-NI); there are no nontarget lesions. In this simplified example, a full imaging assessment is performed at each treatment visit just before the decision about which lesions to inject at that visit. The overall response at each visit was based on the change in sum of diameters (SOD) for all the target lesions together (because there are no nontarget lesions in this example). Once progressive disease (PD) is observed (in this case, because of a new lesion), the overall response assessment thereafter is similar to that of RECIST for immunotherapeutic trials (iRECIST). The injected response is based on the change in SOD of the injected lesions from the assessment immediately before this one. The noninjected response is based on the changes in SOD from baseline and nadir and is considered nonevaluable (NE) once any lesion that was initially selected as T-NI is subsequently injected, as happens in this case with the blue lesion. If this lesion were to grow later, it could contribute to an overall response of PD. iPR, immunotherapeutic partial response; PR, partial response; SD, stable disease.
Decisions at RECIST Progression
At the time of PD as defined in RECIST 1.1, clinical assessment should determine whether continued IT immunotherapy is warranted. If clinical progression is rapid, the decision may be made to discontinue study treatment. If the patient’s condition is clinically stable as defined in iRECIST,25 it may be appropriate to continue treatment. Continuing treatment in the setting of RECIST 1.1 PD is particularly relevant with a mixed response, when injected lesions regress or disappear but a new lesion develops or when existing noninjected lesions enlarge. In such a case, as discussed, the treating physician may reprioritize which lesions to inject, favoring new or enlarging lesions, if they are deemed safe and accessible for injection.
The challenge for IT immunotherapy assessment is not only to avoid misclassification of inflammatory reactions (pseudoprogression) as disease progression but also to account for injection of new or previously noninjected lesions. Additionally, the interval to confirmatory reassessment should allow sufficient time for IT therapy to produce an effect on these lesions; we recommend allowing 4 to 12 weeks (rather than 4 to 8 weeks per iRECIST).
Management at initial radiographic progression (overall response) depends on whether new lesions appear. For clinically stable patients without new lesions, lesions should be injected if they are progressing or were previously injected, and consideration should be given to additional noninjected lesions according to the prioritization guidelines (Fig 4A).
FIG 4.

Management algorithm at initial radiographic progression (A) without and (B) with new lesions. (A) If initial radiographic progression does not involve new lesions, management depends first on whether the patient is clinically stable. If the patient has not experienced clinical decline and the physician and patient decide to continue treatment, the lesions that are enlarging (if they are accessible and can be safely injected) should be injected. Lesions that were previously classified as noninjected may be reclassified as injected at this time, although the target and nontarget categories must be strictly preserved. (B) If progression involves new lesions that are accessible and can be safely injected, they should be prioritized for injection. New lesions that are measurable can contribute to a new lesion sum of diameters for an overall response assessment that resembles RECIST for immunotherapeutic trials (iRECIST). New lesions that are injected can be evaluated as part of the iterative assessment process for injected lesions but may not contribute to the target noninjected (T-NI) tumor burden. PD, progressive disease; NT-I, nontarget injected; NT-NI, nontarget noninjected; T-I, target injected.
New lesions, if present, should be categorized as new target or new nontarget lesions (per iRECIST), and the SOD of the new lesions should be calculated for future overall response assessment. If the new lesions are inaccessible, only existing lesions should continue to be injected, including those that are enlarging and those not yet injected. If the new lesions are accessible, they should be injected according to the principles previously outlined (Fig 4B). Again, the decision to inject should be based on prioritization rules and clinician discretion (described in Guidelines for Prioritization of Lesion Injection for IT Therapy). Regardless of the presence or absence of new lesions, treatment should be discontinued in patients with clinically unstable disease.
Response Assessment After RECIST Progression
Overall response for visits after RECIST progression is determined using a process similar to iRECIST, taking into account target lesions (injected and noninjected combined), nontarget lesions (injected and noninjected combined), and new lesions, to produce overall response categories that include immunotherapeutic CR, immunotherapeutic PR, immunotherapeutic SD, immune unconfirmed PD (iUPD), and immune confirmed PD (iCPD). An additional response category is described in the next section.
Injected lesion assessment after RECIST 1.1 progression uses the same iterative process as before. At each assessment, the current SOD of all target lesions injected at the previous visit (including any new lesions classified as new lesion targets and selected for injection) should be compared with the immediately preceding SOD of the same lesions. Then, based on prioritization rules and clinician discretion, the physician determines which lesions to inject at this visit, and the SOD of these is the new comparator for the next assessment.
Noninjected response after overall progression is also assessed as it was before. As long as the T-NI lesions remain noninjected, the T-NI SOD is compared with baseline and nadir values to determine the noninjected lesion response. If any T-NI lesion must be injected (eg, because of enlargement or because of inaccessibility of other lesions), the maximal noninjected response has been achieved and any subsequent noninjected response is considered nonevaluable.
Management and Response After Confirmed Progression
If RECIST 1.1 PD has been observed and a confirmatory scan shows confirmed PD per iRECIST, it may be appropriate to continue therapy and modify the lesions for injection. As discussed, these are typically mixed responses: injected lesions are responding, but new lesions have appeared or noninjected lesions have enlarged.
For example, if baseline lesions are responding but a new lesion appears, this would be RECIST 1.1 PD (and iUPD by itRECIST). If the new lesion is injected and the next scan shows that this lesion, along with other injected lesions, has responded favorably but an additional new lesion has appeared, this would be considered iCPD by iRECIST, and therapy would be stopped. However, because the injected lesions are responding, the treating physician may decide (if the patient remains clinically stable) that the patient is deriving benefit from continued IT immunotherapy, inject the new lesion, and obtain another confirmatory scan (4-12 weeks later, based on clinical judgment).
We propose a novel response category to describe such situations, designated iTPD (with T representing therapy, which will continue for these patients). This category encompasses situations in which the iRECIST response would have been iCPD (worsening of an existing cause of PD or appearance of a new cause, after an overall response of iUPD) despite the fact that the injected lesions are stable or responding, and the treating physician reprioritizes lesions for injection and continues IT immunotherapy. The response may be designated iTPD, and IT immunotherapy may continue, with imaging every 4 to 12 weeks, until any of the following occurs (at which point the response would become iCPD per itRECIST): clinical progression with worsening signs, symptoms, or performance status; physician and/or patient decision to discontinue therapy because of intolerance; or radiographic progression, particularly in injected lesions (indicating that injection is failing to prevent growth) or physician determines another treatment is clinically indicated (eg, a lesion is impinging on the spinal cord, necessitating urgent intervention).
Final End Points and Outcomes
The proposed itRECIST allows responses after traditionally defined radiographic progression. This aligns with real-life conditions in which treatment is continued for patients exhibiting otherwise good response. Overall responses for each visit are calculated almost identically to RECIST 1.1 (and then iRECIST, after RECIST 1.1 progression) and can be used to calculate traditional end points such as objective response rate, progression-free survival, and duration of response. Maximal quantitative effect on tumor burden can be reported using a waterfall plot that includes all target lesions (injected and noninjected).
For noninjected lesion response, the best categorical response compared with baseline (eg, CR or PR27) can also contribute to a noninjected objective response rate. An additional quantitative measurement is the maximal reduction in the SOD of the T-NI lesions until any such lesion is redesignated for injection. The percentage reduction in SOD for the T-NI lesions is easily visualized in a waterfall plot (Fig 5).
FIG 5.

Sample double waterfall plot. This sample double waterfall plot shows the quantitative best response (maximal reduction in tumor burden) in the target noninjected and target injected lesions for each participant in a trial, in order of the maximal effect on the noninjected lesions.
The proposed end points for injected lesion response are necessarily novel. Because the injected lesions may change at each treatment visit, it is not meaningful to report maximal shrinkage of injected lesions compared with the chronologic baseline. The iterative injected lesion assessment at each visit does not integrate changes across all lesions to capture the maximal effect over time. Similarly, measuring the maximal effect on only the lesions initially chosen for injection may miss critical information. For example, if 1 lesion is initially chosen for injection and shrinks by 90% and 2 other lesions are injected on the next visit and do not shrink at all, it would be misleading to report that injection caused a 90% reduction in injected lesions.
The 2 outcomes that integrate all injected lesions and provide useful comparisons between IT therapies are the maximal size reduction for each injected target lesion from the time of its first injection and the time until IT therapy ceases to provide benefit to injected lesions (lesions enlarge despite injection). Thus, for each T-I lesion, the baseline is its diameter just before it is first injected, and the baseline SOD for injected lesions is the sum of these diameters, which may originate from different time points. The best response SOD is the SOD of these lesions at each lesion’s smallest size after injection (again, possibly at different visits). The maximal percentage reduction in SOD can be represented as a waterfall plot (Fig 5). In addition, the time until the first instance of PD for injected lesions (as previously described) can be reported as a time-to-event end point.
Calculations of injected and noninjected responses can be performed by off-site analysts. Investigators need only record which lesions are chosen as target and nontarget and when they inject each lesion, but they may calculate injected response to inform decisions to discontinue therapy. The Data Supplement provides case examples and case report form design suggestions.
DISCUSSION
As with any criteria modification, we anticipate that itRECIST will likely face issues of understanding and acceptance. The complexity of itRECIST may limit real-world clinical use. Ensuring that recommendations can be adapted into current practice was therefore a focus during development. Effective implementation of itRECIST in clinical trials will depend on ease of use and understanding among investigators, which could be facilitated by software developed to aid response calculations. The criteria were developed by expert consensus and will require empiric validation using historical and newly collected data to correlate itRECIST assessments with clinical outcomes. Consideration should be given to building the correlation between itRECIST and outcomes into clinical trial design. Within the proposed criteria, recommendations were not attempted for decisions lacking broad consensus. Thus, the decision about when to stop therapy in the face of enlarging or new lesions is based on clinical assessment by the treating physician. Although these guidelines were created for IT immunotherapy, similar principles may be applied to response assessment for other focal and IT treatments combined with systemic immunotherapies.
itRECIST represents an important first step toward a standardized method of response assessment for this promising and evolving therapeutic modality. Implementation and validation of itRECIST will allow the standardized evaluation of response to IT therapies while providing data for comparison across clinical trials and correlation with clinical outcomes. The proposed guidelines have been modified from RECIST 1.1 and iRECIST to be easily adopted in trial protocols and routine clinical practice without the need for complex additional assessments by treating physicians, thereby minimizing the burden on clinicians and investigators.
ACKNOWLEDGMENT
Medical writing and/or editorial assistance was provided by Matthew Grzywacz, PhD, and Holly C. Cappelli, PhD, CMPP, of the ApotheCom pembrolizumab team (Yardley, PA). This assistance was funded by Merck Sharp & Dohme (Kenilworth, NJ).
AUTHOR CONTRIBUTIONS
Conception and design: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Response Criteria for Intratumoral Immunotherapy in Solid Tumors: itRECIST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Gregory V. Goldmacher
Employment: Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA
Stock and Other Ownership Interests: Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, Beta Bionics
Anuradha D. Khilnani
Employment: Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA
Stock and Other Ownership Interests: Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA
Robert H. I. Andtbacka
Employment: Novartis (I), Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA (I), Seven and Eight Biopharmaceuticals
Stock and Other Ownership Interests: Novartis (I), Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA (I)
Honoraria: Novartis
Consulting or Advisory Role: Merck/Schering Plough, Novartis, Pfizer
Research Funding: Amgen (Inst), Viralytics (Inst), Takara Bio (Inst), OncoSec (Inst), Provectus (Inst), Idera (Inst), X4 Pharma (Inst), Oncolys BioPharma (Inst)
Jason J. Luke
Stock and Other Ownership Interests: Actym Therapeutics, Mavu Pharmaceutical, Pyxis, Alphamab Oncology, Tempest Therapeutics, Kanaph Therapeutics, Onc.AI
Consulting or Advisory Role: Amgen, Array BioPharma, Bristol Myers Squibb, Merck, EMD Serono, Benevir, Novartis, AstraZeneca/MedImmune, 7 Hills Pharma, Actym Therapeutics, Janssen, Reflexion Medical, Tempest Therapeutics, TTC Oncology, Alphamab Oncology, Mavu Pharmaceutical, Aduro Biotech, Compugen, IDEAYA Biosciences, Spring Bank, Jounce Therapeutics, Vividion, AbbVie, Akrevia, Astellas Pharma, Bayer, Immunocore, Incyte, Leap Therapeutics, Mersana, Partner Therapeutics, Synlogic, Eisai
Research Funding: Merck (Inst), Bristol Myers Squibb (Inst), Boston Biomedical (Inst), MedImmune (Inst), Incyte (Inst), Celldex (Inst), Genentech/Roche (Inst), Pharmacyclics (Inst), Five Prime Therapeutics (Inst), Corvus Pharmaceuticals (Inst), Delcath Systems (Inst), AbbVie (Inst), Immunocore (Inst), Palleon Pharmaceuticals, Checkmate Pharmaceuticals, Macrogenics (Inst), Novartis (Inst), Tesaro (Inst), Evelo Therapeutics, Xencor (Inst), Compugen (Inst), Array BioPharma, FLX Bio
Patents, Royalties, Other Intellectual Property: Serial #15/612,657 (cancer immunotherapy); Serial #PCT/US18/36052 (Microbiome biomarkers for anti-PD-1/PD-L1 responsiveness: Diagnostic, prognostic and therapeutic uses thereof)
Travel, Accommodations, Expenses: Amgen, Bristol Myers Squibb, Array BioPharma, AstraZeneca/MedImmune, Benevir, Castle Biosciences, Checkmate Pharmaceuticals, EMD Serono, Gilead Sciences, Janssen, Merck, Novartis, Reflexion Medical, IDEAYA Biosciences, Jounce Therapeutics, Immunocore, Akrevia, Mersana
F. Stephen Hodi
Employment: Dana-Farber Cancer Institute
Stock and Other Ownership Interests: Apricity, Torque
Consulting or Advisory Role: Merck Sharp & Dohme, Novartis, Genentech/Roche, EMD Serono, Sanofi, Bayer, Aduro Biotech, Pfizer, Verastem, Bristol Myers Squibb, Takeda, Surface, Compass Therapeutics, Partners Therapeutics, Pionyr, 7Hills Pharma, Torque, Rheos, Amgen, Boston Pharmaceuticals
Research Funding: Bristol-Myers Squibb (Inst), Merck Sharp & Dohme (Inst), Genentech/Roche (Inst), Novartis (Inst)
Patents, Royalties, Other Intellectual Property: Patent pending as per institutional policy; patent pending royalties received on MICA-related disorders application to institution per institutional intellectual property policy; angiopoietin-2 biomarkers predictive of anti-immune checkpoint response (Inst); compositions and methods for identification, assessment, prevention, and treatment of melanoma using PD-L1 isoforms; methods of using pembrolizumab and trebananib (Inst)
Travel, Accommodations, Expenses: Novartis, Bristol Myers Squibb
Other Relationship: Bristol Myers Squibb, Genentech/Roche
Aurelien Marabelle
Stock and Other Ownership Interests: Pegascy
Honoraria: Bristol Myers Squibb, AstraZeneca/MedImmune, Oncovir, Merieux
Consulting or Advisory Role: Lytix Biopharma, Eisai, Pierre Fabre, Merck Serono, Novartis, Amgen, Pfizer, Symphogen, AstraZeneca, Servier, Gritstone Oncology, Molecular Partners, Bayer, Partner Therapeutics, Sanofi, Roche, Boehringer Ingelheim, Redx Pharma, Sotio, Innate Pharma, Pfizer, Imcheck, Cerenis Therapeutics, Merck Sharp & Dohme, OSE Immunotherapeutics, Bioncotech
Speakers’ Bureau: Merck Sharp & Dohme, Bristol Myers Squibb, AstraZeneca, Roche
Research Funding: Merus (Inst), Bristol Myers Squibb (Inst), Boehringer Ingelheim (Inst), Transgene (Inst), Merck Sharp & Dohme (Inst)
Patents, Royalties, Other Intellectual Property: Monoclonal antibodies against CD81 (Stanford University)
Travel, Accommodations, Expenses: Bristol Myers Squibb, Merck Sharp & Dohme, Roche, AstraZeneca
Other Relationship: Pegascy
Kevin Harrington
Honoraria: Merck Sharp & Dohme (Inst), Amgen (Inst), Merck Serono (Inst), AstraZeneca/MedImmune (Inst), Boehringer Ingelheim (Inst), Bristol Myers Squibb (Inst), Pfizer (Inst), Replimune (Inst), Oncolys (Inst), Mersana Therapeutics (Inst)
Consulting or Advisory Role: Merck Sharp & Dohme (Inst), Merck Serono (Inst), AstraZeneca/MedImmune (Inst), Boehringer Ingelheim (Inst), Bristol Myers Squibb (Inst), Pfizer (Inst), Replimune (Inst)
Speakers’ Bureau: Merck Sharp & Dohme (Inst), Merck Serono (Inst), Bristol Myers Squibb (Inst)
Research Funding: AstraZeneca (Inst), Merck Sharp & Dohme (Inst), Boehringer-Ingelheim (Inst), Replimune (Inst)
Andrea Perrone
Employment: Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA
Stock and Other Ownership Interests: Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA
Other Relationship: University of Pennsylvania (I)
Archie Tse
Employment: CStone Pharmaceuticals
Leadership: CStone Pharmaceuticals
Stock and Other Ownership Interests: CStone Pharmaceuticals
David C. Madoff
Honoraria: Penumbra
Consulting or Advisory Role: Quantum Surgical, Merck, GE Healthcare
Lawrence H. Schwartz
Consulting or Advisory Role: Novartis, Regeneron
Research Funding: Merck Sharp & Dohme (Inst), Pfizer (Inst), Boehringer Ingelheim (Inst)
Patents, Royalties, Other Intellectual Property: Varian Medical Systems
No other potential conflicts of interest were reported.
REFERENCES
- 1. Amgen: FDA approves IMLYGIC (talimogene laherparepvec) as first oncolytic viral therapy in the US. https://www.amgen.com/media/news-releases/2015/10/fda-approves-imlygic-talimogene-laherparepvec-as-first-oncolytic-viral-therapy-in-the-us/
- 2.Andtbacka RH, Kaufman HL, Collichio F, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33:2780–2788. doi: 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 3.Harrington KJ, Andtbacka RH, Collichio F, et al. Efficacy and safety of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor in patients with stage IIIB/C and IVM1a melanoma: Subanalysis of the Phase III OPTiM trial. OncoTargets Ther. 2016;9:7081–7093. doi: 10.2147/OTT.S115245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Andtbacka RH, Ross M, Puzanov I, et al. Patterns of clinical response with talimogene laherparepvec (T-VEC) in patients with melanoma treated in the OPTiM phase III clinical trial. Ann Surg Oncol. 2016;23:4169–4177. doi: 10.1245/s10434-016-5286-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaufman HL, Andtbacka RHI, Collichio FA, et al. Durable response rate as an endpoint in cancer immunotherapy: Insights from oncolytic virus clinical trials. J Immunother Cancer. 2017;5:72. doi: 10.1186/s40425-017-0276-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun L, Funchain P, Song JM, et al. Talimogene laherparepvec combined with anti-PD-1 based immunotherapy for unresectable stage III-IV melanoma: A case series. J Immunother Cancer. 2018;6:36. doi: 10.1186/s40425-018-0337-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Puzanov I, Milhem MM, Minor D, et al. Talimogene laherparepvec in combination with ipilimumab in previously untreated, unresectable stage IIIB-IV melanoma. J Clin Oncol. 2016;34:2619–2626. doi: 10.1200/JCO.2016.67.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Canton DA, Shirley S, Wright J, et al. Melanoma treatment with intratumoral electroporation of tavokinogene telseplasmid (pIL-12, tavokinogene telseplasmid) Immunotherapy. 2017;9:1309–1321. doi: 10.2217/imt-2017-0096. [DOI] [PubMed] [Google Scholar]
- 9.Andtbacka RHI, Curti BD, Hallmeyer S, et al. Phase II calm extension study: Coxsackievirus A21 delivered intratumorally to patients with advanced melanoma induces immune-cell infiltration in the tumor microenvironment. J Immunother Cancer. 2015;3(suppl 2):P343. [Google Scholar]
- 10.Ribas A, Medina T, Kummar S, et al. SD-101 in combination with pembrolizumab in advanced melanoma: Results of a phase Ib, multicenter study. Cancer Discov. 2018;8:1250–1257. doi: 10.1158/2159-8290.CD-18-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aznar MA, Tinari N, Rullán AJ, et al. Intratumoral delivery of immunotherapy: Act locally, think globally. J Immunol. 2017;198:31–39. doi: 10.4049/jimmunol.1601145. [DOI] [PubMed] [Google Scholar]
- 12.Marabelle A, Tselikas L, de Baere T, et al. Intratumoral immunotherapy: Using the tumor as the remedy. Ann Oncol. 2017;28(suppl 12):xii33–xii43. doi: 10.1093/annonc/mdx683. [DOI] [PubMed] [Google Scholar]
- 13.Pitt JM, Marabelle A, Eggermont A, et al. Targeting the tumor microenvironment: Removing obstruction to anticancer immune responses and immunotherapy. Ann Oncol. 2016;27:1482–1492. doi: 10.1093/annonc/mdw168. [DOI] [PubMed] [Google Scholar]
- 14. Chon H: Effect of STING agonist on tumor immune microenvironment of non-inflamed lung cancer and efficacy of immune checkpoint blockade. J Clin Oncol 36, 2018 (suppl; abstr 178) [Google Scholar]
- 15.Wang S, Campos J, Gallotta M, et al. Intratumoral injection of a CpG oligonucleotide reverts resistance to PD-1 blockade by expanding multifunctional CD8+ T cells. Proc Natl Acad Sci USA. 2016;113:E7240–E7249. doi: 10.1073/pnas.1608555113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. doi: 10.1016/j.cell.2018.07.035. Ribas A, Dummer R, Puzanov I, et al: Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-PD-1 immunotherapy. Cell 170:1109-1119.e10, 2017 [Erratum: Cell 174:1031-1032, 2018] [DOI] [PubMed] [Google Scholar]
- 17.Chesney J, Puzanov I, Collichio F, et al. Randomized, open-label phase II study evaluating the efficacy and safety of talimogene laherparepvec in combination with ipilimumab versus ipilimumab alone in patients with advanced, unresectable melanoma. J Clin Oncol. 2018;36:1658–1667. doi: 10.1200/JCO.2017.73.7379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Andtbacka RHI, Ross MI, Agarwala SS, et al: Efficacy and genetic analysis for a phase II multicenter trial of HF10, a replication-competent HSV-1 oncolytic immunotherapy, and ipilimumab combination treatment in patients with stage IIIb-IV unresectable or metastatic melanoma. J Clin Oncol 36, 2018 (suppl; abstr 9541) [Google Scholar]
- 19.Schwartz LH, Litière S, de Vries E, et al. RECIST 1.1: Update and clarification—From the RECIST committee. Eur J Cancer. 2016;62:132–137. doi: 10.1016/j.ejca.2016.03.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wolchok JD, Hoos A, O’Day S, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: Immune-related response criteria. Clin Cancer Res. 2009;15:7412–7420. doi: 10.1158/1078-0432.CCR-09-1624. [DOI] [PubMed] [Google Scholar]
- 21.Nishino M. Immune-related response evaluations during immune-checkpoint inhibitor therapy: Establishing a “common language” for the new arena of cancer treatment. J Immunother Cancer. 2016;4:30. doi: 10.1186/s40425-016-0134-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 23.Therasse P, Eisenhauer EA, Verweij J. RECIST revisited: A review of validation studies on tumour assessment. Eur J Cancer. 2006;42:1031–1039. doi: 10.1016/j.ejca.2006.01.026. [DOI] [PubMed] [Google Scholar]
- 24.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 25.Seymour L, Bogaerts J, Perrone A, et al. iRECIST: Guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol. 2017;18:e143–e152. doi: 10.1016/S1470-2045(17)30074-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Forde PM, Chaft JE, Smith KN, et al. Neoadjuvant PD-1 blockade in resectable lung cancer. N Engl J Med. 2018;378:1976–1986. doi: 10.1056/NEJMoa1716078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Andtbacka RHI, Dummer R, Gyorki DE, et al: Interim analysis of a randomized, open-label phase 2 study of talimogene laherparepvec (T-VEC) neoadjuvant treatment (neotx) plus surgery (surgx) vs surgx for resectable stage IIIB-IVM1a melanoma (MEL). J Clin Oncol 36, 2018 (suppl; abstr 9508) [Google Scholar]