

Abstract
Sensitivity to allergenic fungi (Alternaria alternata) is associated with acute, severe asthma attacks. Antigen presenting cells (APCs) in the lung sense environmental perturbations that induce cellular stress and metabolic changes and are critical for allergic airway inflammation. However, the mechanisms underlying such environmental sensing by APCs in the lung remains unclear. Here we show that acute Alternaria challenge rapidly induces neutrophil accumulation in airways, and alter expressions of Pyruvate Kinase (PKM2) and hypoxia-inducible factor −1α (Hif-1α) that correlates with proinflammatory mediator release. Blockade of IL33 signaling in vivo led to reduce oxidative stress and glycolysis in lung APCs. Lung-specific ablation of CD11c+ cells abrogates Alternaria-induced neutrophil accumulation and inflammation. Furthermore, administration of Alternaria into the airways stimulated APCs and elevate the expression of Glut-1. Mechanistically, we establish that PKM2 is a critical modulator of lung APC activation in Alternaria-induced acute inflammation. Allosteric activation of PKM2 by a small molecule ML265 or siRNA-mediated knock down correlated negatively with glycolysis and activation of APCs. These results collectively demonstrates that PKM2-mediated glycolytic reprogramming by fungal allergen Alternaria influences lung APC activation, thereby promotes acute airway inflammation. Our data support a model in which Alternaria sensitization in airways induce a circuitry of glycolysis and PKM2 regulation that confers an acute activation of APCs in the lung, whose targeting might represent a strategy for asthma treatment.
Keywords: Fungal asthma, Alternaria, antigen presenting cells, immunometabolism, Pyruvate Kinase
Introduction:
Fungal allergens are proven risk-factor for asthma severity (Denning et al., 2006; Ohollaren et al., 1991). Sensitization to fungal allergens such as Alternaria alternata are associated with fatal asthma exacerbations and hospitalizations (Bush and Prochnau, 2004; Neukirch et al., 1999). Among various common atmospheric mold, Alternaria spores are highly prevalent in grain-growing areas during late summer and/or early autumn period and leads to increased incidents of asthma episodes (Pulimood et al., 2007; Targonski et al., 1995). In clinic, 38.3% of asthmatic children are positive for Alternaria species and manifests symptom with recurrent wheezing and increased airway responsiveness to methacholine (Eggleston et al., 1998; Henderson et al., 1995; Nelson et al., 1999). Previous animal studies have demonstrated various mechanism for unravelling the intricacies of Alternaria-induced allergic asthma (Bankova et al., 2016; Doherty et al., 2012; Murai et al., 2012). An intrinsic protease activity of Alternaria act as effective adjuvant to drive prolonged Th2 type inflammation (Kobayashi et al., 2009; Snelgrove et al., 2014). Alternaria-mediated development of allergic asthma is driven by IL33-ST2 axis in the lung (Cohen et al., 2015; Kouzaki et al., 2011). As such, Alternaria-induced activation of protease activity receptor 2 (PAR-2) in airway epithelial cells is associated with the IL33 release and development of acute airway inflammation (Boitano et al., 2011).
Lung antigen presenting cells (APCs) in the airways are critical innate immune cells at barrier sites and maintains immunity in response to inhaled allergens. In steady state the lung APCs solely fuels from mitochondrial ATP productions via oxidative phosphorylations (OXPHOS), whereas during inflammation the enhance energy demands shifts requirement towards rapid induction of glycolysis (reprogramming) (O’Neill and Pearce, 2016; Wculek et al., 2019). This metabolic reprogramming in lung APCs provides the robust energy required for induction of proinflammatory cytokines, co-stimulatory molecules and effector functions (Everts et al., 2014; Thwe et al., 2019).
The rate-limiting cytosolic enzyme pyruvate kinase (PK) converts the last step of glycolysis from phosphoenol pyruvate to pyruvate that enters into TCA cycle. PKM2 is one of the four isoform of PK enzyme and is induced in activated immune cells thereby regulate inflammation (Palsson-McDermott et al., 2015; Yang et al., 2014), in contrast to PKM1 which is required for basal glycolysis. Upregulation of PKM2 expressions has been linked to inflammatory diseases, such as crohn’s disease, colitis, coronary artery disease and rheumatoid arthritis (Li et al., 2013; Shirai et al., 2016; Tang et al., 2015). PKM2 plays a pivotal role in glycolytic reprogramming by allosteric-switch and interactions with hypoxia-inducible factor −1α (Hif-1α) (Palsson-McDermott et al., 2015). PKM2 in the less active mono/di-meric form translocate into the nucleus and stabilizes a transcriptional complex with Hif-1α, thereby regulating expression of proglycolytic genes and proinflammatory cytokines. PKM2 in its active tetrameric form resides in the cytosol and is unable to further co-activate Hif-1α. The exact mechanism that defines PKM2 regulatory activity in lung inflammation is yet to establish, however, an epigenetic modification of IL33 receptor ST2 expression in group 2 innate lymphoid cells (ILC2) has been identified (Li et al., 2018).
An Alternaria-specific IL33 release and development of robust airway inflammation has previously been demonstrated, however, its impact in promoting metabolic reprograming and activation of lung APCs are still poorly understood. In this study we show that single Alternaria administration rapidly induces robust proinflammatory mediator release and influence glycolytic reprogramming in lung APCs. We specifically show that Alternaria increases oxidative stress in lung APCs and further accelerates metabolic reprograming and activation. Molecularly, we establish that PKM2 is a key regulator in APCs and is required in Alternaria sensitization and airway inflammation. Our findings implicate PKM2 in lung APCs as a pivotal metabolic regulator to initiate and develop Alternaria-induced acute airway inflammation.
Materials and Methods
Reagents
Lyophilized Alternaria alternata and house dust mite (HDM) extracts were purchased from Greer Laboratories (Lenoir, NC, USA). Diphtheria toxin (D0564), 2-Deoxy-D-glucose (D8375) and hydrazine monohydrate (207942) were purchased from Sigma-Aldrich (St Louis, MO). αIL-33 antibody (PA5–47007), anti-mouse IgG (10400C), CM-H2DCFDA (C6827) and 2-NBDG (2-(N-(7-Nitrobenz-2-oxa-1, 3-diazol-4-yl) Amino)-2-Deoxyglucose) (N13195) were all purchased from Thermo Fischer Scientific. Anti-ST2/IL-33R antibody (AF1004-SP) was from R&D Systems. Lactate Dehydrogenase and NAD were from Roche.
Mice and allergen sensitization
Six to eight weeks old naïve C57BL/6J and CD11c-DTR transgenic mice (004509) were purchased from Jackson Laboratory (Bar Harbor, MA). Mice were sensitized intranasally with single administration of Alternaria alternata or HDM extracts (50 μg in 40 μl of PBS). Mice were administered with anti-mouse IL-33 polyclonal antibody (Ser109-Ile266, <0.1 ng/mg endotoxin) or IgG isotype control antibody (1 μg in 40 μl of PBS; i.n.) twice at 12 hours of interval before Alternaria treatment. CD11c-DTR transgenic (Tg) mice were administered with diphtheria toxin or PBS (DT; 50 ng in 40 μl of PBS; i.n) 24 hours before Alternaria challenge and analyzed at 6 hours. All animal procedures and experimental protocols were approved by the Auburn University Animal Care and Use Committee.
Flow cytometry
Flow cytometric antibodies used in this study were purchased from BioLegend or eBiosciences unless indicated and are listed in table 2. Samples containing 2–5 × 106 cells were incubated with mouse Fc block (0.5 μg/test, anti-CD16/CD32) in staining buffer (containing PBS, 3% FBS, 2mM EDTA and 10 mM HEPES buffer) for 15 minutes at 4 °C. Further, detection of surface antigens were performed with a Live/Dead fixable cell stain followed by labeling with antibody cocktails in staining buffer. For intracellular detection of Glut-1 cells were stained with Live/Dead fixable stain and surface markers and immediately fixed with Fix/Perm solution. After two washes, cells were stained with anti-Glut-1-PE antibody (Novus biologicals) and surface and endogenous expression of Glut-1 were determined. Lung alveolar macrophages (AMФ) and APCs were identified as Siglec F+ CD11c+ MHC IIlo F4/80+ and Siglec F− CD11c+ MHC-IIhi CD11b+ cells, respectively. BAL cellularity were characterized as CD3+ CD45R+ expressing lymphocytes (Lym); CD3− CD45R−CD11chi MHC-II+ F4/80+ expressing AMΦ; CD3− CD45R− MHC-II− CCR3− Gr1+ expressing neutrophils (Neu), and CD3−CD45R− MHC-II− CCR3+ Gr1− expressing eosinophils (Eos). Group 2 innate lymphoid cells (ILC2) in lung were identified as Lin− CD45+ KLRG1+ Sca1+ Thy1.2+ cKit+ using lineage-FITC cocktails (CD3, CD11b, CD11c, CD19, B220, Ter-119, and Gr-1). Data were acquired on a LSR-II (BD Biosciences) equipped with 407, 488, 532 and 633 laser lines. Data compensation and downstream analysis was performed with Flow Jo software version 10 (Treestar), using FMO (fluorescence minus one) as controls.
Table 2:
Antibodies used in this study
| Fluorochrome labelled antibodies | Clone | Source | Catalog number |
|---|---|---|---|
| Siglec F-eFluor660 | 1RNM44N | Thermo Fisher Scientific | 50-702-82 |
| SiglecF-BV421 | E50–2440 | BD Biosciences | 562681 |
| CD11c-APC-eFluor780 | N418 | Thermo Fisher Scientific | 47-0114-82 |
| F4/80-eFluor450 | BM8 | BioLegend | 123132 |
| CD11b-PE-TexasRed | M1/70.15 | Thermo Fisher Scientific | RM2817 |
| I-A/I-E (MHC-ll)-PE-Cy7 | M5/114.15.2 | Thermo Fisher Scientific | 25-5321-82 |
| I-A/I-E (MHC-ll)-eFluor450 | M5/114.15.2 | Thermo Fisher Scientific | 48-5321-82 |
| CD103-PerCP-Cy5.5 | M290 | BD Biosciences | 563637 |
| CD3-Alexa Fluor 488 | 145-2C11 | Thermo Fisher Scientific | 53-0031-82 |
| CD45R(B220)-Alexa Fluor 488 | RA3-6B2 | Thermo Fisher Scientific | 53-045-82 |
| CCR3-PE | 83101 | R&D systems | FAB729P |
| Gr1-(Ly6G/Ly6C)-APC | RB6-8C5 | BioLegend | 108412 |
| Lin-FITC Ms | - | BioLegend | 78022 |
| CD25-PE | PC61.5 | Thermo Fisher Scientific | 12-0251-81 |
| KLRG1-PE-Cy7 | 2F1/KLRG1 | BioLegend | 138415 |
| Sca1-BV421 | D7 | BioLegend | 108128 |
| CD45-APC | 30-F11 | BioLegend | 103112 |
| cKit-Alexa fluor700 | ACK2 | Thermo Fisher Scientific | 56-1172-82 |
| CD90.2 (Thy1.2)-APC-Cy7 | 53–2.1 | Thermo Fisher Scientific | 47-0902-82 |
| CD40-FITC | 3/23 | BioLegend | 124608 |
| CD40-APC | 1C10 | Thermo Fisher Scientific | 17-0401-82 |
| CD80-PE-Cy5 | 1610A1 | Thermo Fisher Scientific | 15-0801-82 |
| CD86-PE | PO3 | Thermo Fisher Scientific | MA5-16921 |
| CD209a (DC-SIGN)-eFluor660 | MMD3 | Thermo Fisher Scientific | 50-2094-80 |
| Dectin1-PE | RH1 | BioLegend | 144303 |
| Dectin2-Alexa Fluor 647 | D2.11E4 | Biorad | MCA2415A647T |
| Glut1-PE | Polyclonal | Novus Biologicals | NB110-39113PE |
| Yellow fluorescent stain | - | Thermo Fisher Scientific | L34967 |
ELISA and lactate measurements
Cytokines from BAL, lung homogenate or cell culture supernatant were assessed by two-site sandwich ELISA. IL-6, TNFα, IL-33 (Thermo Fisher Scientific) and mouse CXCL1/KC, IL5, and IL13 was measured using a murine Duoset ELISA kit (R&D systems). Assays were performed according to the manufacturers’ instructions, in 96-well polystyrene MaxiSorp ELISA plates (NUNC).
Lactate concentration in BAL and cell supernatants were measured by a spectrophotometric method using hydrazine media (2X) containing LDH (40 U ml−1), NAD (4 mM), hydrazine monohydrate (400 mM), Tris-buffer (1M) and EDTA (20 mM). Briefly, two-fold serial dilution of standards were prepared (100–3.12 μM) and hydrazine media (100 μl) were added to each well. Reaction was allowed to run for 2 minutes before measuring NADH fluorescence at 340 nm excitation/460 nm emission. Lactate concentrations in the sample was calculated from standard curve.
H&E staining and PKM2 immunofluorescence
Lung left lobe were fixed in 10% formalin for 24 hours, dehydrated through gradient ethanol, and embedded in paraffin. Lung sagittal sections were cut to 4 μm thickness and stained with hematoxylin and eosin. For PKM2 immunostaining paraffin-embedded lung tissues were rehydrated and blocked with 2 % donkey serum in PBS for 60 minutes at room temperature. Lung slides were reacted with rabbit monoclonal PKM2 (1:100 dilution; Cell Signaling Technologies) antibody at 4 °C overnight followed by incubation with PE conjugated anti-rabbit secondary antibody. Signals were enhanced using SA-HRP (1:500 dilution; Jackson Immunoresearch Laboratories) for 10 minutes and TSA (1:100; PerkinElmer) for 45 seconds at room temperature. DAPI as counterstain and Vecta-Shield anti-fade mounting media was used before analyses for confocal microscopy.
Ex vivo assays for reactive oxygen species and glucose uptake
Freshly isolated lung cells were enriched for CD11c+ APCs with positive selection kit (Invitrogen) and incubated with 5 μM of the redox-sensitive probe CM-H2DCFDA, (5-−(and −6) chloromethyl-2’7’-dichlorohydrofluorescein diacetate, acetyl ester; Molecular probes, Life Technologies) for 30 minutes at 37 °C. The stable fluorescent adduct that was produced by oxidation of CM-H2DCFDA in presence of intracellular reactive oxygen species in CD11c+ cells were quantified by an increase in fluorescence in the fluorescein channel by flow cytometry. Lung enriched CD11c+ cells were incubated in glucose-free RPMI medium containing 5 μM of fluorescent d-glucose analogue 2-[N-(7-nitrobenz-2-oxa-1, 3-diazol-4-yl) amino]-2-deoxy d- glucose (Invitrogen) for 60 minutes at 37 °C. After incubation cells were washed with staining buffer and fluorescent intensities were analyzed by flow cytometry.
qRT-PCR
RNA was isolated using trizol reagent (Life Technologies, Grand Island, NY, USA) or PureLink RNA mini kit (Invitrogen). cDNA was prepared using a High Capacity RNA-to-cDNA kit (Applied Biosystems) and 30 ng of cDNA were used with Fast SYBR Green PCR master mix for qRT-PCR (Thermo Fisher Scientific). Gene expression levels were normalized with 36B4 and data were analyzed using Data Assist software (Applied Biosystems). The primers used in this study were listed in Table 1.
Table 1:
Primer sequences used in this study
| Gene | Forward primer sequence | Reverse primer sequence |
|---|---|---|
| Glut-1 | 5’-CATCCTTATTGCCCAGGTGTTT-3’ | 5’- GAAGACGACACTGAGCAGCAGA-3’ |
| HK2 | 5’-TGATCGCCTGCTTATTCACGG-3 | 5’-AACCGCCTAGAAATCTCCAGA-3’ |
| PKM1 | 5’-GCTGTTTGAAGAGCTTGTGC-3’ | 5’-TTATAAGAGGCCTCCACGCT-3’ |
| PKM2 | 5’-TTAGGCCAGCAACGCTTGTAGTGC-3’ | 5’-AGATGCTGCCGCCCTTCTGTGATA-3’ |
| Hif-1α | 5’-GGTTCCAGCAGACCCAGTTA-3’ | 5’-AGGCTCCTTGGATGAGCTTT-3’ |
| Ldha | 5’-CACTGACTCCTGAGGAAGAGGCCC-3’ | 5-’AGCTCAGACGAGAAGGGTGTGGTC-3’ |
| 36B4 | 5’-GGACCCGAGAAGACCTCCTT-3’ | 5’-GCACATCACTCAGAATTTCAATGG-3’ |
In vitro assays
Bone marrow derived dendritic cells (BMDCs) were generated using Iscoves Modified Dulbecco’s medium (IMDM) containing 10% heat-inactivated fetal bovine serum (FBS), penicillin streptomycin solution, recombinant mouse granulocytes macrophage colony stimulating factor (GM-CSF, 20 ng/ml, Peprotech), and recombinant mouse IL-4 (10 ng ml−1, Thermo Fischer Scientific). Non-adherent BMDCs at day 6 culture were collected and plated (0.5 × 106 cells /well) in 24 well plates. Cells were treated with PKM2 allosteric agonist ML265 (100 μM), 2-DG (10 mM), anti-ST2/IL-33R (2 μg ml−1) or control (DMSO) for two hours before Alternaria (2 μg ml−1) treatment. Culture supernatants were collected after 24 hours for ELISA. For knockdown of PKM2, primary bone marrow derived cell line (NR-9456) were obtained from BEI Resources, NIAID, NIH and maintained in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% irradiated fetal bovine serum. Briefly, cells were transfected with either 1 nM anti-PKM2 siRNA or 1 nM scrambled siRNA (Dharmacon) using DharmaFECT. Silencing efficiency were determined after 72 hours by qRT-PCR and Western blot using anti-PKM2 and anti-histone 3 (H3) antibody (Cell Signaling Technology).
Seahorse XFp assays
XFp Extracellular Flux Analyzer (Seahorse Bioscience) was used to analyze real-time changes of ECAR and OCR. BMDCs were plated overnight (120,000 cells /well) in 200 μl of IMDM medium containing 5% FBS and 10 ng ml−1 of GM-CSF at 37 °C. Culture medium was replaced with fresh medium containing 5% FCS and treated with or without ML265 (100 μM) or DMSO for 2 hours and stimulated with Alternaria (2 μg ml−1). Medium was removed after 3 hours and replaced with warmed unbuffered Seahorse stress test glycolysis assay media at pH 7.4 (DMEM without glucose, L-glutamine, phenol red, sodium pyruvate, and sodium bicarbonate) supplemented with 5 mM HEPES and 1% FBS. The cell plate was incubated (non-CO2) for 1 hour at 37 °C. Where indicated, cells were successively treated with glucose (10 mM), oligomycin (1 μM), and 2-DG (50 mM) into the wells by the XF analyzer. In separate experiments, PKM2-siRNA and scrambled-siRNA transfected cells were seeded (80,000 cells /ml) in XFp plates containing DMEM medium with 5% FBS and incubated overnight at 37 °C. ECAR was measured 3 hours after Alternaria (10 μg ml−1) treatment. Data were normalized to total protein and analyzed using Wave software 2.6.1 (Seahorse Bioscience).
Statistics
All results are analyzed using Graph Pad Prism version 7.0 and expressed as means with error bars reflecting sd or sem as indicated. n represents the number of animals per experiment. Differences between two groups were assessed using unpaired two-tailed Student’s t tests. A one-way or two-way ANOVA’s with Tukey’s or Sidak’s multiple comparison post hoc tests were applied for analysis of data from more than two groups.
Results:
Alternaria-induced acute airway inflammation is associated with increase in lung glycolysis
Alternaria is associated with acute exacerbation and sudden asthma severity (Bush and Prochnau, 2004; Pulimood et al., 2007). To elucidate the role of Alternaria in acute airway inflammation, innate mediator secretion and ensuing elevated aerobic glycolysis, we treated naïve mice intranasally with allergen extracts of Alternaria or house dust mite (HDM) and determined the response to challenge after 1 hour and 6 hours (Figure 1). Strikingly, single exposure to Alternaria extract resulted in a robust time-dependent increase in inflammatory cell infiltrate in the airways. This infiltrate was predominantly neutrophilic at 6 hours for both allergens, with an elevation in alveolar macrophages (AMΦ) and eosinophils also apparent (Figure 1A). This was correlated with an increase in BAL level of surrogate neutrophil chemoattractant CXCL1 (C-X-C motif ligand 1) (Figure 1B). Alternaria drives a robust innate mediator release in the airways and is characterized by elevated protein level of IL33, IL6 and TNF-α in the BAL both at 1 hour and 6 hours as compared to HDM (Figure 1C). Interestingly, Th2 cytokines IL5 and IL13 were significantly increased at 6 hours in inflamed lungs exposed to a single dose of Alternaria (Figure 1D). Lung histopathology shows peribronchial inflammatory cell infiltrates predominantly of neutrophils (Figure 1E, top). This correlates with a low pH metabolic shift to aerobic glycolysis and subsequent increase in PKM2 expression (Figure 1E, bottom) and lactate overproduction (Figure 1F) in Alternaria-exposed lungs. Increased expression of glycolytic enzymes including hexokinase, PKM2, and Hif-1α were observed in lung tissues at 6 hours after Alternaria challenge (Figure 1G). Collectively, these findings demonstrate that Alternaria rapidly instigate an aerobic glycolysis and ensue a robust mediator secretion thereby drive acute airway inflammation.
Figure 1. Single administration of Alternaria induces airway inflammation characterized by rapid proinflammatory mediator release and elevated glycolysis.
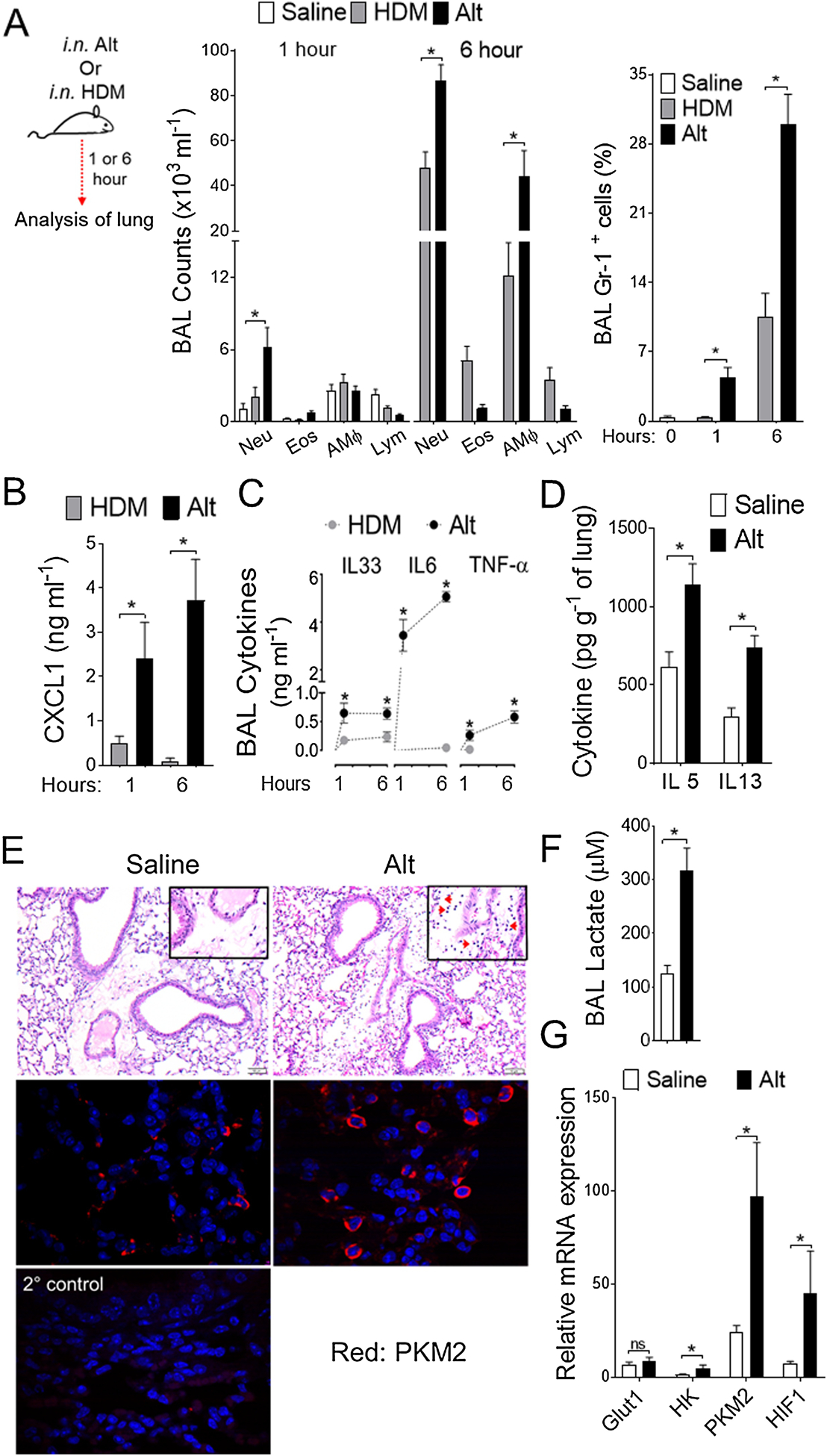
(A) Schematic depicting the time line for allergen challenge (left). Naïve mice were immunized with either HDM or Alternaria, as indicated. i.n., intranasal. Differential cell counts (middle) and percentages of Gr-1+ neutrophils (right) in BAL as measured by flow cytometry. (B) CXCL1 (C) IL33, IL6, and TNF-α levels in BAL and (D) IL5 and IL13 levels in lungs were measured by means of ELISA. (E) Hematoxylin and eosin (H&E)-stained lung sections (top) and PKM2 immunostaining (bottom) in lung tissues at 6 hour (Scale bars 100 μm for 60x images). Red arrows in the inset indicate neutrophils. Alternaria-inflamed lung in which the primary antibody was omitted as a negative (2°) control. (F) BAL lactate levels and (G) qRT-PCR analyses for glucose transporter (Glut-1, Glucose transporter 1), glycolytic enzymes (HK, Hexokinase; PKM2, Pyruvate Kinase M2) and hypoxia-inducing factor (Hif-1α, Hypoxia-inducible factor-1 alpha) in Alternaria-inflamed lungs at 6 hour. Data represents mean ± s.e.m. Two independent experiments; n = 4–8 per group. One-way ANOVA with Sidak’s multiple comparison test *, P < 0.05. HDM, House Dust Mite; Alt, Alternaria; Neu, Neutrophils; Eos, Eosinophils; AMΦ, Alveolar Macrophage; Lym, Lymphocytes.
IL33- drives an early lung cellular infiltrate and induces glycolysis in lung APCs
It is conceivable that Alternaria-induced IL33 drives airway inflammation (Cohen et al., 2015; Snelgrove et al., 2014). To further elucidate the regulatory role of IL33 in mediating glycolysis, we pre-administered IL33 neutralizing antibody before Alternaria treatment and assessed airway inflammation. Anti-IL33 treatment significantly reduced neutrophil infiltration upon Alternaria sensitization manifested by reduced percentages of Gr-1+ neutrophils (Figure 2A). This correlates with CXCL1 and lactate level in BAL (Figure 2B–C). In contrast, the number of ILC2s in lung remains unchanged with anti-IL33 treatment (Figure 2D), suggesting IL33-mediated redundancy of this allergen (McSorley et al., 2014). Next, we sought to determine Alternaria-induced IL33 effect in lung APCs ex vivo. We found a significant decreased level of reactive oxygen species (ROS) in antigen presenting cells isolated from anti-IL33 treated Alternaria-inflamed lung (Figure 2E). In parallel, a similar effect were observed in expression of glycolysis transporter 1 (Glut-1), glycolysis enzyme (pyruvate kinase M2) and hypoxia-inducing factor (Hif-1α) in isolated lung APCs from anti-IL33 treated mice (Figure 2F). Therefore, these results collectively indicate that Alternaria-specific IL33 drives an acute airway inflammation in particular promotes oxidative stress and interfere with lung APCs metabolism.
Figure 2. Alternaria-induced upregulation of glycolysis in lung APCs are IL-33 dependent.
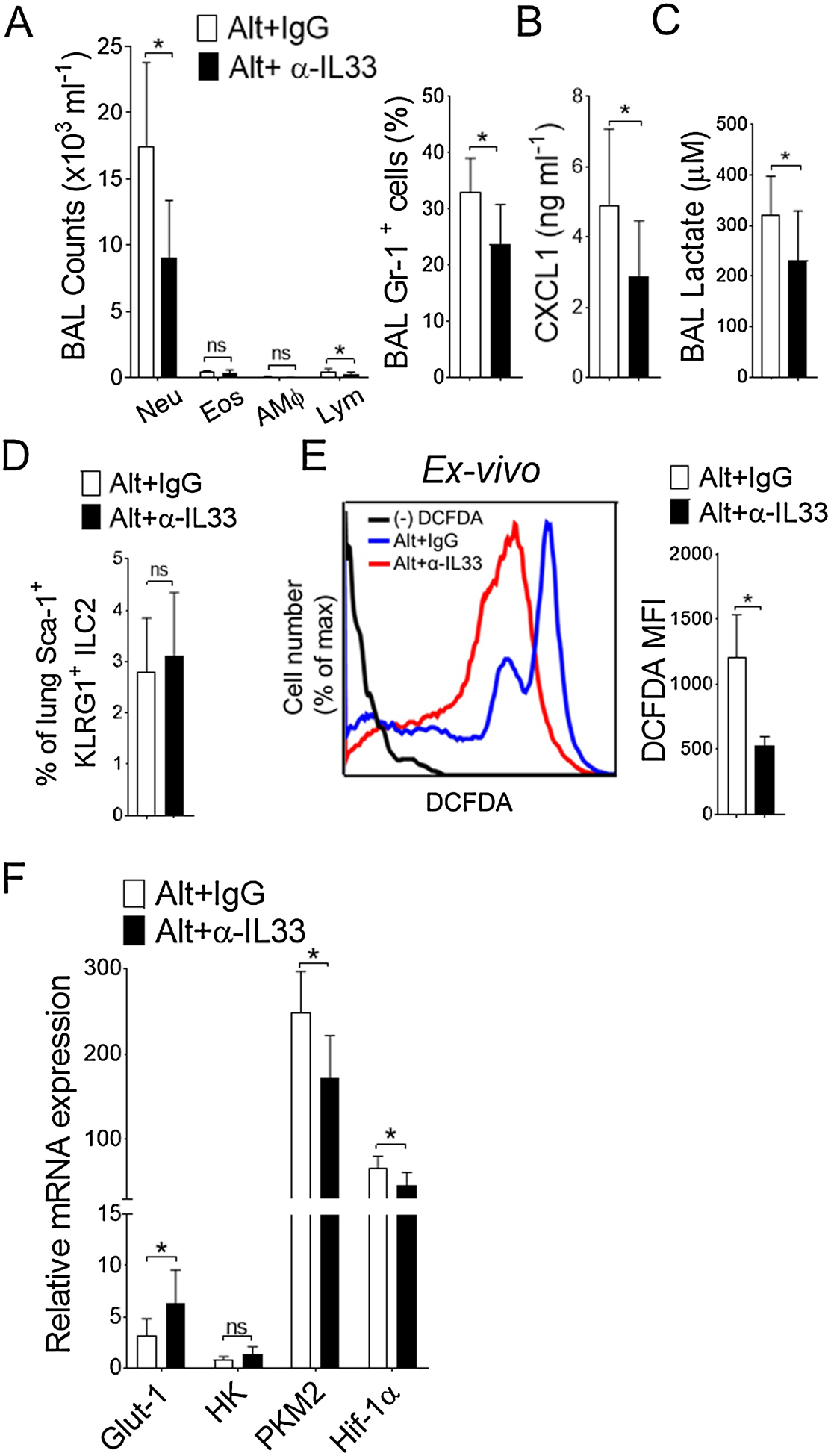
(A) Differential cell counts and percentages of Gr-1+ neutrophils (right) in BAL as measured by flow cytometry. (B) CXCL1 and (C) lactate levels in BAL. (D) Bar graph of mean percentages of lung ILC2 and (E) representative FACS plots (left) and mean fluorescence intensity (MFI) ± s.d. (right) showing endogenous reactive oxygen species (ROS) level in isolated lung CD11c+ cells from isotype IgG or anti-IL33 (50 μg in 40 μl PBS, i.n., administered twice at 12 hour interval) treated mice. Results are from Alternaria-inflamed lungs at 6 hour. (F) qRT-PCR analyses of glycolytic genes in isolated lung CD11c+ cells and expressed as mean ± s.d. n = 5–10 per group. Student’s t test *, P< 0.05. IgG, Isotype IgG; α-IL33, mouse anti- IL33 antibody. Alt, Alternaria; Neu, Neutrophils; Eos, Eosinophils; AMΦ, Alveolar Macrophage; Lym, Lymphocytes.
Activation of lung APCs are critical determinant of Alternaria-mediated acute airway inflammation
Lung dendritic cells play a pivotal role in allergen sensitization to inhaled allergens thereby regulates innate immune response (Lambrecht and Hammad, 2014). To examine more clearly the need for CD11c+ lung APCs in Alternaria-induced acute inflammation, we employed conditional depletion of lung CD11c+ cells by diphtheria toxin (DT) treatment in transgenic DTR mice. FACS analyses of BAL, lung tissues, and draining mediastinal lymph nodes showed a dramatic decrease in CD11c+ cells in transgenic DTR mice receiving DT treatment, but not in peripheral lymph nodes, suggesting lung-specific depletion of CD11c+ cells in these mice (Figure 3A). As expected, the treatment with Alternaria led to a striking decrease in cellular infiltration and Gr-1+ neutrophils in BAL (Figure 3B–C). We observed significant decline in BAL lactate production (Figure 3D), Glut-1 expression and activation of CD11c+ cells (Figure 3E–F) following Alternaria challenge, indicating CD11c+ cells-mediated glycolysis and activation are prerequisite to Alternaria-induced airway inflammation. As such, these results further show an imperative function of antigen presenting cells in Alternaria –mediated acute airway inflammation (Kobayashi et al., 2009).
Figure 3. Lung CD11c+ APCs are critical regulator of Alternaria-induced airway inflammation.
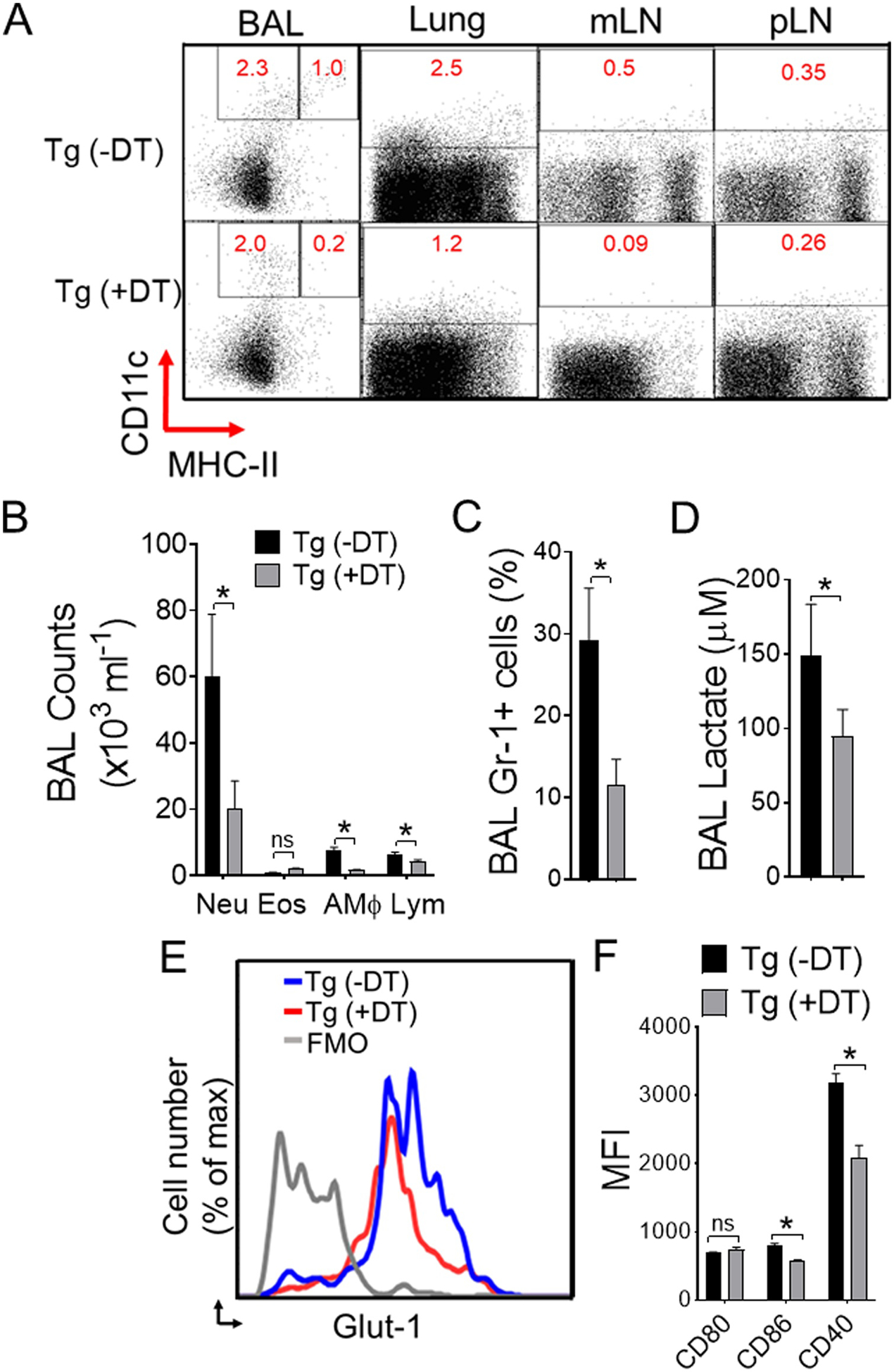
(A) CD11c+ in BAL cells, lungs, lung draining mediastinal lymph node (mLN), and peripheral axillary lymph node (pLN) were analyzed by flow cytometry. Naïve CD11c-DTR transgenic (Tg) mice were injected or left untreated (PBS control) with DT (diphtheria toxin; 50 ng, i.n.) 24-hours before Alternaria challenge and analyzed at 6 hours. (B) Differential cell counts and (C) percentages of Gr-1+ neutrophils in BAL as measured by flow cytometry. (D) BAL lactate and (E) representative flow image of Glut-1 expression. Bar graph of (F) mean fluorescence intensity (MFI) of activation markers gated on lung CD11c+ CD11b+APCs from DTR mice ± DT treatment and receiving Alternaria. Results are expressed as mean ± s.e.m. Two independent experiments; n = 4 per group. Student’s t test *, P< 0.05.
Elevated oxidative stress and PKM2 up-regulation are associated with APCs activation by Alternaria
DCs undergoes rapid metabolic switch to glycolysis with TLR stimulation (Everts et al., 2014). To explore the functional consequences of the specific changes in lung APCs by Alternaria, we investigated activation and glycolysis in inflamed lung CD11c+ APCs ex vivo. Analysis of CD11c+ cells showed a dramatic increase in number of Glut-1 expressing CD11c+ cells in Alternaria lung (Figure 4A). As expected, C-type lectin-1 (Dectin-1), and co stimulatory CD40 and CD86 expression were strikingly increased by Alternaria treatment in CD11c+ lung APCs (Figure 4B). In parallel, we observed increased level of ROS (Figure 4C) and elevated glucose uptake (Figure 4D) by CD11c+ APCs isolated from naïve lungs and restimulated with Alternaria extracts. Expressions of the glycolytic enzyme PKM2 and Hif-1α, but not PKM1 were increased in lung CD11c+ cells (Figure 4E). Therefore, these results indicate that Alternaria promotes oxidative stress and switch to an aerobic glycolysis thereby renders lung CD11c+ cells activation.
Figure 4. Oxidative stress and induction of glycolytic enzyme PKM2 in lung APCs upon Alternaria stimulation.
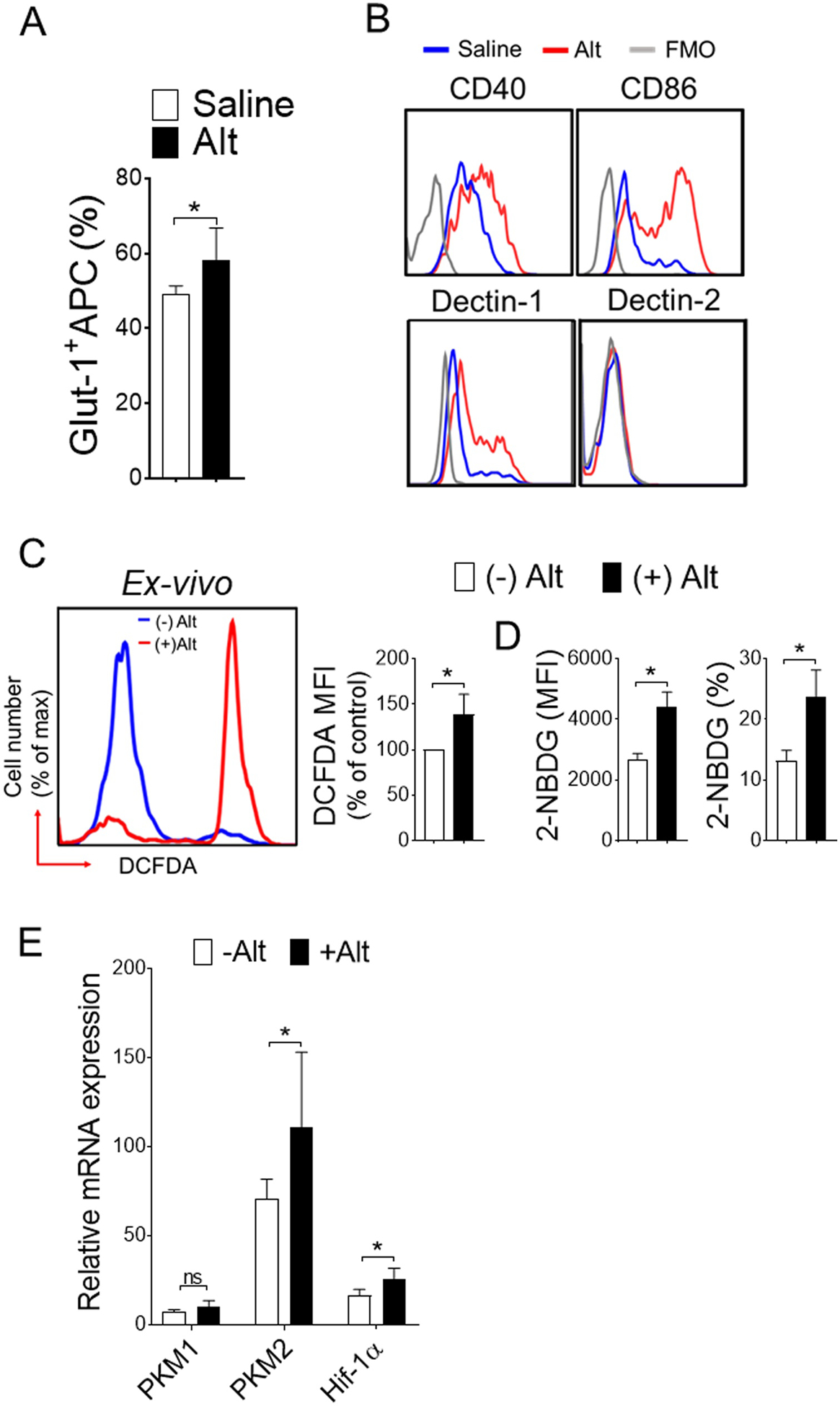
Naïve mice were sensitized, as mentioned in Figure 1. (A) Lung CD11c+ cells were isolated and ex-vivo treated with Alternaria for 3 hours and analyzed for Glut1 expression. Bar graph showing mean percentages of Glut-1+ CD11c+. (B) Representative FACS plot of activation markers gated on live CD11c+ cells. (C) Representative FACS plot (left) and mean fluorescence intensity (MFI) ± s.d. (right) showing endogenous reactive oxygen species (ROS), (D) glucose uptake in lung CD11c+ cells and (E) qRT-PCR analyses. Lung CD11c+ cells were isolated and ex-vivo treated with Alternaria (10 μg ml−1) for 12 hours and incubated with 2-NBDG (a fluorescent D-glucose analog). Results are expressed as mean ± s.d. Two independent experiments; n = 4 per group. Student’s t test *, P< 0.05.
Alternaria-induced increase in glycolysis and inflammation is negatively regulated by PKM2 activation
PKM2 is a key metabolic regulator and stabilizes Hif-1α in response to inflammation by nuclear translocation in its less active mono/dimeric form (Palsson-McDermott et al., 2015). In contrast, more active tetrameric form of PKM2 inhibits binding with Hif-1α, thereby regulates immune cell activation and inflammation (Yang et al., 2014). To determine whether PKM2 up-regulation by Alternaria in CD11c+ APCs directly affects metabolic reprogramming and activation, we employed a well-characterized small molecule activator ML265 and assessed glycolysis in real-time. Normalization of ECAR was found when BMDCs were treated in vitro with ML265 (Figure 5A), suggesting that a direct effect of PKM2 activation attenuates Alternaria-induced glycolytic reprogramming. This correlates with less production of lactate in presence of ML265 (Figure 5B). Similar effect were found in CD11c+ cells on Glut-1 (Figure 5C–D) and CD80, CD86 and CD40 surface expression with PKM2 activation by ML265 (Figure 5E), suggesting PKM2-mediated metabolic regulation is responsive to energy-intensive activation of CD11c+ cells. Although, Alternaria-induced activation of CD11c+ APCs impacted by 2DG, we found minimal effect on ST2 receptor blockade, indicating redundant pathways of IL33-mediated metabolic reprograming (Xu et al., 2019). Notably, the ability to secrete proinflammatory cytokine IL6 and TNF-α were abrogated following PKM2 activation (Figure 5F). Furthermore, ML265 treatment significantly reduced nuclear translocation of PKM2, thereby Hif-1α and Ldha expression in BMDCs (Figure 5G–H). Specific knockdown of PKM2 expression by siRNA (Figure 6A–B) significantly impairs aerobic glycolysis (Figure 6C–D), indicating PKM2 as pivotal players in Alternaria-induced metabolic reprograming. Altogether these results indicate that a tight metabolic control of PKM2 activation is critical in CD11c+ cells particularly during Alternaria-induced inflammation, and show that PKM2 is one of the key determinant of metabolic reprograming in CD11c+ APCs.
Figure 5. PKM2 activation attenuates glycolysis and inflammatory response.
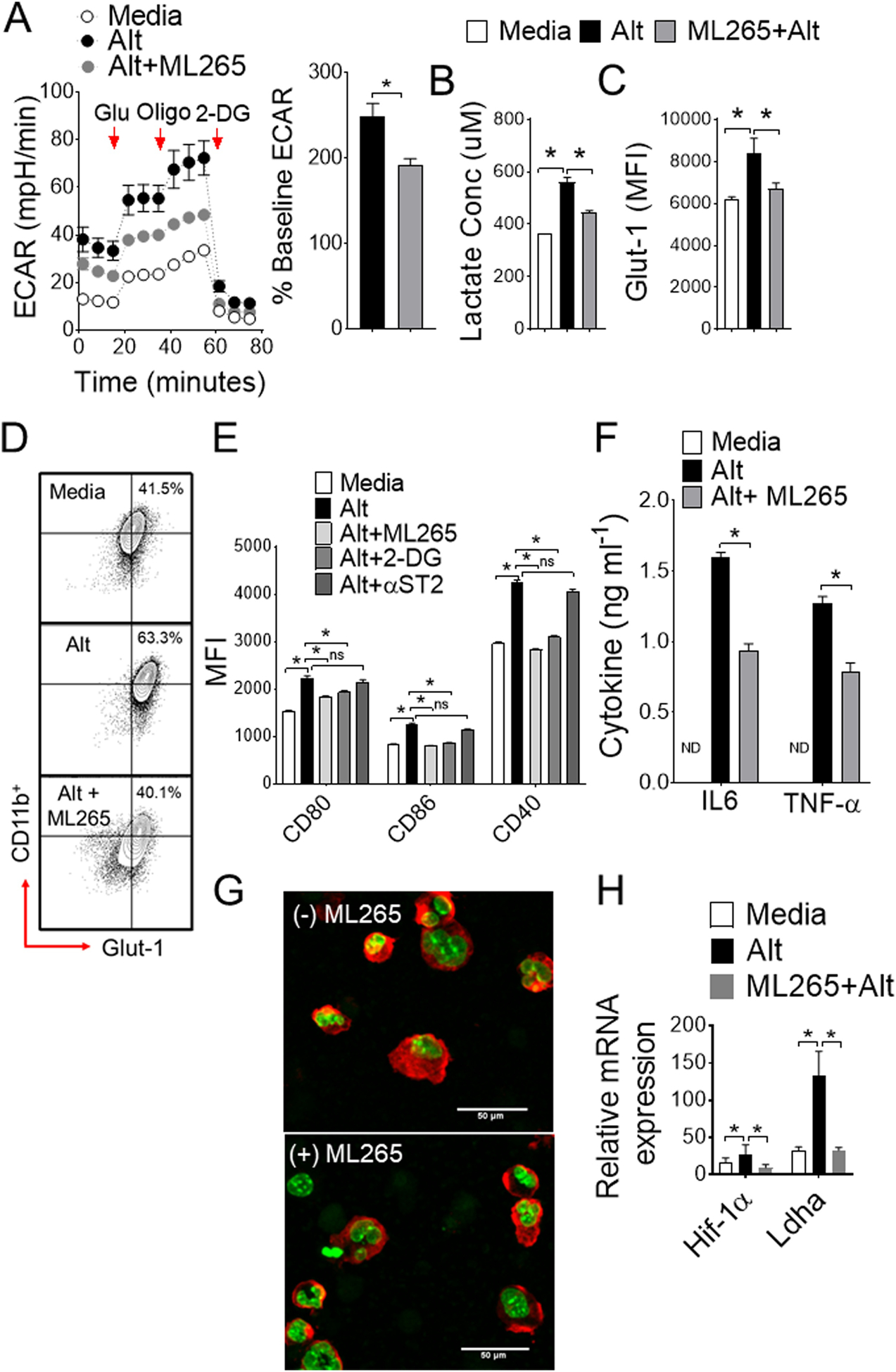
(A) Real-time changes in ECAR (left) and mean percentages of baseline ECAR as assessed by seahorse assay. (B) Lactate production (C) mean fluorescence intensity (MFI) and (D) representative FACS plot of Glut-1 expressions with Alternaria ± ML265. (E) Bar graph shows MFI of CD80, CD86, and CD40 surface expression on CD11b+ BMDCs. Cells were left untreated or treated with Alternaria (10 μg ml−1) for 12 hours with or without PKM2 agonist ML265, glycolysis inhibitor 2-DG or anti-ST2 antibody. (F) IL6 and TNF-α protein levels as measured by ELISA. (G) Nuclear translocation of PKM2 in cytospin cells from Alternaria ± ML265 (Merge confocal image: red- PKM2; green-DAPI) and (H) mRNA expression of Hif-1α and Ldha. One-way ANOVA with Sidak’s multiple comparison test or Student’s t test *, P< 0.05. Results are expressed as mean ± s.e.m at least from three independent experiments.
Figure 6. si-RNA-mediated knock down of PKM2 impairs glycolytic rate.
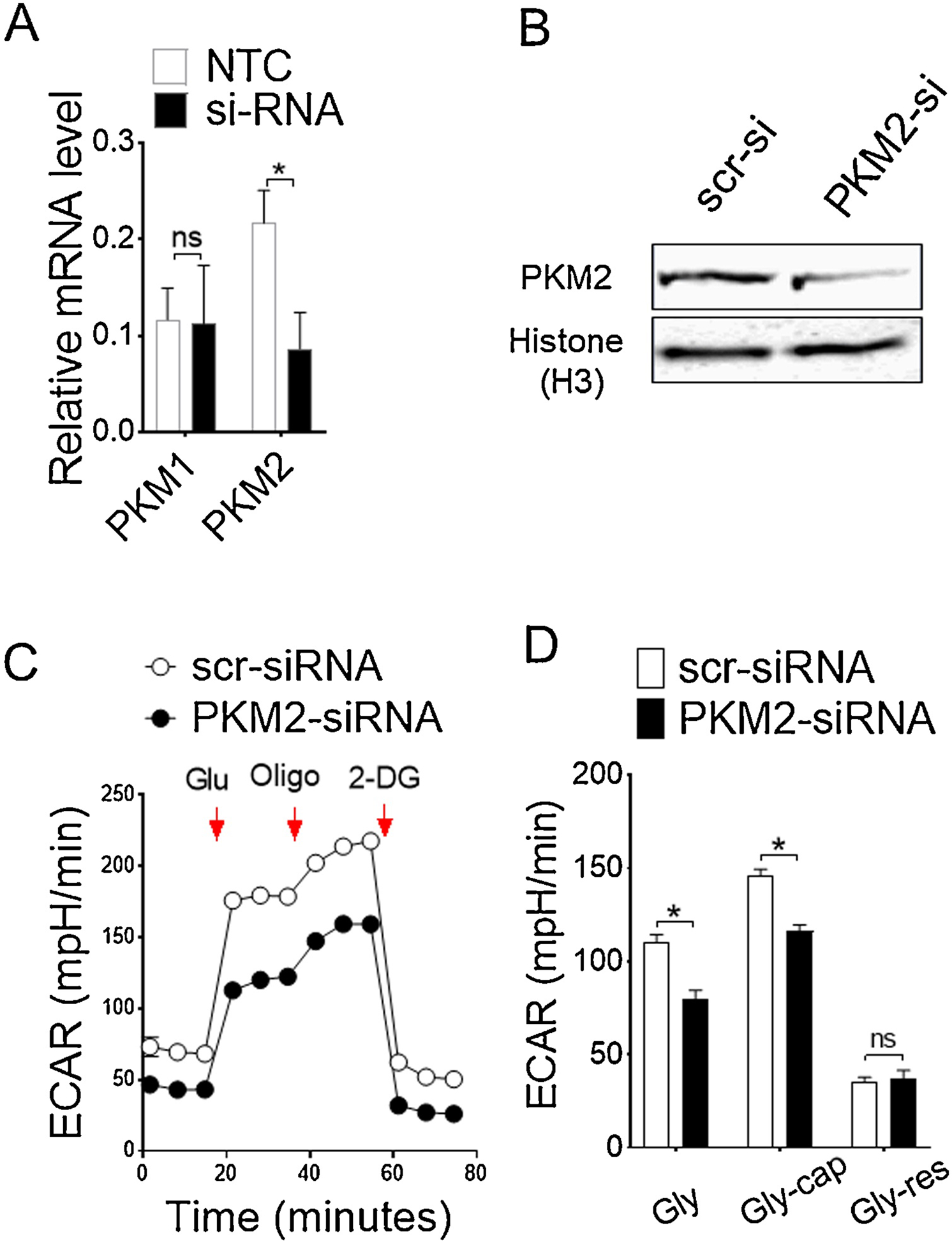
(A) Relative mRNA and (B) protein level for confirmation of knockdown of PKM2 by si-RNA transfection. (C) ECAR and (D) bar graph of mean ECAR ± s.d as assessed by seahorse assay. Glycolysis (Gly) were calculated by subtracting maximum rate measurement before Oligomycin injection from the last rate before glucose injection; Glycolytic capacity (Gly-cap) were determined by subtracting maximum rate measurement after Oligomycin injection from last rate measurement before glucose injection, and Glycolytic reserve (Gly-res) were obtained from the difference between glycolytic capacity and glycolysis. Results are expressed as mean ± s.d., representative of three experiments. Student’s t test *, P< 0.05.
Discussion:
Alterations in glycolysis are implicated in the pathogenesis of several inflammatory lung diseases (Mishra, 2017; Qian et al., 2018; Shi et al., 2015; Sinclair et al., 2017). Sensitivity to fungal allergen Alternaria Alternata develops rapid onset and life-threatening exacerbations of asthma attack (Ohollaren et al., 1991). We show that an Alternaria-specific oxidative stress rapidly increases aerobic glycolysis and effector function of antigen presenting cells, which subsequently directs an early airway inflammatory program characterized by robust proinflammatory mediator release. Mechanistically, we found that Alternaria-induced metabolic reprograming of lung antigen presenting cells are linked to up-regulation of the glycolytic enzyme PKM2 and hypoxia-inducible factor-1α (Hif-1α), a transcription factor that acts as a sensor to low oxygen availability. We show that this metabolic changes in CD11c+ APCs by Alternaria respond unequivocally to activate and release proinflammatory cytokines IL6 and TNF-α in the airways. Finally, we demonstrate that Alternaria-associated changes primes the lung CD11c+ cells with rapid PKM2 activation and increase in glycolysis that could be reversed by allosteric PKM2 activator ML265. In sum, we establish a mechanistic link between CD11c+ cells metabolism and activation in the setting of Alternaria-induced acute exacerbation of allergic asthma.
Alternaria-associated airway changes in allergic asthma is rapidly driven by influx of inflammatory cells into the airways and robust mediator release (Valladao et al., 2016). Epithelium-derived IL33 elicits neutrophil-predominant airway inflammation with Alternaria sensitization that could rationalize the relevance of developing potentially fatal asthma attack in clinics (Causton et al., 2018; Snelgrove et al., 2014). The capacity of early release of IL33 into the airways to promote neutrophilic inflammation is dependent on an Alternaria-specific serine protease activity (Snelgrove et al., 2014), which is lacking from other common aeroallergens such as HDM. As others, we found time-dependent early increase in neutrophil influx and rapid onset of proinflammatory IL33, IL6 and TNF-α cytokine release in the BAL within 6 hours of Alternaria challenge compared to HDM (Hristova et al., 2016; Snelgrove et al., 2014). In parallel, our results demonstrate that Alternaria-induced shift in glycolytic reprograming in APCs drives airway inflammation and is IL33-dependent. We found decline in neutrophil influx and lactate levels in BAL following IL33 neutralization. This is consistent with previous findings of increased lactate levels in patients with neutrophilic asthma compared to less severe eosinophilic asthma (Qian et al., 2018). We subsequently demonstrated that Alternaria-induced oxidative stress in lung CD11c+ APCs promotes glycolytic reprograming and is IL33-mediated. APCs are pivotal players and exacerbate allergic airway inflammation in presence of IL33 (Besnard et al., 2011; Rank et al., 2009). Because ILC2 numbers in lung does not correlate with IL33 neutralization at the time point of our study, we turned to lung CD11c+ cells to further explore how Alternaria controls innate activation of CD11c+ APCs. Furthermore, we show that specific-ablation of lung CD11c+ cells abrogates Alternaria-induced neutrophil influx and reduce Glut-1 expression and activation of CD11c+ APCs.
Early glycolytic changes are fundamental feature of activated APCs that renders immunogenic phenotype. As such, activation of lung APCs in response to TLR triggers are associated with elevated glycolysis that concomitantly shuts mitochondrial oxidative phosphorylation by promoting iNOS expression and NO production. However, the early TLR-driven rapid changes in glycolysis is NO-independent and is led by AKT-mediated activation of key glycolytic enzyme HKII (Everts et al., 2014; Krawczyk et al., 2010; Miyamoto et al., 2008). In contrast, a long-term commitment of glycolysis is NO-dependent, and is supported by induction of Hif-1α. There are substantial precedents to suggest many complementary and synergistic pathways of metabolic reprograming and activation of lung APCs (Lawless et al., 2017; Sinclair et al., 2017), however, its impact on airway inflammation remains poorly understood. We show sustained induction of Glut-1 in lung CD11c+ cells and increased surface expression of activation markers in vivo ensuing Alternaria challenge. We show that metabolic reprogramming in lung CD11c+ cells following Alternaria challenge is Hif-1α and PKM2-dependent. Importantly, elevated glycolytic rate forces PKM2 to enter into the nucleus and co-activate Hif-1α in its less active dimeric form, thereby induces expression of proglycolytic genes including Ldha, Glut-1 as well as proinflammatory IL33 (Li et al., 2018; Palsson-McDermott et al., 2015). As such, stabilizing tetramer formation of PKM2 in the cytosol by small molecule ML265 blocks nuclear translocation and Hif-1α co-activation (Jiang et al., 2010; Shirai et al., 2016). In Alternaria-inflamed lung, we show that PKM2 plays a regulatory role in glycolytic reprograming and activation of APCs. Notably, modulation of PKM2 activity through potent allosteric activator ML265 is effective in restoring Alternaria-induced glycolysis in CD11c+ cells and Glut-1, CD80, CD86, and CD40 expression. However, it remains uncertain whether Alternaria-derived serine protease activity directly linked to allosteric changes of PKM2 and actually contributes to effector function of APCs (Snelgrove et al., 2014). Collectively, these results demonstrate that glycolytic enzyme PKM2 is tightly coupled to CD11c+ APCs activation in Alternaria-associated acute airway inflammation.
In summary, these data demonstrate a mechanistic link between PKM2 activities in lung CD11c+ APCs and Alternaria-induced airway inflammation in the setting of acute exacerbation. We show that amplification of Alternaria-induced proinflammatory responses are directly linked to metabolic adaptations of lung CD11c+ APCs and is controlled by PKM2. Furthermore, metabolic changes in CD11c+ APCs are critical in Alternaria-triggered innate cytokine response and neutrophil recruitment in the lung. Thus, given the central role of CD11c+ APCs in allergic asthma, targeting metabolic regulators might ultimately provide a novel approach to improve Alternaria sensitivity and potentially fatal asthma attack.
Supplementary Material
Acknowledgements:
We thank Dr. Jeff Huang and Francesca Mowry for immunostaining and confocal microscopy and Auburn University Flow cytometry core facilities for the support. We thank BEI Resources, NIAID, NIH for providing the macrophage cell line derived from wild type mice, NR 9456. This study was supported by the Department of Pathobiology, College of Veterinary Medicine, Auburn University and National Heart, Lung, and Blood Institute. The funding support was under Award Number (R00HL131694) to Dr. Mishra.
Financial Support: National Heart, Lung, and Blood Institute of the National Institutes of Health under Award Number (R00HL131694) to Dr. Mishra
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
All the authors have read and approved of the manuscript and do not have any conflicts of interest to declare.
References
- Bankova LG, Lai J, Yoshimoto E, Boyce JA, Austen KF, Kanaoka Y, and Barrett NA (2016). Leukotriene E4 elicits respiratory epithelial cell mucin release through the G-protein-coupled receptor, GPR99. Proc Natl Acad Sci U S A 113, 6242–6247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besnard AG, Togbe D, Guillou N, Erard F, Quesniaux V, and Ryffel B (2011). IL-33-activated dendritic cells are critical for allergic airway inflammation. Eur J Immunol 41, 1675–1686. [DOI] [PubMed] [Google Scholar]
- Boitano S, Flynn AN, Sherwood CL, Schulz SM, Hoffman J, Gruzinova I, and Daines MO (2011). Alternaria alternata serine proteases induce lung inflammation and airway epithelial cell activation via PAR(2). Am J Physiol-Lung C 300, L605–L614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush RK, and Prochnau JJ (2004). Alternaria-induced asthma. J Allergy Clin Immun 113, 227–234. [DOI] [PubMed] [Google Scholar]
- Causton B, Pardo-Saganta A, Gillis J, Discipio K, Kooistra T, Rajagopal J, Xavier RJ, Cho JL, and Medoff BD (2018). CARMA3 Mediates Allergic Lung Inflammation in Response to Alternaria alternata. Am J Respir Cell Mol Biol 59, 684–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen ES, Scott IC, Majithiya JB, Rapley L, Kemp BP, England E, Rees DG, Overed-Sayer CL, Woods J, Bond NJ, et al. (2015). Oxidation of the alarmin IL-33 regulates ST2-dependent inflammation. Nat Commun 6, 8327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denning DW, O’Driscoll BR, Hogaboam CM, Bowyer P, and Niven RM (2006). The link between fungi and severe asthma: a summary of the evidence. Eur Respir J 27, 615–626. [DOI] [PubMed] [Google Scholar]
- Doherty TA, Khorram N, Chang JE, Kim HK, Rosenthal P, Croft M, and Broide DH (2012). STAT6 regulates natural helper cell proliferation during lung inflammation initiated by Alternaria. Am J Physiol Lung Cell Mol Physiol 303, L577–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggleston PA, Rosenstreich D, Lynn M, Gergen P, Baker D, Kattan M, Mortimer KM, Mitchell H, Ownby D, Slavin R, et al. (1998). Relationship of indoor allergen exposure to skin test sensitivity in inner-city children with asthma. J Allergy Clin Immun 102, 563–570. [DOI] [PubMed] [Google Scholar]
- Everts B, Amiel E, Huang SCC, Smith AM, Chang CH, Lam WY, Redmann V, Freitas TC, Blagih J, van der Windt GJW, et al. (2014). TLR-driven early glycolytic reprogramming via the kinases TBK1-IKK epsilon supports the anabolic demands of dendritic cell activation. Nat Immunol 15, 323–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson FW, Henry MM, Ivins SS, Morris R, Neebe EC, Leu SY, and Stewart PW (1995). Correlates of Recurrent Wheezing in School-Age-Children. Am J Resp Crit Care 151, 1786–1793. [DOI] [PubMed] [Google Scholar]
- Hristova M, Habibovic A, Veith C, Janssen-Heininger YM, Dixon AE, Geiszt M, and van der Vliet A (2016). Airway epithelial dual oxidase 1 mediates allergen-induced IL-33 secretion and activation of type 2 immune responses. J Allergy Clin Immunol 137, 1545–1556 e1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Walsh MJ, Brimacombe KR, Anastasiou D, Yu Y, Israelsen WJ, Hong BS, Tempel W, Dimov S, Veith H, et al. (2010). ML265: A potent PKM2 activator induces tetramerization and reduces tumor formation and size in a mouse xenograft model. In Probe Reports from the NIH Molecular Libraries Program (Bethesda (MD)). [PubMed] [Google Scholar]
- Kobayashi T, Iijima K, Radhakrishnan S, Mehta V, Vassallo R, Lawrence CB, Cyong JC, Pease LR, Oguchi K, and Kita H (2009). Asthma-related environmental fungus, Alternaria, activates dendritic cells and produces potent Th2 adjuvant activity. J Immunol 182, 2502–2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzaki H, Iijima K, Kobayashi T, O’Grady SM, and Kita H (2011). The Danger Signal, Extracellular ATP, Is a Sensor for an Airborne Allergen and Triggers IL-33 Release and Innate Th2-Type Responses. Journal of Immunology 186, 4375–4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, Cross JR, Jung E, Thompson CB, Jones RG, et al. (2010). Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 115, 4742–4749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrecht BN, and Hammad H (2014). Allergens and the airway epithelium response: gateway to allergic sensitization. J Allergy Clin Immunol 134, 499–507. [DOI] [PubMed] [Google Scholar]
- Lawless SJ, Kedia-Mehta N, Walls JF, McGarrigle R, Convery O, Sinclair LV, Navarro MN, Murray J, and Finlay DK (2017). Glucose represses dendritic cell-induced T cell responses. Nat Commun 8, 15620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Li D, Zhang X, Wan Q, Zhang W, Zheng M, Zou L, Elly C, Lee JH, and Liu YC (2018). E3 Ligase VHL Promotes Group 2 Innate Lymphoid Cell Maturation and Function via Glycolysis Inhibition and Induction of Interleukin-33 Receptor. Immunity 48, 258–270 e255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XJ, Xu M, Zhao XQ, Zhao JN, Chen FF, Yu W, Gao DY, and Luo B (2013). Proteomic analysis of synovial fibroblast-like synoviocytes from rheumatoid arthritis. Clin Exp Rheumatol 31, 552–558. [PubMed] [Google Scholar]
- McSorley HJ, Blair NF, Smith KA, McKenzie AN, and Maizels RM (2014). Blockade of IL-33 release and suppression of type 2 innate lymphoid cell responses by helminth secreted products in airway allergy. Mucosal Immunol 7, 1068–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra A (2017). Metabolic Plasticity in Dendritic Cell Responses: Implications in Allergic Asthma. J Immunol Res 2017, 5134760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto S, Murphy AN, and Brown JH (2008). Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-II. Cell Death Differ 15, 521–529. [DOI] [PubMed] [Google Scholar]
- Murai H, Qi H, Choudhury B, Wild J, Dharajiya N, Vaidya S, Kalita A, Bacsi A, Corry D, Kurosky A, et al. (2012). Alternaria-induced release of IL-18 from damaged airway epithelial cells: an NF-kappaB dependent mechanism of Th2 differentiation? PLoS One 7, e30280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson HS, Szefler SJ, Jacobs J, Huss K, Shapiro G, Sternberg AL, and Grp CAMPR (1999). The relationships among environmental allergen sensitization, allergen exposure, pulmonary function, and bronchial hyperresponsiveness in the Childhood Asthma Management Program. J Allergy Clin Immun 104, 775–785. [DOI] [PubMed] [Google Scholar]
- Neukirch C, Henry C, Leynaert B, Liard R, Bousquet J, and Neukirch F (1999). Is sensitization to Alternaria alternata a risk factor for severe asthma? A population-based study. J Allergy Clin Immun 103, 709–711. [DOI] [PubMed] [Google Scholar]
- O’Neill LAJ, and Pearce EJ (2016). Immunometabolism governs dendritic cell and macrophage function. J Exp Med 213, 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohollaren MT, Yunginger JW, Offord KP, Somers MJ, Oconnell EJ, Ballard DJ, and Sachs MI (1991). Exposure to an Aeroallergen as a Possible Precipitating Factor in Respiratory Arrest in Young-Patients with Asthma. New Engl J Med 324, 359–363. [DOI] [PubMed] [Google Scholar]
- Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MAR, Sheedy FJ, Gleeson LE, van den Bosch MWM, Quinn SR, Domingo-Fernandez R, Johnston DGW, et al. (2015). Pyruvate Kinase M2 Regulates Hif-1 alpha Activity and IL-1 beta Induction and Is a Critical Determinant of the Warburg Effect in LPS-Activated Macrophages (vol 21, pg 65, 2015). Cell Metab 21, 347–347. [DOI] [PubMed] [Google Scholar]
- Pulimood TB, Corden JM, Bryden C, Sharpies L, and Nasser SM (2007). Epidemic asthma and the role of the fungal mold Alternaria alternata. J Allergy Clin Immun 120, 610–617. [DOI] [PubMed] [Google Scholar]
- Qian X, Aboushousha R, van de Wetering C, Chia SB, Amiel E, Schneider RW, van der Velden JLJ, Lahue KG, Hoagland DA, Casey DT, et al. (2018). IL-1/inhibitory kappa B kinase epsilon-induced glycolysis augment epithelial effector function and promote allergic airways disease. J Allergy Clin Immun 142, 435–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rank MA, Kobayashi T, Kozaki H, Bartemes KR, Squillace DL, and Kita H (2009). IL-33-activated dendritic cells induce an atypical TH2-type response. J Allergy Clin Immunol 123, 1047–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Salamon H, Eugenin EA, Pine R, Cooper A, and Gennaro ML (2015). Infection with Mycobacterium tuberculosis induces the Warburg effect in mouse lungs. Sci Rep 5, 18176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirai T, Nazarewicz RR, Wallis BB, Yanes RE, Watanabe R, Hilhorst M, Tian L, Harrison DG, Giacomini JC, Assimes TL, et al. (2016). The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J Exp Med 213, 337–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair C, Bommakanti G, Gardinassi L, Loebbermann J, Johnson MJ, Hakimpour P, Hagan T, Benitez L, Todor A, Machiah D, et al. (2017). mTOR regulates metabolic adaptation of APCs in the lung and controls the outcome of allergic inflammation. Science 357, 1014–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snelgrove RJ, Gregory LG, Peiro T, Akthar S, Campbell GA, Walker SA, and Lloyd CM (2014). Alternaria-derived serine protease activity drives IL-33-mediated asthma exacerbations. J Allergy Clin Immunol 134, 583–592 e586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Q, Ji Q, Xia W, Li L, Bai J, Ni R, and Qin Y (2015). Pyruvate kinase M2 regulates apoptosis of intestinal epithelial cells in Crohn’s disease. Dig Dis Sci 60, 393–404. [DOI] [PubMed] [Google Scholar]
- Targonski PV, Persky VW, and Ramekrishnan V (1995). Effect of Environmental Molds on Risk of Death from Asthma during the Pollen Season. J Allergy Clin Immun 95, 955–961. [DOI] [PubMed] [Google Scholar]
- Thwe PM, Pelgrom LR, Cooper R, Beauchamp S, Reisz JA, D’Alessandro A, Everts B, and Amiel E (2019). Cell-Intrinsic Glycogen Metabolism Supports Early Glycolytic Reprogramming Required for Dendritic Cell Immune Responses (vol 26, pg 558, 2017). Cell Metab 30, 225–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valladao AC, Frevert CW, Koch LK, Campbell DJ, and Ziegler SF (2016). STAT6 Regulates the Development of Eosinophilic versus Neutrophilic Asthma in Response to Alternaria alternata. J Immunol 197, 4541–4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wculek SK, Khouili SC, Priego E, Heras-Murillo I, and Sancho D (2019). Metabolic Control of Dendritic Cell Functions: Digesting Information. Front Immunol 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Sun L, He Y, Yuan X, Niu J, Su J, and Li D (2019). Deficiency in IL-33/ST2 Axis Reshapes Mitochondrial Metabolism in Lipopolysaccharide-Stimulated Macrophages. Front Immunol 10, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang LC, Xie M, Yang MH, Yu Y, Zhu S, Hou W, Kang R, Lotze MT, Billiar TR, Wang HC, et al. (2014). PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis. Nature Communications 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.