

Abstract
Dendritic cell vaccines are a promising strategy for the treatment of cancer and infectious diseases but have met with mixed success. We report on a lentiviral vector-based dendritic cell vaccine strategy that generates a cluster of differentiation 8 (CD8) T cell response that is much stronger than that achieved by standard peptide-pulsing approaches. The strategy was tested in the mouse lymphocytic choriomeningitis virus (LCMV) model. Bone marrow-derived dendritic cells from SAMHD1 knockout mice were transduced with a lentiviral vector expressing the GP33 major-histocompatibility-complex (MHC)-class-I-restricted peptide epitope and CD40 ligand (CD40L) and injected into wild-type mice. The mice were highly protected against acute and chronic variant CL-13 LCMVs, resulting in a 100-fold greater decrease than that achieved with peptide epitope-pulsed dendritic cells. Inclusion of an MHC-class-II-restricted epitope in the lentiviral vector further increased the CD8 T cell response and resulted in antigen-specific CD8 T cells that exhibited a phenotype associated with functional cytotoxic T cells. The vaccination synergized with checkpoint blockade to reduce the viral load of mice chronically infected with CL-13 to an undetectable level. The strategy improves upon current dendritic cell vaccine strategies; is applicable to the treatment of disease, including AIDS and cancer; and supports the utility of Vpx-containing vectors.
Keywords: LCMV, CD8, lentiviral vector, vaccine, dendritic cell, AIDS, cancer, Vpx, SAMHD1
Graphical Abstract
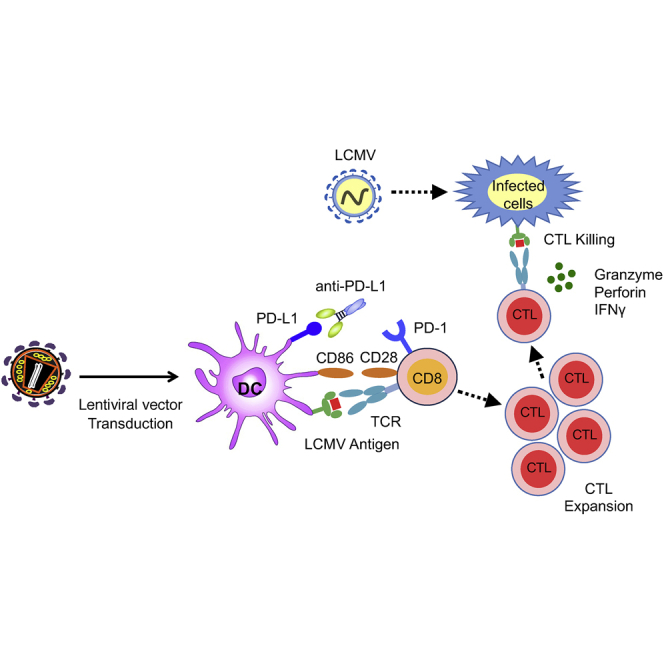
Norton, Tada, and colleagues report on a lentiviral-vector-based dendritic cell vaccine strategy that synergizes with checkpoint inhibitor therapy to induce highly functional CD8 T cells which protect mice against acute lethal viral infection and chronic infection in the lymphocytic choriomeningitis virus mouse model.
Introduction
Therapeutic vaccination offers a promising approach with which to enhance T cell responses in chronic viral infection and cancer. Therapeutic vaccines based on the use of patient dendritic cells (DCs) have been developed for cancer and for HIV infection.1, 2, 3, 4 These rely on the role of DCs as “professional” antigen-presenting cells that take up antigen in the periphery and then migrate to secondary lymphoid organs to present peptide antigens to T cells on major histocompatibility complex (MHC) class I and class II. Cluster of differentiation 80 (CD80) and CD86 accessory proteins on the DCs trigger antigen-specific T cell activation and the release of interleukin 12 (IL-12) and tumor necrosis factor α (TNF-α)which promote a T helper 1 (Th1) phenotype in the responding T cells. In clinical practice, monocytes are purified from the patient’s peripheral blood mononuclear cells (PBMCs), differentiated into monocyte-derived DCs (MDDCs) with granulocyte macrophage-colony-stimulating factor (GM-CSF) and other cytokines, and then pulsed with tumor antigen synthetic peptide or tumor cell lysate or transfected with antigen-encoding RNA or DNA. The DCs are matured with a cocktail of proinflammatory cytokines and Toll-like receptor (TLR) agonist and reinfused.
An alternative approach is to deliver to DC antigen-encoding nucleic acids, either as RNA or DNA, or by transduction with viral vectors. The ability to express full-length proteins in the vectors allows the presentation of multiple epitopes and efficient antigen presentation on MHC class I.5 Viral vectors that have been used to express genes in DCs include adenovirus, adeno-associated virus, onco-retrovirus, vaccinia, and lentivirus. Lentiviral vectors have the advantage that they are not subject to pre-existing immunity and can transduce nondividing cells for stable long-term expression without affecting cellular function.6 Lentiviral vector gene delivery to DCs also provides the opportunity to coexpress immunomodulatory proteins that enhance DC function and increase T cell activation.
Whereas lentiviral vectors have been used for gene delivery to DCs, the efficiency with which they transduce DCs is relatively low due to SAMHD1, a myeloid-restriction factor that blocks lentiviral infection at reverse transcription.7,8 The restriction can be counteracted by packaging the lentiviral accessory protein Vpx into the vectors. Vpx is released into target cells upon virus entry, where it induces the proteasomal degradation of SAMHD1.9,10 Although HIV-1 does not package Vpx, Vpx-containing lentiviral vectors can be produced by cotransfection of 293T cells with an HIV-1 Gag/Pol-packaging plasmid that contains a Vpx-packaging motif introduced into Gag P6 and a codon-optimized Vpx expression plasmid.9, 10, 11 Vectors produced by this method have a two-log increase in titer on DCs.12
We previously reported on the development of lentiviral vectors that express peptide epitopes from HIV-1 and influenza and coexpress CD40 ligand (CD40L).5 CD40L binds to CD40 on DCs, inducing their maturation and activating antigen-presentation pathways. CD40L activated the DCs, as reflected by increased expression of CD83 and the costimulatory protein CD86 and induced the secretion of the Th1 cytokines IL-12p70, TNF-α, and IL-6 without inducing the immunosuppressive cytokine IL-10.5 DCs transduced with lentiviral vector expressing CD40L and the MHC-class-I-restricted M1 epitope of influenza presented the antigen to an M1-specific cytotoxic T lymphocyte (CTL) clone and activated autologous-cocultured anti-influenza CD8 T cells.5 The injection of humanized mice with autologous DCs transduced with a lentiviral expressing human CD40L and the SL9 epitope raised a strong MHC class I T cell response against the epitope and caused a major reduction in HIV-1 virus loads.13 In chronic viral infections, such as HIV, hepatitis C, and lymphocytic choriomeningitis virus (LCMV), T cells become exhausted, contributing to the inability of the immune response to clear the infection.14,15 Similarly, in many cancers, anti-tumor T cells respond to tumor antigens but become exhausted, resulting in failure to prevent tumor growth. The blockade of T cell checkpoints using checkpoint inhibitors, such as antibodies to programmed cell death 1 (PD-1), PD-1 ligand (PD-L1), and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), has been found to reverse T cell exhaustion in chronic viral infection and in cancer, resulting in tumor remissions in melanoma, non-small cell lung cancer, bladder cancer, kidney cancer, and other solid tumors.16, 17, 18, 19, 20, 21, 22 In the case of HIV-infected individuals on long-term combination antiretroviral therapy, the combined lack of viral antigen production and T cell exhaustion contributes to the inability to cure the infection. A means to enhance the immune response and reverse T cell exhaustion could allow patients to reduce the need for life-long therapy.
The LCMV model provides a means to study the T cell response to viral infection and the role of T cell exhaustion in chronic disease.14,23,24 Infection of mice by intraperitoneal (i.p.) injection with the Armstrong strain causes an acute infection that is cleared in 10–12 days, whereas intracranial infection results in lethality in 6–7 days. Infection with the closely related variant, LCMV CL-13, results in a chronic infection that is not cleared for several months, due, in part, to T cell exhaustion resulting from checkpoint activation.25 Antiviral CTLs in the chronically infected mice express increased levels of PD-1 and lymphocyte-activation gene 3 (LAG-3);24 secrete reduced levels of proinflammatory cytokines IL-2, TNF-α, and interferon-γ (IFN-γ); and have diminished cytolytic activity and proliferative potential.25 Adoptive transfer of CD8 T cells from CL-13-infected mice protects the recipient from infection, suggesting that T cell exhaustion can be reversed based on environmental cues.26 Treatment with anti-PD-1 antibody results in reinvigoration of the T cells and a decline in virus load.23 Preclinical studies suggest that checkpoint blockade can synergize with vaccination to enhance T cell cytolytic activity and provide a more durable immune response.27, 28, 29
In this report, we used the LCMV model to test protective and therapeutic lentiviral DC vaccination and compared the responses to those achieved by conventional peptide pulsing. We found that mice immunized with lentiviral-vector-transduced DCs that coexpressed CD40L and the LCMV MHC-class-I-restricted epitope were protected from both acute and lethal LCMV challenge. Transduced DCs induced a more robust protection than that induced by synthetic peptide-pulsed DCs. The transduced DCs induced the expansion of memory CD8 T cells; the addition of an MHC class II epitope to the vaccine vector resulted in CTLs with enhanced cytolytic activity. The combination of DC vaccination with checkpoint blockade using an anti-PD-L1 antibody increased vaccine potency, curing mice chronically infected with LCMV CL-13. The results suggest that a lentiviral vector DC vaccine enhanced by checkpoint blockade may be a promising strategy for the treatment of chronic disease, including cancer and AIDS.
Results
Lentiviral Vector DC Vaccine Establishes a High Degree of Protection against LCMV
An obstacle to the use of lentiviral vectors for gene transfer to DCs is their poor transduction efficiency, a result of the constitutive expression of the myeloid restriction factor SAMHD1, which blocks reverse transcription.7, 8, 9, 10 The block to transduction of human DCs can be circumvented by producing lentiviral vector stocks in which the simian immunodeficiency virus (SIV) Vpx accessory has been packaged into the virions. Vpx-containing virions are produced by cotransfection of 293T cells with a Gag/Pol packaging vector that contains a Vpx-packaging motif and a Vpx expression vector.5 Upon virus entry, Vpx is released from the virions into the cytoplasm, where it degrades SAMHD1. Vpx does not interact with murine SAMHD1 (mSAMHD1), and as a result, the strategy does not relieve the block to the transduction of mouse DCs.30 To model the transduction of human DCs with Vpx-containing lentiviral vectors in mouse DCs, we used DCs derived from SAMHD1 knockout mice for all vaccinations.30 To validate this approach, we generated bone marrow-derived DCs (BMDCs) from wild-type and SAMHD1 knockout mice and transduced them with GFP-expressing lentiviral vector. Analysis of the BMDCs by flow cytometry showed that murine SAMHD1 knockout BMDCs were transduced with a 5-fold increase in titer as compared to wild-type BMDCs (Figure 1), achieving a transduction efficiency similar to that of human DCs transduced with a Vpx-containing lentiviral vector.5 The SAMHD1 knockout did not appear to affect DC or T cell function. The knockout mice display a wild-type distribution of CD3, CD8, and CD4 T cell subsets (Figures S1A and S1B) and respond similarly to LCMV infection (Figure S1C).
Figure 1.

SAMHD1 Knockout BMDCs Are More Permissive to Lentiviral Vector Transduction
Mouse C57BL/6 and SAMHD1 knockout BMDCs were transduced with GFP expressing lentiviral vector (n = 6). After 4 days, the number of GFP+ BMDCs was quantified by flow cytometry. Data represent mean ± SD. Significance determined by Mann-Whitney U tests.
To test the lentiviral vector approach for DC vaccination, we used the LCMV mouse model. For this, we generated mCD40L-GP33, a lentiviral vector that expressed the LCMV MHC-class-I-restricted GP33–41 epitope and mCD40L (Figure 2A, left). It has been shown previously that transduction of human DCs with a CD40L lentiviral expression induces their activation and maturation and causes them to secrete proinflammatory cytokines, thereby enhancing the ability of the transduced DCs to activate T cells.5,13 The GP33–41 epitope was expressed in the vector as a fusion to the carboxy-terminus of CD40L with an intervening self-cleaving P2A amino acid motif, a configuration designed to promote efficient antigen presentation. Because of the type II transmembrane topology of CD40L, we hypothesize that the peptide epitope is released directly into the endoplasmic reticulum (ER) lumen upon biosynthesis, where it assembles with newly synthesized MHC class I molecules (Figure 2A, right). Mouse BMDCs are less responsive to CD40L with regard to the production of proinflammatory cytokines than are human DCs (not shown) but maintained function in the vaccine, as vectors that expressed an inactive, mutated form of CD40L were significantly less protective against LCMV (Figure S2).
Figure 2.

Lentiviral-Vector-Transduced DCs Raise a Protective Response to LCMV Infection
(A) On the left is shown the structure of lentiviral vectors expressing GFP, mCD40L, and mCD40L fused to GP33–41 separated by a picornavirus P2A self-cleaving motif. On the right, the biosynthesis of the CD40L:GP33–41 fusion protein in the ER is diagrammed. Because of the type II topology of CD40L, the peptide is released into the endoplasmic reticulum upon synthesis to assemble with MHC class I proteins. (B) SAMHD1 knockout BMDCs were transduced with lentiviral vectors and 1 × 106 were injected into C57BL/6 mice (n = 5). After 5 days, CD8+ splenic T cells were stained with GP33–41 MHC class I tetramer and analyzed by flow cytometry. (C) Mice were injected with 1 × 106-transduced BMDCs and after 7 days, challenged with LCMV Armstrong (n = 5). After 4 days, the virus load in the spleen was measured by plaque assay. The dotted line represents the lower level of detection. (D) Mice were injected with a serial 5-fold dilution of BMDCs transduced with the mCD40L-GP33 vector or control mCD40L lentiviral vector and then challenged with LCMV Armstrong (n = 4). After 4 days, virus loads were measured by plaque assay. The dotted line represents the lower level of detection. (E) Mice were injected with BMDCs transduced with the lentiviral vector expressing mCD40L or mCD40L and GP33–41. 7 days after immunization, the mice were challenged by intracerebral injection of LCMV Armstrong (n = 5) and monitored for 1 year. After 300 days, the mice were rechallenged with LCMV Armstrong. Data represent mean ± SD. Significance determined by Mann-Whitney U tests.
To vaccinate mice, mCD40L-GP33-transduced DCs were injected i.p. into C57BL/6 mice. To compare the lentiviral vaccine approach to peptide-pulsed DCs, DCs transduced with vector expressing mCD40L alone were pulsed with synthetic GP33–41 peptide and injected into mice. DCs transduced with vectors expressing GFP or mCD40L alone were used as controls. 1 week later, the mice were challenged by i.p injection with LCMV Armstrong. After 4 days, the number of splenic LCMV-specific MHC class I GP33–41 tetramer-binding CD8 T cells was determined by flow cytometry, and the virus load in spleen was determined by plaque assay. The results showed a small number of GP33–41 tetramer+ CD8 T cells in mice injected with control vector or mCD40L vector-transduced BMDCs (Figure 2B). mCD40L-transduced/peptide-pulsed DCs and mCD40L-GP33-transduced DCs both induced about a 10-fold increase in the number of GP33–41 tetramer+ cells, with no significant difference in the number of T cells induced between the peptide-pulsed and lentiviral-vector-transduced BMDCs. Analysis of the splenic virus load showed high titers (>2 × 107 plaque-forming units [PFU]/g) for the control vector and mCD40L-alone vector (Figure 2C). mCD40L-transduced/peptide-pulsed BMDCs resulted in a 2-log reduction in splenic virus load, whereas mCD40L-GP33-transduced DCs decreased the virus load by 4 logs in 4/5 mice. To determine the potency of the transduced BMDCs, we immunized mice with serially diluted mCD40L-GP33–41-transduced BMDCs. As few as 4.0 × 104 mCD40L-GP33-transduced BMDCs were sufficient to protect the mice (Figure 2D).
To determine whether the vaccine would protect against lethal infection, mice were immunized with transduced BMDCs and infected with LCMV Armstrong by intracerebral injection. Mice immunized with control-vector-transduced DCs died by day 8 postinfection, whereas 4/5 mice injected with mCD40L-GP33-transduced BMDCs were protected (Figure 2E). When the surviving mice were rechallenged with LCMV Armstrong by intracerebral injection, 1 year later, all four remained protected. The results demonstrate that the lentiviral vector approach induced a strong T cell-protective response that required very few BMDCs to be fully protective and that T cells induced by endogenously expressed antigen are more functional than those induced by peptide-pulsed DCs.
The CL-13 variant is more resistant to T cell clearance than the Armstrong strain and establishes a chronic infection in mice.14 To test whether the lentiviral vector vaccine approach would protect against CL-13, mice were immunized with transduced BMDCs and then infected with the LCMV CL-13. For these experiments, virus loads were quantified by qRT-PCR, an assay that permits accurate quantification of viral RNA at low virus abundance. The results showed that mCD40L-GP33-transduced BMDCs caused a five-log decrease in blood, with spleen and kidney virus loads falling to undetectable levels (Figure 3A). Expression of the antigen by lentiviral vector transduction suppressed splenic virus load >20-fold greater than peptide-pulsed/mCD40L-transduced BMDCs (Figure 3B).
Figure 3.

Lentiviral Vector DC Vaccine Protects Mice against CL-13 Infection
(A) Mice were immunized with transduced BMDCs and then challenged after 7 days with CL-13 (n = 4). Serum, spleen, and kidney were isolated on day 4 postchallenge, and viral RNA was quantified by qPCR. Serum RNA copy number was normalized to a standard curve generated with plasmid DNA containing an LCMV cDNA. LCMV tissue RNA is represented as fold change from the average RNA copy number in unimmunized and LCMV-challenged mice. The dotted line represents the lower level of detection, as determined using serially diluted plasmid DNA standard. (B) Mice were immunized with BMDCs transduced with lentiviral vector expressing mCD40L and GP33–41 or transduced with mCD40L and pulsed with GP33–41 peptide (n = 3). The mice were challenged with LCMV CL-13, and viral splenic RNA was measured by qPCR. Data represent mean ± SD. Significance determined by Mann-Whitney U tests.
Coexpression of MHC-Class-II-Restricted Epitope Increases CD8 T Cell Functionality
CD4 T cell help is required to promote the expansion and activation of CD8 T cells, and thus, the incorporation of an MHC-class-II-restricted epitope could strengthen the response induced by the transduced BMDCs. To test this possibility, we generated a dual epitope lentiviral vector, mCD40L-GP33.GP66, in which the MHC-II-restricted epitope GP66–77 is expressed downstream of the phosphoglycerate kinase (PGK) promoter, and a single epitope vector, mCD40L-GP66, expressing only the MHC-class II-restricted GP66–77 (Figure 4A). SAMHD1 knockout BMDCs were transduced with the single or double epitope vectors and then injected into C57BL/6 mice, and after 7 days, the mice were challenged with LCMV Armstrong. Both vectors caused a major reduction in virus load. The dual epitope vector appeared to suppress the virus load, 3-fold lower than the single GP33–41 epitope vector, although this difference did not reach statistical significance (Figure 4B). The vector expressing MHC-class-II-restricted GP66–77 epitope alone had only a small effect on virus load.
Figure 4.

Addition of an MHC-Class-II-Restricted Epitope to the Lentiviral Vaccine Vector Enhances Vaccine Efficacy
(A) Structure of the lentiviral vector that expresses MHC-class-I-restricted GP33–41 and MHC-class-II-restricted GP66–77 peptide epitopes is diagrammed. GP66–77 has an amino-terminal ER retention signal. (B) SAMHD1 knockout BMDCs were transduced with control and GP33–41- or dual GP33–41- and GP66–77-expressing lentiviral vectors and then injected into mice. After 5 days, the mice were challenged with LCMV Armstrong, and 4 days postinfection, viral RNA was quantified (n = 5). Data represent mean ± SD. Significance determined by Mann-Whitney U tests.
To compare the functionality of the T cell response induced by the transduced BMDCs with that induced by peptide-pulsed DCs, we analyzed the GP33–41-specific CD8 T cells of vaccinated mice for cell-surface markers associated with CTL function. The markers tested included those associated with cytolytic/effector activity (granzyme B, perforin, CD43, KLRG1, and CX3CR1) and those negatively correlated with cytolytic/effector activity (CD62L and CD27). The analyses did not include control, unvaccinated mice, as these do not have a significant number of GP33–41 tetramer+ cells to analyze. Analysis of the number of GP33–41 tetramer+ cells showed that the lentiviral vector that expressed the class I and class II epitopes induced the largest number of tetramer+ cells, and this was significantly higher than that induced by peptide-pulsed BMDCs (Figure 5A). Of the GP33–41 tetramer+ CD8 T cells from mice vaccinated with lentiviral-vector-transduced BMDCs, the majority expressed granzyme B, perforin, CD43, KLRG1, and CX3CR1, corresponding to an effector or more specifically, short-lived effector cell (SLEC) phenotype. The mice had reduced numbers of cells that expressed CD62L and CD27, a phenotype associated with memory precursor effector cells (MPECs). Overall, the lentiviral vector transduction resulted in more cells expressing markers associated with functional cells as compared to peptide pulsing, and the vector that expressed MHC I and MHC II epitopes generated the highest percentage of T cells with the most functional phenotype (Figure 5B). Whereas the benefit provided by the dual epitope vector did not reach statistical significance, the single epitope vector results in variability in the T cell phenotype as compared to the dual epitope vector, which results in a consistent response with respect to T cell phenotype. The results suggest that vector-transduced BMDCs primed LCMV-specific CD8 T cells with greater activation and cytolytic potential than synthetic peptide-pulsed DCs and that addition of the MHC-class-II-restricted epitope increased with consistency of the response.
Figure 5.

Transduced BMDCs Induce Functional CD8+ T Cells
BMDCs were transduced with lentiviral vectors expressing mCD40L or mCD40L with GP66–77 or transduced with mCD40L lentiviral vector and peptide pulsed. The BMDCs were injected into mice and 5 days later, infected with LCMV. (A) The splenocytes were stained for CD8 and GP33–41 tetramer binding and analyzed by flow cytometry. The results are plotted as the percentage tetramer+ cells of CD8+ cells (n = 3–4). (B) Splenocytes from the mice were stained with GP33–41 tetramer and anti-CD8 antibody and for different markers of CTL function (granzyme B, perforin, CD43, KLRG1, CX3CR1, CD62L, and CD27) and analyzed by flow cytometry. Data represent mean ± SD. Significance determined by Mann-Whitney U tests.
DC Vaccine Synergizes with Checkpoint Blockade in Chronic Infection
In chronic infection by LCMV CL-13, the failure of the immune response to clear the virus is the result, in large part, of checkpoint activation and the ensuing T cell exhaustion. Checkpoint blockade has been shown to partially restore T cell function in LCMV CL-13-infected mice, resulting in a decrease in virus load.31 This finding raised the possibility that checkpoint blockade might synergize with the lentiviral vector DC vaccine to enhance the T cell response to chronic infection. To test this possibility, we established LCMV CL-13 chronically infected mice and injected them with DCs transduced with lentiviral vectors that expressed mCD40L and GP33–41 or mCD40L, GP33–41, and GP66–77. 14 days after infection, the mice were injected with transduced BMDCs and treated with five injections of anti-PD-L1 antibody over 15 days, starting on the day of BMDC immunization, after which, serum virus loads were quantified. The results showed that in mice injected with untransduced DCs, virus load declined 8-fold over 3 weeks, consistent with the natural history of the LCMV CL-13 model (Figure 6). The virus load declined at a similar rate in mice vaccinated with mCD40L-GP33-transduced DCs. Treatment with anti-PD-L1 antibody alone had only a minor effect on virus load. In contrast, vaccination with mCD40L-GP33.GP66-transduced DCs caused a 70-fold reduction in virus load after 3 weeks. The combination of mCD40L-GP33.GP66 vaccination and anti-PD-L1 antibody reduced the viral load >3 logs. The absence of an effect of anti-PD-L1 treatment alone differs from previous findings,23 most likely as a result of the injection here of a limited amount of antibody, as discussed below. Taken together, the results show that neither the single epitope vaccine nor checkpoint blockade had a major effect on virus load. The dual MHC I and MHC II epitope vector, however, was effective at reducing virus load and combined with the anti-PD-L1 blockade, synergized to suppress virus replication dramatically.
Figure 6.

Checkpoint Blockade Synergizes with Transduced BMDCs to Clear Chronic Infection
Mice were infected with LCMV CL-13 for 3 weeks (n = 4). The mice were bled to determine baseline LCMV serum RNA levels. The mice were then infected with transduced or control, untransduced BMDCs and where indicated, injected five times with 50 μg anti-PD-L1 antibody (PD-L1 Ab). Serum RNA levels were measured over 3 weeks. Data represent mean ± SD. Significance determined by Mann-Whitney U tests.
Ahn et al.32 showed that PD-1 blockade, in addition to reviving exhausted CD8 T cells, can act in the primary immune response to modulate naïve-to-effector CD8 T cell differentiation. To determine whether PD-1 blockade would enhance the CD8 T cell response to DC vaccination, we injected wild-type mice with transduced BMDCs and then treated with anti-PD-L1 antibody (Figure 7A). Analysis of the splenocytes after 7 days showed that mice injected with BMDCs transduced with the dual epitope vector (mCD40L-GP33.GP66), and anti-PD-L1 had increased the number of antigen-specific CD8 T cells (Figure 7B). The effect appeared to require the MHC-class-II-restricted epitope, as the increase did not occur with the single epitope vector mCD40L-GP33. Checkpoint blockade also increased the percentage of CD8 T cells expressing markers of CTL function (granzyme B and perforin), particularly for the dual epitope vector. In addition, checkpoint blockade resulted in fewer CD8 T cells expressing CD62L, a marker of naive and central memory T cells that is downregulated in effectors (Figure 7C).
Figure 7.

Checkpoint Inhibitor Induces Primary Immune Response
(A) BMDCs were transduced with control and GP33–41- or dual GP33–41- and GP66–77-expressing lentiviral vectors and then injected into mice (n = 6). The mice were then injected 3 times with 100 μg anti-PD-L1 antibody (PD-L1 Ab). On day 7 after DC injection, the splenocytes were analyzed by flow cytometry. (B) The percentage of GP33–41 tetramer+ CD8+ splenocytes is shown. (C) The percentage of CD8+ splenocytes that expressed markers of CTL function (granzyme B, perforin, and CD62L) is shown. Data represent mean ± SD. Significance determined by Mann-Whitney U tests.
Discussion
We report the development of a therapeutic lentiviral-vector-based DC vaccine strategy that generates antiviral T cell responses that protect against both high-titer and lethal LCMV challenge and cure mice of chronic infection. The protection provided by vectors expressing CD40L and MHC class I LCMV peptide epitope was 100-fold greater than that achieved by peptide epitope-pulsed DCs (Figure 2C), an effect that resulted from an enhanced effector phenotype of the responding CD8 T cells. The addition of an MHC class II epitope to the vector increased the strength of the T cell response in CL-13 chronically infected mice, suggesting that T cell help was required for the restoration of exhausted T cells or for the expansion of naive antiviral CD8 T cells, consistent with the requirement for CD4 T cells to sustain anti-CD8 T cell responses in chronic LCMV infection.33 Vaccination with lentiviral-vector-transduced BMDCs induced antigen-specific CD8 T cells that exhibited a phenotype associated with functional cytotoxic T cells and indicative of a higher degree of function than those induced by peptide-pulsed DCs. The vaccination synergized with checkpoint blockade to cure mice chronically infected with LCMV CL-13. The synergy of the DC vaccination with checkpoint blockade underscores the potential of the approach for the treatment of chronic disease in humans. The results show that lentiviral-vector-transduced BMDCs result in a more pronounced decrease in LCMV load and in more highly functional T cells.
Approaches to DC vaccines have included peptide pulsing, charging with cell lysates, or gene delivery by antigen-encoding RNA or DNA. Gene delivery offers the possibility of expressing more complex polypeptide epitopes, thereby reducing the need to match the antigen to MHC haplotype and providing a greater diversity in the T cell response. In addition, the approach allows for the coexpression of immunomodulatory genes that enhance DC function. Gene delivery to DCs has been achieved by several viral vectors, including adenovirus, adeno-associated virus, gamma retrovirus, and lentiviral vectors. Lentiviral vectors have the advantage that they provide long-term, high-level antigen expression and that they transduce DCs with high efficiency when produced with packaged Vpx. Furthermore, humans generally do not have previous immunity to lentiviruses that would prevent their use, and the vectors do not express any viral proteins. Whereas there have been concerns regarding the use of integrating vectors because of their potential to activate oncogenes or inactivate tumor suppressors,34, 35, 36 to date, such events have not been reported in gene therapy clinical trials of lentiviral vectors. A concern in the injection of transduced DCs is that constitutive expression of CD40L could have the potential to cause inflammation; we did not find this to be the case in the immunized mice. The injection of CD40L expression vector-transduced BMDCs did not cause generalized T cell activation or increased levels of proinflammatory cytokines (Figure S3). Whereas we did not detect an inflammatory response in the mice, we found that mouse BMDCs are considerably less responsive to CD40L than human DCs (data not shown), and thus, potential inflammatory consequences remain a concern for use in humans.
i.p. injection of the mice with lentiviral-vector-transduced BMDCs expressing CD40L and GP33–41 epitope protected the mice against lethal intracranial LCMV infection. As the mice are protected against virus directly injected into the brain, the vaccine-induced protection implies that the immune response penetrated the CNS and that vaccine-induced anti-LCMV CD8 T cells had been recruited to the blood brain barrier to attack the LCMV-infected cells in the meninges. In the absence of vaccination, intracranial infection with LCMV results in lethal neuroinflammation, caused by the migration of CD8 T cells and myeloid cells into the brain that release proinflammatory cytokines. In the vaccinated mice, the CD8 T cells that penetrate the CNS do not cause neuroinflammation. This would suggest that CD8 T cells induced by vaccination differ qualitatively from those induced during infection, such that they secrete fewer proinflammatory cytokines yet maintain the ability to lyse target cells. In addition, in the vaccinated mice, the CD8 T cells may be so effective that they prevent LCMV replication before the virus can spread in the CNS.
In this study, we used SAMHD1 knockout BMDCs to increase lentiviral vector transduction, modeling the effect of Vpx-containing lentiviral vectors on human MDDCs. The SAMHD1 knockout results in about a 5-fold increase in lentiviral vector transduction efficiency. The transduction of human DCs with Vpx-containing virions results in a significantly greater increase in transduction efficiency, providing a 2-log increase in titer on MDDCs and suggesting that human monocyte-derived DCs are more tightly restricted by SAMHD1 than murine BMDCs. High levels of human DC transduction have been achieved with lentiviral vectors lacking packaged Vpx through the use of a high MOI;37,38 however, high MOI requires the production of large amounts of lentiviral vector and can result inactivation of the DCs, due to artifactual TLR signaling. Moreover, high MOI transduction can result in the passive transfer of producer cell proteins into the target DCs, caused by nonspecific virion packaging, a phenomenon termed “pseudotransduction.” Pseudotransduction did not occur in our studies, as transduction of the cells in the presence of an HIV integrase inhibitor completely prevented the detection of vector-encoded protein in the transduced cells.5
The DC vaccine synergized with checkpoint blockade, as demonstrated by the much greater effect on virus load of the combination therapy as compared to checkpoint blockade or DC vaccination alone. The findings further support those of Ha et al.,39 who demonstrated that therapeutic vaccination using a vaccinia viral vector expressing GP33 synergized with anti-PD-L1 blockade. Our findings differed from that of Barber et al.,23 who found that anti-PD-L1 antibody injection alone was associated with a pronounced reduction in virus load. This difference in experimental findings may have resulted from the use of a smaller antibody dose than what was used here.
Previous reports have demonstrated the importance of timing of checkpoint blockade relative to initiation of the immune response.40,41 This could be an important factor to consider in combining the DC vaccine with checkpoint inhibition. In priming of the immune responses to listeria and HIV, signaling through PD-1 is required for an optimal T cell response, and early blockade of PD-L1 can be inhibitory.40,41 In our study, in mice chronically infected with LCMV, PD-L1 blockade, initiated shortly after DC vaccination, strengthened the vaccine-induced anti-LCMV immune response. In this setting, the T cell response has already been primed, and T cells have become exhausted. In uninfected mice, the combination of DC vaccine with anti-PD-L1 antibody injection also resulted in stronger primary T cell responses, arguing against an inhibitory effect of early checkpoint blockade. Thus, the effect of relative timing may be context dependent. The effect of checkpoint blockade on the primary response of mouse T cells to LCMV may differ from that of listeria and HIV. For use in HIV-infected individuals, DC vaccine followed by checkpoint blockade ought to have a positive effect, as it would act on exhausted memory T cells. This would also be the case in cancer immunotherapy.
Interestingly, the injection of a relatively small number of antigen-expressing transduced DCs had a major effect on virus load, despite the presence of resident antigen-presenting cells. CL-13-infected mice maintain a relatively high viral antigen load throughout the course of chronic infection, yet the resident DCs fail to activate a T cell response that suppresses virus replication. Ha et al.39 found that DCs of mice vaccinated with an LCMV antigen-expressing vaccinia virus vector could activate anti-LCMV CD8 T cells in vitro somewhat better than DCs from control mice, suggesting that antigen availability in vivo is insufficient to achieve efficient antigen presentation. In our experiments, the expression of CD40L by the transduced DCs serves to increase DC function, yet DCs transduced with control CD40L expression vector alone do not protect against infection. It is possible that endogenously produced peptide antigen is more efficiently presented by BMDCs than full-length protein take-up and cross-presented. In addition, the responding CD8 T cells appeared phenotypically to be highly functional, expressing markers for cytolytic activity, and the vaccination resulted in the rapid formation of effector memory CD8 T cells (CD62Llo/CD27lo), consistent with the ability of DC vaccination to induce an accelerated memory response.42 The results further support the utility of the vaccine as a means to enhance T cell responses, even in the face of abundant antigen.
The results of our study support the development of Vpx-containing lentiviral vectors, coupled with checkpoint blockade, as an approach to therapeutic vaccination against chronic diseases, such as cancer and AIDS, in which T cell exhaustion plays a role. In cancer, vectors that express tumor antigen or neoantigens, coupled with checkpoint blockade, could enhance T cell responses in individuals that do not respond to checkpoint immunotherapy. In AIDS patients on long-term antiretroviral therapy, the T cell response wanes over time, due to both T cell exhaustion and lack of antigen stimulation. The stimulation of antigen-specific T cells, coupled with checkpoint blockade, could serve to suppress HIV-1 replication, allowing patients to cease antiretroviral therapy.
Materials and Methods
Mice
C57BL/6 mice were purchased from Taconic (NY, USA). SAMHD1 knockout mice were kindly provided by Axel Roers (University of Technology, Dresden, Germany).30
Plasmids
To construct the lentiviral vector plasmid pLenti.mCD40L, murine CD40L cDNA was amplified from lymphocyte RNA by reverse transcriptase PCR using primers with flanking 5′-Xba-I and 3′-Sal-I sites. The amplicon was cleaved with Xba-I and Sal-I and ligated to similarly cleaved pLenti.CMV.GFP.puro to replace GFP (658-5; Addgene, Cambridge, MA, USA). To generate pLenti.mut.mCD40L, an inactivating point mutation (T146N) was introduced into mCD40L, based on the analogous position in human CD40L.43 The mutated cDNA was then amplified by PCR using the primers containing 5′-Xba-I and 3′-Sal-I sites and cloned into pLenti.CMV.GFP.puro. To construct pLenti.mCD40L-GP33, mCD40L was fused to the picornavirus P2A sequence and GP33–41 LCMV epitope sequence by overlapping PCR. The amplicon was cleaved with Xba-I and Sal-I and ligated to pLenti.CMV.GFP.puro. To construct pLenti.mCD40L-GP33.GP66, an amplicon was generated in which the adenovirus E6/gp19K ER signal sequence MRYMILGLLALAAVCSAA was fused in-frame to a sequence encoding codon-optimized MHC II I-A(b6) LCMV GP66–77 peptide DIYKGVYQFKSV, termed GP66, flanked by Pst-I and Xho-I sites and ligated 3′ to the PGK promoter in pLenti.mCD40L-GP33.
Lentiviral Vector Preparation
Lentiviral vector stocks were prepared by calcium phosphate cotransfection of 293T cells with lentiviral vector plasmid, pMDL, pcVSV-G, and pcRev at a mass ratio of 28:10:7:2. Viruses that expressed GFP or mCD40L were titered on 293T cells by flow cytometry as the number of GFP+ or mCD40L+ cells.
Flow Cytometry
Splenocytes were stained with eFluor 450 (eBioscience, Waltham, MA, USA) or Zombie UV viability dye (BioLegend, San Diego, CA, USA) and subsequently stained with indicated antibodies and MHC I tetramers in the presence of CD16/CD32 monoclonal antibodies (mAbs; Fc block; BioLegend): Brilliant Violet (BV)421- or allophycocyanin (APC)-H-2b-KAVYNFATC GP33 tetramer (NIH Tetramer Facility, Emory University) and antibodies for cell-surface proteins Alexa 700-anti-CD3, peridinin-chlorophyll protein (PerCP)-cyanine (Cy)5.5-anti-CD8a, BV785-anti-CD8a APC-Cy7-anti-CD4, APC-CD11c, PerCP-Cy5.5-anti-CD11b, phycoerythrin (PE)-Cy7-anti-CD19, APC-Cy7-anti-I-A/I-E, PerCP-Cy5.5-anti-KLRG1, BV510-anti-CXCR3, BV605-anti-CX3CR1, PE-Dazzle 594-PD-1, APC-Fire 750-anti-CD62L, BV650-anti-CD122, and BV711-anti-CD127 (all BioLegend); BV650-anti-CD122 (BD Biosciences); and PE-Cy7-anti-CD27 (eBioscience). Intracellular perforin and granzyme B were detected following fixation and permeabilization with the FoxP3 transcription buffer kit, as per the manufacturer’s protocol (eBioscience). Antibodies used for intracellular staining were PE-anti-IFN-γ, APC-Cy7-anti-TNF-α, PE-anti-perforin, and AF647-anti-granzyme B (BioLegend). The cells were analyzed on an LSR-II or a Fortessa-X20 (BD Biosciences, Franklin Lakes, NJ, USA), and the data were analyzed with FlowJo software. Serum cytokine levels for IFN-γ, monocyte chemoattractant protein-1 (MCP-1), TNF-α, IL-10, and IL-6 were measured by cytokine bead array from 10 μL of serum using the BD Cytometric Bead Array Mouse Inflammation Kit (BD Biosciences, CA, USA).
Cell Culture
BMDCs were prepared by extracting bone marrow cells from the hind legs of 6- to 12-week-old mice. The cells (5 × 106) were differentiated in a 15-cm Petri dish in RPMI/10% fetal bovine serum (FBS), sodium pyruvate, 2 mM L-glutamine, and 50 μM 2-mercaptoethanol containing 10 ng/mL murine GM-CSF.44 The medium was replenished on days 3 and 6, and the nonadherent cells were harvested on day 8. 293T cells were cultured in DMEM/10% FBS.
BMDC Vaccination and LCMV Infection
For preventative vaccination, SAMHD1 knockout BMDCs (4-6 × 106 cells) were plated in a 10-cm Petri dish and transduced with lentiviral vector at MOI = 5 for 16 h and where indicated, pulsed with 1 μg/mL GP33–41 peptide for 2 h. 1 × 106 BMDCs were then injected into C57BL/6 mice. After 4–7 days, the mice were challenged by i.p. injection of 2.0 × 105 PFU LCMV Armstrong or by intravenous (i.v.) injection of CL-13 (2.0 × 106 PFU). For lethal infections, the mice were injected intracerebrally with LCMV Armstrong (1 × 104 PFU). For therapeutic vaccination, C57BL/6 mice were injected intravenously with CL-13 (5.0 × 106 PFU) and after 3 weeks, injected with transduced BMDCs (1 × 106 cells). For checkpoint blockade, the mice were injected five times (days 0, 3, 6, 9, and 12 post-DC injection), every 3 days, with 50 μg anti-PD-L1 antibody. For fluorescence-activated cell sorting (FACS) analysis, the mice were injected three times (days 0, 3, and 6 post-DC injection), every 3 days, with 100 μg anti-PD-L1 antibody.
Virus-Load Measurement
Blood was collected by submandibular bleeding and then centrifuged to obtain the serum. Spleen and kidney were homogenized with a FastPrep-24 5G homogenizer (MP Biomedicals, Solon, OH, USA) in complete minimal essential medium (MEM) at 10% weight/volume in lysing matrix D tubes (MP Biomedicals). LCMV PFUs were measured in 10-fold serial dilutions on Vero cells seeded in 6-well plates (2 × 105 cells/well) and overlaid with complete Eagle’s MEM (EMEM) containing 0.4% SeaKem moderate electroendoosmotic (ME) agarose (Lonza, Basel, Switzerland). The plates were cultured for 4 days and then fixed with 25% formaldehyde and stained with 0.1% crystal violet.
To measure relative LCMV RNA levels, RNA was prepared from 50 μL serum or 200 μL homogenized spleen and kidney lysates using the Quick-RNA MiniPrep Kit (Zymo Research, Irvine, CA, USA). cDNA was synthesized from RNA derived from 50 μL of serum using LCMV.GP-reverse primer (5′-CTGCTGTGTTCCCGAAACACT-3′). cDNA was synthesized from spleen and kidney RNA (2 μg) using random hexamers. cDNA was synthesized in a reaction containing 1 mM deoxyribonucleotide triphosphates (dNTPs), 1 U RNase inhibitor (Roche, Basel, Switzerland), and 0.5 U reverse transcriptase (Roche) for 30 min at 55°C, followed by 5 min at 85°C. LCMV RNA sequences were quantified by real-time PCR with FastStart Taq DNA polymerase using SYBR Green Master Mix with LCMV.GP forward primer (5′-TGCCTGACCAAATGGATGATT-3′) and LCMV.GP reverse primer. Reactions were amplified for 40 cycles of 95°C/15 s, 60°C/30 s, and 95°C/15 s. Signals were normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and amplified in parallel using primers mGAPDH.forward (5′-GGATCTGACGTGCCGCCTGG-3′) and mGAPDH.reverse (5′-CAGCCCCGGCATCGAAGGTG-3′).
For TaqMan real-time PCR, serum or tissue RNA was mixed with TaqMan Fast Virus 1-Step Master Mix (Applied Biosystems, Waltham, MA, USA), 10 mM forward and reverse primers, and 2 mM probe. PCR was for 5 min at 50°C, followed by 95°C/20 s and 40 cycles 95°C/3 s and 60°C/30 s). Primers were LCMV.GP forward (5′-GGCACATTCACCTGGACTTTG-3′), LCMV.GP reverse (5′-CTGCTGTGTTCCCGAAACACT-3′) LCMV.GP and Fam/ZEN/IBFQ probe (5′-ACTCTTCAGGGGTGGAGAATCCAGGTGGTT-3′). GAPDH was amplified for normalization using mGAPDH.forward (5′-CAATGTGTCCGTCGTGGATCT-3′) and mGAPDH.reverse (5′-GTCCTCAGTGTAGCCCAAGATG-3′) with mGAPDH probe (5′-CGTGCCGCCTGGAGAAACCTGCC-3′). Data from serum were normalized to a standard curve generated using a plasmid containing the LCMV target region (pcDNA6-LCMV.GP). Data from tissue analyses were normalized to mGAPDH. Virus load was determined by the 2−ΔΔCT method.
Statistics
Statistical significance was determined by Shapiro-Wilk and Mann-Whitney U tests and calculated with Graph Pad Prism 6.0e. Significance was calculated based on two-sided testing and is shown in the figures as the mean ± SD with confidence intervals listed as ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
Study Approval
Animal procedures were performed with the written approval of the New York University (NYU) Institutional Animal Care and Use Committee in accordance with all federal, state, and local guidelines.
Author Contributions
T.D.N., D.H., and N.R.L. designed the experiments. T.D.N., T.T., R.L., and V.v.d.H. carried out the experiments. N.R.L. and T.D.N. wrote the manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We thank Rayk Behrendt and Axel Roers for SAMHD1 knockout mice and Hyung Bum Kim for technical assistance. Funding for this study was provided by grants from the NIH (DA046100, AI122390, and AI120898) and Luis Vargas HIV Research, Saperstein Scholar, Weissmann Scholar, and Chairman’s Circle Awards to T.D.N.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.05.018.
Supplemental Information
References
- 1.Constantino J., Gomes C., Falcão A., Neves B.M., Cruz M.T. Dendritic cell-based immunotherapy: a basic review and recent advances. Immunol. Res. 2017;65:798–810. doi: 10.1007/s12026-017-8931-1. [DOI] [PubMed] [Google Scholar]
- 2.Coelho A.V., de Moura R.R., Kamada A.J., da Silva R.C., Guimarães R.L., Brandão L.A., de Alencar L.C., Crovella S. Dendritic Cell-Based Immunotherapies to Fight HIV: How Far from a Success Story? A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2016;17:1985. doi: 10.3390/ijms17121985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.García F., Plana M., Climent N., León A., Gatell J.M., Gallart T. Dendritic cell based vaccines for HIV infection: the way ahead. Hum. Vaccin. Immunother. 2013;9:2445–2452. doi: 10.4161/hv.25876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ellebaek E., Engell-Noerregaard L., Iversen T.Z., Froesig T.M., Munir S., Hadrup S.R., Andersen M.H., Svane I.M. Metastatic melanoma patients treated with dendritic cell vaccination, Interleukin-2 and metronomic cyclophosphamide: results from a phase II trial. Cancer Immunol. Immunother. 2012;61:1791–1804. doi: 10.1007/s00262-012-1242-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Norton T.D., Miller E.A., Bhardwaj N., Landau N.R. Vpx-containing dendritic cell vaccine induces CTLs and reactivates latent HIV-1 in vitro. Gene Ther. 2015;22:227–236. doi: 10.1038/gt.2014.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Esslinger C., Chapatte L., Finke D., Miconnet I., Guillaume P., Lévy F., MacDonald H.R. In vivo administration of a lentiviral vaccine targets DCs and induces efficient CD8(+) T cell responses. J. Clin. Invest. 2003;111:1673–1681. doi: 10.1172/JCI17098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goldstone D.C., Ennis-Adeniran V., Hedden J.J., Groom H.C., Rice G.I., Christodoulou E., Walker P.A., Kelly G., Haire L.F., Yap M.W. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature. 2011;480:379–382. doi: 10.1038/nature10623. [DOI] [PubMed] [Google Scholar]
- 8.Lahouassa H., Daddacha W., Hofmann H., Ayinde D., Logue E.C., Dragin L., Bloch N., Maudet C., Bertrand M., Gramberg T. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol. 2012;13:223–228. doi: 10.1038/ni.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hrecka K., Hao C., Gierszewska M., Swanson S.K., Kesik-Brodacka M., Srivastava S., Florens L., Washburn M.P., Skowronski J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature. 2011;474:658–661. doi: 10.1038/nature10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laguette N., Sobhian B., Casartelli N., Ringeard M., Chable-Bessia C., Ségéral E., Yatim A., Emiliani S., Schwartz O., Benkirane M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature. 2011;474:654–657. doi: 10.1038/nature10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sunseri N., O’Brien M., Bhardwaj N., Landau N.R. Human immunodeficiency virus type 1 modified to package Simian immunodeficiency virus Vpx efficiently infects macrophages and dendritic cells. J. Virol. 2011;85:6263–6274. doi: 10.1128/JVI.00346-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bobadilla S., Sunseri N., Landau N.R. Efficient transduction of myeloid cells by an HIV-1-derived lentiviral vector that packages the Vpx accessory protein. Gene Ther. 2013;20:514–520. doi: 10.1038/gt.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Norton T.D., Zhen A., Tada T., Kim J., Kitchen S., Landau N.R. Lentiviral Vector-Based Dendritic Cell Vaccine Suppresses HIV Replication in Humanized Mice. Mol. Ther. 2019;27:960–973. doi: 10.1016/j.ymthe.2019.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wherry E.J. T cell exhaustion. Nat. Immunol. 2011;12:492–499. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- 15.Bengsch B., Seigel B., Ruhl M., Timm J., Kuntz M., Blum H.E., Pircher H., Thimme R. Coexpression of PD-1, 2B4, CD160 and KLRG1 on exhausted HCV-specific CD8+ T cells is linked to antigen recognition and T cell differentiation. PLoS Pathog. 2010;6:e1000947. doi: 10.1371/journal.ppat.1000947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brahmer J., Reckamp K.L., Baas P., Crinò L., Eberhardt W.E., Poddubskaya E., Antonia S., Pluzanski A., Vokes E.E., Holgado E. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015;373:123–135. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Motzer R.J., Escudier B., McDermott D.F., George S., Hammers H.J., Srinivas S., Tykodi S.S., Sosman J.A., Procopio G., Plimack E.R., CheckMate 025 Investigators Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015;373:1803–1813. doi: 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borghaei H., Paz-Ares L., Horn L., Spigel D.R., Steins M., Ready N.E., Chow L.Q., Vokes E.E., Felip E., Holgado E. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015;373:1627–1639. doi: 10.1056/NEJMoa1507643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Postow M.A., Chesney J., Pavlick A.C., Robert C., Grossmann K., McDermott D., Linette G.P., Meyer N., Giguere J.K., Agarwala S.S. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N. Engl. J. Med. 2015;372:2006–2017. doi: 10.1056/NEJMoa1414428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robert C., Schachter J., Long G.V., Arance A., Grob J.J., Mortier L., Daud A., Carlino M.S., McNeil C., Lotem M., KEYNOTE-006 Investigators Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015;372:2521–2532. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 21.Sharma P., Retz M., Siefker-Radtke A., Baron A., Necchi A., Bedke J., Plimack E.R., Vaena D., Grimm M.O., Bracarda S. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. Lancet Oncol. 2017;18:312–322. doi: 10.1016/S1470-2045(17)30065-7. [DOI] [PubMed] [Google Scholar]
- 22.Weber J.S., D’Angelo S.P., Minor D., Hodi F.S., Gutzmer R., Neyns B., Hoeller C., Khushalani N.I., Miller W.H., Jr., Lao C.D. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375–384. doi: 10.1016/S1470-2045(15)70076-8. [DOI] [PubMed] [Google Scholar]
- 23.Barber D.L., Wherry E.J., Masopust D., Zhu B., Allison J.P., Sharpe A.H., Freeman G.J., Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 24.Wherry E.J., Ha S.J., Kaech S.M., Haining W.N., Sarkar S., Kalia V., Subramaniam S., Blattman J.N., Barber D.L., Ahmed R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27:670–684. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 25.Wherry E.J., Blattman J.N., Murali-Krishna K., van der Most R., Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol. 2003;77:4911–4927. doi: 10.1128/JVI.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brooks D.G., McGavern D.B., Oldstone M.B. Reprogramming of antiviral T cells prevents inactivation and restores T cell activity during persistent viral infection. J. Clin. Invest. 2006;116:1675–1685. doi: 10.1172/JCI26856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bol K.F., Schreibelt G., Gerritsen W.R., de Vries I.J., Figdor C.G. Dendritic Cell-Based Immunotherapy: State of the Art and Beyond. Clin. Cancer Res. 2016;22:1897–1906. doi: 10.1158/1078-0432.CCR-15-1399. [DOI] [PubMed] [Google Scholar]
- 28.Garg A.D., Coulie P.G., Van den Eynde B.J., Agostinis P. Integrating Next-Generation Dendritic Cell Vaccines into the Current Cancer Immunotherapy Landscape. Trends Immunol. 2017;38:577–593. doi: 10.1016/j.it.2017.05.006. [DOI] [PubMed] [Google Scholar]
- 29.Grenier J.M., Yeung S.T., Khanna K.M. Combination Immunotherapy: Taking Cancer Vaccines to the Next Level. Front. Immunol. 2018;9:610. doi: 10.3389/fimmu.2018.00610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Behrendt R., Schumann T., Gerbaulet A., Nguyen L.A., Schubert N., Alexopoulou D., Berka U., Lienenklaus S., Peschke K., Gibbert K. Mouse SAMHD1 has antiretroviral activity and suppresses a spontaneous cell-intrinsic antiviral response. Cell Rep. 2013;4:689–696. doi: 10.1016/j.celrep.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kahan S.M., Zajac A.J. Immune Exhaustion: Past Lessons and New Insights from Lymphocytic Choriomeningitis Virus. Viruses. 2019;11:156. doi: 10.3390/v11020156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ahn E., Araki K., Hashimoto M., Li W., Riley J.L., Cheung J., Sharpe A.H., Freeman G.J., Irving B.A., Ahmed R. Role of PD-1 during effector CD8 T cell differentiation. Proc. Natl. Acad. Sci. USA. 2018;115:4749–4754. doi: 10.1073/pnas.1718217115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aubert R.D., Kamphorst A.O., Sarkar S., Vezys V., Ha S.J., Barber D.L., Ye L., Sharpe A.H., Freeman G.J., Ahmed R. Antigen-specific CD4 T-cell help rescues exhausted CD8 T cells during chronic viral infection. Proc. Natl. Acad. Sci. USA. 2011;108:21182–21187. doi: 10.1073/pnas.1118450109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gaspar H.B., Cooray S., Gilmour K.C., Parsley K.L., Adams S., Howe S.J., Al Ghonaium A., Bayford J., Brown L., Davies E.G. Long-term persistence of a polyclonal T cell repertoire after gene therapy for X-linked severe combined immunodeficiency. Sci. Transl. Med. 2011;3:97ra79. doi: 10.1126/scitranslmed.3002715. [DOI] [PubMed] [Google Scholar]
- 35.Hacein-Bey-Abina S., Garrigue A., Wang G.P., Soulier J., Lim A., Morillon E., Clappier E., Caccavelli L., Delabesse E., Beldjord K. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Invest. 2008;118:3132–3142. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hacein-Bey-Abina S., von Kalle C., Schmidt M., Le Deist F., Wulffraat N., McIntyre E., Radford I., Villeval J.L., Fraser C.C., Cavazzana-Calvo M., Fischer A. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2003;348:255–256. doi: 10.1056/NEJM200301163480314. [DOI] [PubMed] [Google Scholar]
- 37.Breckpot K., Dullaers M., Bonehill A., van Meirvenne S., Heirman C., de Greef C., van der Bruggen P., Thielemans K. Lentivirally transduced dendritic cells as a tool for cancer immunotherapy. J. Gene Med. 2003;5:654–667. doi: 10.1002/jgm.400. [DOI] [PubMed] [Google Scholar]
- 38.Koya R.C., Kasahara N., Favaro P.M., Lau R., Ta H.Q., Weber J.S., Stripecke R. Potent maturation of monocyte-derived dendritic cells after CD40L lentiviral gene delivery. J. Immunother. 2003;26:451–460. doi: 10.1097/00002371-200309000-00008. [DOI] [PubMed] [Google Scholar]
- 39.Ha S.J., Mueller S.N., Wherry E.J., Barber D.L., Aubert R.D., Sharpe A.H., Freeman G.J., Ahmed R. Enhancing therapeutic vaccination by blocking PD-1-mediated inhibitory signals during chronic infection. J. Exp. Med. 2008;205:543–555. doi: 10.1084/jem.20071949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rowe J.H., Johanns T.M., Ertelt J.M., Way S.S. PDL-1 blockade impedes T cell expansion and protective immunity primed by attenuated Listeria monocytogenes. J. Immunol. 2008;180:7553–7557. doi: 10.4049/jimmunol.180.11.7553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garcia-Bates T.M., Palma M.L., Shen C., Gambotto A., Macatangay B.J.C., Ferris R.L., Rinaldo C.R., Mailliard R.B. Contrasting Roles of the PD-1 Signaling Pathway in Dendritic Cell-Mediated Induction and Regulation of HIV-1-Specific Effector T Cell Functions. J. Virol. 2019;93:e02035-18. doi: 10.1128/JVI.02035-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Badovinac V.P., Messingham K.A., Jabbari A., Haring J.S., Harty J.T. Accelerated CD8+ T-cell memory and prime-boost response after dendritic-cell vaccination. Nat. Med. 2005;11:748–756. doi: 10.1038/nm1257. [DOI] [PubMed] [Google Scholar]
- 43.Seyama K., Nonoyama S., Gangsaas I., Hollenbaugh D., Pabst H.F., Aruffo A., Ochs H.D. Mutations of the CD40 ligand gene and its effect on CD40 ligand expression in patients with X-linked hyper IgM syndrome. Blood. 1998;92:2421–2434. [PubMed] [Google Scholar]
- 44.Lutz M.B., Kukutsch N., Ogilvie A.L., Rössner S., Koch F., Romani N., Schuler G. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J. Immunol. Methods. 1999;223:77–92. doi: 10.1016/s0022-1759(98)00204-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.