

Abstract
Satellite repeats are major sequence constituents of centromeres in many plant and animal species. Within a species, a single family of satellite sequences typically occupies centromeres of all chromosomes and is absent from other parts of the genome. Due to their common origin, sequence similarities exist among the centromere-specific satellites in related species. Here, we report a remarkably different pattern of centromere evolution in the plant tribe Fabeae, which includes genera Pisum, Lathyrus, Vicia, and Lens. By immunoprecipitation of centromeric chromatin with CENH3 antibodies, we identified and characterized a large and diverse set of 64 families of centromeric satellites in 14 species. These families differed in their nucleotide sequence, monomer length (33–2,979 bp), and abundance in individual species. Most families were species-specific, and most species possessed multiple (2–12) satellites in their centromeres. Some of the repeats that were shared by several species exhibited promiscuous patterns of centromere association, being located within CENH3 chromatin in some species, but apart from the centromeres in others. Moreover, FISH experiments revealed that the same family could assume centromeric and noncentromeric positions even within a single species. Taken together, these findings suggest that Fabeae centromeres are not shaped by the coevolution of a single centromeric satellite with its interacting CENH3 proteins, as proposed by the centromere drive model. This conclusion is also supported by the absence of pervasive adaptive evolution of CENH3 sequences retrieved from Fabeae species.
Keywords: centromere evolution, satellite DNA, CENH3, ChIP-seq, plant chromosomes
Introduction
Satellite DNA (satDNA) is a class of eukaryotic repetitive DNA characterized by its genomic organization into arrays of tandemly arranged units called monomers. It is most clearly distinguished from other tandemly repeated sequences by its formation of much longer arrays spanning up to megabases in length. Although monomer sizes of tens to a few hundred base pairs are predominant (Macas et al. 2002), satellite monomers can range from lengths typical for microsatellites (2–10 bp) (Heckmann et al. 2013; Talbert et al. 2018) to over 5 kb (Gong et al. 2012). Owing to its rapid sequence turnover, satDNA is the most evolutionarily dynamic component of the genome, as demonstrated by the dramatic variation in its abundance among species and the frequent emergence of species-specific repeat families (Garrido-Ramos 2017). In higher plants, satellite repeats may occur at subtelomeric or interstitial chromosomal regions, but they are preferentially located in, and often confined to, centromeres, especially in species with small genomes (Garrido-Ramos 2015; Oliveira and Torres 2018). Preferential association of satDNA with centromeric loci has also been reported for other lineages of eukaryotes (Plohl et al. 2014; Hartley and O’Neill 2019). However, the significance of this association for centromere maintenance and function, as well as the underlying mechanisms of satDNA accumulation in centromeres, remains incompletely understood.
Centromeres are chromosome regions that facilitate faithful chromosome segregation during cell division. This is achieved by providing an anchor point for assembly of the kinetochore, a protein complex connecting centromeric chromatin to the spindle microtubules (Cheeseman 2014). Consequently, centromeres have a number of features that distinguish them from other parts of the chromosomes. They are marked by the presence of the centromere-specific histone variant CENH3 and other proteins of the constitutive centromere-associated network (Hara and Fukagawa 2017). In addition, centromeres are regions of suppressed meiotic recombination and exhibit characteristic profiles of epigenetic chromatin modifications (Fuchs and Schubert 2012; Zhang, Dong, et al. 2014). It remains controversial whether and how these features drive the evolution of underlying centromeric sequences, especially the satellite repeats. Diverse hypotheses have been proposed on this issue, ranging from the idea that satDNA is a passive hitchhiker to the claim that it is a key determinant of centromere identity.
Perhaps the most influential concept regarding centromere evolution is the centromere drive hypothesis (Henikoff et al. 2001). Centromere drive is proposed to occur in species with asymmetric female meiosis, in which homologous chromosomes compete for inclusion into the egg cell. Although the observed interspecific variation in repeat composition rules out the existence of a universal sequence determinant of centromere identity, this hypothesis still presumes that CENH3 or other kinetochore proteins interact with the centromeric satellites in a sequence-specific manner. Allelic expansion of the satellite array in one of the homologs then results in a stronger centromere, which binds more kinetochore proteins, thus facilitating its preferential transmission to the egg. On the other hand, such asymmetry leads to defects in male meiosis and reduced fertility, which is compensated for by changes in the CENH3 sequence that affect its DNA-binding preferences, resulting in restoration of meiotic parity. This evolutionary arms race between selfish centromeric DNA and its associated kinetochore proteins is predicted to result in diversification of centromeric repeats between species, as well as adaptive evolution of CENH3 or other kinetochore proteins that directly interact with the centromeric sequences (Henikoff et al. 2001; Malik 2009).
In line with the predictions of the centromere drive model, a single centromeric satellite whose sequence has diverged between related species has been reported in Oryza (Lee et al. 2005), Medicago (Yu et al. 2017), and some Brassicaceae species (Lermontova et al. 2014). Adaptive evolution of CENH3 proteins was detected in some of these species (Cooper and Henikoff 2004; Hirsch et al. 2009) as well as in several other taxa with asymmetric female meiosis (Zedek and Bureš 2016). Direct evidence for centromere drive was obtained in the plant genus Mimulus (Finseth et al. 2015), the fly Drosophila melanogaster (Wei et al. 2017), and mouse (Iwata-Otsubo et al. 2017) in which the molecular mechanisms underlying centromere drive have also been elucidated (Akera et al. 2017).
On the other hand, considering the widespread occurrence of centromeric satellites in plant and animal genomes, it is surprising that so few examples of centromere drive have been reported so far. Moreover, some observations are not consistent with the presumed evolutionary arms race between CENH3 and its underlying centromeric satellite(s) (Kawabe et al. 2006; Masonbrink et al. 2014). In addition, maize lines carrying homologous chromosomes with different centromere sizes exhibit no significant distortions in their meiotic segregation (Han et al. 2018), and CENH3 proteins from the phylogenetically distant species Lepidium oleraceum and Zea mays exhibit binding patterns on Arabidopsis thaliana centromeres that were indistinguishable from native CENH3 (Maheshwari et al. 2017). This may indicate that the process is not as common as expected, or that it is active only during limited periods of centromere evolution.
Although the considerations described earlier are mainly based on the presumed sequence-specific interactions of kinetochore proteins with their underlying sequences, it has recently become evident that features other than primary sequence may also be important for the coevolution of satDNA and centromeres. Specifically, it has been proposed that the repeated structure itself is advantageous, as homologous recombination between identical repeat copies generates DNA loops that are required for efficient centromere function (McFarlane and Humphrey 2010). In addition, centromere propagation and function seems to depend upon transcription of its sequences (Duda et al. 2017; Perea-Resa and Blower 2018); thus, the ability of centromeric satellites to produce transcripts at optimal levels may determine their fate in these regions. Finally, in domesticated maize inbred lines, the sequence composition of centromeres can be shaped by inbreeding and selection for centromere-linked genes, a process that may also act during speciation in natural systems (Schneider et al. 2016). Therefore, it is likely that the structure and sequence composition of centromeres in a particular species reflects an interplay of various structural features and evolutionary forces, the nature and importance of which are yet to be determined.
The questions outlined above could be answered by gathering information on centromeric sequences and kinetochore proteins from a wide range of species and examining them in the phylogenetic context. Because it is important to discriminate sequences that are truly associated with centromeric chromatin from surrounding repeats, it would be necessary to perform chromatin immunoprecipitation using antibodies against centromeric proteins coupled with sequencing of retrieved DNA (ChIP-seq). To date, however, relatively few such studies have been conducted in plants, and most have focused on one (Gong et al. 2012; Zhang, Kobližkova, et al. 2014; Kowar et al. 2016) or a small group of species (Gent et al. 2017).
In our previous work, we analyzed centromeric repeats in garden pea (Pisum sativum), a species with peculiar centromere organization consisting of multiple separated domains of CENH3 chromatin arranged along extended primary constrictions of metaphase chromosomes (Neumann et al. 2012). This “meta-polycentric” chromosome organization was later reported in Lathyrus, but not in genera Vicia and Lens, which are phylogenetically related members of the same legume tribe, Fabeae (Neumann et al. 2015). In P. sativum, ChIP-seq experiments with CENH3 antibody revealed unprecedented diversity of centromeric satellites consisting of 13 repeats with different distribution patterns among chromosomes (Neumann et al. 2012). Relative to Vicia and Lens, Pisum and Lathyrus species possess an additional copy of the CENH3 gene, which was speculated to serve as a possible trigger for the expansion of centromeres and the emergence of diverse centromeric satellites. However, subsequent study of Vicia faba, a species with simple centromeres and only one copy of CENH3, also revealed multiple centromeric satellites, three of which are present in the same centromere, whereas the other four are chromosome-specific (Ávila Robledillo et al. 2018). Therefore, Fabeae is a taxon with unusual distribution patterns and possibly highly dynamic turnover of centromeric repeats.
Prompted by these results, in this study, we focused on characterization of centromeric satellites across the whole Fabeae tribe, investigating 15 species in addition to the two analyzed previously. In these experiments, we employed a set of CENH3 antibodies (Neumann et al. 2012, 2015) to perform ChIP-seq in these species and identified centromeric satellites using repetitive sequences characterized by graph-based clustering of genomic reads (Macas et al. 2015) as the reference. As previously demonstrated (Zhang, Kobližkova, et al. 2014; Ávila Robledillo et al. 2018), this approach provides comprehensive information about centromere-associated repeats without the need for an assembled reference genome and as such is suitable for nonmodel species. The identified repeats were further investigated by a combination of fluorescence in situ hybridization (FISH) with immunodetection of CENH3 proteins to confirm their centromeric localization and map their distribution among chromosomes. The experiments revealed an extraordinary diversity of centromeric satellites in Fabeae, their irregular distributions among species, and unexpected localization patterns of some of these repeats on the chromosomes. Finally, we analyzed these findings with respect to the sequence diversity and evolution of CENH3 genes in Fabeae.
Results
ChIP-seq Analysis Reveals Unprecedented Diversity of Centromeric Satellites in Fabeae
To investigate the repeat composition of Fabeae centromeres, we sequenced and analyzed DNA fragments retrieved from centromeric chromatin immunoprecipitated with CENH3 antibody. These experiments were performed with a set of 15 species selected to represent all major evolutionary lineages of Fabeae, as described by Schaefer et al. (2012). To verify the species phylogeny, we calculated a maximum likelihood (ML) tree based on matK–rbcL sequences for the selected set, supplemented with seven additional Fabeae species in which centromeric satellites and/or CENH3 gene sequences have been characterized previously (Neumann et al. 2012, 2015; Ávila Robledillo et al. 2018). The resulting tree topology (fig. 1A) was in general agreement with a previously reported tree (Schaefer et al. 2012), confirming that Pisum and Lathyrus are closely related and form a separate lineage, whereas the other major lineage consists of most Vicia species along with Lens culinaris; in addition, a separate group of two Vicia species (V. ervilia and V. hirsuta) is basal to all Fabeae.
Fig. 1.

Overview of centromeric satellite families identified in Fabeae. Species are arranged based on their phylogenetic distances inferred from a comparison of matK–rbcL sequences using the maximum likelihood algorithm (A). The tree was rooted using five species representing related legume genera as outgroups. Numbers represent estimated node ages in million years ago (MYA), and correspond to the divergence time scale below the tree. The branch leading to the species with meta-polycentric chromosomes is marked with (M). Names of Fabeae species in which satellite repeats were identified using CENH3 ChIP-seq are printed in red, whereas species not analyzed by ChIP but included in the similarity searches are printed in bold. (B and C) The presence of individual satellite families in analyzed species is indicated by squares. Black squares indicate families associated with centromeric chromatin, as revealed by their enrichment in the CENH3 ChIP-seq experiments. The centromeric satellites that simultaneously occur in the genome as additional, noncentromeric loci (revealed by FISH) are marked with gray squares, whereas those present in the respective species but not enriched in ChIP-seq experiments are marked with empty squares. The question mark in FabTR-6 column indicates that this repeat is present in Vicia sepium genome but was not investigated by ChIP-seq in this species. (B) The satellite families from different species displaying sequence similarities are grouped into superfamilies and arranged in columns labeled with the superfamily name. (C) Numbers of species-specific families are symbolized by squares in each row, ranging from one in Lathyrus vernus to seven in V. pisiformis and V. peregrina. Numbers within the squares refer to the family names (FabTR-numbers) listed in table 1.
The ChIP-seq experiments were performed using antibodies raised against CENH3 proteins from V. faba, P. sativum, Lathyrus sativus, and Le. culinaris (supplementary table 1, Supplementary Material online), previously shown to specifically label centromeric chromatin in a number of Fabeae species (Neumann et al. 2012, 2015). The immunoprecipitated DNA fragments were sequenced on the Illumina platform along with control DNA samples extracted from chromatin preparations prior to ChIP (input control). Centromeric repeats were then identified as sequences enriched in the ChIP sample relative to the input. Enrichment of all repeats representing at least 0.01% of the genome was evaluated by similarity-based mapping of ChIP and input reads onto the reference repeat sequences previously generated for individual Fabeae species using the RepeatExplorer pipeline (Macas et al. 2015). In the two species for which a reference was not available (L. niger and L. clymenum), the ChIP and input reads were used directly for comparative RepeatExplorer analysis (Novák et al. 2013), and the enrichment was calculated as a ratio of ChIP to input reads in individual repeat clusters.
Centromeric satellites were identified in 12 out of the 15 investigated species as sequences with ChIP/input ratios between 3 and 333 (table 1). Such enrichment in the ChIPed fraction was revealed for up to eight satellites per species, whereas the majority of investigated repeats exhibited no enrichment (supplementary fig. 1, Supplementary Material online). In five species, one to five additional nontandem sequences, mostly classified as putative LTR-retrotransposons or unknown repeats, were ChIP-enriched (supplementary table 2, Supplementary Material online), whereas in the rest of the species, all enriched repeats represented satellites. In three Vicia species, V. narbonensis, V. ervilia, and V. hirsuta, we identified no ChIP-enriched repeats. Hence, we performed additional experiments to verify that the antibodies used for the ChIP recognize centromeric chromatin. In all three cases, we observed relatively weak but specific immunostaining of primary constrictions of isolated metaphase chromosomes (supplementary fig. 2, Supplementary Material online). These results may reflect a lack of centromere-enriched repetitive sequences in these three species; however, we cannot rule out the possibility that the antibodies failed specifically in the ChIP reaction because the conditions differed from those used for chromosome immunostaining.
Table 1.
Satellite Repeats Associated with Centromeric Chromatin.
| Species | Family | Superfamily | Monomer (bp) | AT (%) | Genome (%) | ChIP Enrichment | Previous Code |
|---|---|---|---|---|---|---|---|
| Lens culinaris | FabTR-35-LNS | 579 | 73.7 | 0.061 | 52.01 | ||
| FabTR-36-LNS | 1,086 | 76.5 | 0.058 | 40.13 | |||
| FabTR-37-LNS | 967 | 74.3 | 0.032 | 34.55 | |||
| FabTR-38-LNS | 1,315 | 74.9 | 0.019 | 40.19 | |||
| Vicia pisiformis | FabTR-17-VPF | 580 | 71.6 | 0.0317 | 5.0 | ||
| FabTR-18-VPF | 2,087 | 63.5 | 0.199 | 20 | |||
| FabTR-63-VPF | 61 | 60.7 | 0.158 | 57.13 | |||
| FabTR-19-VPF | 84 | 69 | 0.101 | 53.78 | |||
| FabTR-20-VPF | 763 | 64 | 0.06 | 79.4 | |||
| FabTR-1-VPF | FabTR-1 | 72 | 62.9 | 0.043 | 3.0 | ||
| FabTR-21-VPF | 778 | 74.3 | 0.039 | 16 | |||
| FabTR-22-VPF | 1,793 | 74.3 | 0.017 | 6.7 | |||
| Vicia peregrina | FabTR-5-VPR | FabTR-5 | 114 | 74.1 | 0.344 | 60 | |
| FabTR-29-VPR | 718 | 73.8 | 0.29 | 67.48 | |||
| FabTR-30-VPR | 898 | 73.8 | 0.267 | 91.59 | |||
| FabTR-32-VPR | 1,189 | 63.6 | 0.115 | 333.2 | |||
| FabTR-33-VPR | 30 | 56.7 | 0.087 | 22.35 | |||
| FabTR-34-VPR | 324 | 76 | 0.083 | 55.74 | |||
| FabTR-61-VPR | 569 | 68.5 | 0.031 | 130.8 | |||
| FabTR-62-VPR | 1,244 | 71.9 | 0.013 | 10.58 | |||
| Vicia faba | FabTR-1-VFB | FabTR-1 | 50 | 64 | 0.132 | 103.6 | VfSat6 |
| FabTR-5-VFB | FabTR-5 | 44 | 70.5 | 0.102 | 103.2 | VfSat7 | |
| FabTR-4-VFB | FabTR-4 | 2,033 | 71.7 | 0.061 | 91.3 | VfSat8 | |
| FabTR-13-VFB | 1,762 | 74.7 | 0.042 | 41.2 | VfSat10 | ||
| FabTR-14-VFB | 47 | 68.1 | 0.036 | 149.2 | VfSat13 | ||
| FabTR-15-VFB | 1,712 | 65.1 | 0.038 | 109.9 | VfSat16 | ||
| FabTR-16-VFB | 1,325 | 73.3 | 0.008 | 81.9 | VfSat23 | ||
| Vicia sativa | FabTR-6-VSA | FabTR-6 | 624 | 67.3 | 0.101 | 62.3 | |
| Vicia villosa | FabTR-27-VVL | 156 | 64.7 | 2.063 | 9.7 | ||
| FabTR-3-VVL | FabTR-3 | 602 | 75.9 | 0.226 | 80.9 | ||
| FabTR-28-VVL | 1,792 | 67.3 | 0.053 | 92.9 | |||
| Vicia tetrasperma | FabTR-24-VTS | 959 | 66.9 | 0.361 | 59.01 | ||
| FabTR-4-VTS | FabTR-4 | 1,614 | 69.8 | 0.254 | 69.62 | ||
| FabTR-25-VTS | 33 | 69.7 | 0.069 | 100 | |||
| FabTR-26-VTS | 470 | 64.5 | 0.055 | 72.11 | |||
| FabTR-5-VTS | FabTR-5 | 44 | 63.6 | 0.045 | 103.21 | ||
| Pisum sativum | FabTR-7-PST | FabTR-7 | 867 | 77 | 0.02 | 51.7 | TR1 |
| FabTR-8-PST | FabTR-8 | 244 | 76.6 | 0.01 | 59.3 | TR6 | |
| FabTR-46-PST | 164 | 72.6 | 0.124 | 49.7 | TR7 | ||
| FabTR-9-PST | FabTR-9 | 658 | 74.5 | 0.01 | 76.3 | TR10 | |
| FabTR-10-PST-A | FabTR-10 | 459 | 75.4 | 0.127 | 65.9–74.9 | TR11-TR19 | |
| FabTR-10-PST-B | FabTR-10 | 1,975 | 76.6 | 0.127 | 65.9–74.9 | TR11-TR19 | |
| FabTR-47-PST | 105 | 69.5 | 0.013 | 5.4 | TR12 | ||
| FabTR-11-PST | FabTR-11 | 1,637 | 74.3 | 0.012 | 82.5 | TR18 | |
| FabTR-12-PST | FabTR-12 | 844 | 78 | 0.179 | 50.7 | TR20 | |
| FabTR-48-PST | 613 | 71.6 | 0.013 | 44 | TR21 | ||
| FabTR-49-PST | 882 | 76.2 | 0.003 | 102.9 | TR22 | ||
| FabTR-50-PST | 1,812 | 69.9 | 0.087 | 10.7 | TR23 | ||
| PisTR-B | PisTR-B | 50 | 72 | 1.26 | 20.5 | ||
| Pisum fulvum | FabTR-7-PFL | FabTR-7 | 864 | 77 | 0.059 | 42.1 | TR1 |
| FabTR-8-PFL | FabTR-8 | 242 | 77.3 | 0.033 | 45.9 | TR6 | |
| FabTR-9-PFL | FabTR-9 | 659 | 74.7 | 0.044 | 73.5 | TR10 | |
| FabTR-10-PFL-A | FabTR-10 | 502 | 76.9 | 0.236 | 53.4 | TR11-TR19 | |
| FabTR-10-PFL-B | FabTR-10 | 2,170 | 76.7 | 0.236 | 53.4 | TR11-TR19 | |
| FabTR-11-PFL | FabTR-11 | 2,979 | 74.2 | 0.009 | 98.3 | TR18 | |
| FabTR-12-PFL | FabTR-12 | 864 | 73.6 | 0.01 | 52.8 | TR20 | |
| Lathyrus clymenum | FabTR-42-LACLM | 36 | 61.1 | 4.119 | 78.31 | ||
| FabTR-43-LACLM | 30/60/70 | 60 | 0.805 | 60.29–98.18 | |||
| FabTR-44-LACLM | 60 | 50 | 0.977 | 57.21 | |||
| FabTR-45-LACLM | 102 | 57.4 | 0.162 | 76.39 | |||
| Lathyrus niger | FabTR-2-LNGER | FabTR-2 | 49/50/100 | 75.5 | 0.069 | 86.6–98.27 | |
| Lathyrus vernus | FabTR-2-LAV | FabTR-2 | 49 | 77.6 | 0.584 | 62.1 | |
| FabTR-3-LAV | FabTR-3 | 972 | 77.8 | 0.022 | 3.08 | ||
| FabTR-41-LAV | 54 | 79.2 | 0.017 | 8.34 | |||
| Lathyrus sativus | FabTR-2-LAS | FabTR-2 | 49 | 73.5 | 1.679 | 38.53 | |
| Lathyrus latifolius | FabTR-2-LAL | FabTR-2 | 49 | 73.5 | 1.228 | 46.54 |
Including the previously reported centromeric satellites from P. sativum (Neumann et al. 2012) and V. faba (Ávila Robledillo et al. 2018), we identified a total of 64 centromeric satDNA families in Fabeae. In most species, we detected multiple centromeric satellites, and none of these repeats was shared across all species. The basic characteristics of centromeric satellites are summarized in table 1 and their consensus monomer sequences are provided in supplementary file 1, Supplementary Material online. Monomer lengths varied considerably (33–2,979 bp), as did their nucleotide composition (50–79% AT). To evaluate sequence similarities that could point to a common origin of centromeric satellites from different species, we compared the monomer sequences using alignment-free similarity measures as defined by statistics (Reinert et al. 2009). We also performed these analyses on the complete set of 430 putative satellite repeats predicted previously for the investigated species (Macas et al. 2015) to detect similarities between centromeric and noncentromeric satellites. The results revealed that most satellite repeat families, regardless of their association with centromeres, were species-specific (supplementary fig. 3A, Supplementary Material online). A subset of the repeats exhibited sequence similarities that led to the definition of 13 superfamilies that included centromeric satellites and consisted of the families present in two to five species (fig. 1B). Although satellites assigned to the same superfamily exhibited significant similarities, some families had sequence variations, especially with respect to monomer size (supplementary fig. 3B–D, Supplementary Material online and table 1). Moreover, some of the centromeric superfamilies included satellites that were ChIP-enriched in only some species, but were not centromeric in the rest (supplementary fig. 3B and C, Supplementary Material online and fig. 1B).
Most Species Possess Multiple Centromeric Satellites That Are Often Species-Specific
The families of centromeric satellites that we identified were unevenly distributed among Fabeae species. A large fraction of families (37 of 64; 58%) were species-specific (fig. 1C), whereas the remaining repeats belonged to satellite superfamilies shared by several species (fig. 1B). In the phylogenetic lineages including Vicia spp. and Le. culinaris, all species but one (V. sativa) possessed multiple (two to eight) centromeric satellites, with up to seven species-specific satellites in V. pisiformis and V. peregrina (fig. 1C). The largest number of centromeric satellites, 12, occurred in the P. sativum genome; however, only six of these satellites were shared with its sister species P. fulvum (fig. 1B and C).
Three of the investigated Lathyrus species, L. sativus, L. latifolius, and L. niger, possessed single centromeric satellites that were classified as members of the same superfamily, FabTR-2. The same centromeric repeat was also identified in closely related L. vernus; however, this species possessed two additional, albeit far less abundant centromeric satellites (table 1). The existence of a single-dominant centromeric satellite in these three Lathyrus species contrasted with the situation in the remaining species, L. clymenum, in which FabTR-2 sequences were also present but were not associated with centromeric chromatin. Instead, four species-specific centromeric satellites were identified in this species (fig. 1).
Next, we used FISH combined with immunodetection of CENH3 proteins to confirm ChIP-seq results and investigate the genome distribution of the selected satellite sequences. Contrasting patterns of centromeric satellite distributions were revealed, some of which are schematically depicted on figure 2. When applied to L. sativus, a species containing FabTR-2 as the single centromeric satellite, these experiments confirmed the location of this repeat in all domains of centromeric chromatin distributed along the primary constrictions of the chromosomes (fig. 2A and supplementary fig. 4A–D, Supplementary Material online). In L. vernus, the experiment revealed identical patterns of FabTR-2 colocalization with CENH3 chromatin (supplementary fig. 4E–H, Supplementary Material online), whereas the two additional ChIP-enriched satellites identified for this species were detected as minor loci overlapping with FabTR-2 signals (data not shown). In species with large numbers of centromeric satellite families, these families were unevenly distributed between the chromosomes, as shown in P. sativum (this work and Neumann et al. [2012]) (fig. 2B). The same pattern was also observed for P. fulvum, as none of its six centromeric satellites occurred on all chromosomes (data not shown). Similar types of distribution patterns are also likely shared by Vicia species with high diversity of centromeric satellites. For example, all seven centromeric satellites in V. faba were chromosome-specific (fig. 2C), and FISH localization of two randomly chosen centromeric satellites in V. peregrina revealed their presence in centromeres of four (FabTR-30) and one (FabTR-32) of the seven pairs of chromosomes (supplementary fig. 4I and J, Supplementary Material online).
Fig. 2.
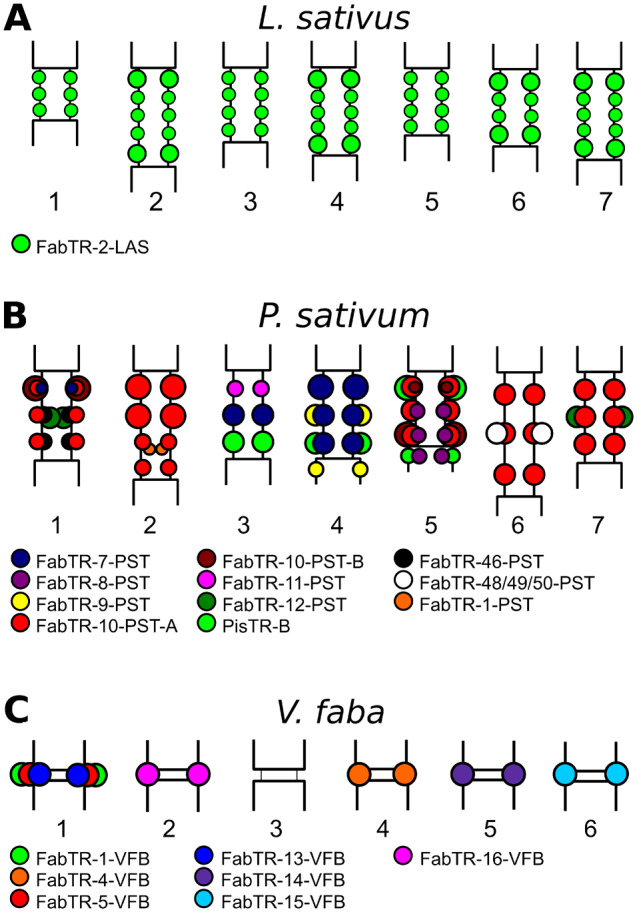
Schematic representation of the satellite repeat distribution in centromeric regions of (A) Lathyrus sativus (n = 7), (B) Pisum sativum (n = 7), and (C) Vicia faba (n = 6) chromosomes. Different families of satellite repeats are distinguished by colors according to the legend provided for each species. In meta-polycentric chromosomes (A and B), the satellite loci associated with CENH3 chromatin are located at the outer periphery of the primary constrictions, whereas those located within the inner regions of P. sativum constrictions lack CENH3.
Association of Some Satellites with Centromeric Chromatin Differs between Species or Even between Chromosomes of the Same Species
A striking feature of some satellite superfamilies was their association with centromeric chromatin in some species, but no enrichment in CENH3 ChIP-seq experiments in the others, suggesting that they were absent from centromeres in these genomes. This pattern was found for five superfamilies: FabTR-1, 2, 11, 12, and PisTR-B (fig. 1). To obtain better insight into their genomic distribution, we performed FISH on metaphase chromosomes, as shown for FabTR-1 repeats in figure 3. FabTR-1 was ChIP-enriched in V. pisiformis and V. faba, and corresponding FISH signals were detected in centromeres of two chromosome pairs in V. pisiformis (fig. 3A) and in the centromere of chromosome 1 of V. faba. An additional minor noncentromeric signal was present within the long arm of chromosome 6 of V. faba (fig. 3B). In the remaining three species, V. tetrasperma, L. vernus, and P. sativum, the repeat is not associated with CENH3 chromatin; in all of them, however, it was found to be located close to the centromeres. In V. tetrasperma, FabTR-1 signals almost entirely overlapped with the primary constrictions on two chromosome pairs, and an additional repeat locus was revealed within the long arm of one of these chromosomes (fig. 3C). One FabTR-1 locus close to the centromere of one chromosome pair was identified in L. vernus (fig. 3D). In P. sativum, the signal was located directly within the extended primary constriction of chromosome 2 (fig. 3E). Detailed examination of metaphase chromosomes employing simultaneous immunodetection of CENH3 revealed that FabTR-1 is located within the inner part of the constriction close to the chromosome axis, whereas the CENH3 chromatin is located on the periphery of the constriction (fig. 3F). These findings confirmed that despite its presence in the centromeric region, the repeat is not associated with centromeric chromatin, consistent with the results of the ChIP-seq experiments.
Fig. 3.

Localization of FabTR-1 repeats on metaphase chromosomes of five Fabeae species. Repeats were detected using FISH (red signals), showing signals within centromeres of two chromosome pairs in Vicia pisiformis (A) and one pair in V. faba (B). A minor noncentromeric signal on V. faba chromosome 6 is marked with an arrow. Two pericentromeric and one interstitial signal were detected in V. tetrasperma (C), whereas Lathyrus vernus (D) and Pisum sativum (E) exhibited signals adjacent to or within primary constrictions of one pair of chromosomes. Closer examination of P. sativum chromosomes using a combination of FISH (red) with immunolabeling of CENH3 proteins (green) revealed that FabTR-1 is located within the inner part of the primary constriction, apart from the CENH3 chromatin located along the constriction periphery (F). Chromosomes counterstained with DAPI are shown in gray.
The existence of additional noncentromeric loci containing centromeric satellites was confirmed for most superfamilies shared by the two Pisum species (fig. 1B). In some cases, this pattern was combined with the presence of the repeat in additional species. For example, FabTR-12 was centromeric in both Pisum species, but noncentromeric in V. faba and L. vernus. In all four species, the repeat was located on two pairs of chromosomes, but was fully associated with CENH3 chromatin only in P. fulvum (fig. 4A and B). In P. sativum, FabTR-12 signals overlapped with CENH3 chromatin only on chromosome 7 (fig. 4D), whereas the FISH signals on chromosome 1 were located within the inner part of the constriction (fig. 4C), similar to FabTR-1 on chromosome 2 (fig. 3F). In V. faba, the repeat was located within long arms of two chromosome pairs (fig. 4E), whereas in L. vernus it was adjacent to primary constrictions (fig. 4F). Yet another interesting example of such distribution is the major Pisum satellite PisTR-B (Neumann et al. 2001) which in P. sativum is associated with centromeric chromatin on chromosomes 3, 4, and 5, whereas most of its loci are distributed in pericentric and subtelomeric regions (fig. 4G–J). Although of similar genomic abundance and chromosomal distribution, it is not associated with centromeres in P. fulvum (figs. 1B and 4K–N).
Fig. 4.

Localization of FabTR-12 and PisTR-B repeats on metaphase chromosomes. Repeats were detected using FISH (red) alone or in combination with immunolabeling of CENH3 (green signals). (A–D) FISH detection of FabTR-12 showing signals overlapping with CENH3 loci on chromosomes 1 and 7 of Pisum fulvum and on chromosome 7 of P. sativum. On the contrary, FabTR-12 signals were located apart from the CENH3 chromatin on P. sativum chromosome 1 (arrow). In Vicia faba (E) and Lathyrus vernus (F), the repeat was also present on two chromosome pairs, but the signals were not centromeric and were instead located within the long chromosome arms. (G–N) Distribution of PisTR-B repeats on chromosomes of the two Pisum species. There are three centromeric PisTR-B loci (arrowheads) that colocalize with CENH3 in P. sativum (G–J); however, this satellite is not associated with the centromeric chromatin in P. fulvum (K–N).
CENH3 Genes Evolved Mainly under Purifying Selection
To determine whether centromeric repeat composition is correlated with the mode of evolution of CENH3 genes, we performed a phylogenetic analysis of CENH3 coding sequences. In our previous study (Neumann et al. 2015), we found two CENH3 variants in Fabeae that differed significantly, particularly in their N-terminal regions. Although the CENH3-2 variant is shared by all species within the tribe, CENH3-1 occurs as an additional gene only in the Pisum/Lathyrus lineage. To better date the CENH3 duplication event, we identified CENH3 sequences in four additional species representing the basal group (V. ervilia, V. hirsuta) or less-represented parts of the Fabeae phylogenetic tree (V. pisiformis, V. tetrasperma) and analyzed them in combination with 32 CENH3-coding sequences identified previously. The results revealed that all four new sequences belong to CENH3-2, and that CENH3-1 and CENH3-2 diverged before radiation of the Fabeae species included in this study (fig. 5). Because V. ervilia and V. hirsuta represent the clade that split earliest from all other Fabeae (Schaefer et al. 2012; fig. 1A), it is likely that the duplication occurred in an ancestor of all Fabeae. Our analysis further suggested that following the duplication, the CENH3-1 gene was lost independently at least three times in: 1) an ancestor of V. hirsuta and V. ervilia, 2) an ancestor of most other Vicia species and Le. culinaris, and 3) in V. tetrasperma or its ancestor (fig. 5). To confirm that CENH3-1 is indeed absent in Vicia species, we sequenced genomic DNA of V. ervilia and V. tetrasperma at 17× and 26× coverage, respectively. CENH3 sequences were either selectively assembled using GRAbB (Brankovics et al. 2016) or identified in super-reads assembled by MaSuRCA (Zimin et al. 2013). Both approaches revealed only a single functional CENH3-2 gene in each species, confirming the absence of CENH3-1. In V. tetrasperma, we detected fragments of an additional CENH3 gene with partial similarity to exon 2, intron 2, exon 3, and intron 3. It was not possible to identify the CENH3 variant from these recovered sequences, but it is likely that they represent remnants of a nonfunctional gene copy (data not shown).
Fig. 5.

Phylogenetic trees of CENH3 sequences. (A) Phylogenetic tree inferred from the alignment of CENH3-coding sequences using the maximum likelihood method, excluding the INDEL region near the 5′ end (see supplementary fig. 5, Supplementary Material online). Bootstrap values are shown only for key nodes. Black dots indicate nodes with low bootstrap support (<50). The scale bar shows genetic distance. (B) Tanglegram showing comparison of the CENH3 tree from the panel (A) with the species tree inferred from matK–rbcL shown in figure 1A. Nodes with low bootstrap support (<50) were collapsed in both trees. The part of the matK–rbcL tree depicted by dashed lines was manually added to the tree to show comparison of phylogenies inferred from matK–rbcL and CENH3-1, and to allow the use of the matK–rbcL tree for analysis of positive selection in CENH3 genes. Red lightning symbols mark three independent losses of CENH3-1 genes. Pisum and Lathyrus species are highlighted by red rectangles. Orange dots indicate CENH3 duplication events.
Protein sequences of CENH3 histones in Fabeae are 119–123 aa in length, share 70.6–100% similarity, and are invariant at only 60 sites (supplementary fig. 5A, Supplementary Material online). To determine whether their divergence was due to positive selection, we analyzed the sequences using BUSTED (Murrell et al. 2015) to detect gene-wide positive selection, FEL (Kosakovsky Pond and Frost 2005) to detect sites under pervasive positive selection, and MEME (Murrell et al. 2012) to detect sites under episodic positive selection. BUSTED found no evidence of gene-wide positive selection of CENH3 genes in Fabeae (table 2). Estimates of ω (ω = Ka/Ks) calculated for the CENH3-1 and CENH3-2 branches were, depending on the tree, 0.374 or 0.375 and 0.254 or 0.269, respectively, suggesting that both CENH3 variants evolved mainly under purifying selective pressure (table 2). FEL and MEME predicted (P < 0.05) a total of eight and two sites that may have evolved under positive selection in CENH3-1 and CENH3-2, respectively, indicating that positive selection explains very little of the variability observed among CENH3 protein sequences in Fabeae (supplementary fig. 5A, Supplementary Material online). We also performed FEL and MEME analyses focusing specifically on CENH3 sequences from the four Lathyrus species possessing FabTR-2 as a single-dominant centromeric satellite, but differing considerably in their centromere sizes (fig. 1B and supplementary fig. 4, Supplementary Material online). The analyses revealed only one positively evolving site in CENH3-1 and none in CENH3-2 (table 2). Pairwise comparison of CENH3 sequences from these species showed one to eight and zero to four amino acid substitutions in CENH3-1 and CENH3-2, respectively (supplementary fig. 5B and C, Supplementary Material online). Of these variable sites, one to three and zero to one appeared to have been predicted (P < 0.05) as positively evolving in the tests performed on the entire branches of CENH3-1 and CENH3-2 or single branches immediately following the CENH3 duplication event (table 2 and supplementary figure 5B and C, Supplementary Material online). These results indicated that the expansion of centromeres in the Lathyrus species was accompanied by very few changes in CENH3 protein sequences and that the positive selection had almost no impact on CENH3 diversification.
Table 2.
Tests for Positive Selection.
| Tested Branches | BUSTED |
FEL |
MEME |
|||
|---|---|---|---|---|---|---|
| CENH3 | matk–rbcL | CENH3 | matk–rbcL | CENH3 | matk–rbcL | |
| All branches |
|
|
— | — |
|
|
| All CENH3-1 branches |
|
|
|
|
|
|
| All CENH3-2 branches |
|
|
— | — | 23 (0.0000) | — |
| All non-Fabeae branches (outgroup) |
|
|
|
|
|
|
| Single CENH3-1 branch after duplication | Not tested | Not tested | Not tested | Not tested |
|
|
| Single CENH3-2 branch after duplication | Not tested | Not tested | Not tested | Not tested | 7 (0.0207) | 7 (0.0265) |
| CENH3-1 in LAS, LAL, LASYL, LNGER, LAV | NA |
|
NA | 105 (0.0145) | NA | 105 (0.0228) |
| CENH3-2 in LAS, LAL, LASYL, LNGER, LAV | NA |
|
NA | — | NA | — |
P values (P) are shown in parenthesis. Sites with P < 0.01 are underlined.
Discussion
In this study, we identified and characterized centromeric satellites in 14 Fabeae species and investigated their distribution with respect to the species phylogeny and the evolution of their CENH3 genes. In terms of the number of included species and newly described centromeric repeats, this is the largest study to date to be conducted on a group of related plants. The methodology employed for the centromeric repeat identification has been proven to be efficient and accurate in a number of studies (Gong et al. 2012; Zhang, Kobližkova, et al. 2014; Kowar et al. 2016; Yang et al. 2018). Compared with an alternative setup in which centromeric sequences are identified by mapping ChIPed and input reads to the genome assemblies (Park 2009), our approach is limited with respect to identification of a single- or low-copy centromeric sequences. However, this limitation is not relevant for repeat-focused studies, as in this case. Moreover, the use of repeated sequences identified by clustering analysis of low-pass Illumina reads as a reference provides several benefits, including unbiased repeat representation and significant reductions in cost and labor relative to building the reference assembly.
Bioinformatic analysis of all highly and moderately repeated sequences revealed CENH3 ChIP-enriched centromeric repeats in all but three species. Except for a small number of retrotransposon and unclassified sequences, all identified centromeric repeats corresponded to families of satDNA (table 1), showing that this class of repeats is dominant in Fabeae centromeres. In three species, no ChIP-enriched sequences were identified, suggesting the absence of abundant repeats in the centromeres of these species. By contrast, most Fabeae species harbor numerous and abundant centromeric repeats, although the FISH mapping of seven centromeric satellites identified in V. faba revealed their absence in the centromere of one chromosome pair (Ávila Robledillo et al. 2018). For practical reasons, our analysis was limited to repeats representing at least 0.01% of the genome, and thus was not exhaustive; hence, an additional analysis targeting individual species with larger volumes of sequencing data would be needed to determine whether their centromeres are truly repeat-free. On the other hand, the negative result of the ChIP-seq analysis obtained for these three species should be interpreted with caution, as it could also have arisen due to the technical issues. This is especially true in V. ervilia and V. hirsuta, which represent ancient phylogenetic lineage of Fabeae and have CENH3 proteins that are relatively divergent from those used to raise the ChIP antibodies (supplementary table 1, Supplementary Material online).
The major finding of this study is the large number and sequence diversity of centromeric satellites within and between Fabeae species which is unique among eukaryotic taxa investigated so far. In many organisms, a single satellite repeat family dominates all centromeres, although it may partially differentiate into chromosome-specific variants or higher order repeats. Although these satellites evolve relatively rapidly, similarities are still detectable between sequences retrieved from related species. Examples of such centromeric satellite superfamilies include the primate alpha satellites (McNulty and Sullivan 2018; Hartley and O’Neill 2019), CentO/CentC in Oryza and Zea (Lee et al. 2005; Bilinski et al. 2015) and cen180 in Arabidopsis and other Brassicaceae (Lermontova et al. 2014). Sequence diversification of such shared centromere-specific superfamilies along with the adaptive evolution of CENH3 proteins found in some taxa, led to the formulation of the centromere drive model (Henikoff et al. 2001; Malik 2009). The model proposes that specific interactions of CENH3 or other inner kinetochore proteins with their underlying centromeric satellites result in stronger centromeres on homologs with expanded satellite arrays that are consequently preferentially transmitted to the germ cells during asymmetric female meiosis. This process is then compensated for by the adaptive evolution of the interacting protein(s), leading to the evolutionary race of arms between selfish centromeric DNA and its associated kinetochore proteins. However, it is unlikely that centromere drive is at work in Fabeae, as the presence of multiple centromeric satellites with different sequences precludes any sequence-dependent coevolution with CENH3 or other kinetochore proteins. This is further supported by the observed lack of pervasive adaptive evolution of Fabeae CENH3 proteins, which was not detected even in the set of Lathyrus species possessing a single centromeric satellite (table 2). Another argument against sequence-dependent deposition of CENH3 to Fabeae centromeres comes from CENH3–YFP fusion protein expression experiments showing that CENH3-2 from V. faba is efficiently deposited onto P. sativum centromeres, and conversely, that CENH3-1 from P. sativum targets centromeres in V. faba (Neumann et al. 2015). Similar results were reported by Maheshwari et al. (2017), demonstrating that CENH3 from evolutionary distant species can replace the native CENH3 in Arabidopsis thaliana.
Another factor to consider when seeking an explanation for the observed diversity of centromeric satellites is the duplication and partial diversification of the two CENH3 gene copies in Pisum and Lathyrus. Coincidentally, species from these genera also exhibit a distinctive type of centromere morphology characterized by extended primary constrictions and occurrence of multiple CENH3 loci (supplementary fig. 4, Supplementary Material online and Neumann et al. 2015). However, neither the CENH3 duplication nor the centromere morphology can be directly linked to the diversity of centromeric satellites, as this group of species includes both of the observed extremes: the single centromeric satellite associated with all CENH3 loci in L. sativus, as well as the most diverse population of centromeric satellites with uneven distribution on P. sativum chromosomes (fig. 2).
Satellite DNA is not necessary or sufficient for centromere establishment and propagation (Piras et al. 2010; Logsdon et al. 2019). In plants, satellite-free centromeres are present on five of the 12 chromosomes of potato (Solanum tuberosum). The remaining seven potato centromeres contain mostly chromosome-specific satellites with exceptionally long monomers originating from recombination of LTR-retrotransposon fragments with other genomic sequences (Gong et al. 2012). This type of centromeric satellites is also present in the closely related S. verrucosum; however, the repeats are mostly species-specific, suggesting their recent and independent origin (Zhang, Kobližkova, et al. 2014). Based on these findings, along with the presence of partially homogenized centromeric satellites in switchgrass species (Yang et al. 2018), it was hypothesized that evolutionarily young centromeres may be repeat-free and only later accumulate random satellites that are subsequently homogenized across different chromosomes, resulting in the selection of a single, structurally favorable repeat to dominate all centromeres (Gong et al. 2012; Zhang, Kobližkova, et al. 2014; Yang et al. 2018). Considering our results in light of this hypothesis, we can see a number of differences suggesting that the diversity of centromeric satellites in Fabeae is not due to their origin in newly formed or relocated centromeres. First, some satellite superfamilies occur in species from different phylogenetic lineages, indicating that their origin dates back to the diversification of Fabeae (FabTR-1 and FabTR-12, fig. 1). Moreover, FISH mapping revealed that some centromeric satellites also occur at additional, noncentromeric loci, suggesting that they might have originated elsewhere in the genome and subsequently invade the centromeres. In addition, we have no evidence of frequent neocentromere formation or chromosome rearrangements in Fabeae, which have relatively stable karyotypes (Badr 2006).
Compared with other plant taxa, most Fabeae species are exceptional in terms of their high diversity of satellite repeats in general (Neumann et al. 2012; Macas et al. 2015; Ávila Robledillo et al. 2018), which might also be reflected in their large numbers of centromeric repeats. This diversity of satellites contrasts even with the closest relatives of Fabeae, genera Trifolium, Medicago, and Cicer, whose species possess one or (rarely) two centromeric satellites (Zatloukalová et al. 2011; Yu et al. 2017; Dluhošová et al. 2018). The molecular or evolutionary processes that made Fabeae so rich in satDNA remain to be fully elucidated, but one possible mechanism was revealed in our recent investigation of L. sativus repeats using ultralong nanopore reads. Most noncentromeric satellites in this species originated relatively recently by amplification of short tandem repeat arrays present in LTR-retrotransposons (Vondrak et al. 2020). The same mechanism was previously proposed for the origin of PisTR-A satellite in P. sativum (Macas et al. 2009); thus, it is likely to contribute to the emergence of species-specific satellites and their high turnover across Fabeae. It is worth noting that the LTR-retrotransposons providing these short array templates belong to the lineage of Ty3/gypsy Ogre elements (Neumann et al. 2019) which represent dominant repeats in the Fabeae genomes (Macas et al. 2015) but are comparably less abundant in the related legume taxa (Macas et al. 2007; Dluhošová et al. 2018), potentially resulting in smaller numbers of Ogre-derived satellite repeats.
Taken together, the results presented in this work, along with the recent data from other species, suggest that the patterns of association and eventual coevolution of satellite repeats with plant centromeres may be far more complex than previously envisioned. It is possible that the mechanisms leading to the centromere drive act only episodically, or in specific cases in which only a single repeat with properties favorable for supporting centromeric chromatin is available. However, should multiple such satellites occur in a genome, they might be co-opted simultaneously or alternatively during centromere evolution, and this seems to have occurred in Fabeae. Several features are thought to be important for “centromere competence” of satellite repeats, including the presence of dyad symmetries (Kasinathan and Henikoff 2018) or WW dinucleotide periodicities in their sequences (Zhang et al. 2013; Yang et al. 2018), as well as a proper level of transcription (Duda et al. 2017; Perea-Resa and Blower 2018). The sequence data acquired in this study will be instrumental in future research of these properties, as it includes diverse satellite sequences and allows for their comparative analysis in species with different modes of association between individual satellite families and centromeric chromatin.
Materials and Methods
Plant Material
Seeds of most Vicia species were obtained from the seed bank of the Leibniz Institute of Plant Genetics and Crop Plant Research (IPK), Gatersleben, Germany. Their accessions in the seed bank are as follows: V. hirsuta (L.) S.F.Gray, VIC728; V. ervilia (L.) Willd., ERV52; V. pisiformis L., VIC36; V. peregrina L., VIC765; V. villosa, VIC876; and V. tetrasperma (L.) Schreb., VIC726. Commercial varieties of V. pannonica “Dětěnická panonská,” V. faba “Merkur,” and P. sativum “Terno” were obtained from Osiva Boršov, Czech Republic; V. sativa “Ebena” from the Agricultural Research Institute Kroměříž, Czech Republic; and Le. culinaris “Eston” from the Nohel garden, Dobříš, Czech Republic. Vicia narbonensis (ICARDA 14) was provided by A. M. Torres (IFAPA Cordoba, Spain). Lathyrus sativus, L. latifolius, and L. niger were purchased from Fratelli Ingegnoli S.p.A., Milano, Italy (Cat. No.: 455), SEMO Smržice, Czech Republic (accession number 1-0040-68867-01), and Arboretum Paseka Makču Pikču, Paseka, Czech Republic, respectively. Lathyrus vernus was collected from a wild population at Vidov, Czech Republic (GPS 48°55′17.401″N, 14°29′44.158″E). Pisum fulvum accession (ICARDA IG64207) was provided by Petr Smýkal, Palacký University, Olomouc, Czech Republic.
Genomic DNA Isolation and Phylogenetic Analysis of Fabeae Species
Genomic DNA was extracted from leaf tissues according to Dellaporta et al. (1983). Sequences of the chloroplast loci (matK and rbcL) used for phylogenetic reconstructions were obtained by PCR amplification of the corresponding DNA fragments from total genomic DNA preparations using the primers MatK-L-F (5′-ATG AAG GAM TAT HMA GTA TAT TTA G-3′) and Matk-L-R (5′-TCA TTC ATC ATG GAC CAG ATC-3′), and rbcL-L_F2 (5′-ATG TCA CCA CAA ACA GAA ACT AAA-3′) and rbcL-L_R2 (5′-TTA CAA AGT ATC CAT TGC TGG G-3′). Alternatively, the matK and rbcL sequences were assembled from previously published NGS data sets (Macas et al. 2015) or retrieved from GenBank, as specified in supplementary table 3, Supplementary Material online. Nucleotide sequences were aligned using Muscle (Edgar 2004). ML phylogenies were estimated using PhyML 3.0 (Guindon et al. 2010) with automatic model selection by SMS (Lefort et al. 2017). Starting trees for ML analysis were calculated using neighbor-joining (NJ) algorithm implemented in SeaView (Gouy et al. 2010). The branch support was evaluated using bootstrap analysis (≥10,000 replicates). Divergence times were estimated using RelTime method implemented in MEGA X (Mello 2018), taking into account that the most recent common ancestor of P. sativum and V. sativa existed 12.9–22.8 Ma (Lavin et al. 2005). Phylogenetic trees were edited using ITOL (Letunic and Bork 2019). Alignment of the concatenated sequences of matK and rbcL used to infer the species tree (fig. 1A) is provided in supplementary file 2, Supplementary Material online.
Identification of Centromeric Repeats Using Chromatin Immunoprecipitation
Chromatin immunoprecipitation was performed on nuclei isolated from fresh leaves as described (Neumann et al. 2012) using custom-made antibodies raised against peptides designed according to the previously identified Fabeae CENH3 protein sequences (Neumann et al. 2012, 2015). A single antibody was always used for ChIP experiments, and it was selected based on 1) the similarity between peptide antigen and the CENH3 sequence in particular species and 2) its performance in in situ immunodetection experiments. Information about the antibodies and their use in individual species is provided in the supplementary table 1, Supplementary Material online and references cited therein. Rabbit polyclonal antibody to CENH3-2 of L. sativus (ID: P60) was produced in the course of this study by Genscript (Piscataway, NJ) using “complete affinity-purified peptide polyclonal package” (Cat. No.: SC1031). ChIPed DNA and input DNA control were sequenced on the Illumina platform in a single-end, 101 nt read mode. The resultant reads were trimmed to 100 nt by removing the first base and quality filtered to exceed the cutoff quality score of 10 over at least 95 nucleotides. Quality-filtered reads were mapped to reference contigs assembled from clusters of genome shotgun sequencing reads representing repetitive sequences of the corresponding species produced and characterized in our previous work (Macas et al. 2015). Similarity-based mapping of reads to repeat contigs was performed using BlastN (Altschul et al. 1997) with the parameters “-m 8 -b 1 -e 1e-20 -W 9 -r 2 -q -3 -G 5 -E 2 -F F,” and was followed by output parsing to ensure that each read was mapped to a maximum of one repeat cluster with the highest similarity score. The proportion of ChIP and input reads mapped to individual clusters was evaluated to identify repeats with a ChIP/input ratio ≥3, which were considered to represent repeats enriched in the ChIP sample. In the two species for which reference contigs were not available (L. niger and L. clymenum), the ChIP and input reads were used directly for comparative RepeatExplorer analysis (Novák et al. 2013) and enrichment was calculated as a ratio of ChIP to input reads in individual repeat clusters.
Sequence Analysis of Satellite Repeats
Putative satellite repeats were identified in the course of our previous study (Macas et al. 2015) via graph-based clustering of genomic shotgun reads using the RepeatExplorer pipeline (Novák et al. 2013). Reconstruction of monomer sequences of selected satellites was performed using TAREAN (Novák et al. 2017). Similarities between satellite sequences were evaluated using alignment-free sequence comparison using distance (Reinert et al. 2009) as implemented in d2-tools (https://code.google.com/archive/p/d2-tools/; last accessed September 23, 2019). Dissimilarity measurement matrices were calculated using shotgun reads from individual satellite clusters for k-mer lengths k from three to nine nucleotides under the zero- to third-order Markov model M. The resultant distance matrix was calculated as arithmetical average of all 27 dissimilarity matrices. The similarity threshold used for visualization was defined as:. This threshold was selected based on the empirical exploration of multiple satellite sequences using dotplot comparisons (Sonnhammer and Durbin 1995).
Alternatively, similarities between centromeric satellites and all other repetitive sequences were detected using BlastN search with default parameters. Contigs assembled from clusters representing repetitive sequences of the corresponding species produced and characterized in our previous work (Macas et al. 2015) were searched against the database of TAREAN-reconstructed satellite centromeric sequences. The top percentile of similarity hits was manually explored using dotplot.
FISH and Immunolabeling
Mitotic chromosomes used for cytogenetic experiments were prepared from root apical meristems synchronized as described previously (Neumann et al. 2015) to increase the proportion of simultaneously dividing cells. The synchronized meristems were processed using different protocols depending on their intended use. For FISH experiments, they were fixed in a 3:1 v/v solution of methanol:glacial acetic acid for 2 days at 4 °C, washed in ice-cold water, and digested in a solution of 4% cellulase (Onozuka R10, Serva Electrophoresis, Heidelberg, Germany), 2% pectinase, and 0.4% pectolyase Y23 (both MP Biomedicals, Santa Ana, CA) in 0.01 M citrate buffer (pH 4.5) for 90 min at 37 °C. One to three digested meristems were transferred to a drop of freshly made 3:1 fixation solution on a glass slide and further macerated using a forceps. The slide was then placed over an alcohol flame to induce chromosome spreading as described by Dong et al. (2000). Following air-drying, the slides were stored at −20 °C. FISH was performed using either oligonucleotide probes that were 5′-labeled with biotin or Rhodamine Red-X during their synthesis (Integrated DNA Technologies, Leuven, Belgium), or using cloned fragments of satellite sequences labeled with biotin using nick-translation (Kato et al. 2006). Nucleotide sequences of the probes are provided in supplementary file 3, Supplementary Material online. FISH was performed as described (Macas et al. 2007) with hybridization and washing temperatures adjusted to account for AT/GC content and hybridization stringency allowing for 10–20% mismatches.
Immunolabeling of CENH3 proteins was performed with chromosomes isolated from the meristems fixed using 4% formaldehyde for 25 min at 23 °C Following fixation, suspensions of purified metaphase chromosomes were prepared as described (Neumann et al. 2002). Alternatively, the meristems were digested in a solution of 2% cellulase and 2% pectinase in phosphate-buffered saline (PBS) for 80–120 min at 28 °C, transferred to a glass slide, and squashed under the coverslip. Immunodetection was performed as follows, slides with chromosome suspensions and squash preparations were treated identically, and all incubations were performed at room temperature unless stated otherwise. Slides were washed in PBS for 5 min, PBS-T1 buffer (1× PBS, 0.5% Triton, pH 7.4) for 25 min, and twice in PBS for 5 min and once in PBS-T2 buffer (1× PBS, 0.1% Tween 20, pH 7.4) for 30 min. The slides were then incubated with the primary CENH3 antibody diluted 1:1,000 in PBS-T2 at 4 °C overnight, and then washed twice in PBS for 5 min and once in PBS-T2 for 5 min. The primary antibodies were detected with anti-rabbit-Rhodamine Red-X-AffiniPure (1:500, Jackson ImmunoResearch, Suffolk, UK; catalog number 111-295-144) or anti-chicken-DyLight488 (1:500, Jackson ImmunoResearch; catalog number 103-485-155) diluted in PBS-T2 buffer for 1 h. After two washes in PBS for 5 min and one wash in PBS-T2 for 5 min, the slides were mounted for observation or processed further if combined detection of DNA sequences by FISH was needed. In such cases, the slides were immediately postfixed in 4% formaldehyde in PBS for 10 min at RT and dehydrated in a series of 70% and 96% ethanol at RT for 5 min each. Chromosomes were denatured by incubation in 1× PCR buffer (Promega, Madison, WI) supplemented with 4 mM MgCl2 for 2 min at 94 °C and used for FISH as described earlier. The slides were counterstained with 4′,6-diamidino-2-phenylindole (DAPI), mounted in Vectashield mounting medium (Vector Laboratories, Burlingame, CA), and examined using a Zeiss AxioImager.Z2 microscope with an AxioCam 506 mono camera. Images were captured and processed using the ZEN pro 2012 software (Carl Zeiss GmbH).
Identification and Analysis of CENH3 Genes
Partial CENH3-coding sequences of V. ervilia, V. hirsuta, V. pisiformis, and V. tetrasperma were identified in Illumina sequence data by BlastN using a query containing all CENH3 sequences identified in Fabeae species previously (Neumann et al. 2012, 2015). Primers designed based on these sequences were then used for RT-PCR and RACE amplification of fragments of CENH3 transcripts, as described by Neumann et al. (2015). Finally, fragments surrounding the 5′ and 3′ end of the coding sequences were used to design primers for amplification of full-length CENH3-coding sequences. Sequences of these primers and details of the amplification conditions are provided in supplementary table 4, Supplementary Material online.
Entire CENH3 genes in V. ervilia and V. tetrasperma were selectively assembled using GRABb (Brankovics et al. 2016) using as input Illumina paired-end reads (2 × 151 nt) and a bait file containing all CENH3-coding sequences available in Fabeae. The CENH3 sequences were also identified in super-reads assembled from the Illumina paired-end reads by MaSuRCA (Zimin et al. 2013). Illumina sequence data used for assembly were custom-produced at Admera Health, LLC (South Plainfield, NJ), and deposited into the SRA database under accessions ERR3523145 and ERR3523144, respectively. Exon/intron structure of the genes and their translation products were predicted using est2genome (Rice et al. 2000) and GeneWise (Birney et al. 2004).
CENH3 sequences were aligned using Muscle (Edgar 2004). Pairwise similarities between CENH3 sequences were inferred from the proportions of variable sites (p-distances) calculated from CENH3 alignment in MEGA (Kumar et al. 2018). Phylogenetic analyses were performed using NJ and ML algorithms implemented in SeaView (Gouy et al. 2010) and PhyML 3.0 (Guindon et al. 2010), respectively. Bootstrap values were calculated from at least 1,000 replications. Phylogenetic trees were drawn and edited using the FigTree program (http://tree.bio.ed.ac.uk/software/figtree/; last accessed May 15, 2017). Tests for positive selection were carried out using the BUSTED (Murrell et al. 2015), FEL (Kosakovsky Pond and Frost 2005), and MEME (Murrell et al. 2012) tools implemented in the software package HyPhy (Kosakovsky Pond et al. 2005).
Availability of Sequence Data
Illumina reads from the ChIPed and control input samples are available in the European Nucleotide Archive (https://www.ebi.ac.uk/ena) under run accession numbers ERR3063140–ERR3063141, ERR3063378–ERR3063383, ERR3063416–ERR3063425, and ERR3063493–ERR3063500. The runs are associated with the study “Repeat characterization in Fabeae genomes” (PRJEB5241) which also includes the corresponding genomic NGS data. Newly identified CENH3 gene sequences are available from GenBank (https://www.ncbi.nlm.nih.gov/genbank/) under accession numbers MK415838–MK415841.
Supplementary Material
Acknowledgments
We thank Ms Vlasta Tetourová and Ms Jana Látalová for their excellent technical assistance. This work was supported by the grants from the Czech Science Foundation (Grant No. 17-09750S), the Czech Academy of Sciences (Grant No. RVO:60077344), and the ELIXIR CZ Research Infrastructure Project (Czech Ministry of Education, Youth and Sports; Grant No. LM2015047), including access to computing and storage facilities.
References
- Akera T, Chmátal L, Trimm E, Yang K, Aonbangkhen C, Chenoweth DM, Janke C, Schultz RM, Lampson MA.. 2017. Spindle asymmetry drives non-Mendelian chromosome segregation. Science 358(6363):668–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ.. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25(17):3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ávila Robledillo L, Koblížková A, Novák P, Böttinger K, Vrbová I, Neumann P, Schubert I, Macas J.. 2018. Satellite DNA in Vicia faba is characterized by remarkable diversity in its sequence composition, association with centromeres, and replication timing. Sci Rep. 8(1):5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badr SF. 2006. Karyotype analysis and chromosome evolution in species of Lathyrus (Fabaceae). Cytologia 71(4):447–455. [DOI] [PubMed] [Google Scholar]
- Bilinski P, Distor K, Gutierrez-Lopez J, Mendoza Mendoza G, Shi J, Dawe RK, Ross-Ibarra J.. 2015. Diversity and evolution of centromere repeats in the maize genome. Chromosoma 124(1):57–65. [DOI] [PubMed] [Google Scholar]
- Birney E, Clamp M, Durbin R.. 2004. GeneWise and Genomewise. Genome Res. 14(5):988–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brankovics B, Zhang H, van Diepeningen AD, van der Lee TAJ, Waalwijk C, de Hoog GS.. 2016. GRAbB: selective assembly of genomic regions, a new niche for genomic research. PLoS Comput Biol. 12(6):e1004753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseman I. 2014. The kinetochore. Cold Spring Harb Perspect Biol. 6(7):a015826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JL, Henikoff S.. 2004. Adaptive evolution of the histone fold domain in centromeric histones. Mol Biol Evol. 21(9):1712–1718. [DOI] [PubMed] [Google Scholar]
- Dellaporta SL, Wood J, Hicks JB.. 1983. A plant DNA minipreparation: version II. Plant Mol Biol Rep. 1(4):19–21. [Google Scholar]
- Dluhošová J, Ištvánek J, Nedělník J, Řepková J.. 2018. Red clover (Trifolium pratense) and zigzag clover (T. medium) – a picture of genomic similarities and differences. Front Plant Sci. 9:724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong F, Song J, Naess SK, Helgeson JP, Gebhardt C, Jiang J.. 2000. Development and applications of a set of chromosome-specific cytogenetic DNA markers in potato. Theor Appl Genet. 101:1001–1007. [Google Scholar]
- Duda Z, Trusiak S, O’Neill R.. 2017. Centromere transcription: means and motive In: Black BE, editor. Centromeres and kinetochores, progress in molecular and subcellular biology. Cham (Switzerland: ): Springer; p. 257–281. [DOI] [PubMed] [Google Scholar]
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32(5):1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finseth FR, Dong Y, Saunders A, Fishman L.. 2015. Duplication and adaptive evolution of a key centromeric protein in Mimulus, a genus with female meiotic drive. Mol Biol Evol. 32(10):2694–2706. [DOI] [PubMed] [Google Scholar]
- Fuchs J, Schubert I.. 2012. Chromosomal distribution and functional interpretation of epigenetic histone marks in plants In: Bass HW, Birchler JA, editors. Plant Cytogenetics. New York: Springer; p. 232–246. [Google Scholar]
- Garrido-Ramos MA. 2015. Satellite DNA in plants: more than just rubbish. Cytogenet Genome Res. 146(2):153–170. [DOI] [PubMed] [Google Scholar]
- Garrido-Ramos MA. 2017. Satellite DNA: an evolving topic. Genes. 8(9):230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gent JI, Wang N, Dawe RK.. 2017. Stable centromere positioning in diverse sequence contexts of complex and satellite centromeres of maize and wild relatives. Genome Biol. 18(1):121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Z, Wu Y, Koblížková A, Torres GA, Wang K, Iovene M, Neumann P, Zhang W, Novák P, Buell CR, et al. 2012. Repeatless and repeat-based centromeres in potato: implications for centromere evolution. Plant Cell 24(9):3559–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouy M, Guindon S, Gascuel O.. 2010. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 27(2):221–224. [DOI] [PubMed] [Google Scholar]
- Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O.. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 59(3):307–321. [DOI] [PubMed] [Google Scholar]
- Han F, Lamb JC, Mccaw ME, Gao Z, Zhang B, Swyers NC, Birchler JA, Anderson L.. 2018. Meiotic studies on combinations of chromosomes with different sized centromeres in maize. Front Plant Sci. 9:785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara M, Fukagawa T.. 2017. Critical foundation of the kinetochore: the Constitutive Centromere-Associated Network (CCAN) In: Black BE, editor. Centromeres and kinetochores, progress in molecular and subcellular biology. Vol. 56 Cham (Switzerland: ): Springer; p. 29–57. [DOI] [PubMed] [Google Scholar]
- Hartley G, O’Neill RJ.. 2019. Centromere repeats: hidden gems of the genome. Genes 10(3):223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckmann S, Macas J, Kumke K, Fuchs J, Schubert V, Ma L, Novák P, Neumann P, Taudien S, Platzer M, et al. 2013. The holocentric species Luzula elegans shows interplay between centromere and large-scale genome organization. Plant J. 73(4):555–565. [DOI] [PubMed] [Google Scholar]
- Henikoff S, Ahmad K, Malik HS.. 2001. The centromere paradox: stable inheritance with rapidly evolving DNA. Science 293(5532):1098–1102. [DOI] [PubMed] [Google Scholar]
- Hirsch CD, Wu Y, Yan H, Jiang J.. 2009. Lineage-specific adaptive evolution of the centromeric protein CENH3 in diploid and allotetraploid Oryza species. Mol Biol Evol. 26(12):2877–2885. [DOI] [PubMed] [Google Scholar]
- Iwata-Otsubo A, Dawicki-McKenna JM, Akera T, Falk SJ, Chmátal L, Yang K, Sullivan BA, Schultz RM, Lampson MA, Black BE.. 2017. Expanded satellite repeats amplify a discrete CENP-A nucleosome assembly site on chromosomes that drive in female meiosis. Curr Biol. 27(15):2365–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasinathan S, Henikoff S.. 2018. Non-B-form DNA is enriched at centromeres. Mol Biol Evol. 35(4):949–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato A, Kato A, Albert PS, Vega JM, Kato A, Albert PS, Vega JM, Birchler JA.. 2006. Sensitive fluorescence in situ hybridization signal detection in maize using directly labeled probes produced by high concentration DNA polymerase nick translation. Biotech Histochem. 81(2–3):71–78. [DOI] [PubMed] [Google Scholar]
- Kawabe A, Nasuda S, Charlesworth D.. 2006. Duplication of centromeric histone H3 (HTR12) gene in Arabidopsis halleri and A. lyrata, plant species with multiple centromeric satellite sequences. Genetics 174(4):2021–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Frost S.. 2005. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol. 22(5):1208–1222. [DOI] [PubMed] [Google Scholar]
- Kowar T, Zakrzewski F, Macas J, Koblížková A, Viehoever P, Weisshaar B, Schmidt T.. 2016. Repeat composition of CenH3-chromatin and H3K9me2-marked heterochromatin in sugar beet (Beta vulgaris). BMC Plant Biol. 16(1):120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K.. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 35(6):1547–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin M, Herendeen PS, Wojciechowski MF.. 2005. Evolutionary rates analysis of Leguminosae implicates a rapid diversification of lineages during the Tertiary. Syst Biol. 54(4):575–594. [DOI] [PubMed] [Google Scholar]
- Lee H-R, Zhang W, Langdon T, Jin W, Yan H, Cheng Z, Jiang J.. 2005. Chromatin immunoprecipitation cloning reveals rapid evolutionary patterns of centromeric DNA in Oryza species. Proc Natl Acad Sci U S A. 102(33):11793–11798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefort V, Longueville J-E, Gascuel O.. 2017. SMS: smart model selection in PhyML. Mol Biol Evol. 34(9):2422–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lermontova I, Sandmann M, Demidov D.. 2014. Centromeres and kinetochores of Brassicaceae. Chromosome Res. 22(2):135–152. [DOI] [PubMed] [Google Scholar]
- Letunic I, Bork P.. 2019. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47(W1):W256–W259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logsdon GA, Gambogi CW, Liskovykh MA, Barrey EJ, Larionov V, Miga KH, Heun P, Black BE.. 2019. Human artificial chromosomes that bypass centromeric DNA. Cell 178(3):624–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macas J, Koblížková A, Navrátilová A, Neumann P.. 2009. Hypervariable 3’ UTR region of plant LTR-retrotransposons as a source of novel satellite repeats. Gene 448(2):198–206. [DOI] [PubMed] [Google Scholar]
- Macas J, Mészáros T, Nouzová M.. 2002. PlantSat: a specialized database for plant satellite repeats. Bioinformatics 18(1):28–35. [DOI] [PubMed] [Google Scholar]
- Macas J, Neumann P, Navrátilová A.. 2007. Repetitive DNA in the pea (Pisum sativum L.) genome: comprehensive characterization using 454 sequencing and comparison to soybean and Medicago truncatula. BMC Genomics 8(1):427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macas J, Novák P, Pellicer J, Čížková J, Koblížková A, Neumann P, Fuková I, Doležel J, Kelly LJ, Leitch IJ.. 2015. In depth characterization of repetitive DNA in 23 plant genomes reveals sources of genome size variation in the legume tribe Fabeae. PLoS One 10(11):e0143424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maheshwari S, Ishii T, Brown CT, Houben A, Comai L.. 2017. Centromere location in Arabidopsis is unaltered by extreme divergence in CENH3 protein sequence. Genome Res. 27(3):471–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik HS. 2009. The centromere-drive hypothesis: a simple basis for centromere complexity. Prog Mol Subcell Biol. 48:33–52. [DOI] [PubMed] [Google Scholar]
- Masonbrink RE, Gallagher JP, Jareczek JJ, Renny-Byfield S, Grover CE, Gong L, Wendel JF.. 2014. CenH3 evolution in diploids and polyploids of three angiosperm genera. BMC Plant Biol. 14(1):383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarlane RJ, Humphrey TC.. 2010. A role for recombination in centromere function. Trends Genet. 26(5):209–213. [DOI] [PubMed] [Google Scholar]
- McNulty SM, Sullivan BA.. 2018. Alpha satellite DNA biology: finding function in the recesses of the genome. Chromosome Res. 26(3):115–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello B. 2018. Estimating TimeTrees with MEGA and the TimeTree Resource. Mol Biol Evol. 35(9):2334–2342. [DOI] [PubMed] [Google Scholar]
- Murrell B, Weaver S, Smith MD, Wertheim JO, Murrell S, Aylward A, Eren K, Pollner T, Martin DP, Smith DM, et al. 2015. Gene-wide identification of episodic selection. Mol Biol Evol. 32(5):1365–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell B, Wertheim JO, Moola S, Weighill T, Scheffler K, Kosakovsky Pond SL.. 2012. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 8(7):e1002764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann P, Navrátilová A, Schroeder-Reiter E, Koblížková A, Steinbauerová V, Chocholová E, Novák P, Wanner G, Macas J.. 2012. Stretching the rules: monocentric chromosomes with multiple centromere domains. PLoS Genet. 8(6):e1002777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann P, Nouzová M, Macas J.. 2001. Molecular and cytogenetic analysis of repetitive DNA in pea (Pisum sativum L.). Génome 44(4):716–728. [PubMed] [Google Scholar]
- Neumann P, Novák P, Hoštáková N, Macas J.. 2019. Systematic survey of plant LTR-retrotransposons elucidates phylogenetic relationships of their polyprotein domains and provides a reference for element classification. Mob DNA. 10:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann P, Pavlíková Z, Koblížková A, Fuková I, Jedličková V, Novák P, Macas J.. 2015. Centromeres off the hook: massive changes in centromere size and structure following duplication of CENH3 gene in Fabeae species. Mol Biol Evol. 32(7):1862–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann P, Požárková D, Vrána J, Doležel J, Macas J.. 2002. Chromosome sorting and PCR-based physical mapping in pea (Pisum sativum L.). Chromosome Res. 10(1):63–71. [DOI] [PubMed] [Google Scholar]
- Novák P, Ávila Robledillo L, Koblížková A, Vrbová I, Neumann P, Macas J.. 2017. TAREAN: a computational tool for identification and characterization of satellite DNA from unassembled short reads. Nucleic Acids Res. 45(12):e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novák P, Neumann P, Pech J, Steinhaisl J, Macas J.. 2013. RepeatExplorer: a Galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinformatics 29(6):792–793. [DOI] [PubMed] [Google Scholar]
- Oliveira LC, Torres GA.. 2018. Plant centromeres: genetics, epigenetics and evolution. Mol Biol Rep. 45(5):1491–1497. [DOI] [PubMed] [Google Scholar]
- Park PJ. 2009. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet. 10(10):669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea-Resa C, Blower MD.. 2018. Centromere biology: transcription goes on stage. Mol Cell Biol. 38:e00263–e00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piras FM, Nergadze SG, Magnani E, Bertoni L, Attolini C, Khoriauli L, Raimondi E, Giulotto E.. 2010. Uncoupling of satellite DNA and centromeric function in the genus Equus. PLoS Genet. 6(2):e1000845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plohl M, Meštrović N, Mravinac B.. 2014. Centromere identity from the DNA point of view. Chromosoma 123(4):313–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pond SLK, Frost SDW, Muse SV.. 2005. HyPhy: hypothesis testing using phylogenies. Bioinformatics 21(5):676–679. [DOI] [PubMed] [Google Scholar]
- Reinert G, Chew D, Sun F, Waterman MS.. 2009. Alignment-free sequence comparison (I): statistics and power. J Comput Biol. 16(12):1615–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A.. 2000. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16(6):276–277. [DOI] [PubMed] [Google Scholar]
- Schaefer H, Hechenleitner P, Santos-Guerra A, de Sequeira MM, Pennington RT, Kenicer G, Carine MA.. 2012. Systematics, biogeography, and character evolution of the legume tribe Fabeae with special focus on the middle-Atlantic island lineages. BMC Evol Biol. 12(1):250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider KL, Xie Z, Wolfgruber TK, Presting GG.. 2016. Inbreeding drives maize centromere evolution. Proc Natl Acad Sci U S A. 113(8):E987–E996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnhammer EL, Durbin R.. 1995. A dot-matrix program with dynamic threshold control suited for genomic DNA and protein sequence analysis. Gene 167(1–2):GC1–GC10. [DOI] [PubMed] [Google Scholar]
- Talbert P, Kasinathan S, Henikoff S.. 2018. Simple and complex centromeric satellites in Drosophila sibling species. Genetics 208(3):977–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vondrak T, Ávila Robledillo L, Novák P, Koblížková A, Neumann P, Macas J.. 2020. Characterization of repeat arrays in ultra-long nanopore reads reveals frequent origin of satellite DNA from retrotransposon-derived tandem repeats. Plant J. 101(2):484–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei K-C, Reddy HM, Rathnam C, Lee J, Lin D, Ji S, Mason JM, Clark AG, Barbash DA.. 2017. A pooled sequencing approach identifies a candidate meiotic driver in Drosophila. Genetics 206(1):451–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Zhao H, Zhang T, Zeng Z, Zhang P, Zhu B, Han Y, Braz GT, Casler MD, Schmutz J, et al. 2018. Amplification and adaptation of centromeric repeats in polyploid switchgrass species. New Phytol. 218(4):1645–1657. [DOI] [PubMed] [Google Scholar]
- Yu F, Dou Q, Liu R, Wang H.. 2017. A conserved repetitive DNA element located in the centromeres of chromosomes in Medicago genus. Genes Genom. 39(8):903–911. [Google Scholar]
- Zatloukalová P, Hřibová E, Kubaláková M, Suchánková P, Šimková H, Adoración C, Kahl G, Millán T, Doležel J.. 2011. Integration of genetic and physical maps of the chickpea (Cicer arietinum L.) genome using flow-sorted chromosomes. Chromosome Res. 19(6):729–739. [DOI] [PubMed] [Google Scholar]
- Zedek F, Bureš P.. 2016. CenH3 evolution reflects meiotic symmetry as predicted by the centromere drive model. Sci Rep. 6:33308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Dong Q, Su H, Birchler JA, Han F.. 2014. Histone phosphorylation: its role during cell cycle and centromere identity in plants. Cytogenet Genome Res. 143(1–3):144–149. [DOI] [PubMed] [Google Scholar]
- Zhang H, Koblížková A, Wang K, Gong Z, Oliveira L, Torres GA, Wu Y, Zhang W, Novák P, Buell CR, et al. 2014. Boom-bust turnovers of megabase-sized centromeric DNA in Solanum species: rapid evolution of DNA sequences associated with centromeres. Plant Cell 26(4):1436–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Talbert PB, Zhang W, Wu Y, Yang Z, Henikoff JG, Henikoff S, Jiang J.. 2013. The CentO satellite confers translational and rotational phasing on cenH3 nucleosomes in rice centromeres. Proc Natl Acad Sci U S A. 110(50):E4875–E4883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimin AV, Marçais G, Puiu D, Roberts M, Salzberg SL, Yorke JA.. 2013. The MaSuRCA genome assembler. Bioinformatics 29(21):2669–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.