

Abstract
Impairment of PINK1/parkin‐mediated mitophagy is currently proposed to be the molecular basis of mitochondrial abnormality in Parkinson's disease (PD). We here demonstrate that PINK1 directly phosphorylates Drp1 on S616. Drp1S616 phosphorylation is significantly reduced in cells and mouse tissues deficient for PINK1, but unaffected by parkin inactivation. PINK1‐mediated mitochondrial fission is Drp1S616 phosphorylation dependent. Overexpression of either wild‐type Drp1 or of the phosphomimetic mutant Drp1S616D, but not a dephosphorylation‐mimic mutant Drp1S616A, rescues PINK1 deficiency‐associated phenotypes in Drosophila. Moreover, Drp1 restores PINK1‐dependent mitochondrial fission in ATG5‐null cells and ATG7‐null Drosophila. Reduced Drp1S616 phosphorylation is detected in fibroblasts derived from 4 PD patients harboring PINK1 mutations and in 4 out of 7 sporadic PD cases. Taken together, we have identified Drp1 as a substrate of PINK1 and a novel mechanism how PINK1 regulates mitochondrial fission independent of parkin and autophagy. Our results further link impaired PINK1‐mediated Drp1S616 phosphorylation with the pathogenesis of both familial and sporadic PD.
Keywords: autophagy, human dermal fibroblasts, mitochondrial dynamics, parkin, Parkinson’s disease
Subject Categories: Autophagy & Cell Death; ; Post-translational Modifications, Proteolysis & Proteomics
PINK1 regulates mitochondrial dynamics by directly phosphorylating Drp1S616.
Introduction
Mutations in PINK1 or parkin cause early‐onset familial form of Parkinson's disease (PD), the second most common neurodegenerative disease 1. PINK1 is a putative serine/threonine kinase located on mitochondria. PINK1 recruits parkin to mitochondria to activate its E3 ligase by phosphorylating both parkin and ubiquitin upon stress 2, 3, 4. This results in activation of parkin E3 ligase and ubiquitination of mitochondrial outer membrane proteins, leading to eventual clearance of damaged mitochondria via mitophagy 5, 6, 7, 8.
Mitochondrial morphology, size, and position within cells are maintained through a balance of fission and fusion. Perturbation of the steady state between these opposing processes has been implicated in several human disorders 9. In general, unopposed fission causes mitochondrial fragmentation that is associated with metabolic dysfunction and disease. Unopposed fusion results in a hyperfused network to counteract metabolic insults, preserve cellular integrity, and protect against autophagy. A number of genes are involved in regulating fission and fusion 10. Among them, mitofusins (Mfn1 and Mfn2) orchestrate mitochondrial outer membrane fusion and are required for the maintenance of a reticular mitochondrial network in cells, while mitochondrial inner membrane protein Opa1 regulates mitochondrial inner membrane fusion. Dynamin‐related protein 1 (Drp1), a cytosolic dynamin GTPase, is the key player for mitochondrial fission. It is well documented that Drp1 phosphorylation plays an important role in controlling mitochondrial fission 11, 12. Phosphorylation of Drp1 at S616 site promotes fission during mitosis and oxidative stress 13, 14. In contrast, PKA‐mediated phosphorylation of Drp1 at S637 site results in fusion under nutrition starvation 15. CDK1/cyclin B 16, PKCdelta 17, CDK5 18, CaMKII, and Erk2 are shown to phosphorylate Drp1 at S616 19, 20. PKA and CaMKI can phosphorylate Drp1 at S637 5, 21. Recent studies have shown that overexpression of PINK1 promotes mitochondrial fission, whereas inactivation of PINK1 leads to excessive fusion 22. In Drosophila, both PINK1 and parkin are shown to genetically interact with multiple genes in mitochondrial fusion/fission machinery, including Drp1, Marf, and Opa1, to regulate mitochondrial fission 22, 23, 24. Nevertheless, the molecular mechanism of this regulation remains unclear.
In this study, we aim to determine the molecular basis of PINK1 regulating mitochondrial dynamics. We demonstrate that PINK1 directly phosphorylates Drp1 at S616 site. PINK1 inactivation, but not parkin inactivation, suppresses Drp1S616 phosphorylation in cells, mouse, and Drosophila tissues. PINK1‐mediated mitochondrial fission is Drp1S616 phosphorylation dependent. Overexpression of wild‐type Drp1 and a phosphorylation‐mimic mutant Drp1S616D, but not a dephosphorylation‐mimic mutant Drp1S616A, can rescue PINK1 deficiency‐associated mitochondrial fission and cell death phenotypes in Drosophila. Moreover, Drp1 rescuing PINK1‐deficient phenotypes is ATG5 and ATG7 independent. Significantly, reduced detection of Drp1S616 phosphorylation is shown in fibroblasts derived from 4 PD patients harboring PINK1 mutants and 4 out of 7 sporadic PD cases. This study identifies a novel mechanism of PINK1 in regulating mitochondrial dynamics that likely contributes to PD pathogenesis.
Results
PINK1 phosphorylates Drp1S616
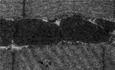
To determine the molecular basis of PINK1/parkin‐mediated mitochondrial dynamics, we immunodetected phosphorylation of Drp1 in PINK1‐null (PINK1KO) and parkin‐null (parkinKO) HEK293 cells (Appendix Fig S1A). Results revealed a ~50% decrease of Drp1 serine 616 phosphorylation (Drp1S616) in PINK1KO, but not in parkinKO HEK293 cells (Fig 1A, B, N and O, Appendix Figs S1A and S2A and B). A significant reduction of Drp1S616 phosphorylation was also detected in PINK1KO mouse embryonic fibroblasts (MEFs), but not in parkinKO MEFs (Fig 1C and D, Appendix Fig S1B) 25. Similar observation was made in 18‐month‐old mouse substantial nigra with PINK1KO, but not in those with parkinKO (Fig 1E and F, Appendix Fig S2C). Moreover, Drp1S616 phosphorylation was colocalized with tyrosine hydroxylase‐positive neuron in substantial nigra, which was markedly reduced in substantial nigra with PINK1KO (Appendix Fig S2C). Interestingly, reduction of Drp1S616 phosphorylation in substantial nigra was in aging‐dependent manner with being detected only in mice with > 12 months (Appendix Fig S2D and E). To further verify that the decreased phosphorylation is PINK1‐dependent, we overexpressed PINK1 variants in PINK1KO HEK293 cells. Results showed that decreased Drp1S616 phosphorylation was effectively recovered with expression of wild‐type PINK1, but not a PINK1 kinase‐dead mutant D384N, a Parkinson's disease pathogenic mutant G309D, nor an N‐terminal 110 aa deletion mutant (Fig 1G and H). CCCP treatment induced 2.5‐fold more increase of Drp1S616 phosphorylation in PINK1WT HEK293 cells than that in PINK1KO HEK293 cells (Appendix Fig S3). A previous study suggests that PINK1 regulates AKAP1/PKA‐mediated phosphorylation at serine 637(Drp1S637) 26. We compared levels of phosphorylation of Drp1S616 and Drp1S637 in PINK1KO cells and mouse tissues. Results showed that down‐regulation of Drp1S616 phosphorylation corresponding to significant elevated Drp1S637 phosphorylation in PINK1KO cells and mouse substantial nigra (Fig 1A–F, Appendix Fig S4A–F). CDK1 and ERK1/2 are also shown to phosphorylate Drp1S616 18, 19. However, little change of their expression and activity‐related phosphorylation was detected in PINK1KO cells (Fig 1I and J). Nevertheless, both CDK1 inhibitor Ro‐3306 and ERK1/2 inhibitor ulixertinib partially inhibited Drp1S616 phosphorylation in PINK1WT and PINK1KO HEK293 cells, although they appeared to be toxic to cause detachment of PINK1KO cells from culture dishes (Appendix Fig S5), further suggesting PINK1 as a kinase of Drp1S616. Together, these results indicate that PINK1 regulates a parkin‐independent phosphorylation of Drp1S616. Sequence alignment revealed that the amino acids surrounding Drp1S616 were evolutionarily conserved (Fig 1K). In vitro, a GST‐Drp1 fusion protein, but not a GST‐Drp1S616A mutation protein, was directly phosphorylated by a recombinant Tribolium castaneum PINK1 (TcPINK1) (Fig 1L and M, Appendix Fig S4G–I). Mass spectrometry analysis and ATPγS in vitro kinase assay unequivocally confirmed phosphorylation of Drp1S616 by TcPINK1 (Appendix Fig S4G–I). Semi‐quantitative mass spectrometry analysis revealed that Drp1S616 phosphorylation was markedly reduced to 50% in PINK1KO HEK293 cells comparing to their wild‐type control cells (Fig 1N and O). Consistently, Phos‐tag gel analysis showed approximately 50% recombinant Drp1 fragment was phosphorylated by TcPINK1 in a 60‐min reaction, while none phosphorylation of Drp1S616A was detected (Appendix Fig S6). The serine 65 of ubiquitin (Ub) is phosphorylated by PINK1 2, 4, 27. Next, PINK1‐mediated phosphorylation of Ub and Drp1 was compared using Phos‐tag gel analysis. Results showed that Km of TcPINK1‐mediated Drp1S616 phosphorylation is 287.5 ± 75.5 μM and Km of TcPINK1‐mediated ubiquitin phosphorylation is 84.4 ± 21.0 μM. Kcat of TcPINK1‐mediated Drp1S616 phosphorylation (4.6 ± 0.7/min) is about fivefold lower than that of TcPINK1‐mediated ubiquitin phosphorylation (20.9 ± 1.9/min) (Fig 1P and Q). Together, results suggest that PINK1 directly phosphorylates Drp1 at serine 616 site.
Figure 1. PINK1 phosphorylates Drp1S616 .

-
A–FDecreased Drp1S616 phosphorylation in PINK1‐null HEK293 cells, MEF cells, and mouse substantial nigra tissues. Lysates of PINK1‐null (PINK1KO) and parkin‐null (parkinKO) HEK293 cells (A), MEF cells (C), mouse substantial nigra tissues (E), and their matched wild‐type controls (WT) were immunodetected with antibodies against either phospho (Ser616)‐Drp1 (pS616), phospho (Ser637)‐Drp1 (pS637), or Drp1. β‐actin was detected as a loading control. Quantitation of pS616/Drp1 for each experiment is shown (B, D, F, respectively). Two independent PINK1KO and parkinKO HEK293 cell lines (1, 2) and substantial nigra from two separate PINK1KO and parkinKO mice (1, 2) were shown. Student's test. *P < 0.05, ***P < 0.001, ns: no significance. Data were presented as mean ± SEM of three independent experiments.
-
G, HOverexpression of PINK1, not PINK1 kinase mutants, reverses Drp1S616 phosphorylation in PINK1KO HEK293 cells. Lysates of PINK1KO and PINK1WT control (WT) HEK293 cells expressing PINK1 variants were immunodetected with an anti‐phospho(Ser616)‐Drp1 antibody (pS616), an anti‐Drp1 antibody (Drp1), and an anti‐PINK1 antibody (PINK1). Cells with mock transfection (Blank) and transfected with an empty plasmid (3.1) were included as controls (G). Quantitation of pS616/Drp1 is shown (H). PINK1: wild‐type PINK1; D384N: PINK1 kinase‐dead mutant; G309D, pathogenic PINK1 mutant; ∆110: PINK1 mitochondrial target‐deficient mutant. Student's test. ***P < 0.001, ns: no significance. Data were presented as mean ± SEM of three independent experiments.
-
I, JPINK1 deficiency does not affect expression and kinase activity of CDK1 and ERK1/2. Lysates of PINK1KO and PINK1WT control (WT) HEK293 cells were immunodetected with an anti‐phospho (Ser616)‐Drp1 antibody (pS616), an anti‐Drp1 antibody (Drp1), an anti‐PINK1 antibody (PINK1), an anti‐CDK1 antibody(CDK1), an anti‐p44/p42 MAPK antibody (ERK1/2), an anti‐phospho (Thr161)‐CDK1 antibody (pThr161), and an anti‐phospho(Thr202/204)‐p44/p42 MAPK antibody (pThr202/204). β‐actin was detected as a loading control (I). Quantitation of CDK1Thr161 phosphorylation (pThr161) and ERK1/2Thr202/204 phosphorylation (pThr202/204) was shown (J). Student's test. ns: no significance. Data were presented as mean ± SEM of three independent experiments.
-
KAlignment of human Drp1S616 in different species. Alignment of human Drp1 (isoform 1) amino acids 612–620 with indicated species. Serine residual (S) corresponding to human Drp1Ser616 was labeled as red.
-
L, MPINK1 phosphorylates Drp1S616 in vitro. Recombinant TcPINK1 was incubated with either GST‐Drp1518‐736 (WT) or GST‐Drp1518‐736, S616A (S616A) in the presence (+)/absence (−) of ATP. After heat inactivation, reactions were treated with (+)/without (−) shrimp alkaline phosphatase (SAP). Phosphorylation of Drp1S616 (pS616) was immunodetected (L, upper panel). Total Drp1 (L, middle panel) and protein loading stained with Coomassie blue staining (L, lower panel) were shown. Quantitation of pS616/Drp1 ratio was shown (M). One‐way ANOVA followed with Tukey's test. ***P < 0.001. Data were presented as mean ± SEM of three independent experiments.
-
N, OLabel‐free relative quantitative mass spectrum analysis of Drp1S616 phosphorylation. Representative spectra for the Drp1 phospho‐peptide (S616) SKPIPMPA(p)SPQK and non‐phospho‐peptide SKPIPMPA(p)SPQK. Spectra obtained for peptides from PINK1WT HEK293 cells are shown (N). The relative ratios of the phospho‐peptides containing phosphor‐Ser616 to peptides containing Ser616 from PINK1WT and PINK1KO HEK293 samples were calculated and plotted (O).
-
P, QKm and Kcat for PINK1‐mediated phosphorylation of ubiquitin and Drp1. In vitro kinase assays were performed with different concentrations of ubiquitin (P) or Drp1 (Q) with TcPINK1 at fixed ATP concentration (500 μM). Results were analyzed on phos‐tag gels and modeled to the Michaelis–Menten equation. The given graphs represent global fits to data collected from 3 sets of reactions for both Ub and Drp1 performed independently. Km: Michaelis constant. Kcat: catalytic constant. Bars represent the mean ± SEM.
PINK1 regulates mitochondrial fission via phosphorylating Drp1S616
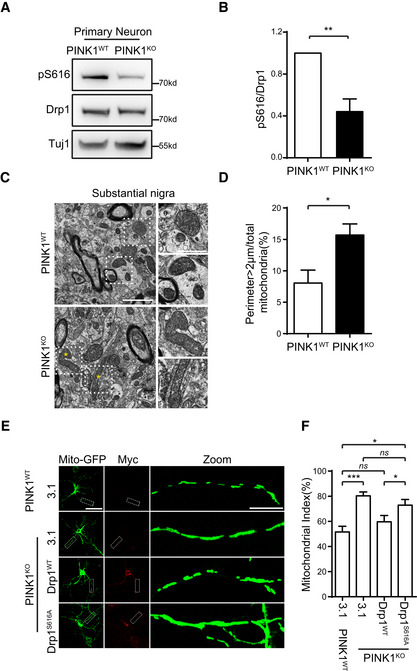
Inactivation of PINK1 resulted in elongated mitochondria in neuron 28. We next examined whether that abnormal mitochondrial morphology in PINK1KO neurons is caused by decreased activity of Drp1 via reducing PINK1‐mediated Drp1S616 phosphorylation (Fig 2A and B, Appendix Fig S1B). Transmission electron microscope (TEM) analysis revealed about a twofold increase of finding large mitochondria with perimeter > 2 μm in substantial nigra of PINK1KO mice than their wild‐type littermates (Fig 2C and D). Consistently, mitochondrial index is significantly increased in primary cortical neuronal cultures generated from PINK1KO mice than that of those neurons generated from their wild‐type littermates (Fig 2E and F, Appendix Fig S1B). Expression of Drp1wt, but not Drp1S616A, led to mitochondrial fragmentation and reduced mitochondrial index in PINK1KO neuronal cultures (Fig 2E and F). Results suggest that PINK1‐mediated Drp1S616 phosphorylation regulates mitochondrial dynamics in neurons.
Figure 2. Drp1, but not Drp1S616A, reverses abnormal dendritic mitochondrial fission in PINK1‐null mouse neurons.
-
A, BDecreased Drp1S616 phosphorylation in PINK1‐null primary neurons. Lysates of DIV6 neurons generated from PINK1KO and PINK1WT control mice were immune‐detected with antibodies against either phospho (Ser616)‐Drp1 (pS616) or Drp1. Tuj1 was detected as a loading control (A). Quantitation of pS616/Drp1 for each experiment is shown (B). Student's test. **P < 0.01. Data were presented as mean ± SEM of three independent experiments.
-
C, DTEM analysis of mitochondria in mouse substantial nigra. Sections of substantial nigra from 18‐month‐old PINK1KO and PINK1WT control mice were analyzed under TEM (C, left panels, scale bar = 2 μm). Amplified images of mitochondria from dot line frames gated region are shown (C, right panels, scale bar = 1 μm). Asterisks indicate abnormal elongated mitochondria. A quantitative analysis of mitochondria with perimeter > 2 μm is shown (D). Over 300 identifiable mitochondria (cristae and/or double membrane) per mice were randomly and blindly selected for perimeter measurement. Student's test. *P < 0.05. Data were presented as mean ± SEM of three independent experiments.
-
E, FOverexpression of Drp1WT, but not phosphorylation mutant Drp1S616A, suppresses mitochondrial fusion in dendrites of PINK1KO primary neurons. (E) DIV4 neurons generated from PINK1WT control or PINK1KO mice were cotransfected mito‐GFP with either pcDNA3.1 (3.1) or Drp1WT or Drp1S616A. Neurons were immunodetected with an antibody against myc‐tag (Myc) (red, to detect exogenous Drp1). Mito‐GFP: green, to detect mitochondria. Representative images were shown, scale bar = 25 μm. Magnified dendritic shafts gated by a dot line frame are shown (Zoom, right panels, scale bar = 5 μm) (F). Quantification of mito index in dendrites of neurons from experiments of (E). Ten neurons were randomly selected and analyzed in each experiment. One‐way ANOVA followed with Tukey's test. *P < 0.05, ***P < 0.001, ns: no significance. Data were presented as mean ± SEM of three independent experiments.
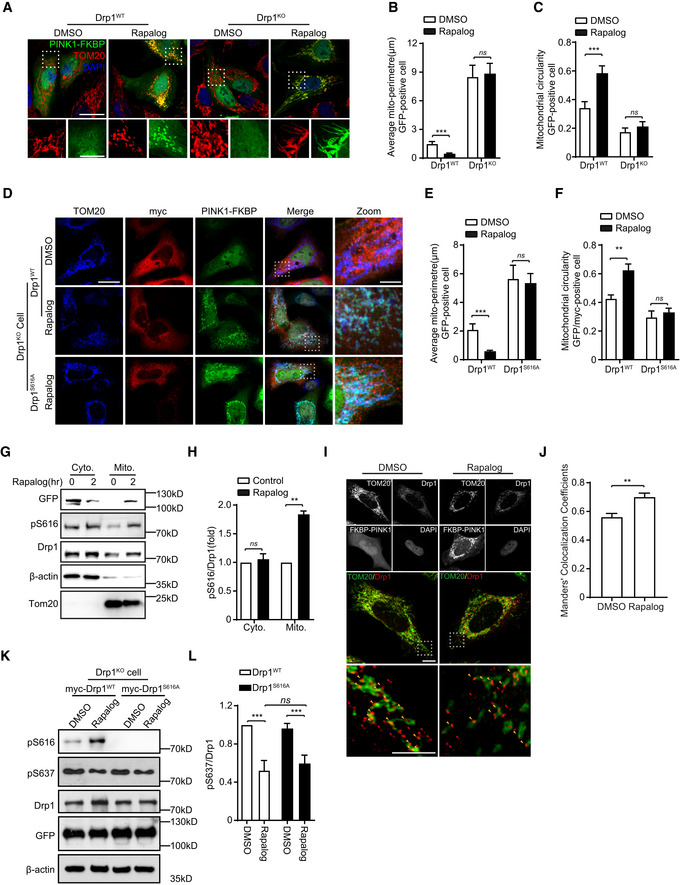
To further confirm roles of PINK1‐mediated Drp1S616 phosphorylation in mitochondrial dynamics, we employed a regulated heterodimerization inducible system 29, 30 (Appendix Fig S7A). HeLa cells are cotransfected FRB‐Fis1 with either ∆110‐PINK1(WT)‐GFP‐FKBP (a PINK1 with kinase activity) or ∆110‐PINK1(D384N)‐GFP‐FKBP (a PINK1 kinase‐dead mutant) (Appendix Fig S7A). After rapalog induction, ∆110‐PINK1(WT)‐GFP‐FKBP was recruited to mitochondria leading to mitochondrial fragmentation. Likewise, kinase‐dead ∆110‐PINK1(D384N)‐GFP‐FKBP mutant was also recruited to mitochondria after induction. However, recruitment of ∆110‐PINK1(D384N) did not cause obvious mitochondrial fragmentation (Appendix Fig S7D–F). Consistently, PINK1WT, but not the kinase‐dead PINK1 mutant, increased Drp1S616 phosphorylation after rapalog induction (Appendix Fig S7B and C). Expression of PINK1 did not induce mitochondrial fragmentation in Drp1KO HeLa cells after induction (Fig 3A–C, Appendix Fig S1E and F). Re‐introduction of Drp1WT, but not Drp1S616A, rescued PINK1‐induced mitochondrial fragmentation phenotype in Drp1KO cells (Fig 3D–F). Consistently, cells expressing PINK1 induced by rapalog resulted in more phosphorylated Drp1S616 to mitochondria than cells did without induction (Fig 3G–J). Furthermore, Drp1S637 phosphorylation was inhibited in Drp1KO HEK293 cells expressing either Drp1WT or Drp1S616A, while mitochondrial fragmentation was observed only in cells expressing Drp1WT but not in cells expressing Drp1S616A (Fig 3K and L). Results indicate that PINK1‐regulated mitochondrial fragmentation is not Drp1S637 phosphorylation dependent. Thus, PINK1 regulates mitochondrial fragmentation via phosphorylating Drp1S616.
Figure 3. Phosphorylation of Drp1S616 is essential for PINK1‐mediated mitochondrial fission.
-
A–CDrp1 is required for mitochondrial fission induced by PINK1. Drp1‐null HeLa cells (Drp1KO) and their wild‐type controls (Drp1WT) were cotransfected Δ110‐PINK1‐GFP‐FKBP with FRB‐MTS. Cells were induced with 250 nM rapalog for 2 h to activate PINK1 kinase, followed by immunodetecting mitochondria (TOM20, red) and Δ110‐PINK1‐GFP‐FKBP/FRB‐MTS (Δ110‐PINK1, green). Nuclei were labeled with DAPI (blue). Cells treated with solvent (DMSO) were used as a treatment control. Representative images were shown (A, upper panel, scale bar = 25 μm). Higher magnification images are also included (A, lower panel, scale bar = 10 μm). Mitochondrial morphology in different transfections was quantified. (B, C). Student's test. ***P < 0.001, ns: no significance. Data were presented as mean ± SEM of three independent experiments. For each condition, > 100 cells were analyzed.
-
D–FDrp1S616 phosphorylation is required for mitochondrial fission induced by PINK1. Drp1KO HeLa cells were cotransfected Δ110‐PINK1‐GFP‐FKBP/FRB‐MTS with either a plasmid encoding Drp1WT or a Drp1 dephosphorylation‐mimic mutant Drp1S616A. Cells were treated with 250 nM rapalog for 2 h to activate PINK1 kinase, followed by immunodetecting mitochondria (TOM20, blue), exogenous Drp1 (myc, red), Δ110‐PINK1‐GFP‐FKBP/FRB‐MTS (PINK1‐FKBP, green). Cells treated with solvent (DMSO) were used as a treatment control. Representative images were shown, scale bar = 25 μm (D). Higher magnification images are also included (D, Zoom, scale bar = 5 μm). Mitochondrial morphology in different transfections was quantified (E, F). Student's test. **P < 0.01, ***P < 0.001, ns: no significance. Data were presented as mean ± SEM of three independent experiments. For each condition, > 100 cells were analyzed.
-
G, HTargeting PINK1 kinase domain to OMM increases mitochondrial localization of phosphorylated Drp1S616. HEK293 cells cotransfected FRB‐MTS with Δ110‐PINK1‐GFP‐FKBP were treated with 250 nM rapalog (Rapalog). After 2 h treatment, cytosol protein (cyto) and mitochondria (Mito) were fractionated and immunoblotted with antibodies against either phospho (Ser616)‐Drp1 (pS616), Drp1 (Drp1), or GFP (PINK1). Tom20 and β‐actin were detected as mitochondrial loading control and cytosol loading control, respectively (G). Quantitation of pS616/Drp1 in various conditions was shown (H). Student's test. **P < 0.01, ns: no significance. Data were presented as mean ± SEM of three independent experiments.
-
I, JTargeting PINK1 kinase domain to the OMM increases mitochondrial localization of Drp1. HeLa cells cotransfected FRB‐MTS with Δ110‐PINK1‐GFP‐FKBP (FKBP‐PINK1) were treated with 250 nM rapalog (Rapalog) for 2 h to activate PINK1 kinase. Cells treated with solvent (DMSO) were included as a control. Cells were immunostained with antibodies against either Tom20, Drp1, and GFP (PINK1‐FKBP). Representative images were shown, scale bar = 2 μm (I). Higher magnification images from gated box are shown (bottom panels). Arrows indicate Drp1 puncta. Mitochondria‐associated (yellow) and non‐associated Drp1(Red) are shown, scale bar = 1 μm. Mitochondria‐associated Drp1 puncta were analyzed by using an ImageJ plugin‐Coloc 2 and expressed as Manders’ colocalization coefficients (MCC) (J). Three different regions from each cell and > 15 cells for each experimental group were analyzed. Student's test. **P < 0.01. Data were presented as mean ± SEM of three independent experiments.
-
K, LPINK1 regulates S616 and S637 phosphorylation of Drp1 via independent mechanisms. DRP1KO HEK293 cells cotransfected FRB‐MTS/Δ110‐PINK1‐GFP‐FKBP (PINK1WT) with either myc‐Drp1WT or myc‐Drp1S616A were treated with 250 nM rapalog (Rapalog) for 2 h to activate PINK1 kinase. Cells treated with solvent (DMSO) were included as a control. Cell lysates were immunoblotting with antibodies against either phospho (Ser616)‐Drp1 (pS616), phospho‐(Ser637)‐Drp1 (pS637), Drp1, and GFP (to detect PINK1‐FKBP) (K). β‐actin was detected as a loading control. Quantitation of pS637/Drp1 in various conditions was shown (L). One‐way ANOVA followed with Tukey's test. ***P < 0.001, ns: no significance. Data were presented as mean ± SEM of three independent experiments.
PINK1 regulates mitochondrial dynamics independent of both parkin and mitophagy
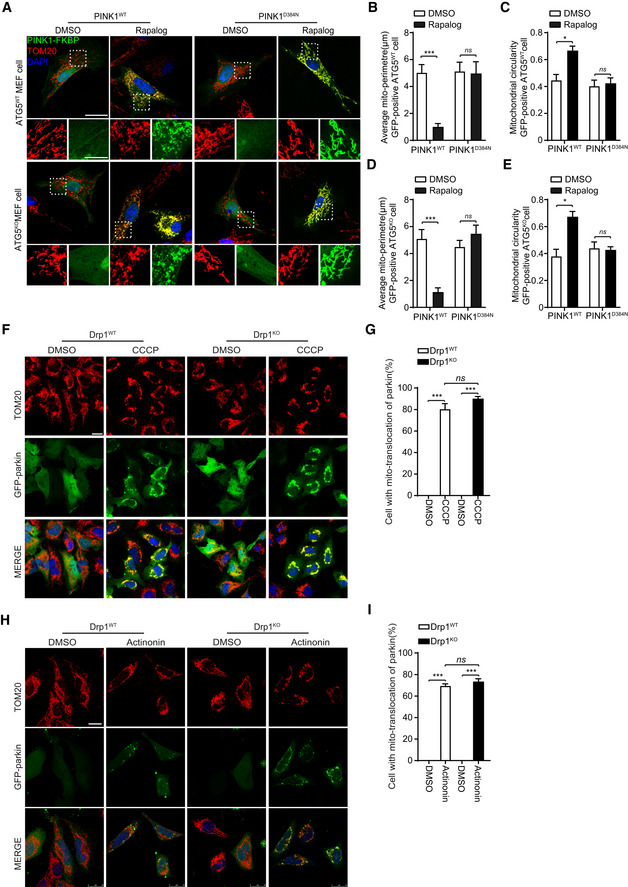
PINK1 regulates mitophagy via recruiting parkin to mitochondria 7, 8, 26, 31, 32. We next asked whether mitophagy was required for PINK1/Drp1 signaling‐mediated mitochondrial dynamics. Phosphorylation of Drp1S616 is not affected in parkinKO cells (Fig 1A–F). Moreover, PINK1, but not the kinase‐dead PINK1, causes Drp1‐dependent mitochondrial fragmentation in HeLa cells, a cell line known to lack of parkin expression (Fig 3A–F). The results suggest that parkin is not essential for PINK1/Drp1 signaling‐mediated mitochondrial dynamics. In MEF cells derived from ATG5KO mouse embryos, rapalog‐induced recruiting of PINK1wt, but not PINK1D384N, led to mitochondrial fragmentation (Fig 4A–E, Appendix Fig S1C and D). Furthermore, parkin was effectively recruited to mitochondria in Drp1KO HeLa cells treated with either CCCP or actinonin (Fig 4F–I). Results imply that Drp1 is unlikely required for PINK1/parkin‐mediated mitophagy. Together, these results indicate that PINK1‐regulated mitochondrial fragmentation is independent of ATG5‐mediated autophagy.
Figure 4. PINK1 phosphorylates Drp1S616 to regulate mitochondrial fission that is independent of mitophagy.
-
A–EATG5 is dispensable for PINK1‐mediated mitochondrial fission. MEF cells derived from ATG5‐null (ATG5KO) and their wild‐type control mice (ATG5WT) were cotransfected Δ110‐PINK1‐GFP‐FKBP/FRB‐MTS with either PINK1WT or a kinase‐dead mutant PINK1D384N. Cells were induced with 250 nM rapalog (Rapalog) for 2 h to activate PINK1 kinase, followed by immunodetecting mitochondria (TOM20, red) and Δ110‐PINK1‐GFP‐FKBP/FRB‐MTS (PINK1‐FKBP, green). Cells treated with solvent (DMSO) were used as a treatment control. Representative images are shown, scale bar = 25 μm (A). Higher magnification images are also included (panels beneath the large cell images, scale bar = 10 μm). Mitochondrial morphology in different transfections was quantified (B–E). Student's test. *P < 0.05, ***P < 0.001, ns: no significance. Data were presented as mean ± SEM of three independent experiments. For each condition, > 100 cells were analyzed.
-
F–IDrp1 is dispensable for recruitment of parkin to mitochondria. Drp1WT and Drp1KO HeLa cells were transfected with GFP‐parkin. Cells were treated with either 20 μM CCCP for 2 h (F) or 150 μM actinonin for 6 h (H), followed by immunodetecting mitochondria (TOM20, red) and parkin (GFP‐parkin, green). Nuclei were labeled with DAPI (blue). Cells treated with solvent (DMSO) were used as a treatment control. Representative images were shown, scale bar = 25 μm. Cell with mitochondrial parkin (%) in different experimental group was quantified (G, I). One‐way ANOVA followed with Tukey's test. ***P < 0.001, ns: no significance. Data were presented as mean ± SEM of three independent experiments. For each condition, > 100 cells were analyzed.
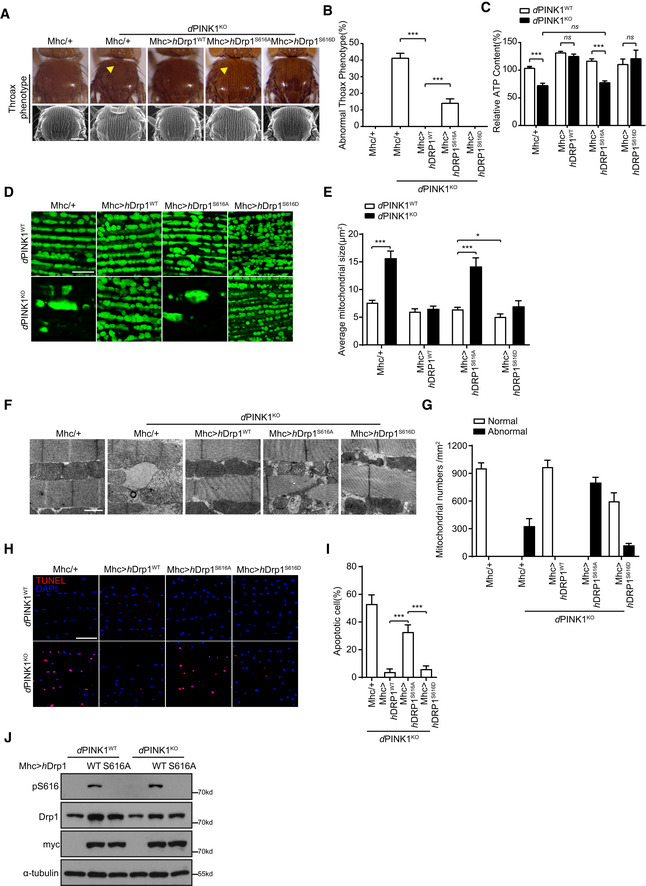
Previous studies suggest a genetic and functional interaction between PINK1 and Drp1 in Drosophila 22, 23, 24, 28. Drosophila with PINK1KO mutation shows crushed thorax and defects in mitochondrial morphology and function. We next expressed human Drp1 variants (hDrp1wt, hDrp1S616A, and hDrp1S616D) driven by muscle‐specific driver mhc‐gal4 in PINK1KO mutant flies (Appendix Fig S1G). No obviously abnormal phenotype was observed when hDrp1wt, hDrp1S616A, or hDrp1S616D was overexpressed in muscles of wild‐type flies. Expression of hDrp1wt and hDrpS616D rescued crushed thorax and reduced ATP production of PINK1‐null mutant flies (Fig 5A–C). Abnormal mitochondrial morphology in indirect flight muscle of PINK1 null mutant was also reversed by expressing hDrp1wt and hDrp1S616D (Fig 5D and E). Expression of hDrp1S616A showed weak rescue of crushed thorax, but had little effect on reduced ATP production and abnormal mitochondrial morphology of PINK1‐null mutant flies (Fig 5A–E). TEM analysis revealed that damaged mitochondrial cristae were restored by overexpressing hDrp1wt and hDrp1S616D, but not hDrp1S616A (Fig 5F and G). It is possible that weak rescue of crushed thorax by hDrp1S616A is via mitochondrial‐independent function of Drp1. Furthermore, expression of hDrp1wt and hDrp1S616D, but not hDrp1S616A, suppressed cells death in PINK1KO flies (Fig 5H and I). Immunoblotting analysis revealed effective expression of hDrp1 variants and phosphorylation of hDrp1S616 in muscle of fly expressing hDrp1wt, but not fly expressing hDrp1S616A (Fig 5J). These results suggested Drp1S616 phosphorylation is essential and sufficient for PINK1‐mediated mitochondrial morphology and function as well as PINK1‐mediated cell survival in Drosophila.
Figure 5. Phosphorylation of Drp1S616 is essential for PINK1‐mediated mitochondrial structures and function in Drosophila .
-
A, BDrp1WT and Drp1S616D, but not Drp1S616A, rescue PINK1‐null induced crushed thorax in Drosophila. Representative thorax images of PINK1WT and PINK1 deficiency (PINK1KO) Drosophila expressing either mhc‐gal4 (Mhc/+), mhc‐gal4‐driven human Drp1wt (Mhc>hDrp1WT), or mhc‐gal4‐driven human Drp1S616A (Mhc>hDrp1S616A) or mhc‐gal4‐driven human Drp1S616D (Mhc>hDrp1S616D) are shown. Images were taken either under light microscopy (A, upper panels, yellow arrowhead indicates the collapsed thorax) or SEM (B, lower panels). Quantitation of crushed thorax is shown (B). > 200 flies for each experiment are analyzed. Scale bar = 200 μm. One‐way ANOVA followed with Tukey's test. ***P < 0.001. Data were presented as mean ± SEM of three independent experiments. For each condition, > 200 flies were quantified.
-
CDrp1WT and Drp1S616D, but not Drp1S616A, restore abnormal ATP production in PINK1‐null flies. ATP production was measured using muscle lysates generated from PINK1WT flies expressing mhc‐gal4 (Mhc/+) (as a control) and PINK1KO flies expressing either mhc‐gal4 (Mhc/+), mhc‐gal4‐driven human Drp1wt (Mhc>hDrp1wt), or mhc‐gal4‐driven human Drp1S616A (Mhc>hDrp1S616A) or mhc‐gal4‐driven human Drp1S616D (Mhc>hDrp1S616D). One‐way ANOVA followed with Tukey's test. ***P < 0.001, ns: no significance. Data were presented as mean ± SEM of three independent experiments.
-
D–GDrp1WT and Drp1S616D, but not Drp1S616A, rescue mitochondrial structures in PINK1KO flies. Representative images of mito‐GFP (green) labeled mitochondria (D) and TEM analysis (F) of IFMs from PINK1WT flies expressing mhc‐gal4 (Mhc/+) (as a control) and PINK1KO flies expressing either mhc‐gal4 (Mhc/+), Mhc‐gal4‐driven human Drp1wt (Mhc>hDrp1wt) or Mhc‐gal4‐driven human Drp1S616A (Mhc>hDrp1S616A) or mhc‐gal4‐driven human Drp1S616D (Mhc>hDrp1S616D) are shown. Scale bar = 10 μm (D) and 1 μm (F). Average mitochondrial size was quantified, and > 3 flies/group and 3–6 pictures of different microscopic fields from each fly were analyzed per repeat. One‐way ANOVA followed with Tukey's test. *P < 0.05, ***P < 0.001. Data were presented as mean ± SEM of three independent experiments (E). Normal and abnormal mitochondrial number per mm2 were quantified. > 3 flies/group and 3–6 pictures of different microscopic fields from each fly were analyzed per repeat. Bars represent the mean ± SEM of three independent experiments (G).
-
H, IDrp1WT and Drp1S616D, but not Drp1S616A, suppress cell death in PINK1KO flies. Representative TUNEL staining (Red) images of IFM sections from PINK1WT and PINK1KO flies expressing either Mhc‐gal4 (Mhc/+), Mhc‐gal4‐driven human Drp1wt (Mhc>hDrp1WT), or Mhc‐gal4‐driven human Drp1S616A (Mhc>hDrp1S616A) or mhc‐gal4‐driven human Drp1S616D (Mhc>hDrp1S616D) are shown. Nuclei were counter‐stained with DAPI (blue), scale bar = 10 μm. Apoptotic cells (%) in different genotype flies were quantified (I). > 5 flies/group and 3–6 pictures of different microscopic fields from each fly were analyzed per repeat. One‐way ANOVA followed with Tukey's test. ***P < 0.001. Data were presented as mean ± SEM of three independent experiments.
-
JExpression and phosphorylation of hDrp1 in Drosophila. Fly muscle lysates generated from PINK1WT or PINK1KO flies expressing mhc‐gal4‐driven hDrp1WT (WT) and hDrp1S616A (S616A) were immunoblotted with antibodies against either phospho(Ser616)‐Drp1(pS616) (to detect phosphorylated hDrp1S616), Drp1 (to detect both endogenous and exogenous Drp1), and myc‐tag (to detect exogenous Drp1). α‐tubulin was detected as a loading control. Flies expressing mhc‐gal4 (−) were included as an expression control.
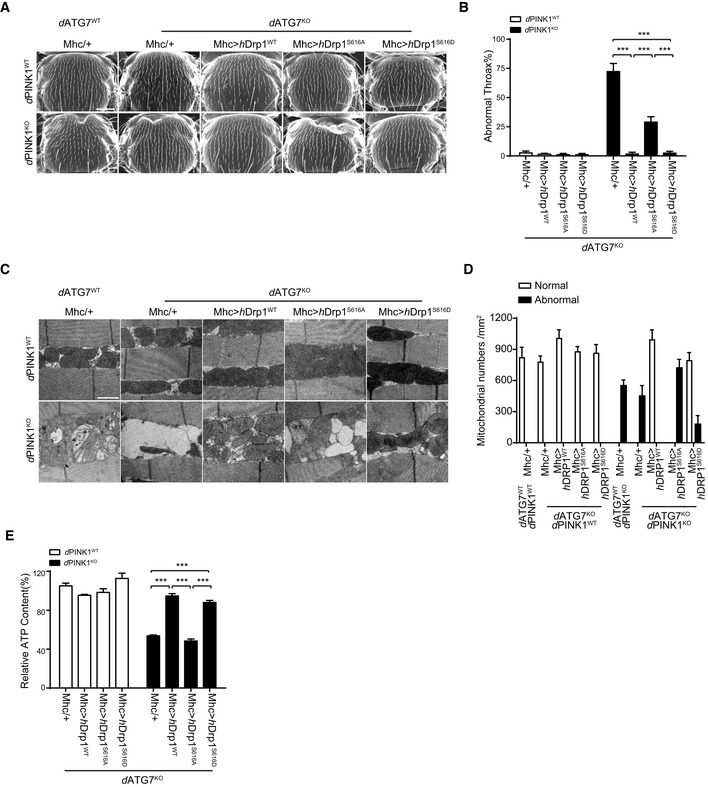
Impaired mitochondrial degradation is hypothesized as mechanism of abnormalities observed in PINK1KO mutant flies 33, 34, 35. We next asked whether mitophagy is required for Drp1 to reverse PINK1KO mutant phenotypes in Drosophila. Flies carrying dATG7‐deficient alleles (d77/d14) 36 showed no obvious phenotype but increase levels in both Triton X‐100 soluble and insoluble ubiquitinated proteins comparing to their wild‐type controls (Appendix Fig S1H and I). Mhc‐gal4‐driven muscle‐specific expression of hDrp1wt and hDrp1S616D, but not hDrp1S616A, in dATG7 and dPINK1/dATG7 double deficient flies effectively rescued PINK1 deficiency‐induced phenotypes, including crushed thorax (Fig 6A and B), mitochondrial abnormality (Fig 6C and D), and reduced ATP production (Fig 6E). These results indicated that Drp1 rescues PINK1‐deficient phenotypes of Drosophila independent of dATG7‐dependent autophagy.
Figure 6. Drp1 rescues PINK1KO induced crushed thorax and mitochondrial abnormality in dATG7‐deficient Drosophila .
-
A, BDrp1 rescues PINK1KO induced collapsed thorax phenotype in dATG7‐deficient Drosophila. Representative SEM images of thorax from PINK1WT, (upper panels) and PINK1KO (lower panels) flies combined with dATP7 deficiency (dATG7KO) or their controls (dATG7WT), followed by expressing either mhc‐gal4 (Mhc/+) alone, or Mhc‐gal4‐driven expression of human Drp1 (Mhc>hDrp1WT), mhc‐gal4‐driven expression of human Drp1S616A (Mhc>hDrp1S616A), or mhc‐gal4‐driven expression of human Drp1S616D (Mhc>hDrp1S616D) are shown (A). Quantitation of crushed thorax in flies with indicated genotypes is presented (B). > 100 flies for each experiment are analyzed. Scale bar = 200 μm. One‐way ANOVA followed with Tukey's test. ***P < 0.001. Data were presented as mean ± SEM of three independent experiments.
-
C, DDrp1 rescues PINK1KO induced mitochondrial defects in dATG7KO Drosophila. Representative TEM images of indirect flight muscle from PINK1WT (upper panels) and PINK1KO (lower panels) flies combined with dATG7KO or their controls (dATG7WT), followed by expressing either mhc‐gal4 (Mhc/+) alone, or Mhc‐gal4‐driven expression of human Drp1(Mhc>hDrp1WT), mhc‐gal4‐driven expression of human Drp1S616A(Mhc>hDrp1S616A), or mhc‐gal4‐driven expression of human Drp1S616D(Mhc>hDrp1S616D) are shown (C). Scale bar = 1 μm. Normal and abnormal mitochondrial number per mm2 from each experimental group were and quantified. > 3 flies/group and 3–6 pictures of different microscopic fields from each fly were analyzed per repeat. Bars represent the mean ± SEM of three independent experiments (D).
-
EDrp1 rescues PINK1KO induced ATP reduction in Drosophila. ATP contents of thorax muscle tissues from the indicated genotypes were measured and normalized against the protein levels One‐way ANOVA followed with Tukey's test. ***P < 0.001, ns: no significance. Data were presented as mean ± SEM of three independent experiments.
Reduced Drp1S616 phosphorylation in dermal fibroblasts of PD patients
To further study Drp1S616 phosphorylation in PD pathogenesis, we obtained patient human dermal fibroblasts (HDF) cells from four patients harboring PINK1 mutations from three different families, including a patient harboring a homozygous c.C1474T (p.R492X) of PINK1 37, a patient harboring a homozygous c.C938T (p.T313M) of PINK1 25, and other two patients harboring compound heterozygous c.C1474T(p.R492X)/c.1501T(p.R501X) of PINK1 (Fig 7A–F). In addition, we collected HDF cells from 7 sporadic PD patients and 5 normal controls individuals with no PINK1 mutations detected by sequencing (Appendix Table S1). Immunoblotting analysis revealed that level of Drp1S616 phosphorylation is markedly lower than that of normal control individuals (Fig 7G and H). To our surprise, level of Drp1S616 phosphorylation in sporadic patients was also significantly lower than that of normal control individuals with 4 out of 7 patients showing a markedly lower level of Drp1S616 phosphorylation (Fig 7I and J). These results suggest that pathogenic PINK1 mutants are likely defective in phosphorylating Drp1S616, indicating that PINK1‐mediated Drp1S616 phosphorylation links to pathogenesis of PD cases harboring PINK1 pathogenic mutations. Our results also suggest that PINK1‐mediated Drp1S616 phosphorylation is likely involved in pathogenesis of a portion of sporadic PD cases.
Figure 7. Decreased Drp1S616 phosphorylation in human fibroblasts derived from PD patients harboring PINK1 mutations.

-
A–FPedigrees of PD families with PINK1 mutations and Sanger sequencing conformation. Pedigrees of three families with four PD patients (A, C, E) and corresponding sequencing chromatogram of four PD patients harboring PINK1 mutations (B, D, F) are shown. Red arrows indicate the mutations in each patient.
-
G, HReduction of Drp1S616 phosphorylation in dermal fibroblasts derived from PD patients with PINK1 mutations. Lysates of cells derived from three unrelated normal control individuals (Control, C) and four familial PD patients with PINK1 mutations (PINK1mutant) were immunoblotted with antibodies recognizing either phosphor‐Drp1S616 (pS616) or Drp1 protein. β‐actin was detected as a loading control (G). A quantitative analysis of phospho‐Drp1S616 (pS616/Drp1) is shown (H). Student's test. **P < 0.01, ns: no significance. Data were presented as mean ± SEM of three independent experiments.
-
I, JDrp1S616 phosphorylation in dermal fibroblasts derived from sporadic PD patients. Lysates of cells derived from five unrelated normal control individuals (Control, C) and seven unrelated sporadic PD patients (Sporadic PD, SP) were immunoblotted with antibodies recognizing either phospho‐Drp1S616 (pS616) or Drp1 protein. β‐actin was detected as a loading control (I). A quantitative analysis of phospho‐Drp1S616 (pS616/Drp1) in sporadic cases is shown (J). Student's test. *P < 0.05. Data were presented as mean ± SEM of three independent experiments.
Discussion
This study demonstrates that PINK1 regulates mitochondrial dynamics via direct phosphorylating Drp1S616. Impairment of this pathway is critical for developing mitochondrial phenotypes of PINK1‐deficient cells and Drosophila, which is independent of both parkin's function and autophagy. Remarkably, Drp1S616 phosphorylation is significantly reduced in HDFs of PD patients harboring PINK1 mutations and a portion of sporadic PD patients. The study identifies Drp1 as a substrate of PINK1 and a novel mechanism of PINK1 in regulating mitochondrial dynamics. The results also suggest that impairment of PINK1‐mediated Drp1S616 phosphorylation is associated with pathogenesis of both familial PD with PINK1 mutations and a large portion of sporadic PD. Furthermore, Drp1S616 phosphorylation is a potential biomarker for PD diagnosis and a target to develop novel PD therapy.
Previous studies have shown that PINK1 plays critical roles in maintaining normal morphology and function of mitochondria. Although the detailed mechanism remains unclear, PINK1/parkin‐mediated mitophagy likely plays a critical role in clearance of damaged mitochondria. Impairment of PINK1/parkin‐mediated mitophagy is believed to contribute to the accumulation of abnormal mitochondria in PINK1‐deficient flies, leading to eventual neurodegeneration. We provide multiple lines of evidence in this study to suggest that PINK1 directly phosphorylates Drp1S616. First, Drp1S616 phosphorylation is significantly reduced in PINK1KO cell lines and mouse tissues. Second, TcPINK1 phosphorylates recombinant Drp1 at S616 but hardly other Ser/Thr sites of recombinant Drp1. Consistently, phos‐tag analysis of in vitro phosphorylation of recombinant Drp1 shows that TcPINK1 phosphorylates only Drp1S616. Third, PINK1‐mediated mitochondrial fragmentation is Drp1S616 dependent. Fourth, the Km of TcPINK1 phosphorylating Drp1S616 is similar to that of TcPINK1 phosphorylating Ub, a bona fide PINK1 substrate.
Our results suggest that neither parkin nor autophagy is required for PINK1‐mediated mitochondrial fission, mitochondrial abnormality, and cell death observed in PINK1‐deficient flies and cultured cells. In contrast, Drp1S616 phosphorylation alone is sufficient to maintain PINK1‐mediated mitochondrial fission and function, as well as cell survival. Other studies suggest that PINK1 or PINK1/parkin interacts with mitochondrial fission/fusion machinery to modulate mitochondrial dynamics 22, 23, 24. Consistent with those findings, we demonstrate that PINK1 promotes fission, via however a different mechanism that is to directly phosphorylate Drp1S616. The facts that PINK1 deficiency caused mitochondrial abnormalities in flies are sufficiently rescued by expressing hDrp1WT and hDrp1S616D, but not a hDrp1S616A phosphorylation mutant, suggest that Drp1 phosphorylation plays an essential role in PINK1 function in flies. Moreover, expression of PINK1 does not promote mitochondrial fission in Drp1‐deficient cells, placing PINK1 to the upstream of Drp1. Thus, this study identifies a novel molecular mechanism of PINK1‐mediated mitochondrial fission. It is possible that PINK1 phosphorylates Drp1S616 to promote fission to separate damaged part and health part of a mitochondrion. Followed fission, the damaged mitochondria are further targeted to degrade by PINK1/parkin‐mediated mitophagy. Therefore, PINK1 likely functions at both fission and mitophagy during the mitochondrial quality control.
Another significance of this study is finding reduced Drp1S616 phosphorylation in HDF cells from patients with PINK1 pathogenic mutations. These results suggest that PINK1 pathogenic mutations are likely with reduced kinase activity. More importantly, the results link PINK1‐mediated Drp1 phosphorylation to PD pathogenesis. To our great surprise, Drp1S616 phosphorylation is also reduced in a portion of sporadic patients although the sample size needs further increased for clinical verification. The results raise at least two important issues related to regulation and pathogenic contribution of PINK1. One is that PINK1 activity is likely impaired in a portion of sporadic patients. It will be important to determine what percentage of sporadic PD is with impaired Drp1S616 phosphorylation in clinic and how this regulation is compromised without finding PINK1 mutations in those PD patients. Another is that PINK1 impairment is likely involved in pathogenesis of a much larger percentage of PD patients than we have originally thought. It may be proven effective to combat PD by developing a Drp1S616 phosphorylation‐based molecular diagnosis and therapies targeting mitochondrial fission machinery.
Materials and Methods
Plasmids, antibodies, and chemicals
pDsRed1‐mito plasmid (6928‐1) was from Clontech (Mountain View, USA), and pGEX4T‐2 plasmid was from General Electric (Connecticut, USA). Plasmids encoding PINK1 variants were described 38. cDNA encoding Drp1 (Human isoform 1) was cloned into pcDNA3.1/myc‐his(B+) (Invitrogen, San Diego, USA). The dephosphorylation‐mimic of Drp1S616 mutants (Drp1S616A) was generated using a QuikChange site‐directed Mutagenesis Kit from Stratagene (La Jolla, USA). cDNA encoding Drp1518‐736 and Drp1518‐736, S616A were subcloned into pGEX4T‐2GE. pC4M‐F2E‐GFP‐FKBP (68058) and pC4‐RhE‐FRB‐Fis1 (68056) were from Addgene (Cambridge, USA). cDNA encoding ▵110‐PINK1 (WT or D384N) with deletion of N‐terminal 1‐110 aa was subcloned into pC4M‐F2E‐GFP‐FKBP to express ▵110‐PINK1 (WT or D384N)‐GFP‐FKBP. All plasmids are sequence confirmed.
Anti‐TOM20 (sc‐17764) antibody and anti‐parkin (sc‐32282) antibody were from Santa Cruz Biotechnology (Santa Cruz, USA). Antibodies against flag tags (F7425) and β‐actin (A5441) were from Sigma‐Aldrich (St. Louis, USA). Anti‐α‐tubulin (ab18251), anti‐thiophosphate ester (ab92570) and anti‐tyrosine hydroxylase (ab76442) antibodies were from Abcam (Cambridge, USA). Antibody for Drp1 (611112) was from Becton Dickinson and Company (Franklin Lakes, USA). Anti‐Drp1 (8570), anti‐Drp1‐pS616 (4494), anti‐Drp1‐pS637 (6319), anti‐PINK1(6946), anti‐myc (2276), anti‐ubiquitin (3936), anti‐CDK1(9116), anti‐pThr161‐CDK1 (9114), anti‐ERK1/2(4695), and anti‐pThr202/Thr204(ERK1/2) (4370) antibodies were from Cell Signaling (Danvers, USA). Antibody for GFP (632381) was from Takara (Minamikusatsu, Japan).
The phosphatase inhibitor cocktail (B15001), CDK1 inhibitor RO‐3306 (S7747), and ERK1/2 inhibitor Ulixertinib (S7854) were from Selleckchem (Houston, USA). Rapalog (635055) was purchased from Takara (Minamikusatsu, Japan). ATPγS(ab138911) and p‐Nitrobenzyl mesylate (ab138910) purchased from Abcam (Cambridge, USA). All other chemicals were from Sigma‐Aldrich (St. Louis, USA).
Cells, Drosophila, and mouse lines
HEK293 and HeLa cells were obtained from American Type Culture Collection (ATCC). PINK1‐null (PINK1KO), parkin‐null (parkinKO) HEK293 cells, and Drp1‐null (Drp1KO) HeLa cells were generated using CRISPR/Cas9 system as described 38, 39. Guiding nucleotides for PINK1: 5′‐ CGCCACCATGGCGGTGCGAC ‐3′ and 5′‐TCTCCGCTTCTTCCGCCAGT ‐3′; For parkin: 5′‐TGTCAGAATCGACCTCCACT‐3′ and 5′‐AGTGCCGTATTTGAAGCCTC‐3′; For Drp1: 5′‐CCACCGTGTTGAAGACGTCC‐3′ and 5′‐GCTGCCTCAAATCGTCGTAG‐3′. At least two independent cell clones for each genotype (PINK1‐null, parkin‐null, and Drp1‐null) were isolated for further experiments.
PINK1‐null fly (PINK1B9) was kindly provided by Dr. Jongkyeong Chung 40, and two ATG7‐deficient (d77 and d14) flies were from Thomas P. Neufeld 36. Flies harboring mhc‐gal4 and uas‐mito‐GFP were obtained from the Bloomington Drosophila Stock Center. Fly strains were grown on standard cornmeal media at 25°C.
The PINK1, parkin, and ATG5‐deficient mouse lines are described previously 25. Mice were genotyped by multiplex PCR on genomic DNA extracted from tail snips. The experimental protocols for using mouse lines were approved by the Ethics Review Committee for Animal Experimentation of Central South University.
Mouse embryonic fibroblasts (MEF) from PINK1, parkin, and ATG5‐deficient mouse embryos and their WT control littermates were generated as described 25.
Cell transfection and treatment
Transfection was done with Lipofectamine 2000 according to the instruction of manufacture. Experiments were performed 36 h after transfection. For the regulated heterodimerization assay, the transfected cells were treated with 250 nM Rapalog for 2 h before immunostaining.
Transfection of primary neuronal cultures was done with calcium phosphate at DIV4 (4 days in vitro). Cells were assayed at 48 h after transfection.
Immunoblotting and immunostaining analysis
Immunoblotting and immunofluorescence were performed as described 38. For immunofluorescence staining of tissue sections, mice were anesthetized with anesthetics and transcardially perfused with phosphate‐buffered saline (PBS) followed by 4% (wt/vol) paraformaldehyde in 0.1 M sodium phosphate buffer. 20‐μm midbrain sections were subjected to immunofluorescence staining for detecting tyrosine hydroxylase and phosphorylated Drp1S616.
In vitro kinase assay
GST‐Drp1518–736 and GST‐Drp1518–736, S616A proteins were generated and purified as described 25. Proteins were dialyzed and concentrated with an Amicon Ultra 30K centrifugal filter (Millipore, Temecula, USA). Recombinant ubiquitin was obtained from R&D System Inc (U‐100H‐10M, Minneapolis, USA). Kinase assay was performed as described 2, 41. Briefly, 1 μg TcPINK1 (R&D system, Minneapolis, USA) was incubated with 2 μg of either GST‐Drp1518–736 or GST‐Drp1518–736, S616A in 50 μl 1 × kinase buffer (50 mM Tris–HCl, pH 7.5, 10 mM DTT, 0.1 mM EGTA, 10 mM Mg(OAc)2, and 100 μM ATP) and incubated at 30°C for 60 min. Reactions were terminated with 50 μl 2 × SDS sample buffer. Proteins were resolved by SDS–PAGE followed by Coomassie Blue (CB) staining or immunoblotting. For ATPγS kinase assay 42, 1 μg TcPINK1 was incubated with 2 μg of either GST‐Drp1518–736 or GST‐Drp1518–736, S616A in 50 μl 1 × kinase buffer (50 mM Tris–HCl, pH 7.5, 10 mM DTT, 0.1 mM EGTA, 10 mM MgOAc, and 500 μM ATPγS) and incubated at 30°C for 20 min or 40 min. After heat inactivation, PNBM (Abcam, Cambridge, USA) was added into each sample for 2 h at 37°C. Samples were resolved with SDS lysis buffer.
Phos‐tag SDS–PAGE and immunoblotting
Phos‐tag SDS–PAGE assay was performed essentially as previously described 2. To detect phospho‐Drp1, 8% (w/v) polyacrylamide tris–glycine gels were prepared with or without addition of phos‐tag acrylamide (10 μM) and MnCl2 (20 μM). To detect phospho‐Ub, 12% (w/v) polyacrylamide tris–glycine gels were prepared with or without addition of phos‐tag acrylamide (20 μM) and ZnCl2 (20 μM). After electrophoresis, phos‐tag gels were soaked in transfer buffer containing 1 mM EDTA, 15 min for 2 times to remove the Mn2+ or Zn2+ before blotting. All other steps were the same to normal SDS–PAGE and Western blotting protocols.
Lc‐ms/ms
LC‐MS/MS was performed essentially as previously described 43. Briefly, the samples after in vitro kinase assay were digested with trypsin (Promega, Madison, USA) at 37°C overnight and desalted by reversed‐phase C18 Sep‐Pak cartridge (Millipore, Temecula, USA). The samples were performed on an EASY‐nLC1000 LC system (Thermo Scientific, San Diego, USA) coupled to the Q‐Exactive mass spectrometer (Thermo Scientific, San Diego, USA). Peptides were separated on an in‐house packed C18‐column (15 cm, 75 μm I.D., and 3‐μm particle size) with a gradient from 12 to 32% buffer B (98% acetonitrile and 0.1% acetic acid) for 120 min. The resolution for MS was set to 70,000 and for MS/MS was set to 17,500. The raw files were processed by MaxQuant software (version 1.4.1.2) with searches against the UniProt human database and a variable modification of serine, threonine, and tyrosine phosphorylation.
K m and K cat measurement
K m and K cat assays were essentially done as previously described 44. Briefly, kinase assays were performed with either ubiquitin (0–400 μM) or GST‐Drp1 (0–300 μM) in the presence of 1 μM TcPINK1 for 0, 10, 20, and 40 min. The assay products were separated on Phos‐tag gels. The intensity of shifted bands was quantified and plotted against reaction time to calculate the initial rate (V0). Then, K m and V max parameters for Drp1 and ubiquitin were obtained from the global fit by using the data analysis software Prism.
Mouse primary neuronal cultures
Mouse primary neuron cultures were done essentially as described 28, 45.
Mitochondrial morphology analysis
For neurons, mitochondrial morphology on 100–150 μm first dendrites was analyzed. The starting position of measurement was at least 25 μm from the soma. The length of each mitochondrial segment and the length of the corresponding dendrite were recorded and calculated. Mitochondrial index = the length of mitochondrial segments/the length of the corresponding dendrite × 100%. Ten neurons were randomly selected and analyzed in each experiment. Three independent experiments are done for each measurement.
For HeLa, HEK293, and HDF cells, mitochondria were labeled with Tom20 antibody. Mitochondrial morphology was quantified using ImageJ as previously described 26. In brief, background was subtracted from raw images, which were subsequently linearly contrast optimized, and then thresholded to generate binary images. The circularity, mean perimeter, and mitochondrial particle number of cells were used to determine morphological changes. > 30 cells in different microscopic fields were analyzed for each experimental group.
For Drosophila muscle, mitochondria were labeled by mito‐GFP. > 10 flies/group and 3–5 pictures of different microscopic fields from each fly were analyzed. The average mitochondrial size (μm2) is used to determine morphological changes. For TEM images, > 3 flies/group and 3–6 pictures of different microscopic fields from each fly were analyzed per repeat. The number of normal and abnormal mitochondria per unit area (mm2) is used to determine mitochondrial ultra‐structural change.
Drp1 puncta analysis
Mitochondrial Drp1 puncta were analyzed essentially as described 46. Briefly, HeLa cells transiently transfected with PINK1‐GFP‐FKBP/FRB‐MTS were fixed and immunostained with an anti‐TOM20 (mitochondrial marker) and an anti‐Drp1 antibodies. Regions of interest with readily resolvable mitochondria and Drp1 were processed as described in Fig 3I and J. Each picture was thresholded mitochondrially associated Drp1 puncta by using ImageJ plugin, Coloc 2, and the value of mitochondrially associated Drp1 means Manders’ colocalization coefficients (MCC). Three different regions from each cell and > 15 cells for each experimental group were analyzed.
Transmission electron microscopy (TEM)
Substantial nigra tissues were dissected from 18‐month‐old mice. Briefly, mice were anesthetized with anesthetics and transcardially perfused with phosphate‐buffered saline (PBS) followed by 2% (wt/vol) paraformaldehyde/2% (wt/vol) glutaraldehyde in 0.1 M sodium phosphate buffer. Substantial nigra sections were removed and post‐fixed in 2.5% (wt/vol) glutaraldehyde in 0.1 M sodium phosphate buffer. Fly thoraces were dissected from 3‐day‐old male flies, fixed in paraformaldehyde (158127, Sigma‐Aldrich, St. Louis, USA)/glutaraldehyde (18462, TED PELLA, Redding, USA), post‐fixed in osmium tetraoxide (19150, Electron Microscopy Sciences, Hatfield, USA), dehydrated in ethanol (46139, Sigma‐Aldrich, St. Louis, USA), and embedded in Epon (45345, Sigma‐Aldrich, St. Louis, USA). After polymerization of Epon, blocks were sectioned to generate 70‐nm‐thick sections using a diamond knife on a microtome (Leica, Wetzlar, Germany). The sections were stained with uranyl acetate (19481, TED PELLA, Redding, USA) and lead citrate (15326, Sigma‐Aldrich, St. Louis, USA). Digital images were obtained on a Tecnai G2 Spirit by FEI equipped with an Eagle 4k HS digital camera. Mitochondria were counted and differentiated depending on diameter. Over 300 identifiable mitochondria (cristae and/or double membrane) randomly and blindly selected per mice for perimeter's measurement. And the numbers of mitochondria (perimeter > 2 μm) in PINK1‐null mice and wild‐type mice (n = 3/group) are compared statistically.
Mitochondrial fractionation
Mitochondrial extraction from HEK293 cells was done essentially as described 38. Briefly, cells were grown on 100‐mm dish until 80–90% confluency, washed twice with ice‐cold PBS, and then scraped into ice‐cold PBS followed by centrifugation at 1,000 × g for 5 min at 4°C. Cell pellets were resuspended in mitochondrial isolation buffer (5 mM Hepes pH 7.4, 3 mM MgCl2, 1 mM EGTA, and 250 mM sucrose) containing protease and phosphatase inhibitors. Lysates were passed through a 5/8‐inch 25‐gauge needle 20 times using a 1‐ml syringe and centrifuged at 1,000 g, 4°C for 20 min. Supernatants were collected, and cytosolic extracts were recovered by centrifugation at 10,000 g, 4°C for 15 min to obtain crude mitochondrial pellets.
Scan electron microscopy (SEM)
Flies at 3‐day‐old were posed and attached to a copper mount followed by analyzed using a scanning electron microscope TM‐1000 (HITACHI, Tokyo, Japan).
Analysis of thorax phenotypes
The percentage of male flies with abnormal crushed thorax was determined under microscopy. > 200 flies were analyzed for each genotype per experiment. Three independent experiments were done for each analysis.
ATP assay
ATP level was quantified using a commercial kit (Promega, Madison, USA). Briefly, lysates from five thoraces of 3‐day‐old flies were prepared for each experiment. Samples were mixed with luminescent solution. The luminescence was measured by an illuminometer (Berthold Technologies, Bad Wildbad, Germany). Values were normalized to protein content.
TUNEL staining
Cell death of 3‐day‐old fly was detected using an in situ cell death detection kit (Roche Applied Science, Penzberg, Germany) protocol. Briefly, fly indirect flight muscles were dissected and fixed in 4% PFA for 30 min at room temperature. Tissues were permeabilized in 0.3% PBST for 1 h followed by 0.1% sodium citrate (0.3% Triton) for 30 min. TUNEL staining was done according to an instruction of the manufacture. Nuclei were counter‐stained by DAPI. Samples were mounted using FluoromountTM and imaged by a confocal microscopy.
Generation of HDF cells
PD patients and normal control individuals (Appendix Table S1) were Han Chinese from Mainland China and recruited from the outpatient neurology clinics of Xiangya Hospital. The skin biopsies were cut into ~1 mm3 pieces and digested with 0.25% trypsin at 37°C for 20 min to separate the dermis from the epidermis. The dermis was collected and minced into small pieces, and then cultured in DMEM medium with 20% FBS. The dermal fibroblasts migrated from the dermal tissues were collected and cultured. The Institutional Ethics Committee of Xiangya Hospital at the Central South University approved the study. Informed consent was obtained from all human subjects recruited for this study.
Statistical analysis
Statistical analysis was performed using Prism 5 software (GraphPad, La Jolla, USA). Two‐tailed Student's t‐test was used to determine the significance of the difference between two groups. Statistical significance between multiple groups was derived using one‐way ANOVA followed by Tukey's test. One‐way ANOVA with Dunnett's tests were used to assess the difference between treatment groups against their controls. All error bars indicate SEM. The quantitation was performed by double‐blinded.
Author contributions
ZZ conceived the project concept and obtained fundings. HH, JT, and ZZ designed experiments. HH, QG, JL, and RT performed in vitro cell and mouse experiments. RW, SS, and FC performed fly experiments. HW, YH, WL, and LL performed mass spectra analysis. XY, JG, and BT generated HDF cell lines and provided the clinical data. HH, QG, JT, and ZZ analyzed data. HH, JT, and ZZ wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Source Data for Appendix
Review Process File
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31730036, 81861138012, 81161120498, 81429002, and 31330031), the Discipline Innovative Engineering Plan (111 Program) of China (B13036) and a key laboratory grant from Hunan province (2016TP1006), the National Key Plan for Scientific Research and Development of China (2016YFC1306000). Science and Technology Major Project of Hunan Provincial Science and Technology Department (2018SK1030).
EMBO Reports (2020) 21: e48686
References
- 1. de Rijk MC, Breteler MM, Graveland GA, Ott A, Grobbee DE, van der Meche FG, Hofman A (1995) Prevalence of Parkinson's disease in the elderly: the Rotterdam Study. Neurology 45: 2143–2146 [DOI] [PubMed] [Google Scholar]
- 2. Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ (2014) PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol 205: 143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vives‐Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS et al (2010) PINK1‐dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USA 107: 378–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T et al (2014) Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510: 162–166 [DOI] [PubMed] [Google Scholar]
- 5. Wauer T, Simicek M, Schubert A, Komander D (2015) Mechanism of phospho‐ubiquitin‐induced PARKIN activation. Nature 524: 370–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F et al (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 189: 211–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8: e1000298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ (2010) Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol 191: 933–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chan DC (2006) Mitochondria: dynamic organelles in disease, aging, and development. Cell 125: 1241–1252 [DOI] [PubMed] [Google Scholar]
- 10. Hoppins S (2014) The regulation of mitochondrial dynamics. Curr Opin Cell Biol 29: 46–52 [DOI] [PubMed] [Google Scholar]
- 11. Bleazard W, McCaffery JM, King EJ, Bale S, Mozdy A, Tieu Q, Nunnari J, Shaw JM (1999) The dynamin‐related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat Cell Biol 1: 298–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chang CR, Blackstone C (2010) Dynamic regulation of mitochondrial fission through modification of the dynamin‐related protein Drp1. Ann N Y Acad Sci 1201: 34–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K (2007) Mitotic phosphorylation of dynamin‐related GTPase Drp1 participates in mitochondrial fission. J Biol Chem 282: 11521–11529 [DOI] [PubMed] [Google Scholar]
- 14. Perdiz D, Lorin S, Leroy‐Gori I, Pous C (2017) Stress‐induced hyperacetylation of microtubule enhances mitochondrial fission and modulates the phosphorylation of Drp1 at 616Ser. Cell Signal 39: 32–43 [DOI] [PubMed] [Google Scholar]
- 15. Gomes LC, Di Benedetto G, Scorrano L (2011) During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 13: 589–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Strack S, Wilson TJ, Cribbs JT (2013) Cyclin‐dependent kinases regulate splice‐specific targeting of dynamin‐related protein 1 to microtubules. J Cell Biol 201: 1037–1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zaja I, Bai X, Liu Y, Kikuchi C, Dosenovic S, Yan Y, Canfield SG, Bosnjak ZJ (2014) Cdk1, PKCdelta and calcineurin‐mediated Drp1 pathway contributes to mitochondrial fission‐induced cardiomyocyte death. Biochem Biophys Res Commun 453: 710–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xie Q, Wu Q, Horbinski CM, Flavahan WA, Yang K, Zhou W, Dombrowski SM, Huang Z, Fang X, Shi Y et al (2015) Mitochondrial control by DRP1 in brain tumor initiating cells. Nat Neurosci 18: 501–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kashatus JA, Nascimento A, Myers LJ, Sher A, Byrne FL, Hoehn KL, Counter CM, Kashatus DF (2015) Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK‐driven tumor growth. Mol Cell 57: 537–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xu S, Wang P, Zhang H, Gong G, Gutierrez Cortes N, Zhu W, Yoon Y, Tian R, Wang W (2016) CaMKII induces permeability transition through Drp1 phosphorylation during chronic beta‐AR stimulation. Nat Commun 7: 13189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Han XJ, Lu YF, Li SA, Kaitsuka T, Sato Y, Tomizawa K, Nairn AC, Takei K, Matsui H, Matsushita M (2008) CaM kinase I alpha‐induced phosphorylation of Drp1 regulates mitochondrial morphology. J Cell Biol 182: 573–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yang Y, Ouyang Y, Yang L, Beal MF, McQuibban A, Vogel H, Lu B (2008) Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci USA 105: 7070–7075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Deng H, Dodson MW, Huang H, Guo M (2008) The Parkinson's disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila . Proc Natl Acad Sci USA 105: 14503–14508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ (2008) The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci USA 105: 1638–1643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xiong H, Wang D, Chen L, Choo YS, Ma H, Tang C, Xia K, Jiang W, Ronai Z, Zhuang X et al (2009) Parkin, PINK1, and DJ‐1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. J Clin Invest 119: 650–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pryde KR, Smith HL, Chau KY, Schapira AH (2016) PINK1 disables the anti‐fission machinery to segregate damaged mitochondria for mitophagy. J Cell Biol 213: 163–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, Alessi DR, Knebel A, Trost M, Muqit MM (2014) Parkin is activated by PINK1‐dependent phosphorylation of ubiquitin at Ser65. Biochem J 460: 127–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yu W, Sun Y, Guo S, Lu B (2011) The PINK1/Parkin pathway regulates mitochondrial dynamics and function in mammalian hippocampal and dopaminergic neurons. Hum Mol Genet 20: 3227–3240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lazarou M, Jin SM, Kane LA, Youle RJ (2012) Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell 22: 320–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Belshaw PJ, Ho SN, Crabtree GR, Schreiber SL (1996) Controlling protein association and subcellular localization with a synthetic ligand that induces heterodimerization of proteins. Proc Natl Acad Sci USA 93: 4604–4607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524: 309–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sarraf SA, Raman M, Guarani‐Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW (2013) Landscape of the PARKIN‐dependent ubiquitylome in response to mitochondrial depolarization. Nature 496: 372–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu S, Lu B (2010) Reduction of protein translation and activation of autophagy protect against PINK1 pathogenesis in Drosophila melanogaster . PLoS Genet 6: e1001237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. de Castro IP, Costa AC, Celardo I, Tufi R, Dinsdale D, Loh SH, Martins LM (2013) Drosophila ref(2)P is required for the parkin‐mediated suppression of mitochondrial dysfunction in pink1 mutants. Cell Death Dis 4: e873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kandul NP, Zhang T, Hay BA, Guo M (2016) Selective removal of deletion‐bearing mitochondrial DNA in heteroplasmic Drosophila . Nat Commun 7: 13100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Juhasz G, Erdi B, Sass M, Neufeld TP (2007) Atg7‐dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila . Genes Dev 21: 3061–3066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yuan XL, Guo JF, Shi ZH, Xiao ZQ, Yan XX, Zhao BL, Tang BS (2010) R492X mutation in PTEN‐induced putative kinase 1 induced cellular mitochondrial dysfunction and oxidative stress. Brain Res 1351: 229–237 [DOI] [PubMed] [Google Scholar]
- 38. Zhang T, Xue L, Li L, Tang C, Wan Z, Wang R, Tan J, Tan Y, Han H, Tian R et al (2016) BNIP3 protein suppresses PINK1 kinase proteolytic cleavage to promote mitophagy. J Biol Chem 291: 21616–21629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sanjana NE, Shalem O, Zhang F (2014) Improved vectors and genome‐wide libraries for CRISPR screening. Nat Methods 11: 783–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM et al (2006) Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441: 1157–1161 [DOI] [PubMed] [Google Scholar]
- 41. Woodroof HI, Pogson JH, Begley M, Cantley LC, Deak M, Campbell DG, van Aalten DM, Whitworth AJ, Alessi DR, Muqit MM (2011) Discovery of catalytically active orthologues of the Parkinson's disease kinase PINK1: analysis of substrate specificity and impact of mutations. Open Biol 1: 110012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hertz NT, Berthet A, Sos ML, Thorn KS, Burlingame AL, Nakamura K, Shokat KM (2013) A neo‐substrate that amplifies catalytic activity of parkinson's‐disease‐related kinase PINK1. Cell 154: 737–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wan H, Tang B, Liao X, Zeng Q, Zhang Z, Liao L (2018) Analysis of neuronal phosphoproteome reveals PINK1 regulation of BAD function and cell death. Cell Death Differ 25: 904–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rasool S, Soya N, Truong L, Croteau N, Lukacs GL, Trempe JF (2018) PINK1 autophosphorylation is required for ubiquitin recognition. EMBO Rep 19: e44981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Deng M, He W, Tan Y, Han H, Hu X, Xia K, Zhang Z, Yan R (2013) Increased expression of reticulon 3 in neurons leads to reduced axonal transport of beta site amyloid precursor protein‐cleaving enzyme 1. J Biol Chem 288: 30236–30245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ji WK, Hatch AL, Merrill RA, Strack S, Higgs HN (2015) Actin filaments target the oligomeric maturation of the dynamin GTPase Drp1 to mitochondrial fission sites. Elife 4: e11553 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Source Data for Appendix
Review Process File