

Abstract
Ertugliflozin, a selective inhibitor of sodium-glucose cotransporter 2 (SGLT2), is approved in the US, EU, and other regions for the treatment of adults with type 2 diabetes mellitus (T2DM). This review summarizes the ertugliflozin pharmacokinetic (PK) and pharmacodynamic data obtained during phase I clinical development, which supported the registration and labeling of this drug. The PK of ertugliflozin was similar in healthy subjects and patients with T2DM. Oral absorption was rapid, with time to peak plasma concentrations (Tmax) occurring at 1 h (fasted) and 2 h (fed) postdose. The terminal phase half-life ranged from 11 to 18 h and steady-state concentrations were achieved by 6 days after initiating once-daily dosing. Ertugliflozin exposure increased in a dose-proportional manner over the tested dose range of 0.5–300 mg. Ertugliflozin is categorized as a Biopharmaceutical Classification System Class I drug with an absolute bioavailability of ~ 100% under fasted conditions. Administration of the ertugliflozin 15 mg commercial tablet with food resulted in no meaningful effect on ertugliflozin area under the plasma concentration–time curve (AUC), but decreased peak concentrations (Cmax) by 29%. The effect on Cmax is not clinically relevant and ertugliflozin can be administered without regard to food. Mild, moderate, and severe renal impairment were associated with a ≤ 70% increase in ertugliflozin exposure relative to subjects with normal renal function, and no dose adjustment in renal impairment patients is needed based on PK results. Consistent with the mechanism of action of SGLT2 inhibitors, 24-h urinary glucose excretion decreased with worsening renal function. In subjects with moderate hepatic impairment, a decrease in AUC (13%) relative to subjects with normal hepatic function was observed and not considered clinically relevant. Concomitant administration of metformin, sitagliptin, glimepiride, or simvastatin with ertugliflozin did not have clinically meaningful effects on the PK of ertugliflozin or the coadministered medications. Coadministration of rifampin decreased ertugliflozin AUC and Cmax by 39% and 15%, respectively, and is not expected to affect efficacy in a clinically meaningful manner. This comprehensive evaluation supports administration to patients with T2DM without regard to prandial status and with no dose adjustments for coadministration with commonly prescribed drugs, or in patients with renal impairment or mild-to-moderate hepatic impairment based on ertugliflozin PK.
Key Points
| This review summarizes ertugliflozin pharmacokinetic (PK) and pharmacodynamic (PD) data obtained during the phase I clinical development program for this drug. |
| The favorable PK/PD profile of ertugliflozin supports administration of ertugliflozin 5 and 15 mg doses as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus. |
| On the basis of these PK data, ertugliflozin can be administered without regard to meals and with no dose adjustments for coadministration with commonly prescribed drugs, or in patients with renal impairment or mild-to-moderate hepatic impairment. |
Introduction
Sodium-glucose cotransporter 2 (SGLT2) inhibitors are a novel class of insulin-independent antihyperglycemic agents for the treatment of type 2 diabetes mellitus (T2DM). SGLT2 is a high-capacity, low-affinity receptor that is highly expressed in the S1 segment of the proximal tubule of the kidney, where it facilitates ~ 90% of glucose reabsorption from the glomerular filtrate [1, 2]. The remaining 10% of glucose reabsorption in the kidney is mediated by SGLT1, a high-affinity, low-capacity receptor expressed in the S3 segment of the proximal renal tubule [1, 2]. SGLT2 inhibition blocks glucose reabsorption within the kidney, resulting in a lowered renal threshold for glucose and increased urinary glucose excretion (UGE), which reduces plasma glucose and glycosylated hemoglobin (HbA1c) levels in patients with hyperglycemia [1, 2]. However, the effect of SGLT2 inhibition to increase UGE is partially offset by compensatory glucose reabsorption by SGLT1 [3]. Additional clinical benefits of this therapeutic class include reductions in weight due to the caloric loss associated with glycosuria, and reductions in blood pressure due to the diuretic and natriuretic effects associated with SGLT2 inhibition [4]. Furthermore, recent clinical trial data have shown that SGLT2 inhibitors can also provide significant renal and cardiovascular (CV) benefits, with observed reductions in renal function decline, kidney-related deaths, hospitalizations for heart failure, and major adverse CV events (CV death, myocardial infarction, or stroke) [5–8].
To date, four SGLT2 inhibitors have received regulatory approval in the US and EU, as well as other countries, for the treatment of T2DM: dapagliflozin, canagliflozin, empagliflozin, and, most recently, ertugliflozin (Fig. 1; Table 1) [9–11]. In addition to ertugliflozin approval as a stand-alone therapy, it has also received separate approvals as a fixed-dose combination (FDC) with metformin and with the dipeptidyl peptidase-4 (DPP4) inhibitor sitagliptin [12–15]. Dapagliflozin, canagliflozin, and empagliflozin also have approved FDCs with metformin [16]. In addition, dapagliflozin and empagliflozin are widely available as FDCs with saxagliptin and linagliptin, respectively [16]; a canagliflozin/teneligliptin FDC has recently been approved in Japan [17]. This review will focus primarily on the phase I pharmacokinetics (PK) and pharmacodynamics (PD) of the ertugliflozin stand-alone therapy.
Fig. 1.

Chemical structure of ertugliflozin, empagliflozin, canagliflozin, and dapagliflozin
Table 1.
Summary of SGLT2 inhibitors currently approved for use in the US and EU and their relative selectivity [9, 26, 59]
| SGLT2 inhibitor | Approval year (US; EU) | SGLT2 IC50 (nM) | SGLT1 IC50 (nM) | Relative selectivity (SGLT2:SGLT1) |
|---|---|---|---|---|
| Canagliflozin | 2013; 2013 | 2.7 | 710 | ~ 260-fold |
| Dapagliflozin | 2014; 2012 | 1.2 | 1400 | ~ 1200-fold |
| Empagliflozin | 2014; 2014 | 3.1 | 8300 | ~ 2700-fold |
| Ertugliflozin | 2017; 2018 | 0.877 | 1960 | ~ 2200-fold |
IC50 50% inhibitory concentration, SGLT1 sodium-glucose cotransporter 1, SGLT2 sodium-glucose cotransporter 2
The efficacy and safety of ertugliflozin has been evaluated in the VERTIS (eValuation of ERTugliflozin effIcacy and Safety) phase III clinical trial program, which consisted of nine trials conducted in ~ 13,000 patients enrolled across more than 40 countries. In these studies, ertugliflozin—when administered once daily either as monotherapy or in conjunction with other antihyperglycemic agents in patients with T2DM—provided clinically meaningful reductions in HbA1c, body weight, and blood pressure, combined with a favorable safety and tolerability profile [18–25]. The design of these phase III studies was supported by the ertugliflozin phase I clinical development program, which included 29 studies (for ertugliflozin as well as the FDC therapies ertugliflozin/metformin and ertugliflozin/sitagliptin) that evaluated the safety, PK, PD, PK/PD relationships, biopharmaceutics, and drug–drug interactions (DDIs) in healthy subjects, subjects with T2DM, or in special populations (subjects with renal or hepatic impairment). This review provides a comprehensive summary of the clinical PK and PD properties of ertugliflozin obtained during the phase I clinical development program.
In Vitro Pharmacology
Structure and Chemical Properties
Ertugliflozin (PF-04971729/MK-8835) belongs to a new subclass of selective SGLT2 inhibitors incorporating a unique dioxa-bicyclo[3.2.1]octane (bridged ketal) ring system [26] (Fig. 1). In the commercial product, ertugliflozin is included as a cocrystal with l-pyroglutamic acid (l-PGA) in a 1:1 ratio, known as ertugliflozin∙l-PGA and described chemically as (1S,2S,3S,4R,5S)-5-[4-Chloro-3-(4-ethoxybenzyl)phenyl]-1-hydroxymethyl-6,8-dioxabicyclo[3.2.1]octane-2,3,4-triol, compound with (2S)-5-oxopyrrolidine-2-carboxylic acid [26]. The corresponding molecular formula for ertugliflozin∙l-PGA is C27H32ClNO10, with a molecular mass of 566.00 g/mol. The commercial formulation of ertugliflozin is an immediate-release tablet for oral administration available in 5 and 15 mg strengths. Ertugliflozin is categorized as a Biopharmaceutical Classification System (BCS) Class I drug based on high solubility and high permeability characteristics [27, 28]. Additionally, ertugliflozin tablets display very rapid in vitro dissolution characteristics (≥ 85% of total drug load dissolved in 15 min) over the gastrointestinal pH range (1.2–6.8) [27, 29].
Selectivity and Inhibition
In vitro, ertugliflozin exhibited high selectivity for SGLT2 over sodium-glucose cotransporter 1 (SGLT1) in a functional assay that detects the inhibition of radiolabeled methyl α-d-glucopyranoside (AMG) uptake via the SGLT1 and SGLT2 transporters expressed in Chinese hamster ovary (CHO) cells [26]. The 50% inhibitory concentration (IC50) values were 0.877 nM for human SGLT2 and 1960 nM for human SGLT1, corresponding to a > 2000-fold selectivity of ertugliflozin for SGLT2 compared with SGLT1 (Table 1) [26]. Among the various SGLT2 inhibitors, ertugliflozin and empagliflozin have the highest selectivity for SGLT2 over SGLT1 (> 2000-fold) compared with dapagliflozin and canagliflozin (Table 1).
Clinical Pharmacokinetics
First-in-Human Studies
Two randomized, placebo-controlled, double-blind, escalating-dose studies were conducted to assess the PK and PD of single oral doses of ertugliflozin in healthy subjects (administered as a solution or suspension following an overnight fast; N = 24; NCT00989079) and the PK/PD of multiple oral doses of ertugliflozin in otherwise healthy overweight/obese subjects (administered as a solution or suspension following a light breakfast; N = 40; NCT01018823) [30]. The ertugliflozin PK data obtained from these initial studies are summarized in Table 2; PD results are described in Sect. 4.1 below. Oral absorption of ertugliflozin was rapid, with median time to maximum plasma concentrations (Tmax) occurring 1.0 h postdose following single-dose administration of ertugliflozin 0.5–300 mg under fasting conditions (Fig. 2a) and 1.5–2.0 h postdose following once-daily administration of ertugliflozin 1–100 mg for 14 days after a light breakfast (Fig. 2b), followed by a biphasic decline. Mean terminal-phase half-life (t½) was consistent across doses (11–17 h across both studies). Steady-state concentrations were achieved by day 6 after initiating once-daily dosing in the multiple-dose study. The accumulation ratio ranged from 1.2–1.4 and was independent of dose [30]. Dose-normalized maximum observed plasma concentration (Cmax) and area under the plasma concentration–time curve (AUC) displayed dose proportionality following single-dose (fasted; Fig. 3a, b) or multiple-dose (fed; Fig. 3c, d) administration.
Table 2.
Summary of plasma and urine ertugliflozin pharmacokinetic parameters following single and multiple dosing [30]a
| Study and dose (mg) | Study day | nb | AUC∞ (ng·h/mL) | AUCτ (ng·h/mL) | Cmax (ng/mL) | Tmax (h) | t½ (h) | CL/F (mL/min) | Ae72% (%) |
|---|---|---|---|---|---|---|---|---|---|
| Single-dose study (single oral dose; fasted) | |||||||||
| 0.5 | 1 | 8 | 45.7 (10) | – | 7.23 (11) | 1.0 (0.5–1.5) | 11.4 (19) | 182 (11) | 0.879 (29) |
| 2.5 | 1 | 8 | 231 (22) | – | 42.8 (21) | 1.0 (0.5–1.1) | 13.1 (24) | 180 (21) | 1.08 (43) |
| 10 | 1 | 8 | 909 (15) | – | 182 (22) | 1.0 (0.5–1.5) | 17.4 (42) | 184 (16) | 0.888 (17) |
| 30 | 1 | 8 | 2810 (18) | – | 545 (24) | 1.0 (0.5–1.5) | 15.2 (33) | 178 (20) | 1.10 (46) |
| 100 | 1 | 8 | 9610 (16) | – | 1620 (16) | 1.0 (0.5–1.5) | 16.2 (36) | 174 (20) | 0.964 (20) |
| 300 | 1 | 7 | 26,400 (16) | – | 4330 (20) | 1.0 (0.5–1.5) | 13.8 (18) | 190 (18) | 1.15 (17) |
| Multiple-dose study (once-daily oral dosing; fed) | |||||||||
| 1 | 1 | 8 | – | 59.46 (12) | 7.154 (15) | 4.00 (0.983–4.02) | – | – | – |
| 14 | 8 | – | 80.85 (15) | 10.19 (15) | 2.00 (1.00–4.00) | NC (n = 0) | 206.1 (13) | – | |
| 5 | 1 | 8 | – | 361.6 (31) | 49.22 (27) | 2.00 (1.00–2.00) | – | – | – |
| 14 | 8 | – | 450.5 (35) | 50.83 (28) | 1.50 (1.00–4.03) | 12.28 (24) | 184.9 (33) | – | |
| 25 | 1 | 8 | – | 1681 (26) | 195.4 (27) | 4.00 (1.00–4.02) | – | – | – |
| 14 | 8 | – | 2045 (26) | 280.8 (28) | 2.00 (1.00–2.00) | 14.81 (41) (n = 7) | 203.7 (23) | – | |
| 100 | 1 | 8 | – | 5647 (16) | 669.2 (15) | 4.00 (1.00–4.02) | – | – | – |
| 14 | 8 | – | 7761 (17) | 1035 (25) | 2.00 (1.00–4.00) | 14.13 (14) | 214.6 (17) | – | |
Ae72% percentage of dose recovered unchanged in urine from 0 to 72 h postdose, AUC area under the plasma concentration–time curve, AUC∞ AUC from time zero extrapolated to infinite time, AUCτ AUC from time zero to time tau, the dosing interval, where tau = 24 h, CL/F apparent clearance, Cmax maximum observed plasma concentration, CV% percentage coefficient of variation, NC not calculated, t½ terminal half-life, Tmax time to maximum plasma concentration
aData are expressed as geometric mean (CV%) for all, except median (range) for Tmax and arithmetic mean (CV%) for t½
bn = number of subjects evaluated against the criteria
Fig. 2.
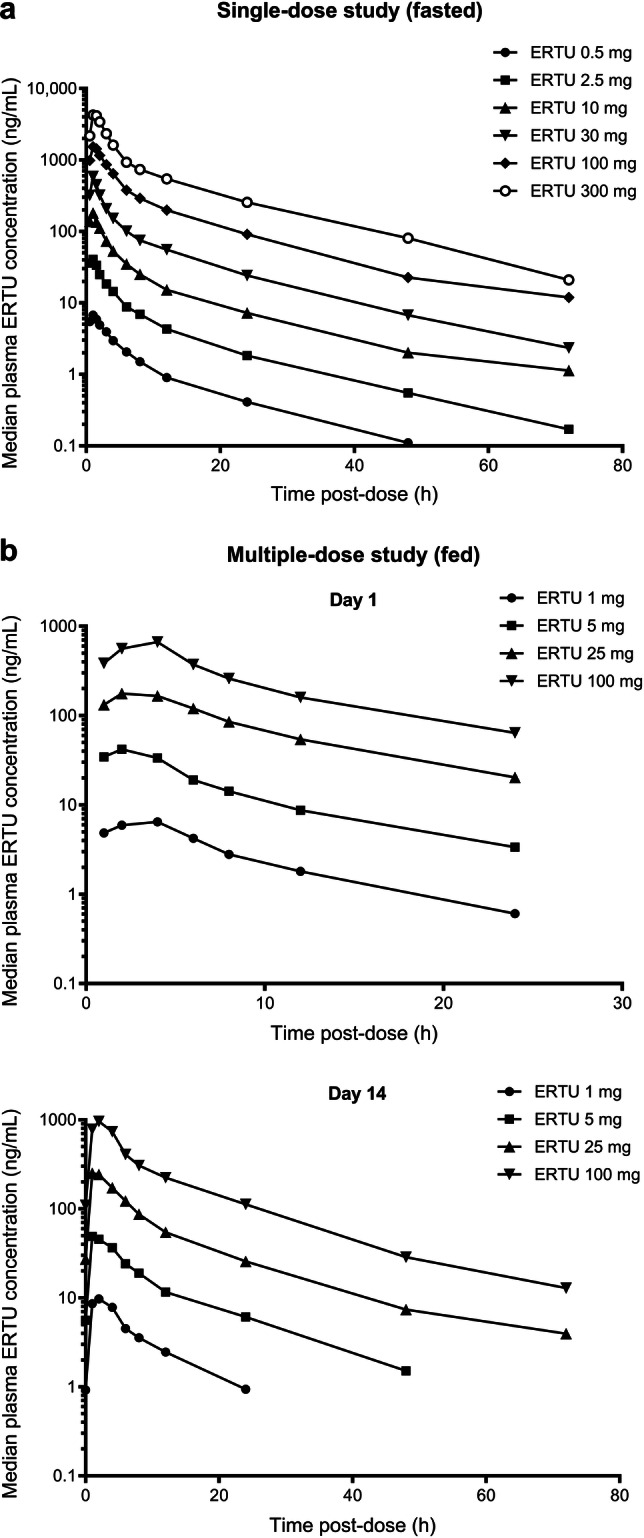
Median plasma ertugliflozin concentration–time curves following a single-dose administration under fasted conditions and b multiple-dose administration under fed conditions [30]. ERTU ertugliflozin
Fig. 3.

Dose-normalized a Cmax and b AUC∞ following single-dose administration under fasted conditions; and c Cmax and d AUCτ at day 14 following multiple-dose administration under fed conditions [30]. Open gray circles identify individual subject data; closed black circles identify arithmetic means. Box plot provides median and 25%/75% quartiles with whiskers extended to the minimum/maximum value. AUC area under the plasma concentration–time curve, AUC∞ AUC from time zero extrapolated to infinite time, AUCτ AUC from time zero to time tau, the dosing interval, where tau = 24 h, Cmax maximum observed plasma concentration, dn dose-normalized, ERTU ertugliflozin
Absorption
The results of PK studies in preclinical species suggested that ertugliflozin was well-absorbed, with an oral bioavailability (F) of 69% in rats and 94% in dogs; the fraction of the oral dose absorbed (Fa) was estimated to be ~ 75% and ~ 100%, respectively, indicating moderate-to-good permeability [31]. However, an initial mass balance study of ertugliflozin in humans estimated ertugliflozin Fa to be at least 50% [32]. Additionally, in this study, the major component in feces was unchanged ertugliflozin, accounting for 33.8% of the administered dose. To address this apparent variability in absorption, absolute oral F and Fa of ertugliflozin in humans were estimated using a two-period study design incorporating 14C-microtracer dosing in each period [27]. In this open-label, nonrandomized, fixed-sequence study (NCT02411929), eight healthy, fasted subjects received a 15 mg oral unlabeled ertugliflozin dose followed 1 h later by a 100 μg (400 nCi) intravenous 14C-ertugliflozin dose in period 1. In period 2, all subjects received a 15 mg oral unlabeled ertugliflozin dose at the same time as a 100 μg oral 14C-ertugliflozin dose. Estimated values for oral F ((AUCoral/14C-AUCiv) × (14C-Doseiv/Doseoral)) and Fa ((14C_Total_Urineoral/14C_Total_Urineiv) × (14C-Doseiv/14C-Doseoral)) were 105% and 111%, respectively, indicating that oral absorption of ertugliflozin under fasted conditions is complete and that ertugliflozin can be considered highly permeable [27].
In vitro studies using Madin–Darby canine kidney (MDCK) cells expressing multidrug resistance 1 (MDR1; also known as permeability glycoprotein [P-gp]) or breast cancer resistance protein (BCRP) genes indicate that ertugliflozin is a substrate for P-gp- and BCRP-mediated efflux [29]. However, as the oral F of ertugliflozin is ~ 100% [27] and dose-proportional increases in ertugliflozin exposure are observed over the 0.5–300 mg dose range [30], neither P-gp nor BCRP are likely to be a limiting factor for oral absorption of ertugliflozin at therapeutic doses, and inhibition of these transporters is unlikely to increase ertugliflozin exposures.
Distribution
In vitro binding studies found that ertugliflozin is extensively bound to plasma proteins in rat (~ 96%), dog (~ 97%), and human (~ 94–95%) plasma, and binding is independent of ertugliflozin concentration [31]. Blood:plasma ratios for ertugliflozin indicated preferential distribution into plasma versus red blood cells [31]. Ertugliflozin PK parameter data from the two-period 14C-microtracer study described above (Sect. 3.2) [27] demonstrated that steady-state volume of distribution (Vss) following intravenous administration of radiolabeled ertugliflozin was 85.5 L, which is indicative of moderate extravascular tissue distribution.
Metabolism
A single-dose study of 14C-ertugliflozin (25 mg/100 μCi suspension) conducted in six healthy males to characterize the metabolic profile and routes of excretion of ertugliflozin following oral administration revealed that the primary clearance (CL) mechanism of ertugliflozin is metabolism: the major metabolic pathway is glucuronidation (~ 86%), with minor contributions from oxidative metabolism (~ 12%) [32]. Two pharmacologically inactive glucuronide metabolites—ertugliflozin-2-O-β-glucuronide (M5a; PF-06685948) and ertugliflozin-3-O-β-glucuronide (M5c; PF-06481944)—are considered the primary circulating metabolites of ertugliflozin (referred to as M4a and M4c, respectively, in the study by Miao et al. [32]). An in vitro assessment of ertugliflozin metabolism indicated that the formation of M5a and M5c is likely catalyzed by the uridine 5′-diphospho-glucuronosyltransferase (UGT) enzyme isoforms UGT1A9 and UGT2B7 [31]. Ertugliflozin underwent minimal phase I metabolism to monohydroxylated metabolites and des-ethyl ertugliflozin [31] via oxidative metabolism by the cytochrome P450 (CYP) isoforms CYP3A4, CYP3A5, and CYP2C8.
Excretion/Elimination
The initial PK/PD study of single-dose oral administration of ertugliflozin 0.5–300 mg in healthy subjects under fasted conditions (Sect. 3.1) [30] found that the percentage of dose recovered unchanged in urine was negligible (Table 2). This was confirmed following a single, oral dose of 14C-ertugliflozin [32], where unchanged ertugliflozin recovered in urine accounted for 1.5% of the administered dose, indicating that renal excretion is not a major CL mechanism for ertugliflozin. The mean total recovery of radioactivity in urine and feces was 91.1% (50.2% in urine; 40.9% in feces), with target recovery (> 90%) occurring ~ 168 h postdose [32]. Glucuronide metabolites of ertugliflozin were the major urinary constituents, together accounting for 43.9% of the dose recovered in urine. The major component in feces was unchanged ertugliflozin, accounting for 33.8% of the administered dose [32]. Oxidative metabolites of ertugliflozin accounted for 4.1% of the recovered dose in feces. As absorption of ertugliflozin after an oral dose was complete in humans [27] and no significant biliary excretion of ertugliflozin was observed in preclinical animal studies [32], the unchanged ertugliflozin recovered in feces is presumed to result from glucuronide metabolites that are excreted in the bile, hydrolyzed back to the parent drug in the intestine, and eliminated via the feces. Hence, the primary CL mechanism for ertugliflozin is metabolism, with glucuronidation being the main biotransformation pathway, with minor contributions from oxidative metabolism [31, 32].
Ertugliflozin PK parameter data from the two-period 14C-microtracer study (Sect. 3.2) [27] revealed a mean CL following intravenous administration of radiolabeled ertugliflozin of 187.2 mL/min. In single- and multiple-dose studies of ertugliflozin under fasted and fed conditions, respectively [30], apparent CL (CL/F) ranged from 174 to 190 mL/min following a single oral dose of ertugliflozin 0.5–300 mg in healthy subjects, and from 185 to 215 mL/min following once-daily oral dosing of ertugliflozin 1–100 mg for 14 days in overweight/obese subjects. Thus, oral CL of ertugliflozin appears to be similar to systemic CL following administration via the intravenous route, consistent with an oral F of ~ 100%.
Effect of Food
The effect of food on the PK of the maximum approved strength of ertugliflozin (15 mg) was evaluated in an open-label, two-period, two-sequence, single-dose, crossover study where 14 healthy subjects were randomized to receive the ertugliflozin commercial tablet administered under both fasted and fed conditions [33]. During the fed phase, subjects received a standard high-fat, high-calorie breakfast, and the study drug was administered ~ 30 min after beginning the meal. Under fed conditions, the median Tmax of ertugliflozin was delayed by 1 h compared with the fasted state (2.0 h postdose fed vs. 1.0 h postdose fasted), and the Cmax for ertugliflozin was decreased by 29% compared with the fasted state (fed:fasted adjusted geometric mean ratio [GMR] 70.7 [90% confidence interval [CI] 61.7–80.9]). However, total exposure (AUC from time zero extrapolated to infinite time [AUC∞]) was comparable between the fasted and fed states for ertugliflozin, with the 90% CI of the adjusted GMR falling within the accepted bioequivalence limits of 80–125% (GMR 91.7 [90% CI 88.0–95.4]). A similar effect of food on ertugliflozin PK was observed in separate studies for the ertugliflozin/sitagliptin (15/100 mg) and ertugliflozin/metformin (7.5/1000 mg) FDC tablets [33].
As ertugliflozin efficacy is linked to total exposure rather than peak plasma concentrations, the effect of food on ertugliflozin Tmax and Cmax is not considered to be clinically relevant [33]. Taken together, these data indicate that ertugliflozin alone or as part of an FDC therapy with sitagliptin can be administered without regard to meals; however, the ertugliflozin/metformin FDC tablet should be given with meals in order to reduce the associated gastrointestinal adverse effects of metformin [34].
Pharmacokinetics (PK) of Twice-Daily Versus Once-Daily Dosing Regimens
Although ertugliflozin is approved for once-daily dosing, the ertugliflozin/metformin FDC contains an immediate-release formulation of metformin, and therefore twice-daily dosing is recommended for this combination. To assess whether steady-state ertugliflozin PK and PD (described in Sect. 4.1 below) were equivalent at the same total daily dose irrespective of whether ertugliflozin is administered twice daily or once daily, an open-label, randomized, multiple-dose, crossover study was conducted where healthy subjects (N = 50) received ertugliflozin 2.5 mg twice daily and 5 mg once daily, or ertugliflozin 7.5 mg twice daily and 15 mg once daily, for 6 days [35]. Oral absorption of ertugliflozin was rapid for both doses and both dose regimens (Table 3). AUC from time zero to 24 h (AUC24) was comparable between the dose regimens for each dose, with the 90% CIs of the adjusted GMRs (twice daily:once daily) falling within accepted bioequivalence limits (80–125%) (Table 3), indicating no clinically meaningful differences in PK between the twice-daily and once-daily regimens for 5 and 15 mg total daily doses of ertugliflozin [35].
Table 3.
Summary of plasma ertugliflozin steady-state pharmacokinetic parameters following twice-daily and once-daily dosing [35]a
| Dose and regimen | N/nb | AUC24 (ng·h/mL) | Cmax1 (ng/mL)c | Tmax1 (h)c | Cmax2 (ng/mL)d | Tmax2 (h)d | AUC24 bid:qd GMR (90% CI)e |
|---|---|---|---|---|---|---|---|
| Ertugliflozin 5 mg total daily dose | |||||||
| 2.5 mg bid | 22/20 | 399.2 (18) | 47.5 (25)f | 1.0 (0.5–1.1)f | 42.8 (28) | 2.0 (1.0–2.1) | 100.8 (98.8 − 102.8) |
| 5 mg qd | 22/22 | 397.9 (18) | 81.3 (29) | 1.0 (0.5–2.1) | – | – | |
| Ertugliflozin 15 mg total daily dose | |||||||
| 7.5 mg bid | 27/26 | 1192 (20) | 154.2 (20) | 1.0 (0.5–2.0) | 140.1 (21) | 1.0 (1.0–2.0) | 99.7 (97.1 − 102.5) |
| 15 mg qd | 28/28 | 1193 (22) | 268.2 (20) | 1.0 (0.5–2.1) | – | – | |
AUC area under the plasma concentration–time curve, AUC24 AUC from time zero to 24 h, bid twice daily, CI confidence interval, Cmax maximum observed plasma concentration, CV% percentage coefficient of variation, GMR geometric mean ratio, qd once daily, Tmax time to maximum plasma concentration
aData are expressed as geometric mean (CV%) for AUC24 and Cmax, and median (range) for Tmax. GMR (90% CI) is expressed as a percentage
bN/n = number of subjects in the treatment group/number of subjects contributing to the summary statistics
cCmax1 and Tmax1 indicate post-morning dosing for the bid regimen
dCmax2 and Tmax2 indicate post-evening dosing for the bid regimen
eAdjusted geometric means were obtained using a mixed-effects model (separate for each cohort) with sequence, period, and treatment as fixed effects and subject within sequence as a random effect. The adjusted mean difference and 90% CI were exponentiated to provide estimates of the GMR (Test:Reference [bid:qd]) and 90% CI for the ratio
fTwenty-one subjects were included in the summary statistics for Cmax1 and Tmax1 for this dose regimen
PK in Patients with Type 2 Diabetes Mellitus (T2DM)
The PK parameters of ertugliflozin were similar between healthy subjects and patients with T2DM. A phase I, open-label study (NCT01948986) in healthy subjects with normal renal function (n = 8), patients with T2DM and normal renal function (n = 6), and patients with T2DM and impaired renal function (mild, n = 8; moderate, n = 8; severe, n = 6) was conducted to assess the effect of renal impairment on the PK and PD of a single oral dose of ertugliflozin [36]; these results are discussed in further detail below (Sect. 5.1). This study also showed that in healthy subjects with normal renal function and patients with T2DM and normal renal function, rapid absorption of ertugliflozin (Tmax, 1.0 h postdose) was followed by similar Cmax, total exposure (AUC∞), and t½ values (Table 4); drug CL was also unaffected in patients with T2DM and normal renal function [36]. PD results in patients with T2DM are described in Sect. 4.2 below.
Table 4.
Summary of plasma and urine ertugliflozin pharmacokinetic parameters following single dosing in patients with renal or hepatic impairment [36, 39]a
| Patient group | nb | AUC∞ (ng·h/mL) | Cmax (ng/mL) | Tmax (h) | t½ (h) | Vz/F (L) | CL/F (mL/min) | CLR (mL/min) | Ae96% (%) | Ae48% (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| Renal impairment study | ||||||||||
| Patients with T2DM | ||||||||||
| Normal RF | 6 | 1199 (42) | 216 (35) | 1.00 (1.00–1.50) | 14.6 ± 6.4 | 240 (53) | 209 (42) | 2.09 (28) | 0.995 (55) | – |
| Mild RI | 8 | 1908 (28) | 313 (30) | 1.50 (1.00–2.00) | 25.9 ± 14.0 | 255 (50) | 131 (28) | 0.99 (45) | 0.720 (54) | – |
| Moderate RI | 8 | 2075 (19) | 306 (23) | 1.50 (0.50–2.00) | 22.9 ± 7.4 | 228 (27) | 120 (19) | 0.80 (34) | 0.646 (21) | – |
| Severe RI | 6 | 1895 (23) | 196 (28) | 1.51 (0.50–3.02) | 24.2 ± 6.0 | 269 (41) | 132 (23) | 0.54 (23) | 0.389 (40) | – |
| Healthy subjects | ||||||||||
| Normal RF | 8 | 1236 (27) | 219 (26) | 1.00 (1.00–2.00) | 17.7 ± 3.5 | 305 (39) | 202 (27) | 1.68 (33) | 0.821 (48) | – |
| Hepatic impairment study | ||||||||||
| Moderate HI patients | 8 | 1430 (39) | 251.1 (27) | 1.25 (0.50–4.00) | 14.56 ± 6.54 | 200.9 (43) | 174.8 (39) | 1.509 (38) | – | 0.8324 (59) |
| Healthy subjects | 8 | 1636 (14) | 319.0 (11) | 1.00 (1.00–2.00) | 13.77 ± 4.51 | 173.1 (40) | 152.7 (14) | 1.365 (33) | – | 0.8519 (32) |
Ae48% percentage of dose recovered unchanged in urine from 0 to 48 h postdose, Ae96% percentage of dose recovered unchanged in urine from 0 to 96 h postdose, AUC area under the plasma concentration–time curve, AUC∞ AUC from time zero extrapolated to infinite time, CL/F apparent clearance, CLR renal clearance, Cmax maximum observed plasma concentration, CV% percentage coefficient of variation, HI hepatic impairment, RF renal function, RI renal impairment, t½ terminal half-life, T2DM type 2 diabetes mellitus, Tmax time to maximum plasma concentration, Vz/F apparent volume of distribution
aData are expressed as geometric mean (CV%) for all, except median (range) for Tmax and arithmetic mean ± standard deviation for t½
bn = number of subjects
Clinical Pharmacodynamics
Effects on Urinary Glucose Excretion (UGE) in Healthy Subjects
Administration of single oral escalating doses of ertugliflozin in healthy subjects under fasted conditions (N = 24; NCT00989079) led to dose-dependent increases in cumulative 24-h UGE (UGE24) values (Fig. 4a) [30]. Similar results were observed following multiple oral escalating doses of ertugliflozin in otherwise healthy overweight/obese subjects under fed conditions (N = 40; NCT01018823), and dose-dependent increases in UGE24 values were similar on day 1 and at steady state (day 14) for the respective ertugliflozin dose groups (Fig. 4b) [30]. In healthy subjects, increases in UGE occurred without changes in serum glucose levels (unpublished data).
Fig. 4.
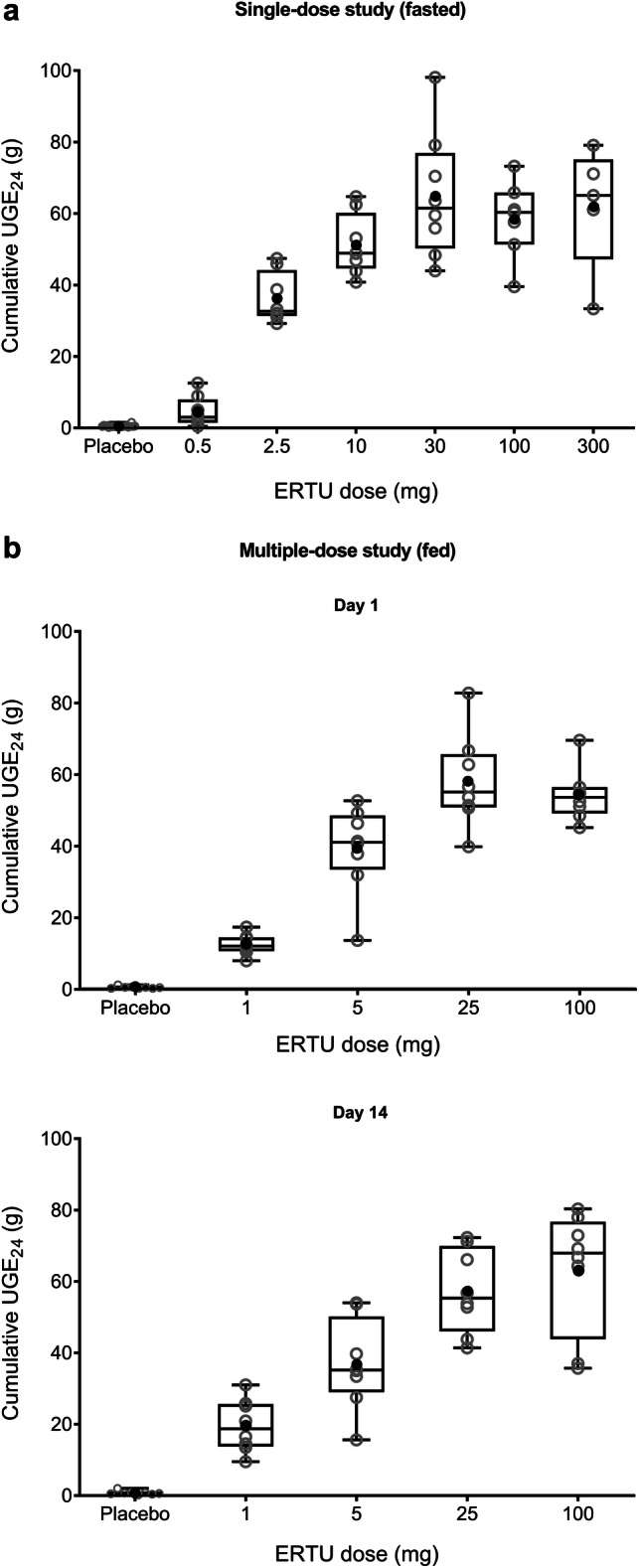
Cumulative UGE24 values following a single-dose administration under fasted conditions and b multiple-dose administration under fed conditions [30]. Open gray circles identify individual subject data; closed black circles identify arithmetic means. Box plot provides median and 25%/75% quartiles with whiskers extended to the minimum/maximum value. ERTU ertugliflozin, UGE24 urinary glucose excretion over 0–24 h
A consistent PD profile was observed for steady-state ertugliflozin irrespective of whether it was administered twice daily or once daily in healthy subjects (N = 50) who received ertugliflozin 2.5 mg twice daily and 5 mg once daily, or ertugliflozin 7.5 mg twice daily and 15 mg once daily, for 6 days [35]. Mean UGE over the 0–6, 6–12, 12–18, and 18–24 h time intervals after the morning dose on day 6 were similar between the twice-daily and once-daily regimens for both total daily dose cohorts (Fig. 5). Mean UGE24 on day 6 ranged from 52.5 to 58.6 g across dose regimens and dose cohorts [35]. The GMR (90% CI) of UGE24 values for twice-daily versus once-daily administration of a total daily dose of 5 or 15 mg were 110.2% (103.0–117.9%) and 102.8% (97.7–108.1%), respectively, with the 90% CIs falling within the accepted range for bioequivalence (80–125%) [35].
Fig. 5.
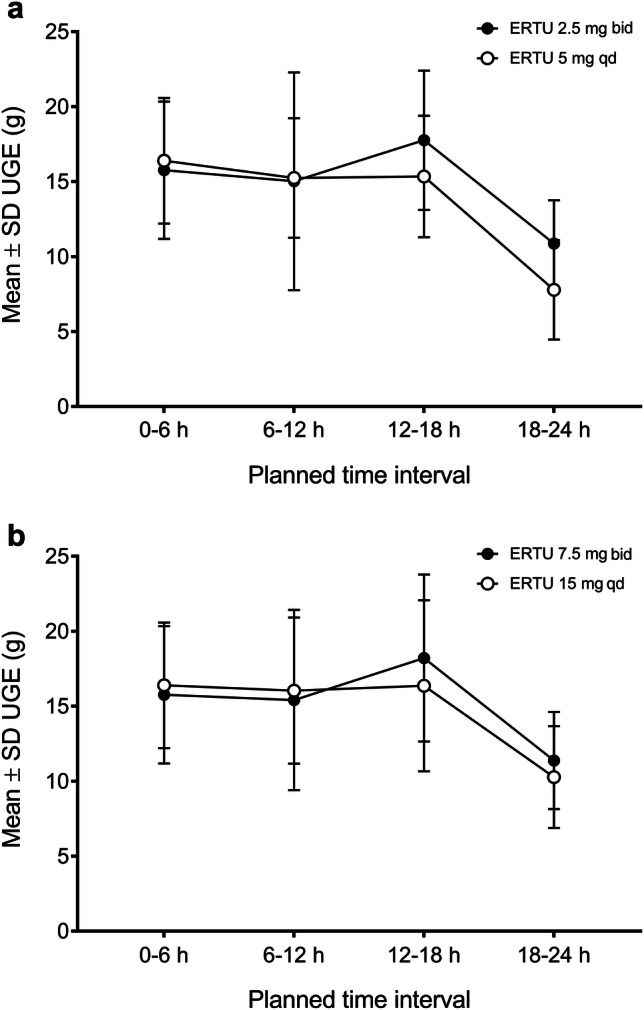
Mean ± SD UGE over time intervals for a ertugliflozin 2.5 mg bid/5 mg qd, and b ertugliflozin 7.5 mg bid/15 mg qd. Figure redrawn from Dawra et al. [35] (licensed under CC BY 4.0). bid twice daily, ERTU ertugliflozin, qd once daily, SD standard deviation, UGE urinary glucose excretion
Effects on UGE in Patients with T2DM
In a phase I, open-label study evaluating the PK, PD, and tolerability of a single oral dose of ertugliflozin 15 mg in healthy subjects with normal renal function and patients with T2DM with or without renal impairment (N = 36; NCT01948986), median change from baseline UGE24 values were lower in healthy subjects (45.8 g; n = 8) than in the subset of patients with T2DM and normal renal function (68.1 g; n = 6) following ertugliflozin administration [36]. These observations were expected based on the higher circulating glucose levels in patients with T2DM. In T2DM patients with normal renal function, the increase in UGE24 was accompanied by a decrease in plasma glucose levels (unpublished data).
Special Populations
Patients with Renal Impairment
As the mechanism of action of SGLT2 inhibitors relies on glucose filtration through the kidney, the effect of renal impairment on the PK and PD of a single oral dose of ertugliflozin 15 mg was assessed in a phase I, open-label study (NCT01948986) in healthy subjects with normal renal function (n = 8), patients with T2DM and normal renal function (n = 6), and patients with T2DM and impaired renal function (mild, n = 8; moderate, n = 8; severe, n = 6) [36]. Renal function was based on estimated glomerular filtration rate calculated using the four-variable Modification of Diet in Renal Disease equation, was not normalized for body surface area, and was defined as normal renal function, ≥ 90 mL/min; mild renal impairment, 60–89 mL/min; moderate renal impairment, 30–59 mL/min; or severe renal impairment, < 30 mL/min. The PK parameters of ertugliflozin were similar between healthy subjects and patients with T2DM and normal renal function (Table 4) [36]. Ertugliflozin was rapidly absorbed across all groups, with a median Tmax of 1.00–1.51 h. In patients with T2DM and impaired renal function, mean t½ values for ertugliflozin were slightly prolonged compared with healthy subjects and patients with T2DM and normal renal function (23–26 h vs. 15–18 h, respectively). The percentage of dose recovered unchanged in urine from 0 to 96 h postdose (Ae96%) was ~ 1% in subjects with normal renal function, and decreased as renal function decreased (Table 4). Based on log-linear regression analyses, predicted mean AUC∞ values for ertugliflozin in patients with T2DM and mild, moderate, or severe renal impairment were ~ 1.2-, 1.4-, and 1.7-fold higher, respectively, compared with subjects with normal renal function; similar results were obtained with a categorical analysis based on one-way analysis of variance [36]. These increases in ertugliflozin exposure with renal impairment are not considered clinically relevant and no dose adjustment is required in patients with renal impairment from a PK perspective.
With respect to PD effects of renal impairment following ertugliflozin administration, change from baseline in UGE24 decreased with decreasing renal function, as expected from the mechanism of action of this drug class [36]. For patients with T2DM and mild, moderate, or severe renal impairment, respective median UGE24 values were ~ 53%, 42%, and 15% of the median UGE24 value in patients with T2DM and normal renal function. Despite these reductions, considerable glycosuria was observed in patients with T2DM and mild or moderate renal impairment, with median UGE24 values of 36.4 g and 28.8 g, respectively. However, it is well-recognized that HbA1c lowering for SGLT2 inhibitors is diminished in patients with moderate or severe renal impairment [37, 38].
Patients with Hepatic Impairment
As glucuronidation, primarily occurring in the liver, is the main biotransformation pathway for ertugliflozin [31, 32], the effect of hepatic impairment on the PK of a single oral dose of ertugliflozin 15 mg was assessed in a phase I, open-label study (NCT02115347) in healthy subjects (n = 8) and patients with moderate hepatic impairment (Child–Pugh score 7–9; n = 8) [39]. The PK parameters of ertugliflozin were similar between healthy subjects and patients with impaired hepatic function (Table 4) [39]. Ertugliflozin was rapidly absorbed, with a median Tmax of 1.00 h in healthy subjects and 1.25 h in patients with hepatic impairment. Mean t½ values for ertugliflozin were similar between groups (~ 14 h), as were CL/F and renal CL (CLR) (Table 4). The percentage of dose recovered unchanged in urine from 0 to 48 h postdose (Ae48%) was < 1% in both groups. Comparing patients with impaired hepatic function versus healthy subjects, adjusted GMR (90% CI) was 87.4% (68.1–112.2%) for AUC∞ and 78.7% (65.7–94.2%) for Cmax. The unbound fraction of ertugliflozin in plasma was similar in healthy subjects (0.034) and patients with impaired hepatic function (0.037), as were the total and peak exposures of unbound ertugliflozin [39]. This small effect of moderate hepatic impairment on ertugliflozin PK is not considered to be clinically relevant, and no adjustments of ertugliflozin dose are required in patients with T2DM and mild or moderate hepatic impairment. There is currently no clinical experience of ertugliflozin use in patients with Child–Pugh class C (severe) hepatic impairment.
Drug–Drug Interaction Studies
Overview
As ertugliflozin is primarily metabolized via glucuronidation by UGT1A9 and UGT2B7, with a minor contribution from oxidation by CYP3A4 and CYP3A5 [31, 32], an open-label, two-period, fixed-sequence study was conducted in healthy subjects to assess the effect of multiple doses (600 mg) of rifampin—an inducer of drug-metabolizing enzymes, including UGT and CYP isozymes—on the PK of a single dose (15 mg) of ertugliflozin [40]. In addition, as SGLT2 inhibitors will likely be used concomitantly with other antidiabetic agents, such as metformin, sitagliptin, and glimepiride, it is important to evaluate possible DDIs between these medications [41]. The potential for DDIs between ertugliflozin 15 mg and sitagliptin 100 mg (N = 12), metformin 1000 mg (N = 18), glimepiride 1 mg (N = 18), or simvastatin 40 mg (N = 18), was assessed in four separate open-label, randomized, single-dose, crossover studies conducted in healthy adults [42]. The results of these DDI studies are summarized below and in Table 5. A brief summary of the in vitro assessment of the potential for DDIs is also given.
Table 5.
| Study and treatment | N/nb | AUC∞ (ng·h/mL)c | AUClast (ng·h/mL)c | Cmax (ng/mL)d | Tmax (h) | t½ (h) | AUC∞ coadmin:alone GMR (90% CI) | Cmax coadmin:alone GMR (90% CI) |
|---|---|---|---|---|---|---|---|---|
| Ertugliflozin–rifampin study | ||||||||
| Ertugliflozin PK | ||||||||
| Ertugliflozin 15 mg sd | 12/12 | 1370 (30) | 1350 (31) | 236.1 (38) | 1.00 (1.00–3.00) | 12.3 ± 2.9 | 61.2 (57.2–65.4) | 84.6 (74.2–96.5) |
| Ertugliflozin 15 mg sd + rifampin 600 mg qd | 12/12 | 838.1 (21) | 828.5 (22) | 199.8 (40) | 1.00 (0.50–3.08) | 9.2 ± 2.8 | ||
| Ertugliflozin–sitagliptin study | ||||||||
| Ertugliflozin PK | ||||||||
| Ertugliflozin 15 mg | 12/12 | 1413 (26) | 1385 (26) | 262.9 (25) | 1.00 (1.00–3.00) | 12.63 ± 5.15 | 102.3 (99.7–104.9) | 98.2 (91.2–105.7) |
| Ertugliflozin 15 mg + sitagliptin 100 mg | 12/12 | 1445 (25) | 1412 (24) | 258.1 (26) | 1.00 (0.50–2.10) | 14.17 ± 4.55 | ||
| Sitagliptin PK | ||||||||
| Sitagliptin 100 mg | 12/12 | 6.882 (21)c | 6.814 (21)c | 792.0 (24)d | 2.00 (1.00–4.00) | 11.00 ± 2.89 | 101.7 (98.4–105.0) | 101.7 (91.7–112.8) |
| Ertugliflozin 15 mg + sitagliptin 100 mg | 12/12 | 6.997 (20)c | 6.912 (21)c | 805.3 (24)d | 3.00 (1.00–6.00) | 11.79 ± 2.98 | ||
| Ertugliflozin–metformin study | ||||||||
| Ertugliflozin PK | ||||||||
| Ertugliflozin 15 mg | 18/17 | 1363 (24) | 1346 (23) | 272.3 (24) | 1.02 (1.00–2.00) | 11.79 ± 2.34 | 100.3 (97.4–103.3) | 97.1 (88.8–106.3) |
| Ertugliflozin 15 mg + metformin 1000 mg | 18/17 | 1388 (23) | 1367 (22) | 264.5 (20) | 1.29 (1.00–3.00) | 13.48 ± 4.65 | ||
| Metformin PK | ||||||||
| Metformin 1000 mg | 18/13 | 12,770 (27) | 12,550 (26) | 1983 (26) | 2.00 (0.50–4.00) | 10.23 ± 2.39 | 100.9 (90.6–112.4) | 94.0 (82.9–106.6) |
| Ertugliflozin 15 mg + metformin 1000 mg | 18/13 | 12,260 (27) | 12,270 (23) | 1835 (26) | 2.00 (1.00–3.00) | 14.47 ± 6.94 | ||
| Ertugliflozin–glimepiride study | ||||||||
| Ertugliflozin PK | ||||||||
| Ertugliflozin 15 mg | 17e/17 | 1225 (19) | 1210 (19) | 143.8 (17) | 2.0 (1.5–3.0) | 10.63 ± 2.44 | 102.1 (97.2–107.3) | 98.2 (92.2–104.6) |
| Ertugliflozin 15 mg + glimepiride 1 mg | 16f/16 | 1272 (19) | 1256 (19) | 144.3 (20) | 2.0 (1.5–3.0) | 11.27 ± 3.28 | ||
| Glimepiride PK | ||||||||
| Glimepiride 1 mg | 18/13 | 202.3 (66) | 174.4 (73) | 29.42 (64) | 3.00 (1.00–12.0) | 5.89 ± 2.79 | 109.8 (98.1–122.9) | 97.4 (71.1–133.5) |
| Ertugliflozin 15 mg + glimepiride 1 mg | 16f/11 | 223.8 (78) | 231.7 (64) | 30.13 (52) | 4.00 (1.50–12.0) | 6.68 ± 4.02 | ||
| Ertugliflozin–simvastatin study | ||||||||
| Ertugliflozin PK | ||||||||
| Ertugliflozin 15 mg | 18/18 | 1371 (24) | 1348 (25) | 267.0 (23) | 1.5 (1.0–2.5) | 12.34 ± 3.07 | 102.4 (99.6–105.3) | 105.2 (98.3–112.5) |
| Ertugliflozin 15 mg + simvastatin 40 mg | 18/18 | 1404 (27) | 1378 (26) | 280.8 (28) | 1.0 (1.0–2.0) | 12.58 ± 3.98 | ||
| Simvastatin PK | ||||||||
| Simvastatin 40 mg | 18/12 | 39.28 (55) | 36.28 (72) | 7.914 (63) | 1.00 (0.50–12.0) | 5.88 ± 1.96 | 123.8 (90.9–168.7) | 119.1 (97.2–145.8) |
| Ertugliflozin 15 mg + simvastatin 40 mg | 18/18 | 46.88 (89) | 45.11 (90) | 9.421 (81) | 1.25 (0.50–12.00) | 7.44 ± 2.72 | ||
| Simvastatin acid PK | ||||||||
| Simvastatin 40 mg | 18/16 | 23.49 (107) | 23.03 (110) | 1.803 (106) | 4.00 (1.50–12.0) | 8.44 ± 6.00 | 130.5 (108.3–157.1) | 115.7 (95.7–139.7) |
| Ertugliflozin 15 mg + simvastatin 40 mg | 18/14 | 38.35 (78) | 29.47 (125) | 2.085 (117) | 4.00 (2.50–8.00) | 8.60 ± 2.91 | ||
AUC area under the plasma concentration–time curve, AUC∞ AUC from time zero extrapolated to infinite time, AUClast AUC from time zero to time of the last quantifiable concentration, CI confidence interval, Cmax maximum observed plasma concentration, coadmin coadministered, CV% percentage coefficient of variation, GMR geometric mean ratio, PK pharmacokinetics, qd once daily, sd single dose, t½ terminal half-life, Tmax time to maximum plasma concentration
aData are expressed as geometric mean (CV%) for all, except median (range) for Tmax and arithmetic mean ± standard deviation for t½. GMR (90% CI) is expressed as a percentage
bN/n = number of subjects contributing to the summary statistics/number of subjects with reportable t½ and AUC∞
cAUC values for sitagliptin are reported in µM·h
dCmax for sitagliptin is reported in nM
eData for one subject were excluded from the analysis due to the occurrence of vomiting within 2 × the median Tmax for the treatment
fData for two subjects were excluded from the analysis due to the occurrence of vomiting close to/within 2 × the median Tmax for the treatments
In Vitro Assessment of Drug Metabolism Enzymes and Transporter Proteins
As oxidative metabolism via the CYP isozymes CYP3A4, CYP3A5, and CYP2C8 plays a minimal role in ertugliflozin biotransformation [31, 32], it is unlikely that coadministration of ertugliflozin with drugs that are CYP inhibitors or inducers will affect the PK of ertugliflozin. In vitro, ertugliflozin did not demonstrate any clinically relevant inhibition or induction of common drug-metabolizing enzymes (CYP and UGT isozymes) [29, 31, 43], or of various efflux/uptake transporters (P-gp, BCRP, organic anion transporter [OAT], organic anion transporting polypeptide [OATP], and organic cation transporter [OCT] isoforms) [29, 31]. Therefore, it is unlikely that coadministration of ertugliflozin will affect the PK of substrates for these enzymes and transporters.
Effect of Coadministered Medications on the PK of Ertugliflozin
The PK parameters of a single oral dose of ertugliflozin 15 mg administered alone or coadministered with multiple doses of rifampin 600 mg are shown in Table 5 [40]. The mean t½ of ertugliflozin was reduced by ~ 3 h in the presence of steady-state rifampin; AUC and Cmax values were also reduced (Table 5). Adjusted GMRs (90% CI) for ertugliflozin AUC∞ and Cmax values were 61.2% (57.2–65.4%) and 84.6% (74.2–96.5%), respectively [40]. Ertugliflozin dose–HbA1c response modeling was used to evaluate the impact of reduced ertugliflozin exposures of this magnitude on glycemic efficacy [40]. The model predicted that meaningful glycemic efficacy would be maintained with ertugliflozin at both doses (5 and 15 mg) despite the reduction in ertugliflozin exposure following coadministration with rifampin [40]. The estimated dose for half-maximal effect (ED50) from the dose–response model was 1.30 mg, with the lowest dose (5 mg) of ertugliflozin predicted to provide a placebo-corrected change in HbA1c from baseline of more than −0.6% even when coadministered with rifampin [40]. Hence, no adjustment of ertugliflozin dose would be required should ertugliflozin be administered concomitantly with a drug that is a known inducer of UGT/CYP enzymes.
The PK parameters of a single oral dose of ertugliflozin 15 mg were unaffected when administered in combination with a single oral dose of either sitagliptin 100 mg, metformin 1000 mg, glimepiride 1 mg, or simvastatin 40 mg (Table 5) [42]. The 90% CIs for the adjusted GMR of ertugliflozin AUC∞ and Cmax were within accepted bioequivalence limits (80–125%), indicating that there was no clinically meaningful effect of coadministration of ertugliflozin with sitagliptin, metformin, glimepiride, or simvastatin on ertugliflozin PK [42].
Effect of Ertugliflozin on the PK of Coadministered Medications
The PK parameters of single oral doses of sitagliptin 100 mg or metformin 1000 mg were unaffected when administered in combination with a single oral dose of ertugliflozin 15 mg (Table 5) [42]. The 90% CIs for the adjusted GMRs of sitagliptin and metformin AUC∞ and Cmax values were within accepted bioequivalence limits (80–125%), indicating that coadministration of sitagliptin or metformin with ertugliflozin had no clinically meaningful effect on their PK [42]. The PK parameters of a single oral dose of glimepiride 1 mg were broadly similar when administered alone or in combination with a single oral dose of ertugliflozin 15 mg (Table 5) [42]. Although the 90% CI for the adjusted GMR of glimepiride AUC∞ fell within accepted bioequivalence limits (109.8% [98.1–122.9%]), the 90% CI for the adjusted GMR of Cmax fell outside these limits (97.4% [71.1–133.5%]). Glimepiride plasma concentration–time profiles exhibited a double peak, resulting in high variability in Cmax values, with median Tmax ranging from 1.00–12.0 h. However, the overall lack of an effect of ertugliflozin on total and peak exposure of glimepiride suggests that ertugliflozin had no clinically meaningful effect on glimepiride PK following coadministration [42]. With respect to the effect of ertugliflozin coadministration on the PK of simvastatin and its active metabolite simvastatin acid, the adjusted GMRs of simvastatin AUC∞ and Cmax values were increased (by ~ 24% and 19%, respectively), as were the adjusted GMRs of simvastatin acid AUC∞ and Cmax (by ~ 30% and 16%, respectively), following concomitant administration of simvastatin and ertugliflozin (Table 5) [42]. The modest increases in simvastatin and simvastatin acid exposure observed following coadministration with ertugliflozin are not considered to be clinically relevant [42].
Safety
General Safety Findings from the Phase I Studies
The ertugliflozin phase I program included 29 studies and a total of ~ 690 subjects who received at least one dose of ertugliflozin (≤ 4 mg up to 300 mg), either alone or in combination with another drug. Ertugliflozin was generally safe and well-tolerated across the phase I program. There were no deaths, serious adverse events (AEs), or severe AEs in healthy phase I subjects. A comprehensive assessment of pooled safety outcomes from the phase III clinical trial program has demonstrated that ertugliflozin is safe and well-tolerated at both the 5 and 15 mg approved doses, with a safety profile that is generally consistent with other members of the SGLT2-inhibitor class [29, 44].
Thorough QTc Study
To evaluate the potential effects of a supratherapeutic dose of ertugliflozin on prolongation of the cardiac QT interval, a randomized, three-treatment, six-sequence, three-period, crossover, placebo- and active-controlled study was conducted in 42 healthy subjects where fasted subjects received a single oral dose of ertugliflozin 100 mg (~ 6.7-fold greater than the highest ertugliflozin dose of 15 mg used in phase III studies), moxifloxacin 400 mg as a positive control, or placebo [45]. Following treatment with ertugliflozin, the maximum least squares mean (90% CI) difference in QT interval corrected for heart rate (QTc) using the Fridericia correction (QTcF) observed between ertugliflozin and placebo was 2.99 ms, which was less than the threshold of potential clinical concern of 5 ms. Moreover, the upper bounds of the two-sided 90% CIs were < 10 ms at all measurements postdose [45]. No clinically significant changes in electrocardiogram parameters were detected in any of the subjects receiving ertugliflozin; therefore, a lack of an effect of ertugliflozin on the QTcF interval was demonstrated in this study. Given the known PK profile of ertugliflozin in healthy subjects, in patients with renal or hepatic impairment, and in the presence of interacting concomitant medications, this supratherapeutic, 100 mg dose of ertugliflozin was expected to adequately cover the extremes of individual exposures that might be obtained at the therapeutic doses of ertugliflozin of 5 and 15 mg.
Summary, Perspectives, and Conclusions
This review summarizes the PK/PD properties of ertugliflozin obtained during the phase I clinical development program. Ertugliflozin has an oral F of ~ 100%, a t½ of 11–18 h, allowing once-daily administration, and dose-proportional and time-independent PK over the 0.5–300 mg single-dose range and 1–100 mg multiple-dose range. Ertugliflozin is rapidly absorbed following oral administration, with Tmax occurring at 1–2 h postdose. Ertugliflozin undergoes minimal renal excretion, with the primary CL mechanism being metabolism via glucuronidation to pharmacologically inactive metabolites. No clinically significant changes in the PK of ertugliflozin alone, or as an FDC therapy with sitagliptin or metformin, were observed following administration with food; however, due to the gastrointestinal adverse effects associated with metformin, it is recommended that the ertugliflozin/metformin FDC be taken with meals. The PK profile of ertugliflozin was similar in healthy subjects and patients with T2DM. A lack of clinically significant changes in ertugliflozin PK indicates that dose adjustment is not necessary in patients with renal impairment or mild-to-moderate hepatic impairment. Coadministration of ertugliflozin with medications commonly prescribed in patients with T2DM did not affect ertugliflozin PK, and ertugliflozin did not produce clinically meaningful alterations in the PK of these coadministered drugs. Dose–response modeling indicates that clinically meaningful glycemic efficacy would be maintained following coadministration of the 5 or 15 mg dose of ertugliflozin with rifampin, or other drug inducers of UGT/CYP enzymes. Ertugliflozin induces dose-dependent increases in UGE in healthy subjects. Change from baseline UGE24 decreased in patients with T2DM as renal impairment increased, which is to be expected from the mechanism of action of this drug class. There is a diminution in HbA1c lowering with SGLT2 inhibitors as renal function declines, with no meaningful HbA1c lowering with this class in patients with severe renal impairment [37, 38]. However, favorable effects on blood pressure lowering, along with improved CV and renal outcomes, have been noted with certain SGLT2 inhibitors in patients with moderate renal impairment [4, 5, 38]. At present, different dosing recommendations for SGLT2 inhibitors exist for patients with renal impairment in various countries, and prescribers are advised to check the approved labeling in their respective regions.
As a therapeutic class, SGLT2 inhibitors have demonstrated additional clinical benefits beyond HbA1c lowering, with particular interest around the potential for a CV benefit in patients with T2DM. In CV outcomes trials, empagliflozin [46], canagliflozin [47], and dapagliflozin [48] significantly reduced the occurrence of major adverse CV events and hospitalizations for heart failure in T2DM patients. Furthermore, in patients with heart failure, dapagliflozin reduced the risk of worsening heart failure or CV death irrespective of the presence or absence of T2DM at baseline [7]. Additional trials in heart failure patients are planned to further assess the potential cardioprotective effect of SGLT2 inhibitors in this high-risk patient group [49, 50], including one study specifically enrolling heart failure patients without diabetes [51]. The ongoing VERTIS CV trial, designed to assess the effect of ertugliflozin treatment on CV and renal outcomes in 8246 patients with T2DM and established CV disease [52], is due to report out in 2020 and will provide additional insight on the effect of this class to reduce CV risk.
The mechanism(s) underlying the observed CV benefit of SGLT2 inhibition in T2DM are unclear, but appear to be independent of reductions in glucose or traditional CV risk factors, such as lipids and blood pressure [53]. Potential hypotheses include inhibition of the sodium–hydrogen exchanger (NHE) in the heart and/or kidney, with associated reductions in cardiac injury as well as diuretic and natriuretic effects [54, 55]; improvements in myocardial energy metabolism leading to enhanced cardiac function [55]; and reduced cardiac inflammation via attenuated activation of the nucleotide-binding domain-like receptor protein 3 (NLRP3) inflammasome [56]. Hemodynamic changes related to plasma volume contraction resulting in decreased circulatory load, and differential regulation of interstitial versus intravascular volume leading to reduced cardiac congestion, have also been postulated as potential mediators of the beneficial effect of SGLT2 inhibition on CV (particularly heart failure) risk [57, 58]. Further investigations are required to elucidate the mechanistic interplay between T2DM, SGLT2 inhibition, and CV risk reduction.
In conclusion, the favorable PK/PD profile of ertugliflozin across the phase I studies described in this review supported the registration and approval of ertugliflozin 5 and 15 mg doses as an adjunct to diet and exercise to improve glycemic control in adults with T2DM.
Compliance with Ethical Standards
Funding
The studies described in this review were sponsored by Pfizer Inc., New York, NY, USA, in collaboration with Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA (MSD). Editorial support was provided by Shirley Smith, PhD, of Engage Scientific Solutions, and was funded by Pfizer Inc. and MSD.
Conflict of interest
Daryl J. Fediuk, Gianluca Nucci, Vikas Kumar Dawra, Neeta B. Amin, Steven G. Terra, and Vaishali Sahasrabudhe are employees of Pfizer Inc. and may own shares/stock options in Pfizer Inc. Rebecca A. Boyd was an employee of Pfizer Inc. at the time the studies described in this review were conducted. David L. Cutler and Rajesh Krishna were employees of MSD at the time the studies described in this review were conducted and may own stock in Merck & Co., Inc., Kenilworth, NJ, USA.
Footnotes
David L. Cutler, Rebecca A. Boyd, Rajesh Krishna: Affiliation at the time of conduct of the studies described in this review.
References
- 1.DeFronzo RA, Davidson JA, Del Prato S. The role of the kidneys in glucose homeostasis: a new path towards normalizing glycaemia. Diabetes Obes Metab. 2012;14(1):5–14. doi: 10.1111/j.1463-1326.2011.01511.x. [DOI] [PubMed] [Google Scholar]
- 2.Ferrannini E, Solini A. SGLT2 inhibition in diabetes mellitus: rationale and clinical prospects. Nat Rev Endocrinol. 2012;8(8):495–502. doi: 10.1038/nrendo.2011.243. [DOI] [PubMed] [Google Scholar]
- 3.DeFronzo RA, Norton L, Abdul-Ghani M. Renal, metabolic and cardiovascular considerations of SGLT2 inhibition. Nat Rev Nephrol. 2017;13(1):11–26. doi: 10.1038/nrneph.2016.170. [DOI] [PubMed] [Google Scholar]
- 4.Thomas MC, Cherney DZI. The actions of SGLT2 inhibitors on metabolism, renal function and blood pressure. Diabetologia. 2018;61(10):2098–2107. doi: 10.1007/s00125-018-4669-0. [DOI] [PubMed] [Google Scholar]
- 5.Zelniker TA, Wiviott SD, Raz I, Im K, Goodrich EL, Bonaca MP, et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. Lancet. 2019;393(10166):31–39. doi: 10.1016/S0140-6736(18)32590-X. [DOI] [PubMed] [Google Scholar]
- 6.Perkovic V, Jardine MJ, Neal B, Bompoint S, Heerspink HJL, Charytan DM, et al. Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N Engl J Med. 2019;380(24):2295–2306. doi: 10.1056/NEJMoa1811744. [DOI] [PubMed] [Google Scholar]
- 7.McMurray JJV, Solomon SD, Inzucchi SE, Kober L, Kosiborod MN, Martinez FA, et al. Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med. 2019;381(21):1995–2008. doi: 10.1056/NEJMoa1911303. [DOI] [PubMed] [Google Scholar]
- 8.Scheen AJ. Implications of the recent CVOTs in type 2 diabetes. Impact on guidelines: the endocrinologist point of view. Diabetes Res Clin Pract. 2020;159:107726. doi: 10.1016/j.diabres.2019.05.005. [DOI] [PubMed] [Google Scholar]
- 9.Haas B, Eckstein N, Pfeifer V, Mayer P, Hass MD. Efficacy, safety and regulatory status of SGLT2 inhibitors: focus on canagliflozin. Nutr Diabetes. 2014;4:e143. doi: 10.1038/nutd.2014.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.US Food and Drug Administration; Merck Sharp & Dohme Corp., Whitehouse Station, NJ, USA. Steglatro™ (ertugliflozin): prescribing information. 2017. Available at: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&applno=209803.
- 11.European Medicines Agency; Merck Sharp & Dohme Ltd, Hoddesdon, UK. Steglatro™ (ertugliflozin): summary of product characteristics. 2018. Available at: https://www.ema.europa.eu/en/medicines/human/EPAR/steglatro.
- 12.US Food and Drug Administration; Merck Sharp & Dohme Corp., Whitehouse Station, NJ, USA. Segluromet™ (ertugliflozin/metformin): prescribing information. 2017. https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&applno=209806.
- 13.US Food and Drug Administration; Merck Sharp & Dohme Corp., Whitehouse Station, NJ, USA. Steglujan™ (ertugliflozin/sitagliptin): prescribing information. 2017. Available at: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&applno=209805.
- 14.European Medicines Agency; Merck Sharp & Dohme Ltd, Hoddesdon, UK. Segluromet™ (ertugliflozin/metformin): summary of product characteristics. 2018. Available at: https://www.ema.europa.eu/en/medicines/human/EPAR/segluromet.
- 15.European Medicines Agency; Merck Sharp & Dohme Ltd, Hoddesdon, UK. Steglujan™ (ertugliflozin/sitagliptin): summary of product characteristics. 2018. Available at: https://www.ema.europa.eu/en/medicines/human/EPAR/steglujan.
- 16.Harris SB. The power of two: an update on fixed-dose combinations for type 2 diabetes. Expert Rev Clin Pharmacol. 2016;9(11):1453–1462. doi: 10.1080/17512433.2016.1221758. [DOI] [PubMed] [Google Scholar]
- 17.Ceriello A, De Nigris V, Iijima H, Matsui T, Gouda M. The unique pharmacological and pharmacokinetic profile of teneligliptin: implications for clinical practice. Drugs. 2019;79(7):733–750. doi: 10.1007/s40265-019-01086-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Terra SG, Focht K, Davies M, Frias J, Derosa G, Darekar A, et al. Phase III, efficacy and safety study of ertugliflozin monotherapy in people with type 2 diabetes mellitus inadequately controlled with diet and exercise alone. Diabetes Obes Metab. 2017;19(5):721–728. doi: 10.1111/dom.12888. [DOI] [PubMed] [Google Scholar]
- 19.Aronson R, Frias J, Goldman A, Darekar A, Lauring B, Terra SG. Long-term efficacy and safety of ertugliflozin monotherapy in patients with inadequately controlled T2DM despite diet and exercise: VERTIS MONO extension study. Diabetes Obes Metab. 2018;20(6):1453–1460. doi: 10.1111/dom.13251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosenstock J, Frias J, Pall D, Charbonnel B, Pascu R, Saur D, et al. Effect of ertugliflozin on glucose control, body weight, blood pressure and bone density in type 2 diabetes mellitus inadequately controlled on metformin monotherapy (VERTIS MET) Diabetes Obes Metab. 2018;20(3):520–529. doi: 10.1111/dom.13103. [DOI] [PubMed] [Google Scholar]
- 21.Dagogo-Jack S, Liu J, Eldor R, Amorin G, Johnson J, Hille D, et al. Efficacy and safety of the addition of ertugliflozin in patients with type 2 diabetes mellitus inadequately controlled with metformin and sitagliptin: the VERTIS SITA2 placebo-controlled randomized study. Diabetes Obes Metab. 2018;20(3):530–540. doi: 10.1111/dom.13116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller S, Krumins T, Zhou H, Huyck S, Johnson J, Golm G, et al. Ertugliflozin and sitagliptin co-initiation in patients with type 2 diabetes: the VERTIS SITA randomized study. Diabetes Ther. 2018;9(1):253–268. doi: 10.1007/s13300-017-0358-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pratley RE, Eldor R, Raji A, Golm G, Huyck SB, Qiu Y, et al. Ertugliflozin plus sitagliptin versus either individual agent over 52 weeks in patients with type 2 diabetes mellitus inadequately controlled with metformin: the VERTIS FACTORIAL randomized trial. Diabetes Obes Metab. 2018;20(5):1111–1120. doi: 10.1111/dom.13194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hollander P, Liu J, Hill J, Johnson J, Jiang ZW, Golm G, et al. Ertugliflozin compared with glimepiride in patients with type 2 diabetes mellitus inadequately controlled on metformin: the VERTIS SU randomized study. Diabetes Ther. 2018;9(1):193–207. doi: 10.1007/s13300-017-0354-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grunberger G, Camp S, Johnson J, Huyck S, Terra SG, Mancuso JP, et al. Ertugliflozin in patients with stage 3 chronic kidney disease and type 2 diabetes mellitus: the VERTIS RENAL randomized study. Diabetes Ther. 2018;9(1):49–66. doi: 10.1007/s13300-017-0337-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mascitti V, Maurer TS, Robinson RP, Bian J, Boustany-Kari CM, Brandt T, et al. Discovery of a clinical candidate from the structurally unique dioxa-bicyclo[3.2.1]octane class of sodium-dependent glucose cotransporter 2 inhibitors. J Med Chem. 2011;54(8):2952–2960. doi: 10.1021/jm200049r. [DOI] [PubMed] [Google Scholar]
- 27.Raje S, Callegari E, Sahasrabudhe V, Vaz A, Shi H, Fluhler E, et al. Novel application of the two-period microtracer approach to determine absolute oral bioavailability and fraction absorbed of ertugliflozin. Clin Transl Sci. 2018;11(4):405–411. doi: 10.1111/cts.12549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.US Food and Drug Administration. Guidance for industry: waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a biopharmaceutics classification system. 2017. Available at: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070246.pdf.
- 29.European Medicines Agency; Committee for Medicinal Products for Human Use. Steglatro™ (ertugliflozin): European public assessment report. 2018. Available at: https://www.ema.europa.eu/documents/assessment-report/steglatro-epar-public-assessment-report_en.pdf.
- 30.Nucci G, Le V, Sweeney K, Amin N. Single- and multiple-dose pharmacokinetics and pharmacodynamics of ertugliflozin, an oral selective inhibitor of SGLT2, in healthy subjects. Clin Pharmacol Ther. 2018;103(S1):S83. [Google Scholar]
- 31.Kalgutkar AS, Tugnait M, Zhu T, Kimoto E, Miao Z, Mascitti V, et al. Preclinical species and human disposition of PF-04971729, a selective inhibitor of the sodium-dependent glucose cotransporter 2 and clinical candidate for the treatment of type 2 diabetes mellitus. Drug Metab Dispos. 2011;39(9):1609–1619. doi: 10.1124/dmd.111.040675. [DOI] [PubMed] [Google Scholar]
- 32.Miao Z, Nucci G, Amin N, Sharma R, Mascitti V, Tugnait M, et al. Pharmacokinetics, metabolism, and excretion of the antidiabetic agent ertugliflozin (PF-04971729) in healthy male subjects. Drug Metab Dispos. 2013;41(2):445–456. doi: 10.1124/dmd.112.049551. [DOI] [PubMed] [Google Scholar]
- 33.Sahasrabudhe V, Fediuk DJ, Matschke K, Shi H, Liang Y, Hickman A, et al. Effect of food on the pharmacokinetics of ertugliflozin and its fixed-dose combinations ertugliflozin/sitagliptin and ertugliflozin/metformin. Clin Pharmacol Drug Dev. 2019;8(5):619–627. doi: 10.1002/cpdd.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bonnet F, Scheen A. Understanding and overcoming metformin gastrointestinal intolerance. Diabetes Obes Metab. 2017;19(4):473–481. doi: 10.1111/dom.12854. [DOI] [PubMed] [Google Scholar]
- 35.Dawra VK, Liang Y, Shi H, Bass A, Hickman A, Terra SG, et al. A PK/PD study comparing twice-daily to once-daily dosing regimens of ertugliflozin in healthy subjects. Int J Clin Pharmacol Ther. 2019;57(4):207–216. doi: 10.5414/CP203343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sahasrabudhe V, Terra SG, Hickman A, Saur D, Shi H, O’Gorman M, et al. The effect of renal impairment on the pharmacokinetics and pharmacodynamics of ertugliflozin in subjects with type 2 diabetes mellitus. J Clin Pharmacol. 2017;57(11):1432–1443. doi: 10.1002/jcph.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scheen AJ. Pharmacokinetics, pharmacodynamics and clinical use of SGLT2 inhibitors in patients with type 2 diabetes mellitus and chronic kidney disease. Clin Pharmacokinet. 2015;54(7):691–708. doi: 10.1007/s40262-015-0264-4. [DOI] [PubMed] [Google Scholar]
- 38.Fioretto P, Zambon A, Rossato M, Busetto L, Vettor R. SGLT2 inhibitors and the diabetic kidney. Diabetes Care. 2016;39(Suppl 2):S165–S171. doi: 10.2337/dcS15-3006. [DOI] [PubMed] [Google Scholar]
- 39.Sahasrabudhe V, Terra SG, Hickman A, Saur D, Raje S, Shi H, et al. Pharmacokinetics of single-dose ertugliflozin in patients with hepatic impairment. Clin Ther. 2018;40(10):1701–1710. doi: 10.1016/j.clinthera.2018.06.015. [DOI] [PubMed] [Google Scholar]
- 40.Dawra VK, Sahasrabudhe V, Liang Y, Matschke K, Shi H, Hickman A, et al. Effect of rifampin on the pharmacokinetics of ertugliflozin in healthy subjects. Clin Ther. 2018;40(9):1538–1547. doi: 10.1016/j.clinthera.2018.07.014. [DOI] [PubMed] [Google Scholar]
- 41.Scheen AJ. Drug-drug interactions with sodium-glucose cotransporters type 2 (SGLT2) inhibitors, new oral glucose-lowering agents for the management of type 2 diabetes mellitus. Clin Pharmacokinet. 2014;53(4):295–304. doi: 10.1007/s40262-013-0128-8. [DOI] [PubMed] [Google Scholar]
- 42.Dawra VK, Cutler DL, Zhou S, Krishna R, Shi H, Liang Y, et al. Assessment of the drug interaction potential of ertugliflozin with sitagliptin, metformin, glimepiride, or simvastatin in healthy subjects. Clin Pharmacol Drug Dev. 2019;8(3):314–325. doi: 10.1002/cpdd.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.US Food and Drug Administration; Center for Drug Evaluation and Research. Steglatro™ (ertugliflozin): clinical pharmacology and biopharmaceutics review. 2016. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/209803,209805,209806Orig1s000ClinPharmR.pdf.
- 44.US Food and Drug Administration; Center for Drug Evaluation and Research. Steglatro™ (ertugliflozin): clinical review. 2016. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/209803,209805,209806Orig1s000MedR.pdf.
- 45.Sahasrabudhe V, Saur D, Matschke K, Terra SG, Hickman A, Huyghe I, et al. A phase 1, randomized, placebo- and active-controlled crossover study to determine the effect of single-dose ertugliflozin on QTc interval in healthy volunteers. Clin Pharmacol Drug Dev. 2018;7(5):513–523. doi: 10.1002/cpdd.421. [DOI] [PubMed] [Google Scholar]
- 46.Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373(22):2117–2128. doi: 10.1056/NEJMoa1504720. [DOI] [PubMed] [Google Scholar]
- 47.Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. 2017;377(7):644–657. doi: 10.1056/NEJMoa1611925. [DOI] [PubMed] [Google Scholar]
- 48.Wiviott SD, Raz I, Bonaca MP, Mosenzon O, Kato ET, Cahn A, et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2019;380(4):347–357. doi: 10.1056/NEJMoa1812389. [DOI] [PubMed] [Google Scholar]
- 49.Anker SD, Butler J, Filippatos GS, Jamal W, Salsali A, Schnee J, et al. Evaluation of the effects of sodium-glucose co-transporter 2 inhibition with empagliflozin on morbidity and mortality in patients with chronic heart failure and a preserved ejection fraction: rationale for and design of the EMPEROR-Preserved Trial. Eur J Heart Fail. 2019;21(10):1279–1287. doi: 10.1002/ejhf.1596. [DOI] [PubMed] [Google Scholar]
- 50.Packer M, Butler J, Filippatos GS, Jamal W, Salsali A, Schnee J, et al. Evaluation of the effect of sodium-glucose co-transporter 2 inhibition with empagliflozin on morbidity and mortality of patients with chronic heart failure and a reduced ejection fraction: rationale for and design of the EMPEROR-Reduced trial. Eur J Heart Fail. 2019;21(10):1270–1278. doi: 10.1002/ejhf.1536. [DOI] [PubMed] [Google Scholar]
- 51.Santos-Gallego CG, Garcia-Ropero A, Mancini D, Pinney SP, Contreras JP, Fergus I, et al. Rationale and design of the EMPA-TROPISM trial (ATRU-4): are the “cardiac benefits” of empagliflozin independent of its hypoglycemic activity? Cardiovasc Drugs Ther. 2019;33(1):87–95. doi: 10.1007/s10557-018-06850-0. [DOI] [PubMed] [Google Scholar]
- 52.Cannon CP, McGuire DK, Pratley R, Dagogo-Jack S, Mancuso J, Huyck S, et al. Design and baseline characteristics of the eValuation of ERTugliflozin effIcacy and Safety CardioVascular outcomes trial (VERTIS-CV) Am Heart J. 2018;206:11–23. doi: 10.1016/j.ahj.2018.08.016. [DOI] [PubMed] [Google Scholar]
- 53.Flores E, Santos-Gallego CG, Diaz-Mejia N, Badimon JJ. Do the SGLT-2 inhibitors offer more than hypoglycemic activity? Cardiovasc Drugs Ther. 2018;32(2):213–222. doi: 10.1007/s10557-018-6786-x. [DOI] [PubMed] [Google Scholar]
- 54.Packer M, Anker SD, Butler J, Filippatos G, Zannad F. Effects of sodium-glucose cotransporter 2 inhibitors for the treatment of patients with heart failure: proposal of a novel mechanism of action. JAMA Cardiol. 2017;2(9):1025–1029. doi: 10.1001/jamacardio.2017.2275. [DOI] [PubMed] [Google Scholar]
- 55.Garcia-Ropero A, Badimon JJ, Santos-Gallego CG. The pharmacokinetics and pharmacodynamics of SGLT2 inhibitors for type 2 diabetes mellitus: the latest developments. Expert Opin Drug Metab Toxicol. 2018;14(12):1287–1302. doi: 10.1080/17425255.2018.1551877. [DOI] [PubMed] [Google Scholar]
- 56.Byrne NJ, Matsumura N, Maayah ZH, Ferdaoussi M, Takahara S, Darwesh AM, et al. Empagliflozin blunts worsening cardiac dysfunction associated with reduced NLRP3 (nucleotide-binding domain-like receptor protein 3) inflammasome activation in heart failure. Circ Heart Fail. 2020;13(1):e006277. doi: 10.1161/CIRCHEARTFAILURE.119.006277. [DOI] [PubMed] [Google Scholar]
- 57.Inzucchi SE, Zinman B, Fitchett D, Wanner C, Ferrannini E, Schumacher M, et al. How does empagliflozin reduce cardiovascular mortality? Insights from a mediation analysis of the EMPA-REG OUTCOME trial. Diabetes Care. 2018;41(2):356–363. doi: 10.2337/dc17-1096. [DOI] [PubMed] [Google Scholar]
- 58.Hallow KM, Helmlinger G, Greasley PJ, McMurray JJV, Boulton DW. Why do SGLT2 inhibitors reduce heart failure hospitalization? A differential volume regulation hypothesis. Diabetes Obes Metab. 2018;20(3):479–487. doi: 10.1111/dom.13126. [DOI] [PubMed] [Google Scholar]
- 59.Grempler R, Thomas L, Eckhardt M, Himmelsbach F, Sauer A, Sharp DE, et al. Empagliflozin, a novel selective sodium glucose cotransporter-2 (SGLT-2) inhibitor: characterisation and comparison with other SGLT-2 inhibitors. Diabetes Obes Metab. 2012;14(1):83–90. doi: 10.1111/j.1463-1326.2011.01517.x. [DOI] [PubMed] [Google Scholar]