

Abstract
Lentivirus vectors (LVs) are efficient tools for gene transfer, but the non-specific nature of transgene integration by the viral integration machinery carries an inherent risk for genotoxicity. We modified the integration machinery of LVs and harnessed the cellular DNA double-strand break repair machinery to integrate transgenes into ribosomal DNA, a promising genomic safe-harbor site for transgenes. LVs carrying modified I-PpoI-derived homing endonuclease proteins were characterized in detail, and we found that at least 21% of all integration sites localized to ribosomal DNA when LV transduction was coupled to target DNA cleavage. In addition to the primary sequence recognized by the endonuclease, integration was also enriched in chromatin domains topologically associated with nucleoli, which contain the targeted ribosome RNA genes. Targeting of this highly repetitive region for integration was not associated with detectable DNA deletions or negative impacts on cell health in transduced primary human T cells. The modified LVs characterized here have an overall lower risk for insertional mutagenesis than regular LVs and can thus improve the safety of gene and cellular therapy.
Keywords: targeted integration, lentivirus vector, genomic safe harbor site, ribosomal DNA, I-Ppl, meganuclease, HIV-1 integrase, protein transduction, all-in-one LV, site-specific
Graphical Abstract
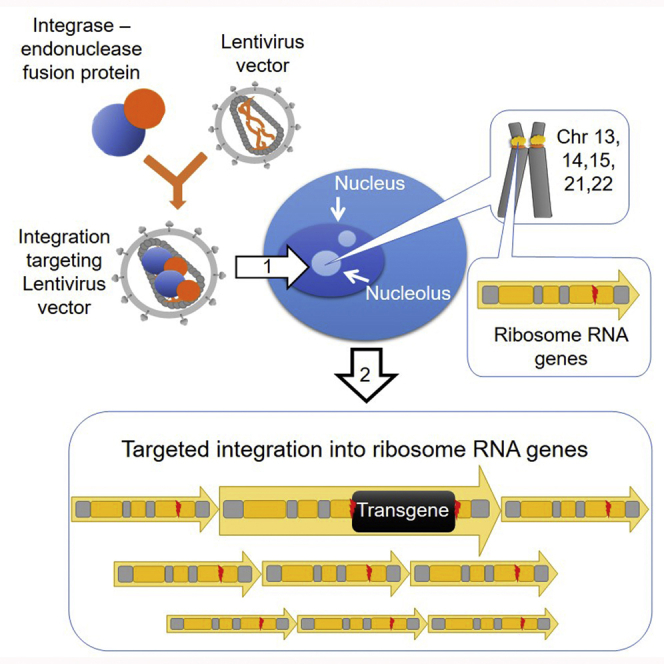
Random integration of therapeutic genes can cause undesired side effects. This study shows that lentivirus vector integration can be efficiently targeted to ribosomal DNA with vectors that carry an endonuclease and the transgene. rDNA cleavage and targeted integration were well tolerated by primary human T cells, and the transgene became transcribed.
Introduction
Human immunodeficiency virus (HIV) 1-based lentivirus vectors (LVs) are increasingly used in different gene therapy trials ranging from the treatment of monogenic diseases to cell therapy of cancer.1,2 Despite being less genotoxic than the more frequently used gammaretrovirus vectors,3 LVs—like all integrating gene transfer systems—possess a risk of causing undesired genomic events that can lead to new malignancies. The genotoxicity risks of LVs are mainly related to aberrant transcriptional activation or inactivation of cellular genes and the induction of new splice variants with potentially oncogenic effects.4
The HIV-1 integrase protein (IN) catalyzes permanent incorporation of vector-carried transgenes into the chromatin of host cells.5 It processes the viral long terminal repeats (LTRs), which flank the viral genome, so that two nucleotides from the LTR's 3′ ends are cleaved off (the 3' guanine-thymine, or GT, dinucleotide). Cellular DNA repair enzymes finish the integration reaction by sealing remaining gaps between the provirus and genomic DNA. Mainly through IN’s interaction with its cellular co-factor PSIP1 (also called lens epithelium-derived growth factor LEDGF/p75) lentiviruses have a strong preference to integrate within coding sequences of actively transcribed protein-encoding genes.6,7 Although no severe adverse effects have been described to date that would result from the typical integration pattern of LVs,2 permanent transgene delivery into target cells would optimally take place in a predefined genomic region that could house transgenes with minimal risks for genotoxicity.
Ribosomal DNA (rDNA) consists of highly repetitive ribosomal RNA (rRNA) genes, of which there are about 400–600 copies in each cell.8 rRNA genes are typically organized as tandem repeats that are separated by intergenic spacer (IGS) regions (Figure 1A). Apart from the 5S rRNA that is encoded from a cluster in chromosome 1, the genes encoding for the RNA components of ribosomes reside in the short arms of the acrocentric human chromosomes 13, 14, 15, 21, and 22 that form the nucleoli.9 Due to the wealth of rRNA genes and the isolated location of nucleolar DNA distant from protein-encoding genes with oncogenic potential, rDNA represents a promising genomic safe harbor for the integration of therapeutic transgenes.
Figure 1.

rDNA and the LVs Generated in This Study to Direct Integration into the I-PpoI Site
(A) An illustration of an acrocentric chromosome (top), the repeating rDNA units (yellow arrows) that contain the rRNA genes and the IGS (middle), and one rRNA gene with its flanking sequences (bottom). Each rRNA gene unit encodes a 45S pre-rRNA that serves as the precursor for the 18S, 5.8S, and 28S rRNAs of mature ribosomes. The I-PpoI site within the 28S rRNA gene is highlighted with a red box. In the current genome version hg38, there are three I-PpoI sites on chromosome 21 that are annotated with a 28S rRNA gene (Table S1). (B) Illustration of the different IN molecule-containing LVs studied in this work, with an enlargement of one IN-fusion protein-containing LV particle. rDNA, ribosomal DNA; rRNA, ribosomal RNA; ETS, external transcribed spacer; ITS, internal transcribed spacer; LV, lentivirus vector; IN, integrase; IND64V, integration deficient IN.
DNA double strand breaks (DSBs) are repaired in cells mainly through two pathways, the non-homologous end joining (NHEJ) and homologous recombination (HR).10 Small insertions or deletions (indel mutations) frequently accompany NHEJ-driven DSB repair, but both pathways have been used successfully for genome editing and to integrate donor DNA molecules into specific sites with the aid of different nucleases.11,12 Most currently available nuclease-based techniques, however, rely on transfection and require using at least two separate vectors or molecules, which can reduce the efficiency of desired modifications and hampers their in vivo use.
We have characterized the full integration site repertoire of LVs that carry an enzymatically weakened homing endonuclease protein that was incorporated into the vectors with the aim of targeting integration to the DSBs it generates. I-PpoI recognizes a 15 bp sequence present in the 28S rRNA genes (RNA28S) of eukaryotes (Figure 1A).13,14 The coupling of LV-transduction with target DNA cleavage enabled an unprecedently high level of transgene integration targeting into rDNA and decreased the genotoxicity risks associated with the use of LVs for gene transfer. These vectors retain the large packaging capacity of LVs and are directly suitable for both ex vivo and in vivo gene transfer applications.
Results
Third-Generation LVs Used for Targeted Integration into rDNA
In order to generate targeted DSBs into rDNA, we used an IN-I-PpoIH78A fusion protein that binds to and cleaves the 28S rRNA gene but affects cellular viability less than the wild-type endonuclease.15 Third-generation LVs containing the IN-I-PpoIH78A were produced with our previously established method that results in the incorporation of both the IN-fusion protein and the integration-deficient IN (IND6V) molecules into vector particles (Figure 1B), which improves their titers and functionality.16 LVs carrying the IN-I-PpoIH78A protein (hereafter called D+H) were characterized side-by-side with LVs carrying the enzymatically inactivated IN-I-PpoIN119A (D+N)16,17 to better delineate the effects of target DNA cleavage on vector integration. Unmodified LVs (INwt) were used as a control. All vectors whose complete integrome was analyzed contained an EGFP transgene construct compatible with both LV-catalyzed and NHEJ-driven integration. The proportion of MRC-5 lung fibroblast cells positive for EGFP expression was 83%–97% at day 2 or 3 post-transduction when genomic DNA was extracted for IS analysis (Table S2).
IN-I-PpoIH78A/N119A Inclusion Changes the Global Integration Pattern and Genotoxicity Risks of LVs
IS were analyzed separately for the non-repetitive and repetitive portions of the human genome (Hg38). The total numbers of IS retrieved for the different vector types were 20,789 for LV-INwt, 7,181 for LV-D+H, and 2,906 for LV-D+N. The proportions of IS that had multiple hits in the genome (MH-IS) of the total data was found to be significantly higher in the IN-modified LVs in comparison to the control LV (Figure 2A). The exactly mappable or unique hit (UH)-IS were used to determine the overall integration pattern for each vector. The chromosomal distribution of IS was similar between the vectors apart from deviations in seven chromosomes (Figure 2B). The distribution of IS within genes was more uniform throughout the coding region for the IN-fusion protein containing LVs than for the INwt LVs, which typically integrate less frequently in the first 10th percentile of a gene’s length (Figure 2C).18 All analyzed LVs favored integration within genes over integration in their upstream regions, but in comparison to INwt LVs, there was a small but statistically significant increase in integration within the first 5 kb upstream of genes with the IN-modified LVs. The IN-fusion protein-containing LVs had fewer intragenic IS than INwt LVs (Figure 2D) and hence a smaller risk to interrupt cellular genes with important functions. A vector’s tendency to integrate into or close to oncogenes is an important parameter of its safety, and HIV is known to integrate into these areas more than would be expected through chance.19 Both IN-fusion protein-containing LVs had fewer IS within and near oncogenes in comparison to INwt-LVs (Figure 2E; Table S3). The IN-fusion protein LVs mainly integrated without IN activity in contrast to INwt LVs, whose LTRs were most frequently processed (Figure S1).
Figure 2.

Effects of IN-I-PpoIH78A/N119A Fusion Protein Inclusion on the Integration Characteristics of LVs
(A) Composition of the integration site data and numbers of unique IS (UH) and multiple-hit IS (MH) retrieved for the different vectors. (B) Chromosomal distribution of integration sites. Chromosome numbers are shown on the x axis. (C) Distribution of integration sites with respect to upstream (US) regions of genes, the gene length (% of within gene) and downstream (DS) of genes. (D) A more detailed illustration of IS distribution within the uniquely mapping (UH; blue) and repetitive (MH; orange) portions of the genome. (E) Integration frequency within oncogenes. A list comprising 2,579 human cancer genes (http://www.bushmanlab.org/links/genelists) was used for the comparison. The statistical differences between the IN-modified LVs and the control LV are shown above the bars (p < 0.0001 for both). Statistical differences between LVs were calculated using two-sided Fisher’s exact test (D+H LVs versus D+N LVs) or with two-sided chi-square test (INwt LV compared to D+H or D+N LVs). ∗∗∗p < 0.001; ∗∗p < 0.01; ∗p < 0.05. In (C), the black asterisks denote differences between the control vector INwt LV and the IN-modified LVs, and gray asterisks denote differences between the D+H and D+N LVs. Intrag., intragenic IS; Interg., intergenic IS; INwt, wild-type integrase; D+H, IND64V and IN-I-PpoIH78A-containing LVs; D+N, IND64V and IN-I-PpoIN119A-containing LVs.
rRNA and tRNA Repeats Are the Most Favored Targets for the IN-Modified LVs within the Repetitive Genome
The MH-IS were used to characterize the vectors’ preferences to integrate within different genomic repeat elements, which were identified using RepeatMasker.20 I-PpoI has 12 perfect recognition sites in the current genome version (Hg38), and all but two of these localize to rRNA repeat-contained sequences placed either on the acrocentric chromosome 21 or in non-acrocentric chromosomes that contain fragments of rRNA genes (Table S1). For D+H LVs, 41.9% of the vector’s MH reads were within rRNA repeats (Figure 3A). In contrast, D+N LV reads were most frequently associated with transfer RNA (tRNA) genes (17.8%), SINE/Alu repeats, and third most with rRNA repeats. tRNA genes were among the top three repeats also for the D+H LVs. INwt LVs preferred SINE/Alu (40.0%) and LINE/L1 repeats (15.5%) and had very few integrations in either rRNA or tRNA genes. Interestingly, also signal recognition particle RNA (srpRNA) and other repetitive non-coding RNA (ncRNA) genes were more frequently targeted for integration by the IN-modified LVs than by the control vector (Figure 3A; Figure S2). Based on the differences between the D+H and D+N LVs, it is evident that the introduction of DSBs increases vector integration into rRNA repeats.
Figure 3.

Characterization of Vector Integration within the Repetitive Genome and rDNA
(A) Integration frequency into different repeat types within the repetitive genome. (B) Total efficiency of integration targeting into an rDNA unit (including the rRNA coding region and the IGS) and within a 235-bp window around the I-PpoI site. For the ddPCR-based quantification of I-PpoI site-directed integration, the mean (with SEM) of six measurements is shown. (C–E) Coverage plots where read coverage on the positive strand (+ve; scale on the right y axis) is shown with a darker shade and on the negative strand (−ve; scale on the left y axis) with a lighter shade for each LV type. (C) A large-scale view of IS read localization within the Chr21 locus containing annotated rRNA genes (window size, 50 kb). (D) A closeup view of IS distribution within the 28S rRNA gene (window size, 1.6 kb). (E) Illustration of the reads mapping within and near the I-PpoI site (shown with purple fonts). Window size, 300 bp. ∗Repeatmasker-identified repeats without manual correction and annotation of additional rRNA gene unit features. ∗∗Repeatmasker-identified repeats. ¤: integration frequency within an area extending 203 bp upstream and 32 bp downstream of the cleaved I-PpoI site (see Figure S3 for details).
28S rRNA Gene Cleavage Enables Highly Efficient Integration Targeting to rDNA
In addition to nucleolus-associated rDNA, rRNA gene segments are also found in the non-nucleolar genome,21 and a fraction of the uniquely mapping IS reads localized to these sites. The compiled IS data comprising both the unique and multiple hit IS reads was therefore analyzed to determine the absolute numbers of rDNA-localized integrations. For the D+H LVs, 21.3% of all IS localized to sequences contained within an rDNA unit (Figure 3B), and the most favored locus within the rRNA gene was the 28S rRNA (Figure 3C). rDNA-localized IS comprised 2.6% and 0.08% of all IS for the vectors D+N and INwt, respectively (Figure 3B), which is well in line with our previous characterizations of these vectors.16 Similar to D+H LVs, the majority of D+N LV proviruses clustered into 28S rRNA but with a much lower frequency (Figure 3C).
To verify the differences between the vectors in catalyzing targeted integration, we used a droplet digital PCR (ddPCR)-based method that detects integrated vector genomes within a 235-bp window around the I-PpoI site in the 28S rRNA gene (Figure S3). At day 9 post-transduction, 20.9% of the D+H LV proviruses were estimated to reside in this locus in transduced MRC-5 cells (Figure 3B; see also Table S4). The proportion of IS reads within the same window was 9.9%. In comparison, for the LVs containing D+N and INwt the proportion of IS reads was 0.8% and 0.02%, respectively, and the ddPCR-based targeting estimates 0.2% and 0.1% (Figure 3B). Integration of the IN-modified LVs occurred more frequently in sense orientation both near the I-PpoI site (66% for D+H and 71% for D+N; Figure 3D) and within it (Figure 3E). Typical for DSB repair through NHEJ, integration into the I-PpoI site involved small indel mutations, which were observed more frequently in the D+H LV-treated than in the D+N LV-transduced cells (Figure S4).
The ddPCR result suggested that for LV D+H, the actual level of integration targeting into the immediate vicinity of the I-PpoI site in the rRNA gene is at least two times higher than that resolved with the IS sequencing method. Next, we used vectors containing a selectable marker for zeocin resistance to test whether the 28S rRNA insertions remained stable through conditions that require expression of the transgene. The proportion of proviruses in and near the I-PpoI site remained similar between selected and unselected hTERT-RPE1 cells, as verified with ddPCR (Table S5). Taken together, when LV transduction is coupled with the cleavage of target DNA by a vector-carried endonuclease, stable and highly efficient targeted integration of transgenes into rDNA is achieved.
Integrase-I-PpoI Fusion Proteins Target Integration into Strong Hotspots That Are Distinct from the Areas Naturally Preferred by HIV-Derived LVs
Specific genomic loci have been identified that recur as preferential integration loci, or integration hotspots, for HIV-1 and LVs.22,23 Such common integration sites (CISs) were identified to see if the inclusion of the IN-I-PpoI-fusion proteins altered the natural preferences of LVs. Significant CISs containing at least three ISs were characterized for their genomic coordinates and for the features they contained. In comparison to the IN-modified LVs, a larger proportion of INwt LVs’ unique ISs were engaged with integration hotspots, but proportionally fewer ISs formed the strongest CIS (Figure S5; Data S1). The majority of the 15 strongest CISs (n = 18 individual CISs) of the LV INwt were localized within protein-encoding genes (77.8%) (Table 1), with many of the hotspots residing in regions previously characterized as preferred integration sites for LVs and HIV-1 (Tables S6 and S7).22, 23, 24, 25, 26 The median CIS positions (CIS foci) of the seven strongest hotspots of the D+H LVs (n = 26) were frequently found in intergenic loci (35%), and in many cases the RefSeq gene within the hotspot or nearest to it was a ncRNA gene (31%) (Table 1; Figure S6A). All together, six D+H LV CIS foci were within an rRNA repeat and five of them localized to I-PpoI cleavage sites on separate non-acrocentric chromosomes (Table 1; Data S1), verifying correct I-PpoI activity and NHEJ-driven insertion at the generated DSBs. The five strongest CIS foci (n = 21 individual CISs) of the D+N LVs revealed a similar preference toward intergenic areas and ncRNA gene proximity as was seen for D+H LVs, but instead of rRNA gene repeats, the hotspots frequently associated with tRNA repeats (29%) (Table 1; Figure S6). All together, 9.5% of all D+N LVs’ unique CIS-associated ISs were within tRNA repeats, whereas neither tRNA nor rRNA repeats were found in the hotspot-contained IS of the INwt LVs (n = 8,450) (Figure S6B). Analysis of all CIS-associated UH-IS confirmed that both IN-modified LVs had significantly more intergenic IS than the control vector (Figure 4A). INwt-LVs’ CIS-associated IS localized into or near protein-encoding genes more frequently than those of D+H LVs, and the latter targeted RNA genes more often than the control vector. Genes and pseudogenes of the ribsomal proteins L and S (RPL and RPS, respectively) contained in the large and small ribosome subunits were also frequently associated with the CIS of the D+H LVs (Table 1).
Table 1.
Characterization of the Strongest Integration Hotspots among the Uniquely Mappable IS
| Rank | IS # | Median Location | Genea | Repeata,b | Nearest RefSeq Gene | Dimension (kB) | |
|---|---|---|---|---|---|---|---|
| INwt (UH) | 1 | 67 | chr16:1633220 | CRAMP1 | SINE/Alu | 524 | |
| 2 | 53 | chr8:144306704 | HSF1 | LINE/L1 | 475 | ||
| 3 | 52 | chr16:2080539 | TSC2 | SINE/Alu | 334 | ||
| 4 | 44 | chr11:66094636 | PACS1 | LINE/L1 | 465 | ||
| 5 | 35 | chr11:65566836 | intergenic | NA | SSSCA1-AS1 | 235 | |
| 6 | 33 | chr16:688665 | WDR24 | NA | 368 | ||
| 7 | 31 | chr1:1334252 | TAS1R3 | NA | 184 | ||
| 8 | 28 | chr19:1199664 | intergenic | NA | STK11 | 223 | |
| 9 | 27 | chr6:30681690 | PPP1R18 | SINE/Alu | 317 | ||
| 10 | 25 | chr17:81593484 | NPLOC4 | DNA/hAT-Charlie | 163 | ||
| 11 | 22 | chr17:82147186 | CCDC57 | simple | 279 | ||
| 12 | 21 | chr9:128599563 | SPTAN1 | SINE/Alu | 311 | ||
| 13 | 19 | chr12:49150673 | intergenic | SINE/Alu | TUBA1B | 247 | |
| 13 | 19 | chr19:49842535 | PTOV1-AS1 | SINE/Alu | 157 | ||
| 14 | 18 | chr6:31687953 | ABHD16A | NA | 182 | ||
| 14 | 18 | chr10:112589294 | VTI1A | LTR/ERV-MaLR | 174 | ||
| 15 | 17 | chr11:65218552 | SLC22A20P | NA | 166 | ||
| 15 | 17 | chr17:81880539 | intergenic | LINE/L1 | ALYREF | 84 | |
| D+H (UH) | 1 | 12 | chr6:27631516 | intergenic | (tRNA) | LINC01012 | 37 |
| 2 | 11 | chr6:28658243 | intergenic | tRNA | LINC00533 | 86 | |
| 3 | 10 | chr5:140711372 | VTRNA1-1 | NA | 8 | ||
| 4 | 9 | chr2:38482053 | LOC101929596 (RPLP0P6) | NA | 1 | ||
| 4 | 9 | chr3:182901763 | ATP11B | NA | 0 | ||
| 4 | 9 | chr20:30512867 | intergenic | LSU-rRNA_Hsa | MLLT10P1 | 1 | |
| 5 | 6 | chr2:131102011 | intergenic | NA | PLEKHB2 | 69 | |
| 5 | 6 | chr2:132279863 | intergenic | LSU-rRNA_Hsa | ANKRD30BL | 0 | |
| 6 | 5 | chr11:65611215 | MAP3K11 | NA | 55 | ||
| 6 | 5 | chr17:81897445 | ANAPC11 | NA | 52 | ||
| 6 | 5 | chr20:44466866 | intergenic (RPL37AP1) | NA | LINC01620 /C20orf62 | 0 | |
| 7 | 4 | chr1:8866735 | ENO1 | NA | 17 | ||
| 7 | 4 | chr1:174904258 | RABGAP1L | SINE/Alu | 48 | ||
| 7 | 4 | chr2:3577177 | RPS7 | SINE/Alu | 19 | ||
| 7 | 4 | chr2:27050883 | intergenic | (tRNA) | AGBL5-AS1 | 30 | |
| 7 | 4 | chr4:145884509 | ZNF827 | NA | 47 | ||
| 7 | 4 | chr5:122352156 | SNCAIP | NA | 37 | ||
| 7 | 4 | chr6:153282725 | intergenic (RPL27AP6) | NA | RGS17 | 32 | |
| 7 | 4 | chr10:125738308 | EDRF1 | NA | 0 | ||
| 7 | 4 | chr11:77886544 | INTS4/AAMDC | LSU-rRNA_Hsa | 15 | ||
| 7 | 4 | chr12:56175248 | SMARCC2 | SINE/Alu | 22 | ||
| 7 | 4 | chr16:685472 | WDR24 | NA | 29 | ||
| 7 | 4 | chr19:1131901 | SBNO2 | NA | 36 | ||
| 7 | 4 | chr19:12894097 | GCDH (RPS6P25) | NA | 36 | ||
| 7 | 4 | chr21:8415028 | intergenic | simple (45S rRNA)c | MIR6724-1 | 39 | |
| 7 | 4 | chrX:135542502 | INTS6L | SINE/Alu | 0 | ||
| D+N (UH) | 1 | 10 | chr6:27631467 | intergenic | tRNA | LINC01012 | 167 |
| 2 | 7 | chr8:144456689 | CYHR1 | NA | 114 | ||
| 3 | 5 | chr11:66348159 | LOC102724064 | tRNA | 7 | ||
| 3 | 5 | chr12:56190397 | intergenic | tRNA | SMARCC2 | 0 | |
| 3 | 5 | chr19:3982952 | EEF2 | NA | 6 | ||
| 4 | 4 | chr5:140711372 | VTRNA1-1 | NA | 8 | ||
| 5 | 3 | chr1:951876 | NOC2L | NA | 6 | ||
| 5 | 3 | chr1:145157237 | intergenic | tRNA | LOC103091866 | 0 | |
| 5 | 3 | chr1:156312177 | CCT3 | NA | 8 | ||
| 5 | 3 | chr2:27050871 | intergenic | tRNA (SINE/Alu) | AGBL5-AS1 | 15 | |
| 5 | 3 | chr5:178204539 | HNRNPAB | NA | 38 | ||
| 5 | 3 | chr5:181236966 | RACK1 | NA | 51 | ||
| 5 | 3 | chr7:5634480 | RNF216 | NA | 39 | ||
| 5 | 3 | chr8:144311250 | HSF1 | NA | 5 | ||
| 5 | 3 | chr9:127972911 | FAM102A | NA | 44 | ||
| 5 | 3 | chr9:136375334 | intergenic | NA | SNAPC4 | 8 | |
| 5 | 3 | chr16:1817574 | HAGH | NA | 18 | ||
| 5 | 3 | chr16:1960749 | NDUFB10 | SINE/MIR | 15 | ||
| 5 | 3 | chr16:67887498 | NRN1L | SINE/Alu | 8 | ||
| 5 | 3 | chr17:8221619 | LINC00324 | tRNA | 6 | ||
| 5 | 3 | chr20:63678092 | RTEL1 | NA | 4 |
UH, unique hits; NA, not applicable; LSU-rRNA_Hsa, large subunit (28S) rRNA repeat.
Gene and repeat family in CIS median locus.
Repeat is shown in parenthesis if it is found in > 50% of the reads, but not in the exact CIS median locus.
ISs are placed into the IGS (UCSC genome browser Hg38).
Figure 4.

Characterization of CIS-Associated IS
(A) All unique IS associated with CIS were analyzed for their occurrence in intergenic loci, pseudogenes, ncRNA genes (“RNA genes”) and protein-encoding genes. The proportions of IS within each feature are shown as a percentage of all CIS-associated UH-IS. The numbers of CIS-contained IS are as follows: 8,450 for LV INwt, 333 for LV D+H, and 81 for LV D+N. (B) Characterization of the proportion of IS localizing to protein-encoding genes, pseudogenes, ncRNA genes, and ribosomal protein-encoding genes (RPL and RPS genes) of all CIS-associated IS (UH-MH-CIS). The numbers of all CIS-associated IS are as follows: 2,506 for LV D+H, 498 for LV D+N, and 10,367 for LV INwt. The differences between the vectors were analyzed with two-sided Fisher’s exact test (D+H LVs versus D+N LVs) or with two-sided chi-square test (INwt LV compared to D+H or D+N LVs). ∗∗∗p < 0.001; ∗p < 0.05. In (B), the asterisks are shown only for INwt LV, whose difference to each IN-modified LVs was similar. Ribosomal prot., genes encoding for the protein constituents of mature ribosomes.
The repeat-associated ISs make up at least one-third of the total IS number in the IN-fusion protein LVs, and a more accurate representation of genomic features and gene types preferentially targeted for integration by these vectors could be obtained by analyzing CIS in a combined dataset containing both the UH- and the MH-IS. In this analysis, the D+H LVs’ strongest CIS was now identified in the 28S rRNA gene, and it contained 19% (n = 1367 IS) of all ISs (Table 2; Figure S7A). The strongest CIS of the D+N vectors also localized into the 28S rRNA gene with 2.5% of all IS. Integration targeting to the most preferred locus was again the weakest for LV INwt, as only 0.3% (n = 68 ISs) of the vector’s IS localized to the strongest CIS (Table 2; Figure S7A). Inclusion of the MH data into the CIS analysis enabled the detection of new repetitive gene types, such as 5S rRNA (RNA5S) and srpRNA genes, in the integration hotspots of the IN-modified LVs (Table 2). The characteristic preferences of these LVs to integrate into tRNA and rRNA repeats and intergenic loci remained the same but became more pronounced (Table 2; Figure S7B). Similarly, the differences between the IN-modified LVs and the control LV in targeting protein-encoding genes, RNA genes and the multiple ribosome subunit genes grew stronger (Figure 4B). Finally, a clear increase in the IS numbers per strongest CIS was observed, owing to the large proportion of MH-IS forming them (Table 2). For the INwt LV, the differences between the two analysis types were much subtler and mainly related to slightly higher IS numbers per identified CIS (Tables 1 and 2). Taken together, the integration hotspots of the IN-modified LVs strongly associate with repetitive RNA-encoding genes and show very little resemblance to the well-characterized hotspots near protein-encoding genes of unmodified LVs.
Table 2.
Characterization of the Strongest Integration Hotspots among All IS
| Rank | IS # | Median Location | Genea | Repeata,b | Nearest RefSeq Gene | Dimension (kB) | MH% | |
|---|---|---|---|---|---|---|---|---|
| INwt (UH+MH) | 1 | 68 | chr16:1639939 | CRAMP1L | NA | 524 | 1.5 | |
| 2 | 61 | chr16:2083750 | TSC2 | NA | 334 | 14.8 | ||
| 3 | 57 | chr8:144321862 | DGAT1 | NA | 475 | 7.0 | ||
| 4 | 51 | chr11:66093177 | PACS1 | LINE/L1 | 465 | 13.7 | ||
| 5 | 38 | chr11:65553655 | LTBP3 | NA | 258 | 7.9 | ||
| 6 | 34 | chr1:1336483 | DVL1 | NA | 184 | 8.8 | ||
| 7 | 33 | chr16:688665 | WDR24 | NA | 368 | 0.0 | ||
| 8 | 31 | chr19:26670953 | centromeric | Satellite/centromere | LINC00662 | 544 | 100.0 | |
| 9 | 30 | chr6:30665510 | DHX16 | NA | 317 | 10.0 | ||
| 10 | 28 | chr19:1199664 | intergenic | NA | STK11 | 223 | 0.0 | |
| 11 | 26 | chr17:81592393 | NPLOC4 | NA | 163 | 3.8 | ||
| 12 | 25 | chr17:82142721 | CCDC57 | NA | 371 | 12.0 | ||
| 12 | 25 | chr22:50382044 | PPP6R2 | LINE/L1 | 279 | 28.0 | ||
| 13 | 23 | chr9:128606115 | SPTAN1 | SINE/Alu | 311 | 8.7 | ||
| 14 | 22 | chr12:49147302 | intergenic | SINE/Alu | TUBA1A | 247 | 13.6 | |
| 15 | 21 | chr19:49849663 | PTOV1-AS1 | NA | 157 | 9.5 | ||
| D+H (UH+MH) | 1 | 1367 | chr21:8444914 | RNA28SN1 | LSU-rRNA_Hsa | MIR6724-4 | 135 | 99.7 |
| 2 | 130 | chr14:49862760 | RN7SL2 | srpRNA/7SLRNA | 9 | 100.0 | ||
| 3 | 53 | chr14:49586605 | RN7SL1 | srpRNA/7SLRNA | 0 | 100.0 | ||
| 4 | 37 | chr20:30512867 | intergenic | LSU-rRNA_Hsa | MLLT10P1 | 0 | 75.7 | |
| 5 | 34 | chr2:132279864 | intergenic | LSU-rRNA_Hsa | ANKRD30BL | 0 | 82.4 | |
| 6 | 33 | chr11:77886544 | INTS4/AAMDC | LSU-rRNA_Hsa | 30 | 87.9 | ||
| 7 | 29 | chr6:27631414 | intergenic | (tRNA) | LINC01012 | 163 | 48.3 | |
| 7 | 29 | chr17:8187235 | intergenic | tRNA | MIR4521 | 111 | 93.1 | |
| 8 | 26 | chr1:228646038 | RHOU/DUSP5P1/RNA5S17 | 5S rRNA | 3 | 100.0 | ||
| 9 | 23 | chr19:50128415 | SNAR-A11 | NA | 11 | 100.0 | ||
| 10 | 21 | chr6:125780266 | intergenic | tRNA | NCOA7 | 43 | 90.5 | |
| 11 | 19 | chr1:237603123 | RYR2 | LSU-rRNA_Hsa | 0 | 84.2 | ||
| 12 | 15 | chrX:109054236 | intergenic | LSU-rRNA_Hsa | MIR6087 | 0 | 100.0 | |
| 13 | 14 | chr16:3191572 | intergenic | tRNA | OR1F1 | 83 | 85.7 | |
| 13 | 14 | chr21:17454798 | intergenic | tRNA | LINC01549 | 0 | 100.0 | |
| 13 | 14 | chr22:32039474 | intergenic (RPS17P16) | NA | SLC5A1 | 0 | 78.6 | |
| 14 | 13 | chr8:69690270 | SLCO5A1 | LSU-rRNA_Hsa | 0 | 76.9 | ||
| 15 | 12 | chr6:28863698 | intergenic | tRNA | LINC01623 | 192 | 66.7 | |
| D+N (UH+MH) | 1 | 73 | chr21:8444904 | intergenic | LSU-rRNA_Hsa | MIR6724-4 | 12 | 100.0 |
| 2 | 47 | chr17:8125819 | intergenic | (tRNA) | HES7 | 108 | 91.5 | |
| 3 | 29 | chr14:49862666 | RN7SL2 | srpRNA/7SLRNA | 0 | 100.0 | ||
| 4 | 22 | chr6:27618534 | intergenic | SINE/Alu (tRNA) | LINC01012 | 167 | 54.5 | |
| 5 | 15 | chr5:181207830 | intergenic | tRNA | TRIM7 | 68 | 80.0 | |
| 5 | 15 | chr16:3191501 | intergenic | tRNA | OR1F1 | 16 | 93.3 | |
| 6 | 11 | chr6:125780305 | intergenic | tRNA | NCOA7 | 0 | 100.0 | |
| 6 | 11 | chr14:49586625 | RN7SL1 | srpRNA/7SLRNA | 0 | 90.9 | ||
| 6 | 11 | chr19:50128411 | SNAR-A11 | NA | 16 | 100.0 | ||
| 7 | 9 | chr19:46811031 | intergenic | SINE/Alu | SNAR-E | 20 | 77.8 | |
| 8 | 8 | chr1:145287841 | intergenic | tRNA | NBPF20 | 0 | 100.0 | |
| 8 | 8 | chr1:161425134 | intergenic | LINE/L1 (tRNA) | CFAP126 | 85 | 87.5 | |
| 8 | 8 | chr17:82494740 | intergenic | tRNA | NARF | 0 | 100.0 | |
| 8 | 8 | chr19:1021625 | RNU6-2 | snRNA/U6 | 59 | 87.5 | ||
| 9 | 7 | chr1:228646036 | RHOU/DUSP5P1 | (5S rRNA) | 2 | 100.0 | ||
| 9 | 7 | chr5:140711373 | VTRNA1-1 | NA | 15 | 42.9 | ||
| 9 | 7 | chr8:144456689 | CYHR1 | NA | 114 | 0.0 | ||
| 9 | 7 | chr12:56190397 | intergenic | tRNA | SMARCC2 | 0 | 28.6 | |
| 9 | 7 | chr14:58239894 | intergenic | tRNA | ACTR10 | 0 | 100.0 | |
| 9 | 7 | chr15:45201222 | intergenic | tRNA | SHF | 0 | 100.0 | |
| 9 | 7 | chr19:1383594 | intergenic | tRNA | NDUFS7 | 4 | 85.7 | |
| 9 | 7 | chr21:17454808 | intergenic | tRNA | LINC01549 | 0 | 100.0 | |
| 10 | 6 | chr9:133020150 | intergenic (EEF1A1P5) | NA | SNORD141A | 1 | 100.0 | |
| 10 | 6 | chr11:66348155 | LOC102724064 | tRNA | 7 | 16.7 | ||
| 10 | 6 | chr16:68742482 | CDH1 | 5S rRNA | 0 | 100.0 | ||
| 10 | 6 | chr19:4724132 | intergenic | tRNA | DPP9 | 0 | 100.0 |
MH%, fraction of the multiple hit (MH)-IS of all CIS-forming IS; UH, unique hits; NA, not applicable; LSU-rRNA_Hsa, large subunit (28S) rRNA repeat.
Gene and repeat family in CIS median locus.
Repeat is shown in parenthesis if it is found in >50% of the reads, but not in the exact CIS median locus.
I-PpoI Protein Inclusion Increases Vector Integration in Genomic Features That Are Enriched in Nucleolus-Associated Domains
Nucleolus-associated domains (NADs) are defined chromatin domains that dynamically interact with nucleoli.27 Enrichment of pseudogenes in NADs has been characterized in plants,28 and the ribosomal protein encoding genes are known to have multiple processed pseudogenes in the human genome. Also, specific gene families and genes, such as those encoding for tRNAs and the protein constituents of the ribosomes, are enriched in NADs.29, 30, 31, 32, 33, 34 Since these gene types were frequently hit by the IN-modified LVs (Figures 3A and 4B) and identified in their integration hotspots (Tables 1 and 2; Figures S6 and S7), we asked whether additional similarities would exist between the identified CIS loci and NAD-contained regions. After annotating the ISs of the different LVs with pseudogenes, we found that integration in pseudogenes occurred more frequently with the IN-modified LVs than with the control LV (Figure 5A). When the pseudogene-annotations were used in place of the original NCBI Reference Sequence Database (Refseq) gene annotations, integration was found to be more frequent also in RPL and RPS gene-derived sequences with the IN-modified LVs than with the INwt LVs (Figure 5A). In addition to these structural proteins of the ribosomes, also larger groups of genes related to ribosome biogenesis contained more integrations with the IN-modified LVs than with the control LV (Figure 5B).
Figure 5.

Characterization of Preferential LV Integration in Specific Gene Sets and Gene Ontology Terms
(A) Integration frequency within pseudogenes and ribosomal protein genes or pseudogenes derived of them. (B) Integration frequency in gene sets involved in ribosome biogenesis (ribosome biogenesis in eukaryote SuperPath35) and rRNA processing (rRNA processing in the nucleus and cytosol SuperPath35). (C–E) Enrichment heatmaps of the most overrepresented pathways and processes among genes present in the CIS-engaged integration sites, colored by p values. Heatmap in (C), for D+H LVs; in (D), for D+N LVs; and in (E), for INwt LVs. RPL/RPS genes, large subunit ribosomal proteins/small subunit ribosomal proteins, respectively, or pseudogenes derived of these genes. In (A) and (B), the differences between the datasets were calculated with two-sided chi-square tests. ∗∗∗p < 0.001; ∗p < 0.05.
Significantly enriched Gene Ontology (GO) terms among NAD genes include ribosome, mitochondrion, cytosolic large/small ribosomal subunit and nucleolus.29 A GO-analysis of the CIS-engaged genes revealed that several pathways and processes related to ribosome structure and function were enriched among the genes preferentially targeted for integration by the IN-fusion protein LVs and that similar GO-terms were enriched as among NAD-associated genes (Figures 5C and 5D; Data S2). Interestingly, also mitochondria-related terms were enriched for D+N LVs, but not for D+H LVs. For the INwt LV, no enrichment of ribosomal structure or function-related terms was observed (Figure 5E). In line with previous studies,36 the most enriched pathways and processes were instead related to cell cycle and its control as well as chromatin organization. The similarities between NAD-associated features and the gene types preferentially targeted for integration by the IN-fusion protein LVs indicates that the localization of a chromosomal region close to nucleoli is an additional determinant of the vectors’ preferential integration, in addition to the primary sequence recognized by I-PpoI.
Integration Targeting and Cellular Responses to Transduction in Primary Human T Cells
Having confirmed rDNA-targeted integration in both the slowly and finitely dividing lung fibroblast cells (MRC-5) and in the non-cancerous but immortalized retinal pigment epithelium cells (hTERT-RPE1), we asked how the IN-modified vectors would perform in the transduction of primary human T cells, which represent a relevant cell type for clinical gene and cell therapy. For this aim, T cells from two individuals were enriched, transduced with the different LVs, and assayed for targeted integration and different indicators of cell health and cytotoxicity. Estimation of targeted integration at day 10 post-transduction with the ddPCR-based method showed that up to 8% of the D+H LV integration events reside in the immediate vicinity of the I-PpoI site in the 28S rRNA gene, the mean targeting efficiencies ranging from 2.6% to 5.7% (Figures 6A and 6B; Tables S8 [day 2] and S9 [day 10]). With the INwt control LVs, the mean targeting efficiencies were 0.0%–0.1%.
Figure 6.

Quantification of Targeted Integration in the 28S rRNA Gene and Detection of Potential Deletions in the rRNA Gene and in the Short Arms of the Acrocentric Chromosomes
(A and B) The proportion of vectors integrated near the I-PpoI site in the 28S rRNA gene was quantitated with ddPCR. The vector dose used (5k and 10k vp/cell) is shown in parenthesis after the LV abbreviation. The values of the two analyzed wells per vector and vp-dose combinations are shown (mean with SEM from duplicate measurements per sample; see also Table S9) with the results from T cells extracted from donor 1 shown in (A) and T cells from donor 2 in (B). (C and D) The copy number of the 18S rRNA gene (C) and the DJ region (D) were quantitated from T cells transduced with 10k vp/cell at day 2 post-transduction. The same sample replicates were used as in (A) and (B). These four measurements per vector group (Table S10) are shown with their mean and SEM. The differences in copy numbers were analyzed with one-way ANOVA by comparing the vector-groups’ values to the same donor’s NTD control with Dunnett’s multiple comparison test. NTD, non-transduced cells; DJ, distal junction sequence; p.td, post-transduction; rRNA, ribosomal RNA.
The number of metabolically active live cells was determined to study if T cells transduced with the D+H-containing LVs proliferate similarly to cells transduced with the control LV. In a test using 5,000 vector particles (5k vp) per cell, the number of viable cells was the highest in the INwt LV group, and no differences between the groups were observed that could be specifically addressed to the IN content of the modified LVs (Figures S8A and S8B). When using a higher vector dose of 10k vp/cell, the only test group having significantly fewer metabolically active cells in comparison to the INwt control at the last time point assayed was the D+H LV group, whose mean cell numbers were 81%–85% of those of the control vector (Figures S8C and S8D).
Next, it was studied whether the cleavage of rRNA genes and subsequent transgene integration would cause direct cytotoxicity or induce apoptosis that is followed by secondary necrosis. Of the three LVs tested, a statistically significant increase in the apoptosis signal in relation to untreated cells was observed only for LV D+N at day 3 post-transduction (5k vp/cell, p < 0.05) (Figure S9). An elevated necrosis signal was observed for INwt LVs in altogether three time points (p < 0.05, p < 0.01, and p < 0.001), and for D+H LV at one time point (p < 0.05) in comparison to non-transduced cells (Figure S10). Etoposide-treated cells were positive for apoptosis induction at day 1 and for necrosis at days 2 and 3 post-treatment (Figures S9 and S10). Since there was no increase of necrosis in T cells that would be clearly attributable to the D+H content of the vectors, it is likely that the decrease in cell numbers we observed in the viability test results from a moderate slowdown of division and/or metabolism in LV D+H-transduced cells.
As learned from studies using the Cas nucleases, target DNA cleavage can cause different types of mutations and rearrangements of genomic DNA, including large deletions.37, 38, 39, 40 rDNA represents a recombination hotspot in meiotic cells and in cancer, hence the number of rRNA genes can vary substantially both between and within individuals.8,41, 42, 43, 44 To see if the number of rRNA genes would be affected by the use of D+H LVs, we quantitated the 18S rRNA gene (RNA18S) copies in transduced T cells at day 2 post transduction. Consistent with previous studies,8 the mean gene copy numbers or rRNA genes varied between 478 and 701 per cell, and no statistically significant differences were observed between the non-transduced cells and D+H or INwt LV-transduced cells (Figure 6C; Table S10). To address the occurrence of larger deletions potentially affecting whole acrocentric chromosome arms, we studied the copy number of the distal junction (DJ) sequence that flanks the rRNA array at the telomeric side.45 Similar to the rRNA genes, no statistically significant differences were observed between the three groups, and 13 to 18 copies of these sequences were detected per cell (Figure 6D). In conclusion, transduction with the 28S rRNA gene-cleaving D+H LVs does not cause detectable variations in the rRNA gene nor in the DJ sequence copy numbers in T cells.
Cleavage of the rRNA gene and transgene integration into it can affect the transcription of both the rDNA and the provirus. To address the question of whether vectors integrated into the I-PpoI site become transcribed, we analyzed total RNA extracted from D+H and INwt LV-transduced T cells at days 2 and 10 post-transduction with site-specific RT-ddPCR. Vector sequence-containing rRNA transcripts were detected at both time points and only in the D+H LV group, confirming that proviruses within the targeted 28S rRNA gene become transcribed (Tables S11 and S12).
Discussion
In this study, we show that LV integration can be directed to the rDNA of normal human cells with an unprecedently high efficiency when transduction is coupled with target site cleavage. In non-selected MRC-5 cells, the vectors carrying an endonuclease with reduced DNA cleaving activity integrated 266 times more frequently into rDNA than the control vectors and 8.2 times more than LVs whose IN-endonuclease content can only bind the target DNA. Other researchers have attempted to direct the integration of recombinant adeno-associated virus vectors (rAAVs) to the same locus but achieved only modest efficiencies: the increase in targeted integration was 8- to 13-fold in comparison to control vectors,46 and 2%–3% of selected hepatocytes were estimated to have the intended integration event within the 28S rRNA gene.47 The LVs characterized in our study promote much higher rDNA targeting, but further comparisons with the rAAVs are challenging due to profound differences in the study designs, IS analysis methods, and in the numbers of IS retrieved (n = 12–176 for the rAAVs).46,47 In addition to rAAVs, also non-viral vectors have been developed to target integration into the rDNA genomic safe harbor locus.48,49 However, in these studies, the levels of both transfection and targeted integration were low, and the analysis lacked thorough examination of the potential off-target integration events.
Our primary focus was to characterize both the complete integrome and the integration targeting efficiency of two IN-modified LVs as comprehensively as possible, which was achieved through the analysis of all ISs at an early time point where minimal clonal expansion of transduced cells had occurred. Analysis of LV D+H-transduced MRC-5 cells at later time points with ddPCR revealed that the efficiency of integration targeting into the 28S rRNA gene-contained I-PpoI site is at least two times higher than resolved through IS sequencing, reaching 21% of all proviruses. When comparing unselected and Zeocin-selected hTERT-RPE1 cells, we found that the proportion of proviruses remained stable in this repetitive DNA locus over time. Transduction tests with primary human T cells confirmed that integration within the I-PpoI site is increased also in this clinically relevant cell type, albeit to a lower degree than observed in the MRC-5 cells.
Subsampling and partitioning errors are known sources for variability in ddPCR, and its precision is decreased at the extremes.50,51 Other factors that can have contributed to the observed differences between the tested cell types include inherent differences in their replication kinetics and susceptibilities to transduction with LVs, lot-to-lot variability between the produced vectors, and a limited number of replicates analyzed per sample. On the other hand, with the IS sequencing method, the number of unique integrations within a highly targeted locus is easily underestimated due to saturation of potential unique MuA transposition sites and read lengths that were used to differentiate individual integrations from PCR-borne replicates. Despite the differences in efficiencies that likely originated from subsampling-related issues, the ddPCR-based method clearly demonstrated that D+H LVs catalyze targeted integration in both primary and cultured cells.
Cleavage of the 28S rRNA gene, its subsequent repair, and simultaneous insertion of proviruses into it could cause genomic rearrangements in this highly repetitive locus, including large deletions. We tested for this possibility and found no signs of gross deletions in the acrocentric chromosomes or in the rRNA genes after transduction with the D+H LVs. A moderate reduction in viable cell numbers was observed in LV D+H-transduced T cells at day 4 after transduction, but no clear indications of cytotoxicity were evident. rRNA gene transcription is halted upon DSB introduction into rDNA, which causes the formation of specific nucleolar cap structures and facilitates repair of the lesions (reviewed in Larsen and Stucki52). The observed reduction in the numbers of metabolically active cells may hence have resulted from the decreased production of the building blocks for ribosomes, which directly affects the metabolic activity of the cell. At days 2 and 10 post-transduction, we were able to detect provirus-containing transcripts from the 28S rRNA gene, which proves that transcription of this locus and the genetic material inserted into it is recommenced after DSB repair.
By analyzing the complete integrome of the modified LVs in MRC-5 cells, we found that proviruses residing outside of the targeted rDNA locus had a lower tendency to integrate within genes and oncogenes but showed a higher preference toward genomic features that are also enriched in NADs, chromatin domains that co-localize with rRNA gene arrays in the three-dimensional organization of the genome.28, 29, 30, 31, 32, 33, 34 One explanation for the preferential targeting to these loci could be that nicks or DSBs occurring randomly in NAD-containing chromosomes capture a proportion of vector genomes that were tethered to nucleolar proximity by the LV-contained I-PpoI protein. For the D+N LVs, the localization of genomic regions in NADs seems to be a stronger determinant of integration hotspot site selection than the distance to an I-PpoI site. The transcriptional status of transgenes inserted into NADs and further verification of this phenomenon remain to be addressed with additional techniques in the future. To our knowledge, this is this first description of distinct genomic regions that are distant from one another on the linear axis of DNA but near in the three-dimensional genome to become jointly affected when site-specific transgene integration was pursued based on primary DNA sequence recognition. This observation may have utility in the prediction of possible off-target sites also when using other nucleases for genome editing, such as the CRISPR/Cas system.
The most desired integrating vectors in gene therapy are those that can direct transgenes into genomic safe-harbor sites to minimize the risks related to insertional mutagenesis. LVs have many benefits as vectors, but their integration profile may endanger normal cellular gene function. First attempts to direct LV integration to specific sites were based on IN-fusion proteins,53 and more recent approaches relied on new chromatin binding preferences assigned for the IN-tethering LEDGF proteins.54, 55, 56, 57 After our first report of using LVs for protein transduction without the previously necessary Vpr-protein fusions,58 many studies have described different LV- or retrovirus vector (RV)-based virus-like particles, or nanoparticles, to transport desired proteins into cells often with the aim of delivering DNA-editing or integration-targeting enzymes.59, 60, 61, 62, 63, 64, 65, 66, 67, 68 In addition, LVs and RVs can deliver these components into cells as transgenes (reviewed in Chen and Gonçalves69) or messenger RNA.70, 71, 72 Systems in which single-vector particles contain both the donor DNA and the enzymes required for targeted integration are superior to multi-construct approaches that may suffer from decreased efficiency if only a fraction of the intended components reach target cells. The majority of recent studies aiming for genome editing and targeted integration utilize the CRISPR/Cas system. With the help of different technical advances and the discovery of alternative Cas variants, it has been possible to improve the specificity of targeted genome modifications (reviewed in Broeders et al.73), but major concerns related to the safety37, 38, 39, 40 and efficacy of the CRISPR-based approaches remain, precluding their wide utility in the clinic at the moment.
In comparison to most genomic safe harbor (GSH) site candidates, rDNA is unique, owing to its repetitive gene context. This feature could pose challenges to both the cells upon transgene integration and to the stability of the transgene itself, but our results in primary human T cells did not support such concerns nor point to major adverse effects. The most important safety features of rDNA as a GSH include its isolated location from potentially oncogenic protein-encoding genes, and the high number of rRNA genes that remain intact despite transgene integration into the locus. rDNA is typically ruled by RNA polymerase I, but it is also accessible to the RNA polymerase II machinery.74, 75, 76, 77 We show that integration can be targeted to the rRNA gene array with an unprecedented efficiency using modified LVs that carry both the donor DNA molecules and the integration-targeting enzyme within single-vector particles. These LVs can deliver large transgenes, are easy to produce with minor modifications to standard protocols, and are suitable for both ex vivo and in vivo gene transfer applications, hence potentially advancing the development next generation applications to treat human diseases.
Materials and Methods
Generation of Third-Generation LVs
Vesicular stomatitis virus G glycoprotein (VSV-G)-pseudotyped third-generation HIV-1-based LVs containing the IN-fusion proteins were produced as described earlier.15,16,58,78 In brief, monolayers of 293T cells were transfected with the production plasmids using calcium phosphate transfection. The plasmids used were pRSV-Rev (encoding for HIV-1 Rev), pCMV-VSVG (encoding for VSV-G), pLV1 (vector construct that contains a PGK promoter-driven EGFP transgene), or pLV1-ZeoR (vector construct carrying a PGK promoter-driven Sh ble gene), and either one or two of the packaging plasmids encoding for the wild-type integrase (pMDLg/pRRE), the integration-deficient integrase (pMDLg/pRRE-IND64V), the IN-fusion protein with DNA cleavage-disabled I-PpoI (pMDLg/pRRE-IN-I-PpoIN119A), or the IN-fusion protein with DNA cleavage-proficient I-PpoI that carries an activity-reducing mutation (pMDLg/pRRE-IN-I-PpoIH78A). Culture supernatants were collected 48 hr after transfection, filtered, suspended in phosphate-buffered saline (PBS), and stored at −70°C until use. Functional vector titers (transducing units [TU]/mL) were estimated through EGFP expression in transduced HeLa cells approximately 68 hr post-transduction, and particle titers were determined based on the level of HIV-1 p24 capsid (CA) antigen using an enzyme-linked immunosorbent assay (PerkinElmer Life and Analytical Sciences, Waltham, MA, USA).
Cells, Transductions, and Cell Health Assays
All transductions were carried out by diluting the LVs into cell culture medium immediately before use or alternatively by pipetting undiluted LVs directly into cell culture medium. On the day after transduction, vector-containing medium was replaced with fresh medium. All cells were incubated at 37°C in a 5% CO2-containing humidified atmosphere.
For the IS sequencing experiment, human MRC-5 lung fibroblasts (ATCC CCL-171) were used. The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; high-glucose, Sigma D6429) supplemented with 1% penicillin-streptomycin (Sigma, P0781), 1% MEM non-essential amino acids without L-Glutamine (Biowest, cat. X0557-100), 1% sodium pyruvate (Biowest cat. L0642-100), and 10% fetal bovine serum (FBS; Sigma, F7524). On the day before transduction, MRC-5 cells were seeded onto 6-well plates at a density of 2 × 10e5 cells per well. An MOI of 4 was used for transduction with the IN-modified LVs (56k–120k vp/cell) and an MOI 1 for transduction with the INwt LV (1k vp/cell). Cells were pelleted at days 2 and 3 post-transduction and stored at −70°C until used for DNA extraction and integration site analysis. To study the proportion of IS occurring near the I-PpoI site with ddPCR, MRC-5 cells were seeded as above and transduced in two separate experiments with the EGFP-LVs using 7.5k vp per cell, which equaled MOI 19 for LV INwt. Cells were collected for analysis at day 9 post-transduction.
For the study of targeted integration in unselected and phleomycin D1 selected cells, hTERT-RPE1 cells (ATCC CRL-4000) were used. Cells were cultivated in 1× Dulbecco's Modified Eagle Medium: Nutrient Mixture F-12 (DMEM/F-12) (Gibco, 31330-038) supplemented with 10% FBS and 0.01 mg/mL of hygromycin B. On the day before transduction, the cells were seeded onto 6-well plates at a density of 4 × 10e5 cells per well. Transduction was carried out with the Sh ble antibiotic resistance gene containing vectors (ZeoR LVs) at a concentration of 5k vp/cell. At day 1 post-transduction, cells to undergo selection were given culture medium supplemented with Zeocin (Invivogen, ant-zn-05) at a final concentration of 300 μg/mL and thereafter subcultivated as necessary. Cell pellets were collected for DNA extraction at days 13 and 15 post-transduction and stored at −70°C until use.
Peripheral blood mononuclear cells (PBMCs) were enriched from two leukoreduction system (LRS) chambers (Finnish Red Cross Blood Service, Helsinki, Finland) using the prefilled Leucosep centrifuge tubes (Greiner Bio-One, #227288). Untouched human T cells were isolated from the PBMCs by using the pan T cell isolation kit (Miltenyi Biotech, #130-096-535Y). 2.5 × 10e7 T cells from both donors were activated with Dynabeads human T-activator CD3/CD28 (Gibco, #11132D) according to the kit protocol. T cells were cultivated in X-Vivo 15 (Lonza, #BE02-060F) supplemented with 5% human AB serum (Biowest, #S4190) and 20 U/mL of human recombinant interleukin-2 (IL-2) (Prospec-Tany Technogene, #CYT-209-b) for 4 days before LV transductions. All transductions were done in triplicate for T cells of both donors using the ZeoR LVs at vector doses of 5k and 10k vp per cell, which equaled MOIs of 5 and 10 of LV INwt-EGFPs, respectively. Cells to be studied for targeted integration with ddPCR were transduced on 24-well plates (1.5 × 10e6 cells per well) and sampled for analysis at days 2 and 10 post-transduction. For the cells analyzed for viability, apoptosis, and necrosis, the activation beads were removed, and then the cells were seeded on white 96-well plates with clear bottoms (PerkinElmer, View-Plate-96-TC, #6005181) at densities of 6,000 cells per well for the viability assay and 10,000 cells per well for the apoptosis/necrosis assay. After vector removal at day 1 post-transduction, the cells were given fresh medium and the assay reagents according to kit protocols. Etoposide (Cayman Chemical, #12092) was used as a positive control for apoptosis induction and necrosis at a final concentration of 8 μM. The viability of transduced cells was monitored with daily luminescence recording for 4 days (days 1, 2, and 4 post-transduction) using the RealTime-Glo MT cell viability assay (Promega, # G9711). Apoptosis and necrosis were examined with the RealTime-Glo annexin V apoptosis and necrosis assay (Promega, #JA1011) that simultaneously measures annexin V exposure and DNA release to differentiate secondary necrosis occurring during late apoptosis from necrosis caused by other cytotoxic events. Annexin V binding (luminescence) and loss of membrane integrity (fluorescence) were recorded at days 1, 2, and 3 post-transduction.
Integration Site Extraction and EGFP Expression Analysis
MRC-5 cells were transduced with an MOI of 1 for the control vector (LV INwt) and 4 for the IN-modified LVs (Table S2). Separate wells were transduced for genomic DNA extraction and for fluorescence-activated cell sorting (FACS) analysis of EGFP expression. Genomic DNA was extracted 2 or 3 days post-transduction using the NucleoSpin tissue kit (Macherey-Nagel, ref. 740952.250) from two separate wells per vector. Vector ISs were extracted with the MuA transposon-based protocol described in Brady et al.,79 using BtsαI for genomic DNA digestion (NEB #R0667S) and primers and linkers listed in the Supplemental Materials and Methods. Primers and oligonucleotides used in the study were ordered from Integrated DNA Technologies, and the MuA transposon used was from Thermo Scientific (F-750, lot # 00383099). Digested DNA was purified before the MuA reactions using Speedbead magnetic carboxylate modified particles (GE Healthcare, part no. 65152105050250). Each of the two individual genomic DNA extractions analyzed per vector were tagged with unique sequence identifiers in both the linker oligo and in the primer (molecular identifier, MID) to minimize sequence carryover between samples and to maximize the resolution of integration sites occurring near each other (Table S2). Amplification of the integration sites was carried out using Phusion Flash PCR master mix (Thermo Scientific, F-548) in two rounds of PCR. In the first PCR, 2 μL of the MuA reaction was used as template. The first PCR program was as follows: 98°C for 10 s, seven cycles of 98°C for 1 s and 72°C for 15 s, 37 cycles of 98°C for 1 s, 57°C for 5 s and 72°C for 15 s, with a final extension at 72°C for 1 min. The amplicons from the first round of PCR were diluted 1:50 with nuclease-free water, and 1 μL of the dilution was used as template for the second round of PCR. The second PCR program was as follows: 98°C for 10 s, seven cycles of 98°C for 1 s, 67°C for 5 s, and 72°C for 15 s, 37 cycles of 98°C for 1 s and 72°C for 15 s, with a final extension at 72°C for 1 min. The amplicons were sequenced in Biocenter Oulu Sequencing Center with an IonTorrent PGM instrument (University of Oulu, Finland). EGFP expression was analyzed with flow cytometry from triplicate wells per vector at the day of genomic DNA extraction from cells fixed with 4% paraformaldehyde in PBS.
ddPCR
The primers, assays, materials, and PCR programs used in the different ddPCR reactions are listed in the Supplemental Materials and Methods. ddPCR was carried out according to Bio-Rad’s recommended protocol. For the study of integration in the immediate vicinity of the I-PpoI site in MRC-5 cells, genomic DNA was extracted for analysis from cells collected at day 9 post-transduction using QIAGEN’s DNeasy blood and tissue kit (ref. 69506) and digested with BsuRI (Thermo Fisher, ref. ER0151) at a concentration of 1 unit/1 μg DNA. Digested genomic DNA was used as template in ddPCR to measure the copy numbers of all vector genomes, episomal vector forms, production plasmid carryover, and integration near the I-PpoI recognition site in the 28S rRNA gene in both sense and antisense orientation.
For the ddPCR analysis of targeted integration in Zeocin selected cells, genomic DNA was extracted from hTERT-RPE1 cells pelleted at day 13 (unselected) and 15 (selected) post-transduction and processed for ddPCR as described above. ddPCR analysis consisted of assays measuring the copy numbers of all vector genomes, episomal vector forms, and vectors integrated in sense orientation near the I-PpoI recognition site in the 28S rRNA gene.
For the detection of targeted integration in primary human CD3+ T cells, genomic DNA was extracted from cells pelleted at days 2 and 10 post-transduction using the AllPrep DNA/RNA mini kit (QIAGEN, #80204). DNA was processed and analyzed with ddPCR as described for MRC-5 cells above. ddPCR was carried out for two replicate wells of non-transduced cells, INwt-transduced cells, and D+H-transduced cells. Each well’s DNA was sampled twice for ddPCR.
Analysis of site-specifically integrated provirus transcription at days 2 and 10 post-transduction was carried out with RT-ddPCR using total RNA extracted from T cells with the AllPrep DNA/RNA mini kit (QIAGEN, #80204) and the protocol established for the detection of targeted integration. One microgram of RNA was treated with DNase I (Thermo Scientific, ref. EN0521) and cDNA synthesis was carried out with a RevertAid RT reverse transcription kit (Thermo Scientific, ref. K1691) with random hexamer primers according to the kit’s protocol. Depending on the assay, 0.5–2.0 μL of the RT reaction was used as template for RT-ddPCR.
The presence of deletions in the rRNA gene array and in the acrocentric chromosome arms was assayed with ddPCR using genomic DNA extracted from T cells transduced with 10k vp/cell and extracted at day 2 post-transduction. Probes binding to the DJ region, which flanks the rRNA gene array on the telomeric side,45 and to the 18S rRNA gene were designed and used for the quantification of the respective areas.
Bioinformatics Data Analysis
Integration Site Analysis
Single-end FASTQ data files were quality filtered and trimmed by Skewer.80 The reads were processed to check for the presence of the linker cassette (LC) sequence that was specific for each sample, and for the transposon-linker sequence. After trimming of LC sequences, the set of reads was aligned with vector sequence by BLAT (BLAST-like alignment tool)81 aligner to subtract potential vector-only reads and to avoid any false positive vector read detection. The reads were then mapped with the LV 3′ LTR sequence using a minimum identity threshold of 95%. The LTR mapped part was trimmed, and the rest of the read region was mapped with human genome reference hg38 with minimum identity of 95%. The reads that mapped uniquely or at multiple sites within the genome were separated in the subsequent steps. A threshold of 90% was employed between the ratio of the BLAT score for primary and secondary mapped reads so that reads with a score ratio greater than this were designated as MH-ISs and others as UH-ISs. To simplify analysis of integration within rDNA, the reads mapping to chromosome 21 (chr21) that had exactly same primary and secondary mapping scores were preferred for their alignment positions in the region between chr21:8433222-8446572. Exact sequence duplicates were removed, and reads were filtered using multiple criteria in order to filter out potential duplicates of a single original integration event. Filtering involved restricting the number of non-mapping base pairs before the start of the genomic region (i.e., between LTR and the region mapping to the genome) using a threshold of 4 bp: the reads that had non-mapping base pairs less than or equal to this threshold were further processed to next steps. Next, only reads that had three or fewer base pairs of non-mapping nucleotides at their 3′ end were considered. The reads were compared to one another, and only those reads that had a difference in the number of deleted base pairs at their LTR ends of ≥2, and whose ISs and “shear sites” (transposition sites) were at least 3 bp apart from other reads were further processed. The collision sequences among samples were subtracted from each sample, and the final reads were mapped against the pLV1 plasmid sequence to remove remaining artifacts. Finally, the genomic positions were annotated according to the RefSeq from the University of California Santa Cruz (UCSC),82 and the RepeatMasker rmblast web version20 was used to annotate repeat regions. To identify integration into pseudogenes, ISs were also annotated with the retro genes table (Retroposed Genes V9, Including Pseudogenes) obtained from UCSC. Additionally, the oncogenes table (v4 May 2018) was retrieved (http://www.bushmanlab.org/links/genelists) and final set of genes obtained from clustered result files were annotated with this set. The plots shown in Figure 3 were generated for rRNA reads by creating bed and bedgraph files using bedtools83 that were processed by in-house script and R packages (karyoploteR and regioneR).84,85
Analysis of the Integration Frequency in Selected Gene Sets
Integration frequency in gene sets involved in the SuperPaths35 of ribosome biogenesis in eukaryotes and rRNA processing in the nucleus and cytosol were conducted using single genes (each IS-tagged gene represented once in the gene list comparison) using the IS datasets where pseudogene annotations were used in place of the initial RefSeq gene annotation.
Analysis of CISs (Integration Hotspot Analysis)
CIS analysis was performed using a graph-based framework for CIS identification86,87 with a threshold of 50 kb between individual ISs. For the analysis of hotspots, only CISs with a p value of less than 0.05 and with a minimum of three IS were accepted. The CIS analysis was performed separated for the IS datasets containing only uniquely mappable ISs (UH-IS dataset) and for the complete IS datasets (UH and MH IS data). The features in the median CIS positions in Tables 1 and 2 were annotated using the RepeatMasker, RefSeq-gene, and RetrogenesV9 tracks of the UCSC genome browser.
GO Analysis of the CIS-Associated IS
Analysis of the most overrepresented pathways and processes among genes present in the CIS-engaged IS was performed using Metascape88 (http://metascape.org/gp/index.html#/main/step1) that uses the following ontology sources: KEGG pathway, GO biological processes, Reactome gene sets, canonical pathways, and CORUM (the comprehensive resource of mammalian protein complexes). In the analysis, all genes in the genome are used as the enrichment background and terms with a p value <0.01, a minimum count of 3, and an enrichment factor >1.5 are collected and grouped into clusters based on their membership similarities. Each cluster is represented with the most statistically significant term within that cluster. The analyzed gene lists contained all genes (both hit genes and nearest genes) from the identified CIS using the complete IS data (UH and MH IS).
Comparison of Recurrent Integration Gene (RIG) Loci with the CIS Foci of INwt LVs
The genomic coordinates from RIG and “hotter zone” (HZ) loci listed by Marini and others22 were converted to the current genome version (Dec. 2013 [GRGh38/hg38]) assembly using the “LiftOver” tool from the UCSC genome browser database.89 The average positions of the RIGs/HZs and the INwt LV CIS were compared, and the RIGs and CIS foci that fell within a 100 kb distance from one another were listed in Table S7.
Statistics
Statistical differences in the integration preferences between LV groups were calculated using a two-sided Fisher’s exact test and with a two-sided Chi-square test. Statistical comparisons between groups in the viability and necrosis assays were done with repeated-measures analysis of variance (ANOVA) followed by the Bonferroni post-test to compare replicate means by row to the control. In the apoptosis assay, each time point was analyzed separately with one-way ANOVA followed by Dunnett’s multiple comparison test. The differences in copy numbers of 18S and DJ sequences were analyzed with one-way ANOVA by comparing the vector-groups’ values to the same donor’s non-transduced cell control with Dunnett’s multiple comparison test. All statistical analysis was done with GraphPad Prism version 5.03 for Windows, GraphPad Software, San Diego, CA USA, https://www.graphpad.com/.
Data Availability
The final IS datasets generated and analyzed in this study are available upon a reasonable request.
Author Contributions
D.S. conceived the study, designed the experiments, conducted cell culture experiments, performed bioinformatics analysis, analyzed the data, and wrote the manuscript. S.A. designed bioinformatics analysis strategy, performed the data analyses, and contributed to writing the analysis method section. A.N. conducted cell culture experiments, performed the integration site extractions, designed and executed the ddPCR assays and performed the analysis, analyzed the data, and participated in writing the manuscript. M.S. and S.Y.-H. supervised and financed the study and edited the manuscript.
Conflicts of Interest
M.S. is co-founder and CEO of GeneWerk GmbH, Heidelberg, Germany.
Acknowledgments
This work was supported by the Finnish Academy Centre of Excellence (307402), the European Research Council (GA670951), and by the Eemil Aaltonen Foundation (to D.S.). This work also got support from the National Virus Vector Laboratory/A.I. Virtanen Institute, University of Eastern Finland, Kuopio, and from the Kuopio Center for Gene and Cell Therapy (KCT). Anssi Kailaanmäki, Elina Koli, Annu Luostarinen and Tanja Kaartinen are acknowledged for their help with T cell extraction and culture-related methods.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.05.019.
Supplemental Information
References
- 1.Cavazzana M., Bushman F.D., Miccio A., André-Schmutz I., Six E. Gene therapy targeting haematopoietic stem cells for inherited diseases: progress and challenges. Nat. Rev. Drug Discov. 2019;18:447–462. doi: 10.1038/s41573-019-0020-9. [DOI] [PubMed] [Google Scholar]
- 2.Milone M.C., O’Doherty U. Clinical use of lentiviral vectors. Leukemia. 2018;32:1529–1541. doi: 10.1038/s41375-018-0106-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Montini E., Cesana D., Schmidt M., Sanvito F., Ponzoni M., Bartholomae C., Sergi Sergi L., Benedicenti F., Ambrosi A., Di Serio C. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat. Biotechnol. 2006;24:687–696. doi: 10.1038/nbt1216. [DOI] [PubMed] [Google Scholar]
- 4.Cavazza A., Moiani A., Mavilio F. Mechanisms of retroviral integration and mutagenesis. Hum. Gene Ther. 2013;24:119–131. doi: 10.1089/hum.2012.203. [DOI] [PubMed] [Google Scholar]
- 5.Craigie R., Bushman F.D. HIV DNA integration. Cold Spring Harb. Perspect. Med. 2012;2:a006890. doi: 10.1101/cshperspect.a006890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schröder A.R.W., Shinn P., Chen H., Berry C., Ecker J.R., Bushman F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002;110:521–529. doi: 10.1016/s0092-8674(02)00864-4. [DOI] [PubMed] [Google Scholar]
- 7.Ciuffi A., Llano M., Poeschla E., Hoffmann C., Leipzig J., Shinn P., Ecker J.R., Bushman F. A role for LEDGF/p75 in targeting HIV DNA integration. Nat. Med. 2005;11:1287–1289. doi: 10.1038/nm1329. [DOI] [PubMed] [Google Scholar]
- 8.Stults D.M., Killen M.W., Pierce H.H., Pierce A.J. Genomic architecture and inheritance of human ribosomal RNA gene clusters. Genome Res. 2008;18:13–18. doi: 10.1101/gr.6858507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schöfer C., Weipoltshammer K. Nucleolus and chromatin. Histochem. Cell Biol. 2018;150:209–225. doi: 10.1007/s00418-018-1696-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scully R., Panday A., Elango R., Willis N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019;20:698–714. doi: 10.1038/s41580-019-0152-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamamoto Y., Gerbi S.A. Making ends meet: targeted integration of DNA fragments by genome editing. Chromosoma. 2018;127:405–420. doi: 10.1007/s00412-018-0677-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Urnov F.D. Ctrl-Alt-inDel: genome editing to reprogram a cell in the clinic. Curr. Opin. Genet. Dev. 2018;52:48–56. doi: 10.1016/j.gde.2018.05.005. [DOI] [PubMed] [Google Scholar]
- 13.Muscarella D.E., Vogt V.M. A mobile group I intron in the nuclear rDNA of Physarum polycephalum. Cell. 1989;56:443–454. doi: 10.1016/0092-8674(89)90247-x. [DOI] [PubMed] [Google Scholar]
- 14.Ellison E.L., Vogt V.M. Interaction of the intron-encoded mobility endonuclease I-PpoI with its target site. Mol. Cell. Biol. 1993;13:7531–7539. doi: 10.1128/mcb.13.12.7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Turkki V., Schenkwein D., Timonen O., Husso T., Lesch H.P., Ylä-Herttuala S. Lentiviral protein transduction with genome-modifying HIV-1 integrase-I-PpoI fusion proteins: studies on specificity and cytotoxicity. BioMed Res. Int. 2014;2014:379340. doi: 10.1155/2014/379340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schenkwein D., Turkki V., Ahlroth M.K., Timonen O., Airenne K.J., Ylä-Herttuala S. rDNA-directed integration by an HIV-1 integrase--I-PpoI fusion protein. Nucleic Acids Res. 2013;41:e61. doi: 10.1093/nar/gks1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mannino S.J., Jenkins C.L., Raines R.T. Chemical mechanism of DNA cleavage by the homing endonuclease I-PpoI. Biochemistry. 1999;38:16178–16186. doi: 10.1021/bi991452v. [DOI] [PubMed] [Google Scholar]
- 18.Mitchell R.S., Beitzel B.F., Schroder A.R.W., Shinn P., Chen H., Berry C.C., Ecker J.R., Bushman F.D. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2004;2:E234. doi: 10.1371/journal.pbio.0020234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brady T., Agosto L.M., Malani N., Berry C.C., O’Doherty U., Bushman F. HIV integration site distributions in resting and activated CD4+ T cells infected in culture. AIDS. 2009;23:1461–1471. doi: 10.1097/QAD.0b013e32832caf28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smit A.F.A., Hubley R., Green P. 2015. RepeatMasker Open-4.0.http://www.repeatmasker.org [Google Scholar]
- 21.Robicheau B.M., Susko E., Harrigan A.M., Snyder M. Ribosomal RNA genes contribute to the formation of pseudogenes and junk DNA in the human genome. Genome Biol. Evol. 2017;9:380–397. doi: 10.1093/gbe/evw307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marini B., Kertesz-Farkas A., Ali H., Lucic B., Lisek K., Manganaro L., Pongor S., Luzzati R., Recchia A., Mavilio F. Nuclear architecture dictates HIV-1 integration site selection. Nature. 2015;521:227–231. doi: 10.1038/nature14226. [DOI] [PubMed] [Google Scholar]
- 23.Biffi A., Bartolomae C.C., Cesana D., Cartier N., Aubourg P., Ranzani M., Cesani M., Benedicenti F., Plati T., Rubagotti E. Lentiviral vector common integration sites in preclinical models and a clinical trial reflect a benign integration bias and not oncogenic selection. Blood. 2011;117:5332–5339. doi: 10.1182/blood-2010-09-306761. [DOI] [PubMed] [Google Scholar]
- 24.Aiuti A., Biasco L., Scaramuzza S., Ferrua F., Cicalese M.P., Baricordi C., Dionisio F., Calabria A., Giannelli S., Castiello M.C. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013;341:1233151. doi: 10.1126/science.1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Biffi A., Montini E., Lorioli L., Cesani M., Fumagalli F., Plati T., Baldoli C., Martino S., Calabria A., Canale S. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341:1233158. doi: 10.1126/science.1233158. [DOI] [PubMed] [Google Scholar]
- 26.Cartier N., Hacein-Bey-Abina S., Bartholomae C.C., Veres G., Schmidt M., Kutschera I., Vidaud M., Abel U., Dal-Cortivo L., Caccavelli L. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326:818–823. doi: 10.1126/science.1171242. [DOI] [PubMed] [Google Scholar]
- 27.Németh A., Längst G. Genome organization in and around the nucleolus. Trends Genet. 2011;27:149–156. doi: 10.1016/j.tig.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 28.Pontvianne F., Carpentier M.-C., Durut N., Pavlištová V., Jaške K., Schořová Š., Parrinello H., Rohmer M., Pikaard C.S., Fojtová M. Identification of Nucleolus-Associated Chromatin Domains Reveals a Role for the Nucleolus in 3D Organization of the A. thaliana Genome. Cell Rep. 2016;16:1574–1587. doi: 10.1016/j.celrep.2016.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu S., Lemos B. The long-range interaction map of ribosomal DNA arrays. PLoS Genet. 2018;14:e1007258. doi: 10.1371/journal.pgen.1007258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Németh A., Conesa A., Santoyo-Lopez J., Medina I., Montaner D., Péterfia B., Solovei I., Cremer T., Dopazo J., Längst G. Initial genomics of the human nucleolus. PLoS Genet. 2010;6:e1000889. doi: 10.1371/journal.pgen.1000889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dillinger S., Straub T., Németh A. Nucleolus association of chromosomal domains is largely maintained in cellular senescence despite massive nuclear reorganisation. PLoS ONE. 2017;12:e0178821. doi: 10.1371/journal.pone.0178821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Koningsbruggen S., Gierlinski M., Schofield P., Martin D., Barton G.J., Ariyurek Y., den Dunnen J.T., Lamond A.I. High-resolution whole-genome sequencing reveals that specific chromatin domains from most human chromosomes associate with nucleoli. Mol. Biol. Cell. 2010;21:3735–3748. doi: 10.1091/mbc.E10-06-0508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu S., Lemos B. A Portrait of Ribosomal DNA Contacts with Hi-C Reveals 5S and 45S rDNA Anchoring Points in the Folded Human Genome. Genome Biol. Evol. 2016;8:3545–3558. doi: 10.1093/gbe/evw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Diesch J., Bywater M.J., Sanij E., Cameron D.P., Schierding W., Brajanovski N., Son J., Sornkom J., Hein N., Evers M. Changes in long-range rDNA-genomic interactions associate with altered RNA polymerase II gene programs during malignant transformation. Commun. Biol. 2019;2:39. doi: 10.1038/s42003-019-0284-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Belinky F., Nativ N., Stelzer G., Zimmerman S., Iny Stein T., Safran M., Lancet D. PathCards: multi-source consolidation of human biological pathways. Database (Oxford) 2015;2015:6. doi: 10.1093/database/bav006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang G.P., Ciuffi A., Leipzig J., Berry C.C., Bushman F.D. HIV integration site selection: analysis by massively parallel pyrosequencing reveals association with epigenetic modifications. Genome Res. 2007;17:1186–1194. doi: 10.1101/gr.6286907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kosicki M., Tomberg K., Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018;36:765–771. doi: 10.1038/nbt.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cullot G., Boutin J., Toutain J., Prat F., Pennamen P., Rooryck C., Teichmann M., Rousseau E., Lamrissi-Garcia I., Guyonnet-Duperat V. CRISPR-Cas9 genome editing induces megabase-scale chromosomal truncations. Nat. Commun. 2019;10:1136. doi: 10.1038/s41467-019-09006-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simeonov D.R., Brandt A.J., Chan A.Y., Cortez J.T., Li Z., Woo J.M., Lee Y., Carvalho C.M.B., Indart A.C., Roth T.L. A large CRISPR-induced bystander mutation causes immune dysregulation. Commun. Biol. 2019;2:70. doi: 10.1038/s42003-019-0321-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu S., Kim J., Tang Q., Chen Q., Liu J., Xu Y., Fu X. CAS9 is a genome mutator by directly disrupting DNA-PK dependent DNA repair pathway. Protein Cell. 2020;11:352–365. doi: 10.1007/s13238-020-00699-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stults D.M., Killen M.W., Williamson E.P., Hourigan J.S., Vargas H.D., Arnold S.M., Moscow J.A., Pierce A.J. Human rRNA gene clusters are recombinational hotspots in cancer. Cancer Res. 2009;69:9096–9104. doi: 10.1158/0008-5472.CAN-09-2680. [DOI] [PubMed] [Google Scholar]
- 42.Gibbons J.G., Branco A.T., Godinho S.A., Yu S., Lemos B. Concerted copy number variation balances ribosomal DNA dosage in human and mouse genomes. Proc. Natl. Acad. Sci. USA. 2015;112:2485–2490. doi: 10.1073/pnas.1416878112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu B., Li H., Perry J.M., Singh V.P., Unruh J., Yu Z., Zakari M., McDowell W., Li L., Gerton J.L. Ribosomal DNA copy number loss and sequence variation in cancer. PLoS Genet. 2017;13:e1006771. doi: 10.1371/journal.pgen.1006771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Killen M.W., Stults D.M., Adachi N., Hanakahi L., Pierce A.J. Loss of Bloom syndrome protein destabilizes human gene cluster architecture. Hum. Mol. Genet. 2009;18:3417–3428. doi: 10.1093/hmg/ddp282. [DOI] [PubMed] [Google Scholar]
- 45.Floutsakou I., Agrawal S., Nguyen T.T., Seoighe C., Ganley A.R.D., McStay B. The shared genomic architecture of human nucleolar organizer regions. Genome Res. 2013;23:2003–2012. doi: 10.1101/gr.157941.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lisowski L., Lau A., Wang Z., Zhang Y., Zhang F., Grompe M., Kay M.A. Ribosomal DNA integrating rAAV-rDNA vectors allow for stable transgene expression. Mol. Ther. 2012;20:1912–1923. doi: 10.1038/mt.2012.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Z., Lisowski L., Finegold M.J., Nakai H., Kay M.A., Grompe M. AAV vectors containing rDNA homology display increased chromosomal integration and transgene persistence. Mol. Ther. 2012;20:1902–1911. doi: 10.1038/mt.2012.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Y., Zhao J., Duan N., Liu W., Zhang Y., Zhou M., Hu Z., Feng M., Liu X., Wu L. Paired CRISPR/Cas9 Nickases Mediate Efficient Site-Specific Integration of F9 into rDNA Locus of Mouse ESCs. Int. J. Mol. Sci. 2018;19:3035. doi: 10.3390/ijms19103035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu B., Chen F., Wu Y., Wang X., Feng M., Li Z., Zhou M., Wang Y., Wu L., Liu X., Liang D. Enhanced tumor growth inhibition by mesenchymal stem cells derived from iPSCs with targeted integration of interleukin24 into rDNA loci. Oncotarget. 2017;8:40791–40803. doi: 10.18632/oncotarget.16584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Basu A.S. Digital Assays Part I: Partitioning Statistics and Digital PCR. SLAS Technol. 2017;22:369–386. doi: 10.1177/2472630317705680. [DOI] [PubMed] [Google Scholar]
- 51.Quan P.L., Sauzade M., Brouzes E. DPCR: A technology review. Sensors (Basel) 2018;18:1271. doi: 10.3390/s18041271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Larsen D.H., Stucki M. Nucleolar responses to DNA double-strand breaks. Nucleic Acids Res. 2016;44:538–544. doi: 10.1093/nar/gkv1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bushman F.D. Tethering human immunodeficiency virus 1 integrase to a DNA site directs integration to nearby sequences. Proc. Natl. Acad. Sci. USA. 1994;91:9233–9237. doi: 10.1073/pnas.91.20.9233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meehan A.M., Saenz D.T., Morrison J.H., Garcia-Rivera J.A., Peretz M., Llano M., Poeschla E.M. LEDGF/p75 proteins with alternative chromatin tethers are functional HIV-1 cofactors. PLoS Pathog. 2009;5:e1000522. doi: 10.1371/journal.ppat.1000522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Silvers R.M., Smith J.A., Schowalter M., Litwin S., Liang Z., Geary K., Daniel R. Modification of integration site preferences of an HIV-1-based vector by expression of a novel synthetic protein. Hum. Gene Ther. 2010;21:337–349. doi: 10.1089/hum.2009.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gijsbers R., Ronen K., Vets S., Malani N., De Rijck J., McNeely M., Bushman F.D., Debyser Z. LEDGF hybrids efficiently retarget lentiviral integration into heterochromatin. Mol. Ther. 2010;18:552–560. doi: 10.1038/mt.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ferris A.L., Wu X., Hughes C.M., Stewart C., Smith S.J., Milne T.A., Wang G.G., Shun M.C., Allis C.D., Engelman A., Hughes S.H. Lens epithelium-derived growth factor fusion proteins redirect HIV-1 DNA integration. Proc. Natl. Acad. Sci. USA. 2010;107:3135–3140. doi: 10.1073/pnas.0914142107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schenkwein D., Turkki V., Kärkkäinen H.-R., Airenne K., Ylä-Herttuala S. Production of HIV-1 integrase fusion protein-carrying lentiviral vectors for gene therapy and protein transduction. Hum. Gene Ther. 2010;21:589–602. doi: 10.1089/hum.2009.051. [DOI] [PubMed] [Google Scholar]
- 59.Voelkel C., Galla M., Maetzig T., Warlich E., Kuehle J., Zychlinski D., Bode J., Cantz T., Schambach A., Baum C. Protein transduction from retroviral Gag precursors. Proc. Natl. Acad. Sci. USA. 2010;107:7805–7810. doi: 10.1073/pnas.0914517107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.He C., Gouble A., Bourdel A., Manchev V., Poirot L., Paques F., Duchateau P., Edelman A., Danos O. Lentiviral protein delivery of meganucleases in human cells mediates gene targeting and alleviates toxicity. Gene Ther. 2014;21:759–766. doi: 10.1038/gt.2014.51. [DOI] [PubMed] [Google Scholar]
- 61.Uhlig K.M., Schülke S., Scheuplein V.A.M., Malczyk A.H., Reusch J., Kugelmann S., Muth A., Koch V., Hutzler S., Bodmer B.S. Lentiviral Protein Transfer Vectors Are an Efficient Vaccine Platform and Induce a Strong Antigen-Specific Cytotoxic T Cell Response. J. Virol. 2015;89:9044–9060. doi: 10.1128/JVI.00844-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Choi J.G., Dang Y., Abraham S., Ma H., Zhang J., Guo H., Cai Y., Mikkelsen J.G., Wu H., Shankar P., Manjunath N. Lentivirus pre-packed with Cas9 protein for safer gene editing. Gene Ther. 2016;23:627–633. doi: 10.1038/gt.2016.27. [DOI] [PubMed] [Google Scholar]
- 63.Prel A., Caval V., Gayon R., Ravassard P., Duthoit C., Payen E., Maouche-Chretien L., Creneguy A., Nguyen T.H., Martin N. Highly efficient in vitro and in vivo delivery of functional RNAs using new versatile MS2-chimeric retrovirus-like particles. Mol. Ther. Methods Clin. Dev. 2015;2:15039. doi: 10.1038/mtm.2015.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cai Y., Bak R.O., Krogh L.B., Staunstrup N.H., Moldt B., Corydon T.J., Schrøder L.D., Mikkelsen J.G. DNA transposition by protein transduction of the piggyBac transposase from lentiviral Gag precursors. Nucleic Acids Res. 2014;42:e28. doi: 10.1093/nar/gkt1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cai Y., Bak R.O., Mikkelsen J.G. Targeted genome editing by lentiviral protein transduction of zinc-finger and TAL-effector nucleases. eLife. 2014;3:e01911. doi: 10.7554/eLife.01911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Skipper K.A., Nielsen M.G., Andersen S., Ryø L.B., Bak R.O., Mikkelsen J.G. Time-Restricted PiggyBac DNA Transposition by Transposase Protein Delivery Using Lentivirus-Derived Nanoparticles. Mol. Ther. Nucleic Acids. 2018;11:253–262. doi: 10.1016/j.omtn.2018.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lyu P., Javidi-Parsijani P., Atala A., Lu B. Delivering Cas9/sgRNA ribonucleoprotein (RNP) by lentiviral capsid-based bionanoparticles for efficient ‘hit-and-run’ genome editing. Nucleic Acids Res. 2019;47:e99. doi: 10.1093/nar/gkz605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mangeot P.E., Risson V., Fusil F., Marnef A., Laurent E., Blin J., Mournetas V., Massouridès E., Sohier T.J.M., Corbin A. Genome editing in primary cells and in vivo using viral-derived Nanoblades loaded with Cas9-sgRNA ribonucleoproteins. Nat. Commun. 2019;10:45. doi: 10.1038/s41467-018-07845-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen X., Gonçalves M.A.F.V. Engineered viruses as genome editing devices. Mol. Ther. 2016;24:447–457. doi: 10.1038/mt.2015.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mock U., Riecken K., Berdien B., Qasim W., Chan E., Cathomen T., Fehse B. Novel lentiviral vectors with mutated reverse transcriptase for mRNA delivery of TALE nucleases. Sci. Rep. 2014;4:6409. doi: 10.1038/srep06409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Knopp Y., Geis F.K., Heckl D., Horn S., Neumann T., Kuehle J., Meyer J., Fehse B., Baum C., Morgan M. Transient Retrovirus-Based CRISPR/Cas9 All-in-One Particles for Efficient, Targeted Gene Knockout. Mol. Ther. Nucleic Acids. 2018;13:256–274. doi: 10.1016/j.omtn.2018.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lu B., Javidi-Parsijani P., Makani V., Mehraein-Ghomi F., Sarhan W.M., Sun D., Yoo K.W., Atala Z.P., Lyu P., Atala A. Delivering SaCas9 mRNA by lentivirus-like bionanoparticles for transient expression and efficient genome editing. Nucleic Acids Res. 2019;47:e44. doi: 10.1093/nar/gkz093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Broeders M., Herrero-Hernandez P., Ernst M.P.T., van der Ploeg A.T., Pijnappel W.W.M.P. Sharpening the Molecular Scissors: Advances in Gene-Editing Technology. iScience. 2020;23:100789. doi: 10.1016/j.isci.2019.100789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kuroki-Kami A., Nichuguti N., Yatabe H., Mizuno S., Kawamura S., Fujiwara H. Targeted gene knockin in zebrafish using the 28S rDNA-specific non-LTR-retrotransposon R2Ol. Mob. DNA. 2019;10:23. doi: 10.1186/s13100-019-0167-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Johansen S.D., Haugen P., Nielsen H. Expression of protein-coding genes embedded in ribosomal DNA. Biol. Chem. 2007;388:679–686. doi: 10.1515/BC.2007.089. [DOI] [PubMed] [Google Scholar]
- 76.Bierhoff H., Schmitz K., Maass F., Ye J., Grummt I. Noncoding transcripts in sense and antisense orientation regulate the epigenetic state of ribosomal RNA genes. Cold Spring Harb. Symp. Quant. Biol. 2010;75:357–364. doi: 10.1101/sqb.2010.75.060. [DOI] [PubMed] [Google Scholar]
- 77.Bierhoff H., Dammert M.A., Brocks D., Dambacher S., Schotta G., Grummt I. Quiescence-induced LncRNAs trigger H4K20 trimethylation and transcriptional silencing. Mol. Cell. 2014;54:675–682. doi: 10.1016/j.molcel.2014.03.032. [DOI] [PubMed] [Google Scholar]
- 78.Follenzi A., Naldini L. Generation of HIV-1 derived lentiviral vectors. Methods Enzymol. 2002;346:454–465. doi: 10.1016/s0076-6879(02)46071-5. [DOI] [PubMed] [Google Scholar]
- 79.Brady T., Roth S.L., Malani N., Wang G.P., Berry C.C., Leboulch P., Hacein-Bey-Abina S., Cavazzana-Calvo M., Papapetrou E.P., Sadelain M. A method to sequence and quantify DNA integration for monitoring outcome in gene therapy. Nucleic Acids Res. 2011;39:e72. doi: 10.1093/nar/gkr140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jiang H., Lei R., Ding S.W., Zhu S. Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinformatics. 2014;15:182. doi: 10.1186/1471-2105-15-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kent W.J. BLAT--the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Karolchik D., Hinrichs A.S., Furey T.S., Roskin K.M., Sugnet C.W., Haussler D., Kent W.J. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004;32:D493–D496. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gel B., Serra E. KaryoploteR: An R/Bioconductor package to plot customizable genomes displaying arbitrary data. Bioinformatics. 2017;33:3088–3090. doi: 10.1093/bioinformatics/btx346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gel B., Díez-Villanueva A., Serra E., Buschbeck M., Peinado M.A., Malinverni R. regioneR: an R/Bioconductor package for the association analysis of genomic regions based on permutation tests. Bioinformatics. 2016;32:289–291. doi: 10.1093/bioinformatics/btv562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fronza R., Vasciaveo A., Benso A., Schmidt M. A Graph Based Framework to Model Virus Integration Sites. Comput. Struct. Biotechnol. J. 2015;14:69–77. doi: 10.1016/j.csbj.2015.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vasciaveo A., Velevska I., Politano G., Savino A., Schmidt M., Fronza R. Common integration sites of published datasets identified using a graph-based framework. Comput. Struct. Biotechnol. J. 2015;14:87–90. doi: 10.1016/j.csbj.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhou Y., Zhou B., Pache L., Chang M., Khodabakhshi A.H., Tanaseichuk O., Benner C., Chanda S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019;10:1523. doi: 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hinrichs A.S., Karolchik D., Baertsch R., Barber G.P., Bejerano G., Clawson H., Diekhans M., Furey T.S., Harte R.A., Hsu F. The UCSC Genome Browser Database: update 2006. Nucleic Acids Res. 2006;34:D590–D598. doi: 10.1093/nar/gkj144. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The final IS datasets generated and analyzed in this study are available upon a reasonable request.