

Abstract
Purpose:
In research settings, circulating tumor DNA (ctDNA) shows promise as a tumor-specific biomarker for pancreatic ductal adenocarcinoma (PDAC). This study aims to perform analytical and clinical validation of a KRAS ctDNA assay in a Clinical Laboratory Improvement Amendments (CLIA) and College of American Pathology–certified clinical laboratory.
Experimental Design:
Digital-droplet PCR was used to detect the major PDAC-associated somatic KRAS mutations (G12D, G12V, G12R, and Q61H) in liquid biopsies. For clinical validation, 290 preoperative and longitudinal postoperative plasma samples were collected from 59 patients with PDAC. The utility of ctDNA status to predict PDAC recurrence during follow-up was assessed.
Results:
ctDNA was detected preoperatively in 29 (49%) patients and was an independent predictor of decreased recurrence-free survival (RFS) and overall survival (OS). Patients who had neoadjuvant chemotherapy were less likely to have preoperative ctDNA than were chemo-na€ ve patients (21% vs. 69%; P < 0.001). ctDNA levels dropped significantly after tumor resection. Persistence of ctDNA in the immediate postoperative period was associated with a high rate of recurrence and poor median RFS (5 months). ctDNA detected during follow-up predicted clinical recurrence [sensitivity 90% (95% confidence interval (CI), 74%–98%), specificity 88% (95% CI, 62%–98%)] with a median lead time of 84 days (interquartile range, 25–146). Detection of ctDNA during postpancreatectomy follow-up was associated with a median OS of 17 months, while median OS was not yet reached at 30 months for patients without ctDNA (P = 0.011).
Conclusions:
Measurement of KRAS ctDNA in a CLIA laboratory setting can be used to predict recurrence and survival in patients with PDAC.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) continues to be a devastating disease, with 133,300 estimated deaths for 2018 in the United States and Europe alone (1, 2). Poor survival is mostly attributed to a lack of effective screening methods, late diagnosis, propensity for early metastatic spread, and minimally effective systemic therapies (3–6). Even after seemingly successful resection of a localized tumor, occult minimal residual local or distant disease causes up to 80% of patients to develop disease recurrence (7).
Currently, nine different chemotherapeutic drugs are approved by the FDA. These are increasingly administered in more effective multidrug regimens, such as FOLFIRINOX (5-fluorouracil, leucovorin, oxaliplatin, and irinotecan) and gemcitabine/nab-paclitaxel (8–11). The optimal evaluation of current and future treatment options emphasizes the need for biomarkers that can reliably identify residual or progressive PDAC, so that refractory patients can be started or switched on to second- or third-line regimens in a timely manner (5, 12). Currently used protein serum markers have several limitations that render them of limited use to help guide treatment decisions (13, 14). Carbohydrate antigen (CA) 19–9, for instance, can be elevated in extra-pancreatic malignancies and benign conditions (13). Furthermore, approximately 6% of the Caucasian population and 22% of the African American population in the United States are Lewis-antigen negative and do not produce CA 19–9 (15).
Cell-free circulating tumor DNA (ctDNA) has shown promise as a biomarker to improve early tumor detection, prognostic stratification, and monitoring of tumor dynamics (16–19). In addition, ctDNA could be useful clinically to detect PDAC recurrence during postpancreatectomy follow-up (17, 20, 21). A robust method for quantifying low-abundance point mutations in cell-free circulating DNA is digital-droplet PCR (ddPCR; refs. 17, 22). Of the major PDAC driver genes (3), KRAS is mutated in more than 90% of PDACs, and over 95% of these mutations are at the hot spots G12D, G12V, G12R, or Q61H (23, 24). Several research studies have reported on the prognostic significance of KRAS ctDNA detected in the metastatic and perioperative setting (Supplementary Table S1; refs. 25, 26). However, few, if any, ctDNA assays for PDAC have been validated in a Clinical Laboratory Improvement Amendments (CLIA)-certified laboratory (Supplementary Table S1; ref. 27).
In this prospective study, we performed analytic and clinical validation of KRAS ctDNA as a prognostic marker using ddPCR in a CLIA laboratory setting as part of a prospective longitudinal single-institution study (NCT02974764; ref. 28), of patients undergoing resection of PDAC.
Materials and Methods
Study design and population
Patients with localized PDAC eligible for pancreatectomy were recruited from a prospective single-institution cohort study (NCT02974764; ref. 28). Approval by the Johns Hopkins Medicine Institutional Review Board (Baltimore, MD; IRB#00092443) was obtained for this prospective study, which was conducted in accordance with the U.S. Common Rule ethical guidelines. Written consent was obtained for all included patients. This study was conducted and presented according to the most recent REporting recommendations for tumor MARKer prognostic studies (REMARK; ref. 29).
Blood samples for ctDNA analysis were drawn prior to surgical incision and postoperatively prior to discharge or at the 1-month postsurgery clinic visit. Follow-up was performed in accordance with National Comprehensive Cancer Network guidelines (30) and consisted of regular clinical postoperative follow-up appointments every 2 or 3 months, often in conjunction with scheduled treatment plans. Longitudinal blood samples were obtained during these clinic or treatment visits, until the patient’s death, loss of follow-up, or consent revocation. At each timepoint, 40 mL of peripheral blood was collected in 10-mL ethylenediaminetetraacetic acid vacutainers (BD Biosciences). Methods and definitions of diagnosing clinical PDAC recurrence after pancreatectomy at the Johns Hopkins Hospital (Baltimore, MD) have been described previously (6, 7, 31). Relevant demographics, clinicopathologic, and treatment data of included patients were collected prospectively. Decisions regarding adjuvant treatment or treatment recurrence were made by the local or institutional treating clinician, who was blinded to the ctDNA results.
Next-generation sequencing of primary tumor
To determine KRAS mutational status, the surgically resected primary tumor was sequenced using next-generation sequencing (NGS; see Supplementary Materials).
CtDNA extraction
Thirteen different extraction kits from various companies were tested to compare yields of ctDNA (see Supplementary Materials). The manual QIAamp Circulating Nucleic Acid Kit (Qiagen) resulted in the highest yield of DNA and was thus used for this study. Within 6 hours of collection, blood samples were centrifuged with a first run at 2,000 × g for 10 minutes and a second run at 4,000 × g for 10 minutes in the CLIA-certified molecular diagnostics laboratory of the Johns Hopkins Hospital (Baltimore, MD). All isolated plasma was aliquoted and stored in −80°C for later use.
Digital droplet PCR and analysis
Emulsion-based ddPCR was performed using the RainDrop Digital PCR system (RainDance Technologies; see Supplementary Materials). Each assay used a single primer set and two TaqMan probes, one specific to the wild-type allele and a second specific to the mutant allele. RainDrop Analyst II (RainDance Technologies) was used to determine a positive signal for either wild-type (VIC) or mutant KRAS (FAM). Positive control (cell line mixtures at 1:1,000 and 1:100), negative control (gDNA), negative control (plasma), and nontemplate control were run additionally on each ddPCR chip. Results were presented as mutant KRAS droplets per 10,000 total genomic droplets (per 10k).
Analytic validation of KRAS ddPCR
Four KRAS assays for G12V, G12D, G12R, and Q61H (c.183A>C) were developed using ddPCR on the RainDance system (see Supplementary Materials). A linearity study was performed using DNA mixtures from cell lines containing known mutations (32, 33), serially diluted into wild-type DNA, to measure accuracy and determine the limit of detection. Mutant droplets were consistently detected in the 1:10,000 samples for all four cell line mixes, consistent with an analytic limit of detection of 1:10,000.
Baseline noise was determined using a set of deidentified normal plasma samples from healthy donors (fresh frozen plasma from donors age less than 25, and frozen within 8 hours of collection) and discarded plasma-24 (PF24, plasma frozen within 24 hours) from the Johns Hopkins Hospital (Baltimore, MD). Using these normal plasma samples, 50 assays were performed sampling >50,000 wild-type droplets for each assay. The baseline noise was exceedingly low for G12V and Q61H, while slightly higher for G12R and bases susceptible to cytosine deamination such as G12D (34). Qualitatively similar findings were found with male normal plasma sampled in 171 separate runs to a total depth of more than 9 million wild-type droplets.
We required three (G12V, Q61H) or four (G12D, G12R) positive droplets for a sample to be called positive and because most plasma samples tested had approximately 5,000–20,000 amplifiable KRAS molecules, the limit of detection for clinical samples was approximately 1:2,000–1:6,000, depending on the number of genomes tested and the specific mutation detected.
Definitions and statistical analysis
Continuous variables were reported as median interquartile range (IQR) and compared with the Mann–Whitney rank-sum test. Categorical variables were summarized as frequency (%) and compared with the χ2 or Fisher exact tests as appropriate. Sensitivity, specificity, positive predictive value, and negative predictive value of KRAS ctDNA detection for prediction of recurrence throughout the follow-up period were calculated using contingency tables. Disease recurrence was characterized using Response Evaluation Criteria in Solid Tumors version 1.1 guidelines.
To assess the prognostic value of ctDNA, recurrence-free survival (RFS) and overall survival (OS) from the time of surgery were the primary endpoints of interest. RFS was defined as the time interval between the date of surgery and the date of recurrence. Patients were censored at the date of last cross-sectional imaging if recurrence did not occur. OS was defined as the time from the date of surgery to death from any cause. Patients were censored at the date of the last follow-up visit if no date of death was available. Kaplan–Meier curves were used to estimate median survival with corresponding 95% confidence intervals (CI). The log-rank test was utilized for subgroup comparison. Univariable Cox proportional-hazard analysis was performed to identify potential preoperative predictors for RFS and OS. In the preoperative setting, proven prognosticators such as N-stage, T-stage, and other variables obtained from the pathologic specimen are not yet available, and for that reason not included in the model. All factors with a P <0.10 in univariable analysis were included as a covariate in a multivariable regression model to identify independent preoperative prognostic factors with corresponding hazard ratios (HRs). Statistical analysis was performed using SPSS version 25.0 (IBM Corporation) and R version 3.3.3 (R Foundation).
Results
Analytic validation of KRAS ddPCR
Analytic validation of ddPCR for ultrasensitive detection of the most common KRAS mutations in PDAC, G12V, G12D, G12R, and Q61H was performed. Normal plasma was used to define baseline noise, and cell line mixes were used to determine the accuracy, limit of detection, and precision. Baseline noise was higher for G12D and G12R; however, patient samples with 1–2 droplets could be followed by a negative result or a high positive result. Accordingly, samples were categorized into three categories: negative, borderline (1 or 2 positive droplets), and positive (see Supplementary Materials).
Study cohort and primary tumor KRAS status
A CONSORT diagram showing study design, patient selection, and cohort analysis is presented in a Supplementary Fig. S1. The final case cohort consisted of 59 patients with resected PDAC. Primary tumor KRAS status was G12V in 28 of 59 (48%), G12D in 16 of 59 (27%), G12R in 12 of 59 (20%), and Q16H in 3 of 59 (5%) patients. Baseline demographics, clinicopathologic, and treatment characteristics are given in Table 1.
Table 1.
Demographic, clinicopathologic, and treatment characteristics of included patients Abbreviations: AJCC, American Joint Committee on Cancer; DP-CAR, distal pancreatectomy with celiac axis resection.
| Variable | All patients (n = 59) | ctDNA Negative (n = 30) | ctDNA Borderline & positive (n = 29) | P |
|---|---|---|---|---|
| Male, n (%) | 31 (53%) | 19 (63%) | 12 (41%) | 0.091 |
| Caucasian, n (%) | 49 (83%) | 24 (80%) | 25 (86%) | 0.731 |
| Age, median y (IQR) | 70 (61–77) | 65 (59–72) | 71 (66–80) | 0.014 |
| Treatment status, n (%) | <0.001 | |||
| Chemo-naïve surgery | 35 (59%) | 11 (37%) | 24 (83%) | |
| Postneoadjuvant surgery | 24 (41%) | 19 (63%) | 5 (17%) | |
| Presurgery CA 19–9, median U/mL (IQR)a | 95 (29–244) | 63 (26–128) | 200 (61–609) | 0.007 |
| Type of operation, n (%) | NA | |||
| Pancreatoduodenectomy | 39 (66%) | 23 (77%) | 16 (55%) | |
| Distal pancreatectomy | 10 (17%) | 3 (10%) | 7 (24%) | |
| Total pancreatectomy | 7 (12%) | 1 (3%) | 6 (21%) | |
| DP-CAR | 3 (5%) | 3 (10%) | 0 (0%) | |
| Location of tumor, n (%) | 0.211 | |||
| Head/uncinate | 43 (73%) | 24 (80%) | 19 (66%) | |
| Body/tail | 16 (27%) | 6 (20%) | 10 (35%) | |
| R0 margin (>1 mm), n (%) | 46 (78%) | 24 (80%) | 22 (76%) | 0.701 |
| Tumor size, median cm (IQR) | 3.0 (2.5–4.0) | 2.5 (2.1–3.5) | 3.5 (2.7–5.1) | 0.002 |
| T-stage (AJCC 8th edition), n (%) | 0.015 | |||
| T1–T2 | 43 (73%) | 26 (87%) | 17 (59%) | |
| T3–T4 | 16 (27%) | 4 (13%) | 12 (41%) | |
| N-stage (AJCC 8th edition), n (%) | <0.001 | |||
| N0 status | 23 (39%) | 19 (63%) | 4 (14%) | |
| N1 status | 20 (34%) | 6 (20%) | 14 (48%) | |
| N2 status | 16 (27%) | 5 (17%) | 11 (38%) | |
| Lymph node positive, n (%) | 37 (63%) | 12 (40%) | 25 (86%) | <0.001 |
| Tumor differentiation, n (%) | 0.138 | |||
| Well/moderate | 40 (68%) | 23 (77%) | 17 (59%) | |
| Poor | 19 (32%) | 7 (23%) | 12 (41%) | |
| Microscopic perineural invasion, n (%) | 49 (83%) | 23 (77%) | 26 (90%) | 0.184 |
| Microscopic lymphovascular invasion, n (%) | 29 (49%) | 10 (35%) | 19 (66%) | 0.002 |
| Tumor cellularity on NGS, median % | 11.7 (6.7–21.0) | 8.3 (3.4–16.0) | 18.2 (10.9–26.9) | <0.001 |
| KRAS mutation | 0.928 | |||
| G12V | 28 (48%) | 15 (50%) | 13 (45%) | |
| G12D | 16 (27%) | 8 (27%) | 8 (28%) | |
| G12R | 12 (20%) | 6 (20%) | 6 (21%) | |
| Q61H | 3 (5%) | 1 (3%) | 2 (7%) |
Forty-seven patients had documented presurgery CA 19–9 values, of whom four patients were nonexpressors of CA 19–9 and excluded from analysis.
Preoperative ctDNA status and association with clinicopathologic features
Prior to incision, 49% of patients (n = 29) had detectable ctDNA, consisting of either a borderline (n = 13; 22%) or a positive sample (n = 16; 27%). In these 29 patients, median number of total KRAS-mutant droplets was 4 (IQR 2–13) and median number of mutant KRAS droplets per 10k was 1.7 (IQR, 0.8–8.5). Several significant associations were observed between presence of preoperative ctDNA and historic clinicopathologic risk factors such as larger tumor size, lymph node positivity, presence of microscopic lymphovascular invasion, and elevated preoperative CA 19–9 (Table 1).
Preoperative ctDNA as predictor for recurrence and survival
After a median follow-up of 16 months (95% CI, 13–19 months), disease recurrence was observed in 41 (70%) of 59 patients and 26 of 59 (44%) had died. For the entire cohort, median RFS was 10 months (95% CI, 8–11 months), while median OS was 22 months (95% CI, 0–not reached). Patients with detectable preoperative ctDNA had significantly decreased median RFS (8 months; 26 of 29 (90%) recurred) when compared with patients negative for ctDNA [19 months; 15/30 (50%) recurred; P < 0.001; Fig. 1A]. Similarly, median OS was not yet reached in the ctDNA-negative group [8/30 (27%) died], compared with a median OS of 14 months [18/29 (62%) died] for patients with preoperative borderline or positive ctDNA (P <0.001; Fig. 1B).
Figure 1.
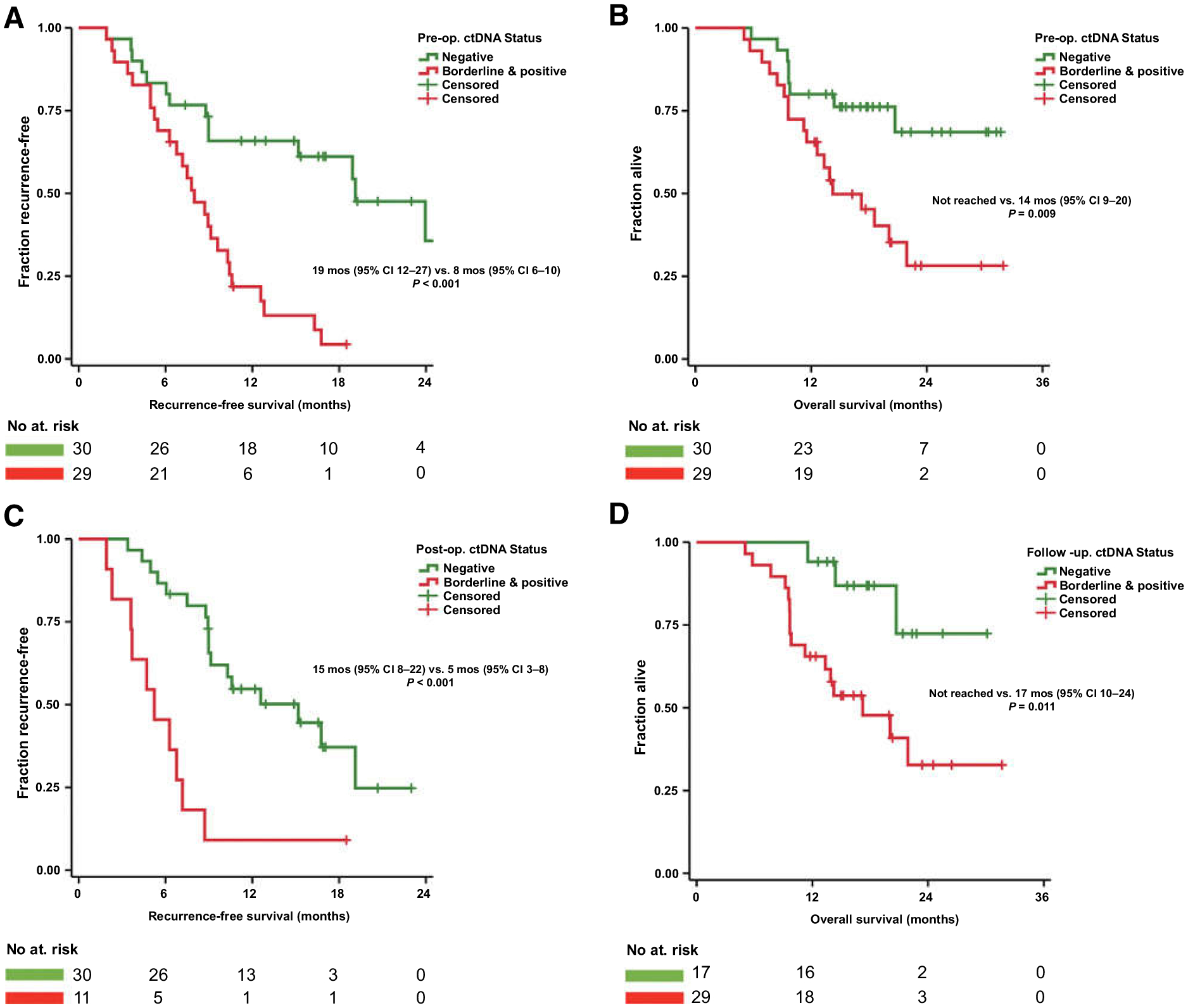
A and B, Patients with preoperative detectable KRAS ctDNA had decreased RFS and OS. C, Patients with detectable KRAS ctDNA in the immediate postoperative period had decreased RFS. D, Patient with detectable KRAS ctDNA during follow-up had decreased OS.
In multivariable Cox regression models of potential preoperative risk factors, detection of ctDNA was an independent preoperative predictor for both RFS (HR = 2.67; P = 0.011) and OS (HR = 2.37; P = 0.048; Supplementary Table S2).
Impact of treatment on ctDNA status
Several interactions between treatment for localized PDAC and ctDNA dynamics were observed. For instance, patients who had prior neoadjuvant chemotherapy were significantly less likely to have ctDNA in their preoperative sample than were chemo-naïve patients (21% vs. 69%; P < 0.001). Also, both preoperative median number of mutant KRAS droplets per 10k [0 (IQR = 0–0.5) vs. 1.2 (IQR = 0–5.4); P = 0.002] and median number of total KRAS-mutant droplets [0 (IQR = 0–1) vs. 2 (IQR = 0–8); P = 0.003] were significantly lower in postneoadjuvant patients than in untreated patients (Supplementary Fig. S2A and S2B). Outcomes were relatively poor for the five patients with detectable preoperative ctDNA after neoadjuvant therapy: all recurred after surgery (median RFS 5 months) and two of the five were deceased after a median follow-up of 12 months following surgery. Anecdotally, one patient with extensive postneoadjuvant treatment effect seen in the surgical specimen (College of American Pathologists score 1) was negative for KRAS ctDNA in both the pre- and postoperative sample. Of 24 patients who had neoadjuvant treatment prior to surgery, the majority received FOLFIRINOX (15/24; 63%). Gemcitabine/nab-paclitaxel was administered in four patients (17%) while five patients (21%) received both FOLFIRINOX and gemcitabine/nab-paclitaxel. Possibly due to small patient numbers, no statistically significant differences in ctDNA positivity were observed on the basis of neoadjuvant chemotherapy regimen or duration.
Forty-six patients had blood samples collected at one or more timepoint after pancreatic resection. Of these, five patients did not have a sample collected in the immediate postoperative period (defined as the period before the start of adjuvant treatment or within 1 month after pancreatectomy when no adjuvant was given). Consequently, 41 patients had an immediate postoperative sample available (sample at median postoperative day 24). In patients with detectable preoperative ctDNA, surgery resulted in a drop of median number of mutant KRAS droplets per 10k from 1.0 (IQR = 0.7–8.3) to 0 (IQR = 0–1.3; P < 0.001), and a drop of median number of total KRAS-mutant droplets from 3 (IQR = 2–30) to 0 (IQR = 0–3; P < 0.001; Fig. 2). Persistence or emergence of ctDNA in the immediate postoperative period was detected in 11 patients (Fig. 2D). Patients with ctDNA detected shortly after surgery prior to adjuvant treatment were highly likely to recur (10/11, 91%) and had a median RFS of only 5 months, significantly shorter than the 15 months (P < 0.001) for patients without detectable postoperative ctDNA [17/30 (57%); recurred; Fig. 1C].
Figure 2.
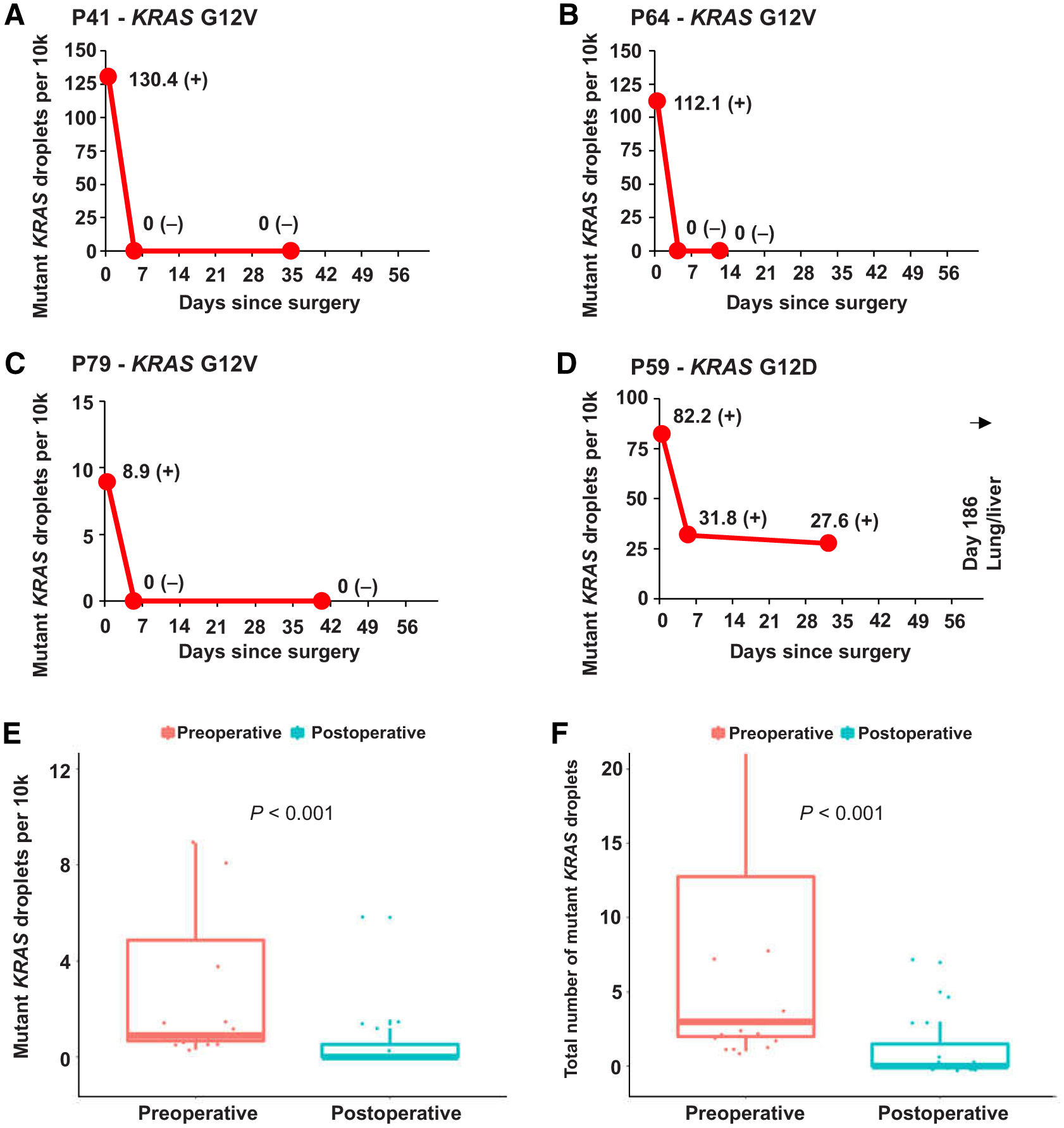
Examples of individual patient graphs showing disappearance (A–C) or persistence (D) of detectable KRAS ctDNA in the 2-month period following surgical resection of the primary tumor. E and F, Boxplots of comparison between KRAS ctDNA-mutant droplets per 10k and total amount of mutant KRAS droplets of preoperative and postoperative samples. (−) signifies a negative KRAS ctDNA result; (±) signifies a borderline KRAS ctDNA result; (+) signifies a positive KRAS ctDNA result. Sample at timepoint 0 represents the preoperative samples.
Longitudinal ctDNA monitoring for prediction of recurrence
To assess the clinical utility of ctDNA for monitoring patients with PDAC for recurrence after their surgery, serial follow-up plasma samples were collected. At total of 277 samples from 46 patients were analyzed (median five samples per patient). Median follow-up for this subcohort of patients was 15 months, during which 30 of 46 (65%) patients recurred and 19 of 46 (41%) died. Table 2 shows that 27 of 30 (90%) patients that recurred at the time of last follow-up had detectable ctDNA during postoperative follow-up (Fig. 3A and B). In contrast, 14 of 16 (88%) patients free of recurrence never had ctDNA detected in any follow-up sample (Fig. 3C and D). In these 16 patients without recurrence, preoperative samples were positive in one patient, borderline in two, and negative in 13 patients. The sensitivity of any detectable ctDNA during postpancreatectomy follow-up for predicting recurrence was 90% (95% CI, 74%–98%), while specificity for predicting disease recurrence was 88% [95% CI, 62%–98%; positive predictive value was 93% (95% CI, 79%–98%) and negative predictive value 82% (95% CI, 61%–93%)]. The diagnostic accuracy of the ctDNA assay for predicting postoperative recurrence was 89% (95% CI, 76%–96%).
Table 2.
Contingency table comparing KRAS ctDNA detection during postpancreatectomy follow-up to clinical disease recurrence
| No disease recurrence | Disease recurrence | Total | |
|---|---|---|---|
| KRAS ctDNA negative during follow-up | 14 (11, 23, 26, 27, 44, 47, 51, 66, 68, 72, 77, 78, 84, 87) | 3 (60, 85, 86) | 17 (37%) |
| KRAS ctDNA borderline during follow-up | 2 (61, 88) | 5 (19, 22, 74, 81, 83) | 7 (15%) |
| KRAS ctDNA positive during follow-up | 0 () | 22 (5, 10, 15 20, 25, 29, 32, 34, 35, 38, 40, 41, 46, 59, 63, 64, 65, 75, 79, 89, 90, 91) | 22 (48%) |
| Total | 16 (35%) | 30 (65%) | 46 (100%) |
NOTE: Individual patient numbers displayed in brackets.
Figure 3.
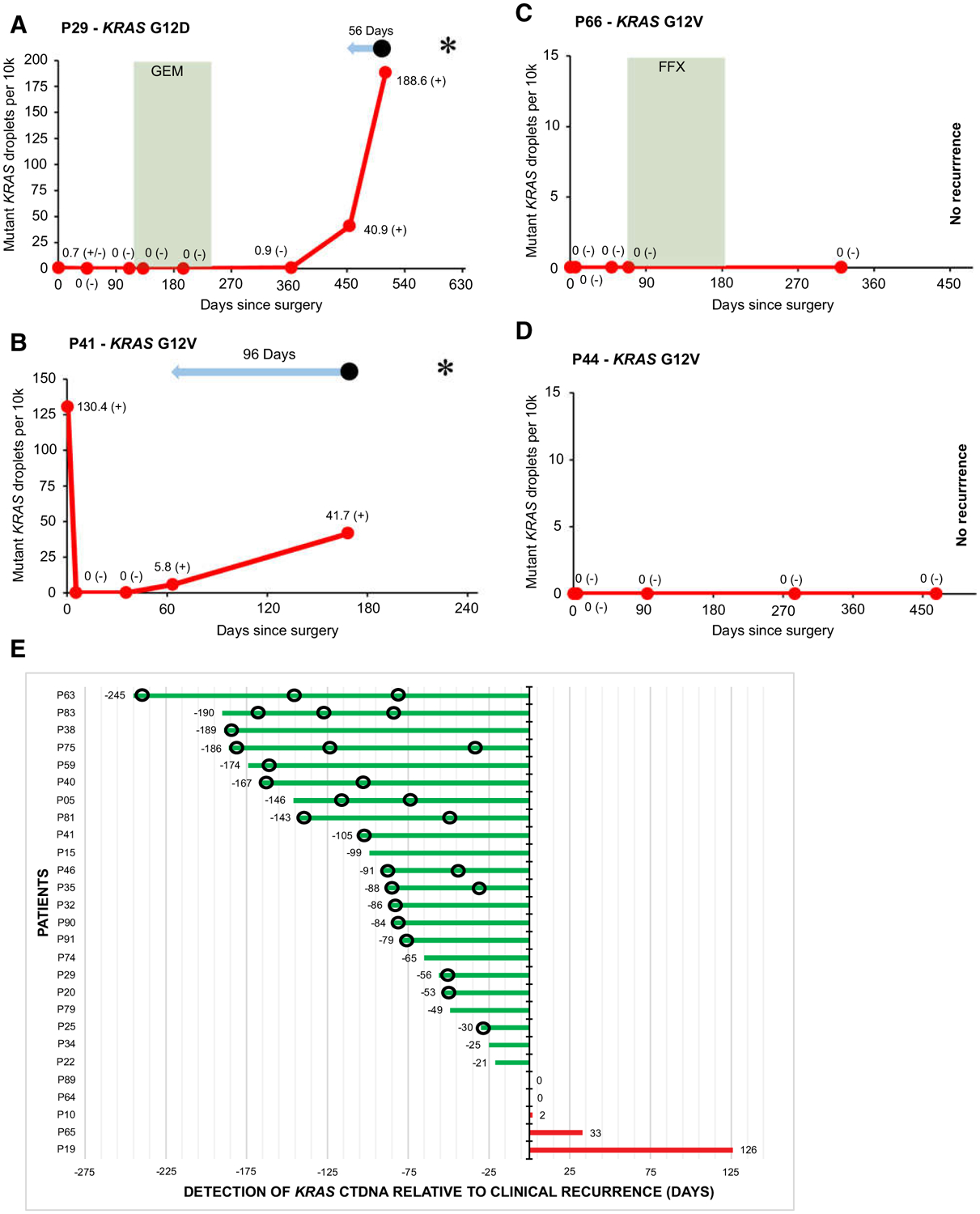
Examples of individual patient graphs showing detectable KRAS ctDNA during follow-up in patients with recurrence (A and B), and no KRAS ctDNA detected during follow-up in patients without recurrence (C and D). E, Plot showing timing of detection of KRAS ctDNA relative to manifestation of clinical recurrence in 27 patients with detectable KRAS ctDNA and recurrence during postpancreatectomy follow-up. In A–D,  signifies chemotherapy treatment. FFX, FOLFIRINOX; GEM, gemcitabine.
signifies chemotherapy treatment. FFX, FOLFIRINOX; GEM, gemcitabine.  Signifies lead time of KRAS ctDNA detection relative to clinical recurrence. • signifies clinical disease recurrence; * signifies patient death; (−) signifies a negative KRAS ctDNA result; (±) signifies a borderline KRAS ctDNA result; (+) signifies a positive KRAS ctDNA result. Sample at timepoint 0 represents the preoperative sample. In E,
Signifies lead time of KRAS ctDNA detection relative to clinical recurrence. • signifies clinical disease recurrence; * signifies patient death; (−) signifies a negative KRAS ctDNA result; (±) signifies a borderline KRAS ctDNA result; (+) signifies a positive KRAS ctDNA result. Sample at timepoint 0 represents the preoperative sample. In E,  signifies imaging without disease recurrence.
signifies imaging without disease recurrence.
Figure 3E shows that in the majority of the 27 patients in whom ctDNA correctly predicted recurrence of their PDAC, the detection of ctDNA preceded the detection of disease recurrence by imaging [22/27 (81%)]. The median lead time of ctDNA relative to imaging was 84 days (IQR = 25–146). Although CA 19–9 was not collected as part of this study, several patients had serial clinical CA 19–9 values available. In some patients, CA 19–9 and ctDNA levels had matching dynamics (Fig. 4A and B). In other patients, the additional value of ctDNA was evident when CA 19–9 remained low despite disease recurrence (Fig. 4C), or when CA 19–9 was undetectable (Fig. 4D).
Figure 4.
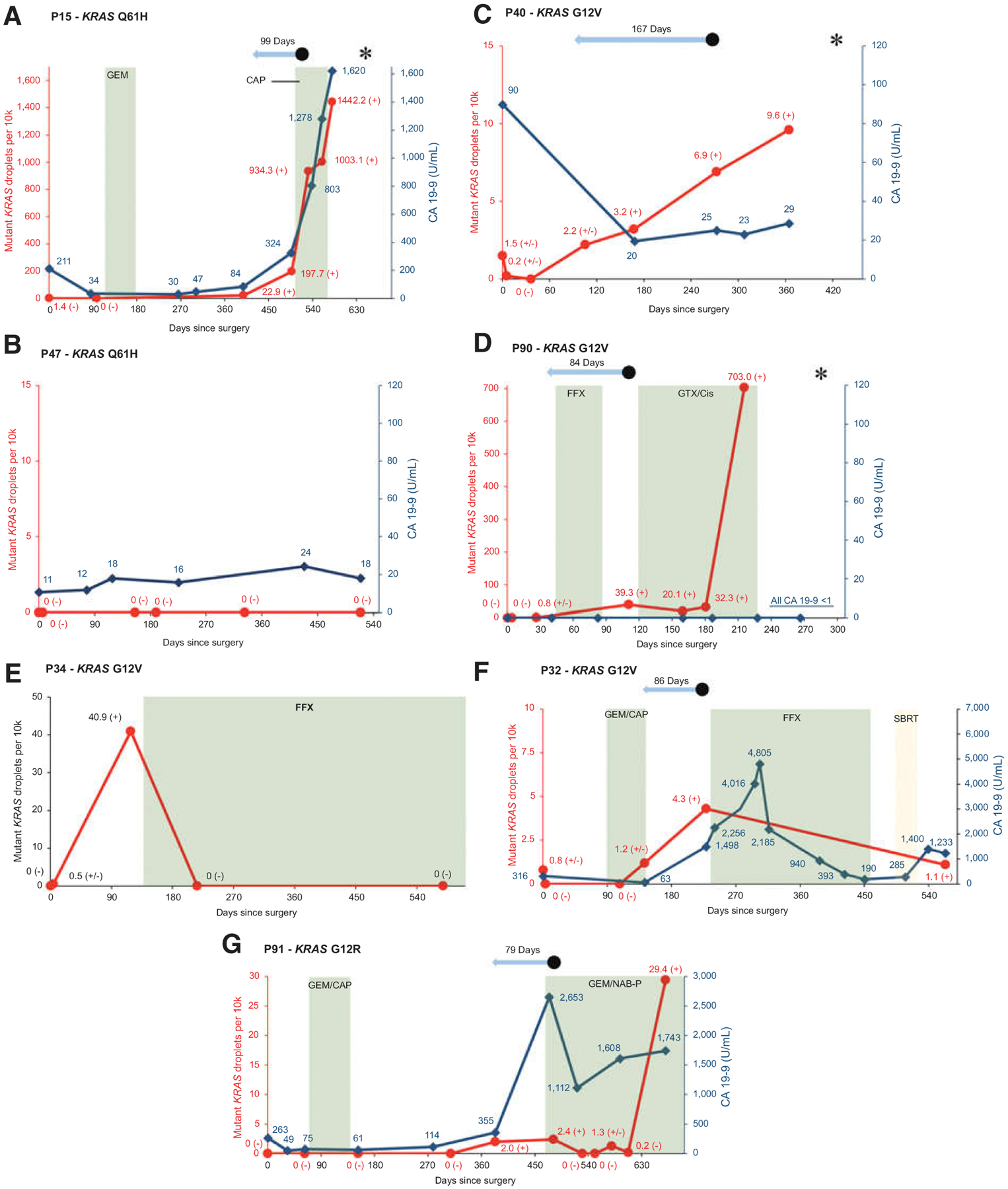
Examples of individual patient graphs showing matching rise in CA 19–9 and KRAS ctDNA in a patient with recurrence (A), similar stability in CA 19–9 and KRAS ctDNA in a patient without recurrence (B), rise in KRAS ctDNA with low and stable CA 19–9 in a patient with recurrence (C), rise in KRAS ctDNA in nonexpressor of CA 19–9 with recurrence (D), KRAS ctDNA response to treatment (E and F), and a patient in whom KRAS ctDNA might have helped clinical decision-making (G).  Red line signifies mutant KRAS droplets per 10k total droplets over time.
Red line signifies mutant KRAS droplets per 10k total droplets over time.  Dark blue line signifies CA 19–9 levels in U/mL over time.
Dark blue line signifies CA 19–9 levels in U/mL over time.  Green plane signifies chemotherapy treatment. CAP, capecitabine; FFX, FOLFIRINOX; GEM, gemcitabine; GTX/Cis, gemcitabine, docetaxel, capecitabine and cisplatin; NAB-P; nab-paclitaxel.
Green plane signifies chemotherapy treatment. CAP, capecitabine; FFX, FOLFIRINOX; GEM, gemcitabine; GTX/Cis, gemcitabine, docetaxel, capecitabine and cisplatin; NAB-P; nab-paclitaxel.  Yellow plane signifies radiotherapy. SBRT, stereotactic body radiation therapy.
Yellow plane signifies radiotherapy. SBRT, stereotactic body radiation therapy.  Light blue arrow signifies lead time of KRAS ctDNA detection relative to clinical recurrence. • signifies clinical disease recurrence; * signifies patient death; (−) signifies a negative KRAS ctDNA result; (±) signifies a borderline KRAS ctDNA result; (+) signifies a positive KRAS ctDNA result. Sample at time point 0 represents the preoperative sample.
Light blue arrow signifies lead time of KRAS ctDNA detection relative to clinical recurrence. • signifies clinical disease recurrence; * signifies patient death; (−) signifies a negative KRAS ctDNA result; (±) signifies a borderline KRAS ctDNA result; (+) signifies a positive KRAS ctDNA result. Sample at time point 0 represents the preoperative sample.
Detection of ctDNA during follow-up was associated with a median OS of 17 months, while median OS was not yet reached at 30 months for patients without detectable ctDNA during follow-up (P = 0.011; Fig. 1D). At the conclusion of the study period, 16 of 29 (55%) patients with detectable ctDNA during follow-up had died (Fig. 3A and B), compared with three of 17 (18%) patients who had consistently negative plasma samples (P = 0.013; Fig. 3C and D).
ctDNA dynamic
sctDNA dynamics correlated with a response to treatment of PDAC recurrence in several patients. For instance, patient 34 developed several hepatic metastases 138 days after pancreatoduodenectomy (Fig. 4E). FOLFIRINOX was started and the metastases showed a dramatic response on CT imaging. Correspondingly, KRAS ctDNA became undetectable and the patient currently continues on chemotherapy with stable disease on imaging. In another example, a patient developed local recurrence and was subsequently treated with FOLFIRINOX followed by radiotherapy (Fig. 4F). A radiographic response was observed, consistent with a decrease in both ctDNA and CA 19–9 levels.
In this observational study, treatment decisions were made independent from ctDNA results. The clinical course of patient 91 illustrates several instances where information on ctDNA might have made a difference in a patient’s treatment (Fig. 4G). This patient, initially diagnosed with borderline resectable PDAC, was treated with neoadjuvant gemcitabine/nab-paclitaxel followed by a pancreatoduodenectomy. An increase in both ctDNA and CA 19–9 was observed on postoperative day 383, coinciding with questionable signs of local recurrence on CT. Repeat imaging performed 3 months later definitively showed local recurrence and new metastases in both lungs. Palliative gemcitabine/nab-paclitaxel was initiated with an observed drop in CA 19–9 and ctDNA. Patient 91 currently remains on treatment with stable disease on imaging. However, the patient’s latest plasma sample showed a sudden increase in ctDNA. In this patient alone, additional evidence provided by ctDNA could have led to an earlier diagnosis of recurrence, and subsequent start of palliative treatment by 3 months. In addition, after being on palliative gemcitabine/nab-paclitaxel for an extended period of time, the recent spike in ctDNA might indicate refractory disease and the need to switch to a different chemotherapeutic regimen.
Discussion
ctDNA testing offers the potential to significantly improve clinical decision making for patients with PDAC (35). However, patients with PDAC often have very low levels of ctDNA (often in the range of 0.1% mutated DNA relative to wild-type DNA) necessitating ultrasensitive and reproducible approaches for clinical testing (16, 17, 36). In this prospective study performed in a CLIA-laboratory setting, analytic validation of a ddPCR KRAS ctDNA assay showed a high degree of accuracy, sensitivity, and precision, with sufficiently low background noise and limit of detection. In addition, this study confirmed the promise of ctDNA as a clinically useful prognostic biomarker in three clinical situations: (i) detectable ctDNA levels as a bad prognostic marker in either presurgery samples or (ii) when persisting in the immediate postoperative period, and (iii) detection of ctDNA during postpancreatectomy follow-up as an accurate predictor of PDAC recurrence. As a result, the KRAS ctDNA assay is now offered as an in-house clinical test in the Johns Hopkins Hospital (Baltimore, MD).
Several research studies have established the detrimental prognostic impact of detecting pretreatment ctDNA in patients with advanced PDAC (25). One recent report of 105 resected patients with PDAC found that the preoperative detection of KRAS ctDNA by ddPCR was associated with both decreased median RFS (6 vs. 16 months; P < 0.001) and decreased OS (14 vs. 28 months; P <0.0001; ref. 37). In recent years, it has been argued that selected patients with resectable PDAC might benefit from a neoadjuvant chemotherapy-first approach (38, 39). However, currently available preoperative risk factors for predicting early PDAC recurrence after resection, such as CA 19–9, have relatively poor predictive value (6). ctDNA analysis might help identify those patients with a high likelihood of early recurrence, which could greatly aid physicians and patients alike in selecting the appropriate sequence of therapies (26).
Surgical resection resulted in a drop of ctDNA in most patients, indicating that most preoperative ctDNA in patients with early-stage PDAC originates from the primary tumor. In a subpopulation of patients, however, ctDNA persisted in the immediate postoperative period. These patients had a recurrence rate of 91% and median RFS of only 5 months, providing evidence of ctDNA reflecting clinically occult residual disease after pancreatectomy. This strong association between postresection ctDNA and disease recurrence has also been shown in recent studies on resected colon and rectal cancer by Tie and colleagues (21, 40). On the basis of those results, the DYNAMIC trial is currently being conducted, randomizing patients with stage II colon or rectal cancer to adjuvant or no adjuvant therapy based on postresection ctDNA results (ACTRN12615000381583). Adjuvant therapy is an essential part of the standard of care for patients with resected PDAC and adequate performance status. Patients with postoperative detectable ctDNA appear to represent a group for whom systemic therapy is necessary.
Postpancreatectomy surveillance for the detection of recurrent PDAC currently includes serial assessment using CT imaging and CA 19–9 testing (30). However, recent meta-analyses have shown that CT and CA 19–9 only have moderate diagnostic accuracy for the diagnosis of recurrent PDAC (14, 41). This study found that postoperative follow-up with KRAS ctDNA predicts recurrence with high sensitivity (90%) and specificity (88%) and predicts recurrence with a median lead time of almost 3 months over imaging. Similarly, a recent study from our institution found that detection of epithelial and mesenchymal circulating tumor cells during postpancreatectomy follow-up accurately identified disease recurrence within the next 2 months (42). These results indicate that the addition of KRAS ctDNA and other liquid biopsies can facilitate accurate and early recognition of PDAC recurrence postpancreatectomy. The theoretical advantage of early detection of disease recurrence is that treatment can be initiated at a time when tumor burden is still limited, possibly resulting in eradication of recurrent disease or delay of progression (27).
Dynamic monitoring of therapeutic response to cancer therapies is another potential use for ctDNA assays. Evidence of correlations between changes in ctDNA levels and tumor responses or outcomes have been demonstrated in other cancers, such as breast cancer (43, 44), lung cancer (20, 45), and colorectal cancer (46, 47). Although this study was neither designed nor powered to assess the value of KRAS ctDNA for monitoring of therapeutic response, observed ctDNA dynamics in several patients suggested a correlation between ctDNA levels and response to cancer treatment. Current evidence for the use of ctDNA for therapeutic tracking in PDAC is limited to several smaller series in patients with metastatic disease (Supplementary Table S1). For instance, in a recent study of patients with metastatic PDAC treated with FOLIRINOX, serial testing found concomitantly reduced ctDNA for 12 patients with partial response or stable disease (48). Another study showed that reduction in KRAS ctDNA was associated with tumor responses observed by CT scan in 10 of 13 patients with metastatic PDAC (49). Future, large prospective interventional trials are needed to demonstrate whether changing treatment based on ctDNA dynamics can improve patient outcomes. In PDAC, an example of such a trial could encompass starting patients on either FOLFIRINOX or gemcitabine/nab-paclitaxel, and switching to the other regimen following a sustained spike in ctDNA indicating refractory disease.
Perhaps no other liquid biopsy has generated more current enthusiasm than tumor-derived cell-free circulating DNA. Consequently, methods for ctDNA detection are constantly expanding and evolving, resulting in different assays with varying advantages and drawbacks regarding sensitivity, limit of detection, and breadth of coverage. With ddPCR as performed on the RainDance instrument, the number of positive droplets is relatively low (~10,000) compared with the number of droplets analyzed (typically 10 million). Accordingly, the probability of a KRAS-positive droplet is about 1/1,000 and therefore the probability of a droplet receiving and amplifying two KRAS molecules is only approximately 1/1,000,000. Furthermore, because the gates for a positive droplet are tight around the positive cluster, various false positives should be eliminated, including a mixed droplet from a wild-type and mutant alleles, a droplet containing a nucleotide mispair from cytosine deamination, or even an early round PCR error. This is a more sensitive and specific technology compared with NGS-based methods that typically have error rates at the 0.1%–1% level (50), although methods, such as the Safe-Sequencing System, have been developed to overcome this problem (51). For clinical purposes, the in-house turnaround time of the current KRAS ddPCR assay is 1–2 weeks.
The results of this prospective study should be interpreted within the context of its limitations. One of these limitations is that direct comparison of CA19–9 with ctDNA in simultaneously collected samples was not possible in this study. Another important limitation is that this KRAS ctDNA assay can only be used to measure tumor dynamics in the +90% of patients with a KRAS-mutated PDAC and could potentially underestimate tumor burden if subclones harboring a different mutation start to expand. However, a recent study that included PDAC showed that there is minimal functional driver gene heterogeneity among metastases (52). Furthermore, the analytic limit of detection of 1:10,000 that was established for this KRAS ddPCR, is difficult to achieve with current multigene panel or NGS approaches (35). Also, the study cohort had relatively high percentages of T1–T2 (73%) and well- to moderately differentiated tumors (68%). It is possible these percentages were higher due to the inclusion of patients undergoing neoadjuvant treatment, because these patients often have smaller tumors on pathology as a result of preoperative chemotherapy treatment. Finally, this study was limited by the relatively small number of patients.
Despite these limitations, this study had important strengths that should also be acknowledged. First, this was a prospective study conducted in a CLIA-certified laboratory setting. Second, a robust analytic validation was performed that included the assessment of background noise in plasma samples from healthy donors. Finally, a relatively large number of longitudinally collected plasma samples was available for clinical validation.
Conclusions
KRAS ctDNA measured in a clinical laboratory provides useful prognostic information for patients with PDAC undergoing preoperative evaluation and postoperative surveillance. Future prospective and interventional clinical trials will determine whether treatment decisions based on ctDNA dynamics can improve outcomes for patients with PDAC.
Supplementary Material
Translational Relevance.
A clinical validation of a KRAS-circulating tumor DNA (ctDNA) assay was performed in a Clinical Laboratory Improvement Amendments-certified and College of American Pathology-inspected clinical laboratory. This study confirmed the clinical utility of ctDNA as a tumor-specific biomarker for prognostic stratification and monitoring of tumor dynamics in patients with pancreatic cancer. First, detection of mutant KRAS ctDNA prior to surgery or in the immediate postoperative period predicts a higher likelihood of tumor recurrence and poorer survival. Second, rising mutant KRAS ctDNA during postoperative follow-up anticipates radiographic/clinical recurrence with a lead time of 84 days. As a result of this study, this KRAS ctDNA test is now live at the Johns Hopkins Hospital (Baltimore, MD; go live date, 12/4/2018). Future prospective and interventional trials will determine whether treatment decisions based on ctDNA dynamics can improve patient outcomes.
Acknowledgments
This study was supported by the Stringer Foundation, the Michael Rolfe Foundation, the Mary Lou Wootton Pancreatic Cancer Research Fund, the Joseph C. Monastra Foundation, and The Sol Goldman Pancreatic Cancer Research Center. The authors also thank the Foundation “De Drie Lichten”, Prins Bernhard Cultuurfonds, VSBfonds, Prof. Micha€el-van Vloten Fonds, the Nijbakker-Morra Foundation, and the Living With Hope Foundation (all from the Netherlands) for providing grants for a research fellowship by V.P. Groot. The authors would also like to acknowledge Drs. Bert Vogelstein, Kenneth Kinzler, Nick Papadopoulos, Jill Phallen, Ben Ho Park, Paul Ness, Paula Hurley, Rena Xian, Lori Sokoll, and Josh Lauring for insightful discussions. In addition, we thank Lisa Shifflet, Steven Bass (RainDance), and Emily Adams for helpful discussions. Finally, we thank the Dayton Blood Center for providing fresh frozen plasma for negative controls.
Disclosure of Potential Conflicts of Interest
A. Narang reports receiving commercial research grants from Boston Scientific. L. Zheng has ownership interests (including patents) at Mingruizhiyao, Alphamab, and Aduro, is a consultant/advisory board member for Foundation Medicine, Oncorus, Novarock, Alphamab, Mingruizhiyao, Biosynergics, and QED, and reports receiving commercial research grants from Bristol-Myers Squibb, Merck, Halozyme, Novarock, iTEOS, Amgen, and Inxmed. V.E. Velculescu has ownership interests (including patents) at and is a consultant/advisory board member for Personal Genome Diagnostics and Ignyta. J.R. Eshleman is a consultant/advisory board member for Promega. No potential conflicts of interest were disclosed by other authors.
Footnotes
Publisher's Disclaimer: The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
References
- 1.Siegel RL, Miller KD, Jemal A Cancer statistics, 2018. CA Cancer J Clin 2018;68:7–30. [DOI] [PubMed] [Google Scholar]
- 2.Malvezzi M, Carioli G, Bertuccio P, Boffetta P, Levi F, La Vecchia C, et al. European cancer mortality predictions for the year 2018 with focus on colorectal cancer. Ann Oncol 2018;29:1016–22. [DOI] [PubMed] [Google Scholar]
- 3.Wolfgang CL, Herman JM, Laheru DA, Klein AP, Erdek MA, Fishman EK, et al. Recent progress in pancreatic cancer.CA Cancer J Clin 2013;63: 318–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neoptolemos JP, Palmer DH, Ghaneh P, Psarelli EE, Valle JW, Halloran CM, et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): a multicentre, open-label, randomised, phase 3 trial. Lancet 2017;389:1011–24. [DOI] [PubMed] [Google Scholar]
- 5.Chin V, Nagrial A, Sjoquist K, O’Connor CA, Chantrill L, Biankin AV, et al. Chemotherapy and radiotherapy for advanced pancreatic cancer. Cochrane Database Syst Rev 2018;3:CD011044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Groot VP, Gemenetzis G, Blair AB, Rivero-Soto RJ, Yu J, Javed AA,et al. Defining and predicting early recurrence in 957 patients with resected pancreatic ductal adenocarcinoma. Ann Surg 2019;269:1154–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Groot VP, Rezaee N, Wu W, Cameron JL, Fishman EK, Hruban RH, et al. Patterns, timing, and predictors of recurrence following pancreatectomy for pancreatic ductal adenocarcinoma. Ann Surg 2018;267:936–45. [DOI] [PubMed] [Google Scholar]
- 8.Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364:1817–25. [DOI] [PubMed] [Google Scholar]
- 9.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus Gemcitabine. N Engl J Med 2013;369:1691–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Castillo CFF.A changing landscape in pancreatic cancer.Ann Surg 2018; 268:9–10. [DOI] [PubMed] [Google Scholar]
- 11.Conroy T, Hammel P, Hebbar M, Ben Abdelghani M, Wei AC, Raoul JL, et al. FOLFIRINOX or gemcitabine as adjuvant therapy for pancreatic cancer. N Engl J Med 2018;379:2395–406. [DOI] [PubMed] [Google Scholar]
- 12.Krantz BA, O’Reilly EM. Biomarker based therapy in pancreatic ductal adenocarcinoma: an emerging reality? Clin Cancer Res 2018;24:2241–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Poruk KE, Gay DZ, Brown K, Mulvihill JD, Boucher KM, Scaife CL, et al. The clinical utility of CA 19–9 in pancreatic adenocarcinoma: diagnostic and prognostic updates. Curr Mol Med 2013;13:340–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Daamen LA, Groot VP, Heerkens HD, Intven MPW, van Santvoort HC, Molenaar IQ. Systematic review on the role of serum tumor markers in the detection of recurrent pancreatic cancer.HPB 2018;20: 297–304. [DOI] [PubMed] [Google Scholar]
- 15.Tempero MA, Uchida E, Takasaki H, Burnett DA, Steplewski Z, Pour PM. Relationship of carbohydrate antigen 19–9 and Lewis antigens in pancreatic cancer. Cancer Res 1987;47:5501–03. [PubMed] [Google Scholar]
- 16.Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med 2008;14: 985–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sausen M, Phallen J, Adleff V, Jones S, Leary RJ, Barrett MT, et al. Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat Commun 2015;6:7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Husain H, Velculescu VE.Cancer DNA in the circulation: the liquid biopsy. JAMA 2017;318:1272–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cohen JD, Li L, Wang Y, Thoburn C, Afsari B, Danilova L, et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018;359:926–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abbosh C, Birkbak NJ, Wilson GA, Jamal-Hanjani M, Constantin T, Salari R, et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 2017;545:446–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tie J, Cohen JD, Wang Y, Li L, Christie M, Simons K, et al. Serial circulating tumour DNA analysis during multimodality treatment of locally advanced rectal cancer: a prospective biomarker study. Gut 2019; 68:663–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem 2011;83: 8604–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.The Cancer Genome Atlas Research Network. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 2017;32: 185–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qian ZR, Rubinson DA, Nowak JA, Morales-Oyarvide V, Dunne RF, Kozak MM, et al. Association of alterations in main driver genes with outcomes of patients with resected pancreatic ductal adenocarcinoma. JAMA Oncol 2018;4:e173420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Creemers A, Krausz S, Strijker M, van der Wel MJ, Soer EC, Reinten RJ, et al. Clinical value of ctDNA in upper-GI cancers: a systematic review and meta-analysis. Biochim Biophys Acta 2017;1868:394–403. [DOI] [PubMed] [Google Scholar]
- 26.Bernard V, Kim DU, San Lucas FA, Castillo J, Allenson K, Mulu FC, et al. Circulating nucleic acids associate with outcomes of patients with pancreatic cancer. Gastroenterology 2019;156:108–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Merker JD, Oxnard GR, Compton C, Diehn M, Hurley P, Lazar AJ, et al. Circulating tumor DNA analysis in patients with cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. J Clin Oncol 2018;36:1631–41. [DOI] [PubMed] [Google Scholar]
- 28.ClinicalTrials.gov/National Library of Medicine (US). NCT02974764. Changes in biomarkers from blood over time in patients with pancreatic adenocarcinoma. Available from:https://clinicaltrials.gov/ct2/show/NCT02974764.
- 29.Sauerbrei W, Taube SE, McShane LM, Cavenagh MM, Altman DG. Reporting recommendations for tumor marker prognostic studies (REMARK): an abridged explanation and elaboration. J Natl Cancer Inst 2018;110:803–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tempero MA, Malafa MP, Al-Hawary M, Asbun H, Bain A, Behrman SW, et al. Pancreatic adenocarcinoma, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2017;15: 1028–61. [DOI] [PubMed] [Google Scholar]
- 31.Groot VP, Gemenetzis G, Blair AB, Ding D, Javed AA, Burkhart RA, et al. Implications of the pattern of disease recurrence on survival following pancreatectomy for pancreatic ductal adenocarcinoma. Ann Surg Oncol 2018;25:2475–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones S, Zhang X, Parsons DW, Lin JC-H, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008;321:1801–06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Norris AL, Roberts NJ, Jones S, Wheelan SJ, Papadopoulos N, Vogelstein B, et al. Familial and sporadic pancreatic cancer share the same molecular pathogenesis. Fam Cancer 2015;14:95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen G, Mosier S, Gocke CD, Lin MT, Eshleman JR. Cytosine deamination is a major cause of baseline noise in next-generation sequencing. Mol Diagn Ther 2014;18:587–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Corcoran RB, Chabner BA.Application of cell-free DNA analysis to cancer treatment.N Engl J Med 2018;379:1754–65. [DOI] [PubMed] [Google Scholar]
- 36.Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med 2014;6:224ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hadano N, Murakami Y, Uemura K, Hashimoto Y, Kondo N, Nakagawa N, et al. Prognostic value of circulating tumour DNA in patients undergoing curative resection for pancreatic cancer. Br J Cancer 2016;115: 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Geus SWL, Evans DB, Bliss LA, Eskander MF, Smith JK, Wolff RA, et al. Neoadjuvant therapy versus upfront surgical strategies in resectable pancreatic cancer: a Markov decision analysis. Eur J Surg Oncol 2016;42: 1552–60. [DOI] [PubMed] [Google Scholar]
- 39.Versteijne E, Vogel JA, Besselink MG, Busch ORC, Wilmink JW, Daams JG, et al. Meta-analysis comparing upfront surgery with neoadjuvant treatment in patients with resectable or borderline resectable pancreatic cancer. Br J Surg 2018;105:946–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tie J, Wang Y, Tomasetti C, Li L, Springer S, Kinde I, et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci Transl Med 2016;8:346ra92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Daamen LA, Groot VP, Goense L, Wessels FJ, Borel Rinkes IH, Intven MPW, et al. The diagnostic performance of CT versus FDG PET-CT for the detection of recurrent pancreatic cancer: a systematic review and meta-analysis. Eur J Radiol 2018;106:128–36. [DOI] [PubMed] [Google Scholar]
- 42.Gemenetzis G, Groot VP, Yu J, Ding D, Teinor JA, Javed AA, et al. Circulating tumor cells dynamics in pancreatic adenocarcinoma correlate with disease status. Ann Surg 2018;268:408–20. [DOI] [PubMed] [Google Scholar]
- 43.Dawson SJ, Tsui DWY, Murtaza M, Biggs H, Rueda OM, Chin SF, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med 2013;368:1199–209. [DOI] [PubMed] [Google Scholar]
- 44.Riva F, Bidard FC, Houy A, Saliou A, Madic J, Rampanou A, et al. Patient-specific circulating tumor DNA detection during neoadjuvant chemotherapy in triple-negative breast cancer. Clin Chem 2017;63: 691–99. [DOI] [PubMed] [Google Scholar]
- 45.Mok T, Wu YL, Lee JS, Yu CJ, Sriuranpong V, Sandoval-Tan J, et al. Detection and dynamic changes of EGFR mutations from circulating tumor DNA as a predictor of survival outcomes in NSCLC patients treated with first-line intercalated erlotinib and chemotherapy. Clin Cancer Res 2015;21:3196–203. [DOI] [PubMed] [Google Scholar]
- 46.Tie J, Kinde I, Wang Y, Wong HL, Roebert J, Christie M, et al. Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Ann Oncol 2015;26:1715–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Siravegna G, Mussolin B, Buscarino M, Corti G, Cassingena A, Crisafulli G, et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med 2015;21:795–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wei T, Zhang Q, Li X, Su W, Li G, Ma T, et al. Monitoring tumor burden in response to FOLFIRINOX chemotherapy via profiling circulating cell-free DNA in pancreatic cancer. Mol Cancer Ther 2019;18:196–203. [DOI] [PubMed] [Google Scholar]
- 49.Cheng H, Liu C, Jiang J, Luo G, Lu Y, Jin K, et al. Analysis of ctDNA to predict prognosis and monitor treatment responses in metastatic pancreatic cancer patients. Int J cancer 2017;140:2344–50. [DOI] [PubMed] [Google Scholar]
- 50.Lin MT, Mosier SL, Thiess M, Beierl KF, Debeljak M, Tseng LH, et al. Clinical validation of KRAS, BRAF, and EGFR mutation detection using next-generation sequencing. Am J Clin Pathol 2014;141:856–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kinde I, Wu J, Papadopoulos N, Kinzler KW, Vogelstein B Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci U S A. 2011;108:9530–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reiter JG, Makohon-Moore AP, Gerold JM, Heyde A, Attiyeh MA, Kohutek ZA, et al. Minimal functional driver gene heterogeneity among untreated metastases. Science 2018;361:1033–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.