

Abstract
Staphylococcus aureus is a notorious pathogen causing significant morbidity and mortality worldwide. The ability of S. aureus to survive and replicate within phagocytes such as macrophages represents an important facet of immune evasion and contributes to pathogenesis. The mechanisms by which S. aureus acquires nutrients within host cells to support growth remain poorly characterized. Here, we demonstrate that macrophages infected with S. aureus maintain their dynamic ruffling behavior and consume macromolecules from the extracellular milieu. To support the notion that fluid‐phase uptake by macrophages can provide S. aureus with nutrients, we utilized the pharmacological inhibitors PIK‐III and Dynasore to impair uptake of extracellular macromolecules. Inhibitor treatment also impaired S. aureus replication within macrophages. Finally, using a mutant of S. aureus that is defective in purine biosynthesis we show that intracellular growth is inhibited unless the macrophage culture medium is supplemented with the metabolite inosine monophosphate. This growth rescue can be impaired by inhibition of fluid‐phase uptake. In summary, through consumption of the extracellular environment macrophages deliver nutrients to phagolysosomal S. aureus to promote bacterial growth.
Keywords: macrophage, nutrient, phagolysosome, pinocytosis, Staphylococcus
Subject Categories: Membrane & Intracellular Transport; Metabolism; Microbiology, Virology & Host Pathogen Interaction
Fluid‐phase uptake by infected macrophages via pinocytosis and macropinocytosis enables the delivery of macromolecules to intraphagosomal S. aureus. Macrophage‐driven delivery of extracellular material provides S. aureus with nutrients that support intracellular growth.
Introduction
Staphylococcus aureus is a notorious bacterial pathogen and a global cause of significant morbidity and mortality 1. Treatment of S. aureus infection can be complicated due to the emergence of drug‐resistant strains such as methicillin‐resistant S. aureus (MRSA) 2, 3 that are highly pathogenic and cause nosocomial 4, 5, 6 and community‐acquired infections 7, 8. The profound capacity of S. aureus to cause infection and colonize virtually any tissue from the human body can, in part, be attributed to the vast repertoire of virulence factors that can be deployed by S. aureus to extract nutrients, adhere to host tissues, and to circumvent immunity (reviewed in 9, 10, 11).
It is generally accepted that S. aureus is a facultative intracellular pathogen 12 that can survive and grow within immune cells such as neutrophils and macrophages 13, 14. Indeed, macrophages represent a critical bottleneck during S. aureus infection. This is exemplified by murine bacteremia models in which bloodborne S. aureus bacteria are quickly captured by Kupffer cells. Here, a subset of the bacterial population survives, replicates, and emerges from within Kupffer cells and subsequently peritoneal macrophages to efficiently infect other visceral organs 15, 16. Previous work from our group and others has demonstrated that within macrophages, S. aureus commences growth while confined to LAMP‐1‐positive acidic phagolysosomes 13, 15. Moreover, onset of bacterial replication is significantly delayed and occurs hours (> 6 h) after infection 13, 15, 17. The mechanisms by which S. aureus obtains nutrients to support growth within the macrophage phagolysosome are unknown. Given the plethora of host proteins (e.g., SLC11A1 and SLC38A9) that transport nutrients (i.e., Mg2+ or amino acids) out of the phagosome lumen 18, 19, 20, 21, 22, 23, it might be expected that S. aureus occupies an intracellular niche that is nutrient deplete. Despite this, S. aureus must acquire nutrients as the bacteria proliferate within the phagolysosome of host phagocytes 13.
The ability of macrophages to constitutively elaborate dynamic membrane ruffles endows this cell type with the ability to consume copious amounts of the fluid‐phase environment through pinocytosis and/or macropinocytosis 24, 25. Previous work has demonstrated that this dynamic behavior is essential for macrophage immune surveillance by facilitating the capture of phagocytic targets 26 and delivery of pathogen‐associated ligands to the cytosolic pattern recognition receptors NOD1/2 24. Fluid‐phase uptake also functions as a cellular nutrient retrieval system to support eukaryotic cell growth and repair 27, 28, 29. Given our observation that S. aureus resides within a phagolysosome prior to replication inside macrophages, we posited that S. aureus might usurp ongoing macrophage fluid‐phase uptake for nutrient acquisition. Herein, we provide evidence demonstrating S. aureus‐infected macrophages maintain their dynamic ruffling behavior which leads to the consumption of macromolecules from the extracellular milieu; this also demonstrates that intracellular S. aureus are not overtly intoxicating the phagocytes before replication proceeds. Moreover, we demonstrate that this fluid‐phase material can be consumed, degraded, and delivered to intracellular S. aureus. Through pharmacologic inhibition of membrane ruffling and endocytosis, which ablates fluid‐phase uptake, we demonstrate that proliferation of S. aureus within macrophages can be suppressed. Finally, using this inhibitory strategy we demonstrate that the intracellular growth of an S. aureus mutant that is defective in purine biosynthesis can be controlled by the ability of macrophages to consume the metabolite inosine monophosphate from the extracellular milieu.
Results and Discussion
S. aureus‐infected macrophages consume macromolecules from the extracellular milieu
Consumption of the fluid‐phase environment by macrophages through endocytosis, pinocytosis, and micropinocytosis enables these cells to ingest a plethora of macromolecules that can be delivered to lysosomes 28, 30. Upon discovering that S. aureus resides within macrophage phagolysosomes prior to bacterial replication 13, 17, we questioned whether ongoing fluid‐phase uptake by infected macrophages could provide phagolysosomal cocci with nutrients for growth. To begin to address this question, we first sought to determine whether macrophages infected with S. aureus USA300 displayed the constitutive ruffling behavior that is characteristic of these cells 24, 25, 26. Through live‐cell time‐lapse video microscopy, it was evident that RAW 264.7 macrophages infected with S. aureus maintained the ability to form membrane ruffles and cytoplasmic vacuoles consistent with pinosome/macropinosome formation; this feature was indistinguishable between macrophages infected with either live or heat‐killed S. aureus (Fig 1A and Movies EV1 and EV2). Similar experiments performed in primary murine bone marrow‐derived macrophages established that sustained ruffling was not unique to RAW macrophages (Movies EV3 and EV4). This dynamic macrophage behavior is dependent on rearrangement of the actin cytoskeleton that underlies the plasma membrane, and perturbation of the cellular machinery that coordinates actin remodeling inhibits membrane ruffling and fluid‐phase uptake 25, 26. The phosphoinositide phosphatidylinositol 4,5‐bisphosphate (PI(4,5)P2) is crucial for the formation of phosphatidic acid (PA) at the macrophage plasma membrane. This constitutive production of PA localizes RhoGTPase guanine nucleotide exchange factors (e.g., Tiam1) to the cytoplasmic membrane to effect actin rearrangement 25. Furthermore, depletion of PI(4,5)P2 or diacyl glycerol kinase inhibition perturbs PA production and suppresses membrane ruffling 25. In accordance with our observation that macrophage ruffling is unaffected by S. aureus, we find that macrophages infected with live S. aureus USA300 for at least 8 h maintain their plasmalemmal pool of PI(4,5)P2. This is revealed by the accumulation of the lipid biosensor PH‐Plcδ‐RFP, in both infected and uninfected cells, to the cytoplasmic membrane (Fig 1B). In contrast, depletion of PI(4,5)P2 by treatment of macrophages with the calcium ionophore ionomycin displaces RFP fluorescence from the plasmalemma to the cytoplasm (Fig 1B). Taken together, these data reveal that S. aureus‐infected macrophages sustain their dynamic ruffling behavior.
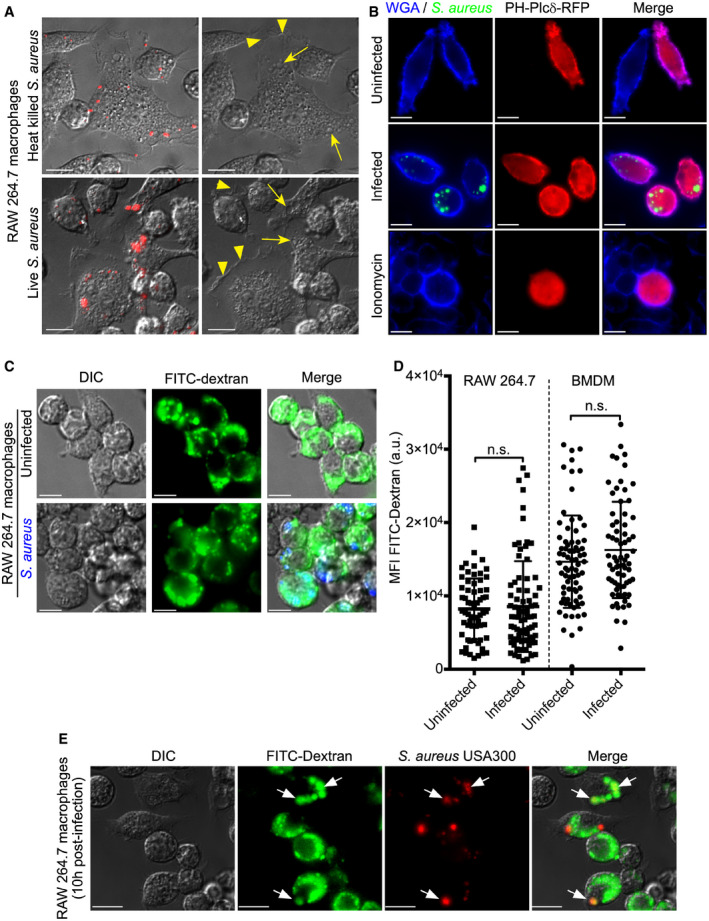
Figure 1. Staphylococcus aureus‐infected macrophages display continuous membrane ruffling and internalize the fluid‐phase environment.
-
ARAW 264.7 macrophages infected with heat‐killed (top panels) or live (bottom panels) S. aureus USA300 (in red) were imaged by DIC time‐lapse microscopy at 6 h post‐infection. The images on the left are of time zero in the image sequence and show the presence of S. aureus. The images on the right are of the same cells, and the presence of membrane ruffles and dynamic filopodia is denoted by the yellow arrowheads. Candidate pinosomes are denoted by yellow arrows.
-
BRAW 264.7 macrophages expressing the lipid biosensor for PI(4,5)P2, PH‐Plcδ‐RFP (in red), were left uninfected or were infected with live GFP‐expressing S. aureus USA300 (in green) for 8 h prior to fixation. The macrophage plasmalemma and extracellular bacteria are indicated by Cy5‐conjugated wheat germ agglutinin (WGA, in blue). As a control, to demonstrate displacement of PH‐Plcδ‐RFP from the macrophage membrane, transfected cells were treated with 10 μM ionomycin in the presence of 1 mM CaCl2 for 20 min prior to fixation. These images are representative of three independent experiments.
-
C, DInternalization of FITC–dextran by uninfected (top row) and S. aureus (in blue)‐infected (bottom row) RAW 264.7 macrophages at 10 h post‐infection. These images are representative of three independent experiments. The quantitation of FITC–dextran uptake by either uninfected or infected RAW 264.7 and primary bone marrow‐derived macrophages is shown in (D). For each experiment, macrophages were treated with 1 μM concanamycin A for 30 min prior to image acquisition. Shown is the mean fluorescence intensity (MFI) in arbitrary units (a.u.) ± standard deviation where each symbol represents the measurement of a single cell. The data are derived from three independent experiments, and n.s. indicates the data are not significant as determined by an unpaired two‐tailed t‐test (P > 0.05).
-
EPotential for co‐localization of FITC–dextran (in green) with phagocytosed S. aureus expressing mCherry (in red). The micrographs depict representative macrophages infected with S. aureus at 10 h post‐infection, and the white arrows point to mCherry positive bacteria that are co‐localizing with dextran. Macrophages were treated with 1 μM concanamycin A prior to imaging live.
Next, we sought to determine whether S. aureus‐infected macrophages maintain the ability to internalize material from the extracellular milieu. To accomplish this, we monitored the uptake of 10 kDa FITC‐conjugated dextran which was used as a fluorescent fluid‐phase tracer. Here, macrophages were left uninfected or were infected with live S. aureus USA300 at a multiplicity of infection (MOI) of 30 to create a high intracellular bacterial burden where it might be expected that dextran uptake would be perturbed. Remarkably, live‐cell fluorescence microscopy demonstrated that at 10 h post‐infection the internalization of FITC–dextran appeared unaffected by the presence of intracellular S. aureus (Fig 1C). To ensure accurate detection of FITC fluorescence, which can be quenched in acidic lysosomes and phagolysosomes 31, macrophages were treated with concanamycin A, an inhibitor of the vacuolar ATPase, for at least 30 min prior to fluorescence imaging. Quantitation of the cellular FITC fluorescence intensity for uninfected and S. aureus‐infected cells confirmed that fluid‐phase consumption of dextran was indistinguishable (Fig 1D). Similar experiments were performed using primary bone marrow‐derived macrophages and confirmed that intracellular S. aureus infection did not perturb fluid‐phase uptake in primary macrophages (Fig 1D). Interestingly, analysis of dextran localization in infected cells revealed that in some instances FITC–dextran appeared to co‐localize with phagocytosed S. aureus USA300 (Fig 1E). This might be expected if macromolecules derived from the extracellular milieu are delivered to S. aureus‐containing phagosomes (SaCP). Moreover, this would indicate that phagolysosomal S. aureus can remain in contact with the extracellular environment through ongoing fluid‐phase uptake by the phagocyte. To confirm this assertion and establish that dextran delivery to intracellular S. aureus was not an artifact of an elevated MOI, dextran internalization experiments were also performed using a MOI of 10. Importantly, even with a reduced MOI it was evident that live S. aureus could indeed co‐localize with dextran ingested by macrophages after phagocytosis of the bacteria (Fig 1E). Taken together, these data reveal that S. aureus‐infected macrophages consume macromolecules from the extracellular milieu that can be delivered to intracellular S. aureus.
If S. aureus is to derive nutrition from macromolecules internalized by macrophages, it stands to reason that this material should be degraded and delivered to the SaCP. To determine whether S. aureus‐infected macrophages are indeed capable of degrading material ingested from the fluid‐phase environment, we utilized the fluorogenic substrate DQ™ Green BSA that fluoresces after proteolytic processing 32. Macrophages were either uninfected or infected at a MOI of 30 with live S. aureus USA300, and at 1.5 h post‐infection, after gentamicin treatment, DQ™ Green BSA was added to the tissue culture medium. At 10 h post‐infection, macrophages were imaged by live‐cell fluorescence microscopy revealing that both uninfected and infected macrophages exhibited green fluorescence consistent with the ability of these cells to internalize and degrade DQ™ Green BSA during the infection (Fig 2A). Quantitation of the fluorescence intensity of uninfected and S. aureus‐containing macrophages revealed that degradative capacity of these phagocytes was indistinguishable (Fig 2B). Presumably, DQ‐BSA uptake and degradation continue over time as uninfected and infected RAW cells exhibit minimal fluorescence with only short (< 30 min) DQ‐BSA incubation. The resulting images at 10 h post‐infection revealed that similar to FITC–dextran, fluorescent (i.e., proteolyzed) DQ™ Green BSA can also co‐localize with phagocytosed S. aureus (Fig 2A). Moreover, using an established fluorescence‐based assay to identify intracellularly proliferating S. aureus 33 we found that replicating bacteria can also co‐localize with DQ™ Green BSA within macrophages (Fig 2C). Taken together, these data indicate that macrophages can degrade macromolecules internalized from the extracellular milieu and this degraded material can be delivered to the SaCP where S. aureus can proliferate.
Figure 2. Infected macrophages can internalize and deliver degraded extracellular macromolecules to intracellular S. aureus .
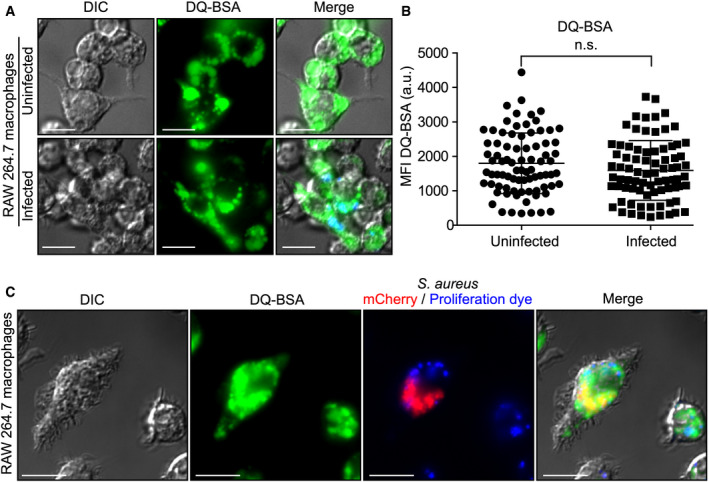
- Internalization and processing of DQ™ Green BSA by uninfected and S. aureus (in blue)‐infected RAW 264.7 macrophages at 10 h post‐infection. These images are representative of three independent experiments.
- Quantitation of DQ‐BSA green fluorescence displayed by either uninfected or S. aureus (in blue)‐infected RAW 264.7. Shown is the mean fluorescence intensity (MFI) in arbitrary units (a.u) ± standard deviation where each symbol represents the measurement of a single cell. The data are derived from three independent experiments, and n.s. indicates the data are not significant as determined by an unpaired two‐tailed t‐test (P > 0.05).
- Fluorescence‐based proliferation assays were performed in conjunction with the probe DQ™ Green BSA. Processed DQ™ Green BSA could be found co‐localizing with intracellular mCherry‐expressing S. aureus (in red) and with intracellular S. aureus that has replicated (i.e., proliferation dye negative) at 10 h post‐infection as depicted here. Data information: Scale bars equal 10 μm.
Inhibition of fluid‐phase uptake by macrophages can alter S. aureus growth within macrophages
Next, we sought to determine whether compounds added to the extracellular milieu could impact the physiology of S. aureus residing within the macrophage phagolysosome. Therefore, we devised a method that would allow us to block the collective fluid‐phase uptake by macrophages. We utilized the pharmacologic agents Dynasore, an inhibitor of the GTPases dynamin 1 and 2, and PIK‐III, an inhibitor of the type III phosphatidylinositol 3‐kinase (PI3K) vacuolar protein sorting 34 (Vps34) 34, 35. The importance of dynamin proteins to receptor‐mediated endocytosis and to catalyzing membrane scission events is well established, and Dynasore has previously been shown to inhibit receptor‐mediated endocytosis of transferrin 34, 36. Control experiments monitoring endocytosis of Alexa Fluor 647‐conjugated transferrin by RAW 264.7 macrophages in the presence of the Dynasore inhibitor reaffirmed this assertion causing Alexa Fluor 647 transferrin to be retained at the plasmalemma (Fig 3A). In contrast, PIK‐III did not adversely affect transferrin endocytosis which resembled vehicle control‐treated cells (Fig 3A). Remarkably, PIK‐III, which has been shown to inhibit autophagy‐related processes 35, had the unexpected effect on macrophages where treatment ablated the formation of dynamic membrane ruffles and protrusions which were displayed by vehicle control‐treated cells (Movies EV5 and EV6). Due to the pronounced effect of PIK‐III on membrane ruffling, we speculated that drug treatment would also severely impair dextran uptake thorough macropinosome/pinosome formation. To test this notion, we monitored the uptake of fluorescent dextran by macrophages treated with vehicle only, PIK‐III, Dynasore, and PIK‐III with Dynasore. This analysis revealed that while in the presence of PIK‐III, macrophages are devoid of membrane ruffles and are severely impaired in their ability to internalize the fluorescent fluid‐phase tracer (Fig 3B and C). Interestingly, Dynasore treatment reduced, but did not block, dextran uptake (Fig 3C). This indicates dynamin‐dependent and dynamin‐independent processes could be involved in dextran ingestion or dynamin inhibition is not absolute (Fig 3C). Regardless, given these results we utilized PIK‐III and Dynasore simultaneously to effectively inhibit fluid‐phase uptake through endocytic and pinocytic pathways.
Figure 3. The uptake of macromolecules from the extracellular environment can be inhibited pharmacologically.
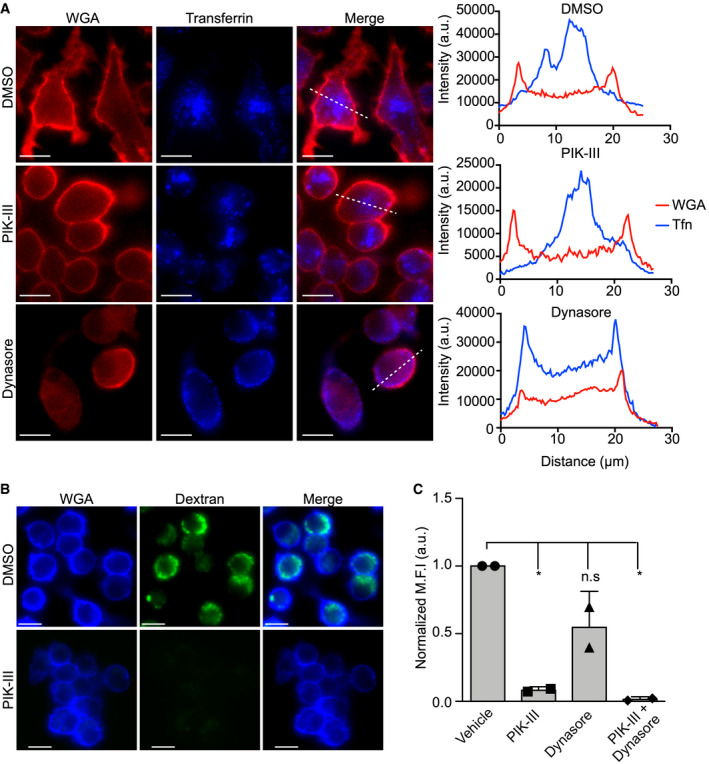
- Uptake of Alexa® Fluor 647‐conjugated transferrin by RAW 264.7 macrophages in the presence and absence of PIK‐III and Dynasore. The fluorescence micrographs show transferrin (in blue) and the macrophage plasmalemma demarcated by TMR‐conjugated wheat germ agglutinin (in red). The dashed line indicates the region of the cell for which the line scan analysis, shown graphically on the right, was done. These micrographs are representative images obtained from three independent experiments. Line scan analysis was performed to indicate the distribution of fluorescent transferrin (blue line) relative to the plasmalemma (red line) for the DMSO‐, PIK‐III and Dynasore‐treated cells presented in the fluorescence micrographs. Scale bars equal 10 μm.
- RAW macrophages were pre‐treated with vehicle (DMSO) or the vps34 inhibitor PIK‐III prior to the addition of 10 kDa FITC–dextran (100 μg/ml) for 2 h. Prior to imaging live, macrophages were also stained with Cy5‐conjugated WGA. The representative micrographs depict inhibited dextran uptake indicating PIK‐III treatment can perturb fluid‐phase uptake. Scale bars equal 10 μm.
- The intensity of dextran fluorescence was quantified for RAW cells pre‐treated with vehicle, PIK‐III (10 μM), Dynasore (100 μM), or PIK‐III and Dynasore (10 and 100 μM), respectively. The data shown are the mean fluorescence intensity ± standard deviation for each experiment calculated from at least 30 cells from two independent experiments. * indicates P < 0.05, as determined by an ordinary one‐way ANOVA with a Bonferroni multiple comparisons test.
Next, we sought to demonstrate that compounds added to the extracellular milieu after S. aureus had been phagocytosed could impact intracellular growth and/or survival of the bacteria. We utilized the aminoglycoside antibiotic gentamicin, which does not diffuse directly across biological membranes but can be internalized through fluid‐phase uptake 13, 37. In addition, we employed the large (1.45 kDa) glycopeptide‐based antibiotic vancomycin that poorly penetrates eukaryotic cells 15, 38 but should also be internalized via fluid‐phase uptake akin to dextran and DQ™ Green BSA. To determine the impact of these compounds on intracellular bacteria, macrophages were allowed to phagocytose S. aureus and the extracellular bacteria were inactivated by treatment with 100 μg/ml gentamicin. After this initial antibiotic treatment, macrophages were maintained in either tissue culture medium alone or medium supplemented with gentamicin (300 μg/ml) or vancomycin (300 μg/ml) until 10 h post‐infection. High concentrations of antibiotics were purposefully employed here in an attempt to promote intracellular killing over a period of several hours. Next, infected cells were washed extensively, and the intracellular bacterial burden was determined (Fig 4A). This analysis revealed that macrophages without prolonged antibiotic treatment failed to kill intracellular S. aureus and prevent bacterial growth (Fig 4A). In contrast, in the presence of gentamicin or vancomycin the intracellular burden of S. aureus was reduced as compared to the untreated control (Fig 4A). Given the chemical properties of these compounds and the likelihood they are entering infected macrophages via fluid‐phase uptake, we speculated that treatment of macrophages with PIK‐III and Dynasore should, in part, rescue intracellular S. aureus survival. As a proof of principle experiment, we performed macrophage infections as described above; however, macrophages were treated with either a vehicle control or PIK‐III/Dynasore prior to the addition of gentamicin at the high concentration which was maintained throughout the experiment. At 10 h post‐infection, macrophage lysates were plated to determine whether the presence of PIK‐III/Dynasore could increase intracellular survival of phagocytosed S. aureus when gentamicin is maintained in the extracellular environment as compared to infected cells without PIK‐III/Dynasore treatment. We found that treatment of S. aureus‐infected macrophages with the inhibitors increased the number of bacteria recovered from within host cells by 2.1‐fold as compared to vehicle control‐treated cells despite the very high concentration of gentamicin employed (Fig 4B). This is consistent with the notion that gentamicin is entering the cells via fluid‐phase uptake, which can be impaired upon treatment with PIK‐III and Dynasore.
Figure 4. Perturbation of fluid‐phase uptake can modulate growth of S. aureus within macrophages.
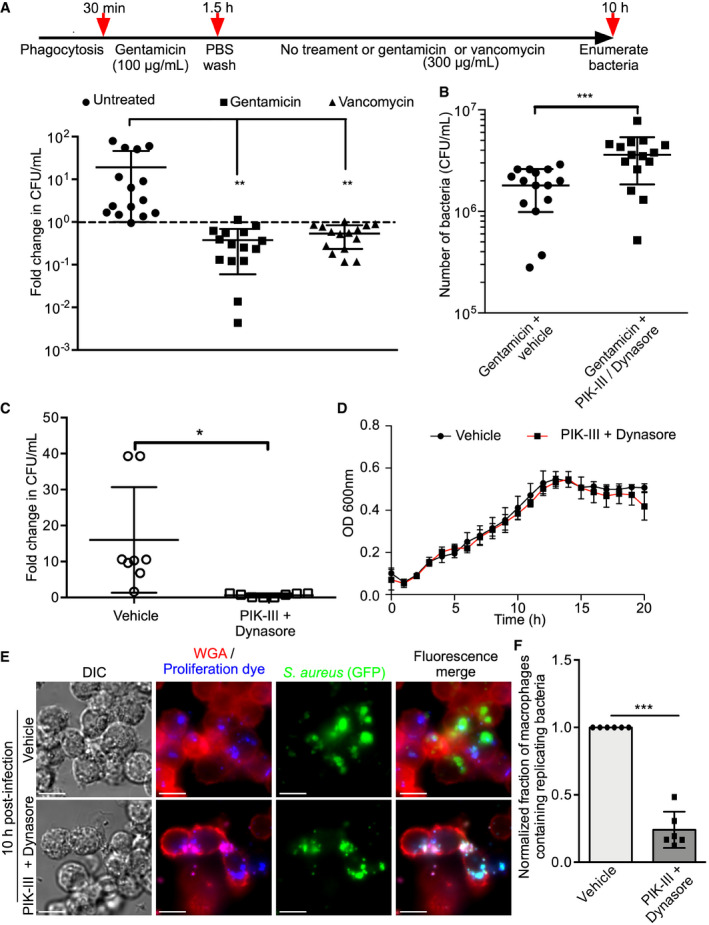
- The effects of prolonged exposure to gentamicin (300 μg/ml) or vancomycin (300 μg/ml) on the survival of phagocytosed S. aureus USA300 in RAW macrophages are shown. Depicted above the graph is a timeline detailing the treatment regime. The data are presented as the mean fold change in CFU/ml ± standard deviation for 15 biological replicates of S. aureus, represented by each symbol, from five independent experiments. For each replicate, the fold change was calculated by dividing the bacterial count at 10 h post‐infection by the counts at 1.5 h post‐infection. The dashed line indicates a fold change value equal to 1 (i.e., no change in bacterial burden). **P < 0.01 and statistical significance was determined by a one‐way ANOVA with Dunnett's post hoc test.
- The ability to protect S. aureus USA300 from intracellular killing by gentamicin is shown. Here, experiments were performed as in (A) except during the initial gentamicin treatment (100 μg/ml for 1 h) to kill extracellular bacteria macrophages were also treated with either DMSO or PIK‐III/Dynasore to inhibit fluid‐phase uptake. At 1.5 h post‐infection, cells were washed and treated with gentamicin (300 μg/ml) in the presence of either DMSO (vehicle) or PIK‐III + Dynasore which were maintained throughout the experiment. The data are presented as the mean number of surviving bacteria in CFU/ml at 10 h post‐infection ± standard deviation. These data are derived from a total of 15 biological replicates of S. aureus from at least three independent experiments. ***P < 0.001 and statistical significance was determined by an unpaired two‐tailed t‐test with Welch's correction.
- The effect of PIK‐III/Dynasore treatment on the ability of S. aureus USA300 to replicate within RAW macrophages is shown. After gentamicin treatment at 1.5 h, RAW 264.7 macrophages having phagocytosed S. aureus were lysed and plated to determine the number of gentamicin protected bacteria. Alternatively, infected samples treated with either DMSO as a vehicle control or with PIK‐III/Dynasore which was added at 1.5 h post‐infection after gentamicin. At 10 h post‐infection, macrophages exposed to DMSO or inhibitors, that were maintained throughout the experiment, were lysed and plated to determine the bacterial burden. The data shown are the mean fold change in CFU/ml at 10 h where the data are normalized to the bacterial burden obtained at 1.5 h. Each symbol represents a biological replicate (n = 8) of S. aureus and derives from three independent experiments. The data presented are the mean ± standard deviation. * indicates P < 0.05 as determined by an unpaired two‐tailed t‐test with Welch's correction.
- The effect of PIK‐III + Dynasore or DMSO on the growth of S. aureus USA300 in RPMI with 5% (v/v) FBS is shown. The data are the mean OD600 nm ± standard deviation of four biological replicates of S. aureus for each condition and are representative of three independent experiments.
- Analysis of the effects of PIK‐III/Dynasore treatment on S. aureus replication using a fluorescence‐based bacterial proliferation assay is shown. The micrographs depict S. aureus USA300 expressing GFP that were co‐labeled with proliferation dye. The top row of micrographs depict vehicle‐treated macrophages where GFP‐positive, yet proliferation dye‐negative bacteria were present. The bottom row depict inhibitor treated macrophages where the bacteria fail to replicate and remain proliferation dye positive. The macrophage plasmalemma and extracellular bacteria are demarcated by TMR‐WGA (in red). Images were acquired at 10 h post‐infection and are representative of three independent experiments. Scale bars equal 10 μm.
- The normalized fraction of macrophages containing replicating bacteria is shown. The graph represents the mean ± standard deviation of the number of macrophages that contain replicating bacteria where the data are normalized to the vehicle control condition. Each symbol represents a biological replicate (n = 6) of S. aureus and derives from three independent experiments. ****P < 0.0001 and statistical significance was determined by an unpaired two‐tailed t‐test using Welch's correction.
Previous work from our laboratory and others has shown that S. aureus commences proliferation within the macrophage phagolysosome both in vitro and in vivo 13, 15. Considering that material ingested from the extracellular milieu is delivered to intracellular S. aureus, we hypothesized that bacteria residing within the macrophage might exploit this phenomenon and use nutrients derived from the tissue culture medium and/or the bovine serum used as a supplement for intraphagosomal growth. To test this notion, we performed macrophage infections where after inactivation of extracellular S. aureus host cells were treated with either a vehicle control or were exposed to PIK‐III/Dynasore until 10 h post‐infection. Analysis of the bacterial burden at this time revealed that untreated macrophages failed to curtail S. aureus growth as indicated by the increase in bacterial counts as compared to 1.5 h post‐infection (Fig 4C). In contrast, pharmacologic treatment of S. aureus‐infected macrophages suppressed growth of intracellular S. aureus, as bacterial numbers remain unchanged (i.e., the fold change is ~1) over the course of the infection (Fig 4C). Importantly, PIK‐III and Dynasore are without effect on the ability of S. aureus to grow in tissue culture medium alone, indicating the observed growth impairment within macrophages is due to the effects of PIK‐III and Dynasore on the phagocytes (Fig 4D). To visualize the impact of PIK‐III and Dynasore treatment on S. aureus replication within macrophages, we also performed fluorescence‐based bacterial proliferation assays 33. Congruent with our determination of the cellular bacterial burden, we find that GFP‐positive, yet proliferation dye‐negative S. aureus (i.e., replicating bacteria) can readily be detected at 10 h post‐infection within vehicle control‐treated cells. In contrast, inhibitor treatment reduces the fraction of macrophages that contain replicating bacteria as indicated by retention of the proliferation dye by intracellular S. aureus (Fig 4E and F). Taken together, these data reveal that S. aureus residing within macrophages can be affected by components (e.g., antibiotics or conceivably nutrients) found in the extracellular environment and indicate that, in the absence of extracellular antibiotics, inhibition of fluid‐phase uptake can impair S. aureus growth intracellularly.
Nutrients derived from the extracellular milieu support the growth of intracellular S. aureus in macrophages
The compounds PIK‐III and Dynasore employed to inhibit fluid‐phase uptake will have alternative effects on cellular processes that could impact bacterial growth. Indeed, PIK‐III, an inhibitor of Vps34, perturbs autophagy‐related processes which could also impact nutrient availability within infected macrophages 35. In epithelioid cells, autophagy‐related processes have been reported to be required for intracellular S. aureus growth 39; however, in RAW 264.7 macrophages it has previously been shown that the SaCP is devoid of the autophagy‐related protein LC3 13. To demonstrate that these “off‐target” effects of PIK‐III treatment cannot explain the observed impediment to S. aureus growth inside macrophages in response to PIK‐III/Dynasore treatment, we performed infections using a purine biosynthesis mutant of S. aureus; growth of this mutant requires an exogenous source of purine (e.g., inosine monophosphate (IMP)) (Fig 5A). The mutant employed, derived from S. aureus USA300, carries a transposon in the purK gene which encodes an enzyme involved in the conversion of 5‐aminoimidazole ribonucleotide to 4‐carboxy‐5‐aminoimidazole ribonucleotide, an intermediate in the purine biosynthesis pathway 40. As PurK catalyzes an intermediate step in the biosynthesis of IMP in S. aureus, provision of IMP should enable purK‐deficient bacteria to overcome growth impairment when purines are limited. Indeed, when grown in RPMI with 5% (v/v) FBS the purK mutant of S. aureus demonstrates significant growth impairment as compared to wild‐type S. aureus USA300 (Fig 5A). Furthermore, supplementation of purK mutant cultures with 50 μM IMP restores growth of the mutant back to wild‐type levels (Fig 5A). Because growth of purK‐deficient S. aureus can be rescued by IMP, we utilized this strain for macrophage infections. Data from those experiments revealed these bacteria fail to grow intracellularly under routine culture conditions where IMP is not added to the tissue culture medium (Fig 5B). In contrast, growth of purK S. aureus can be rescued upon supplementation of the tissue culture medium with 50 μM IMP which was added after inactivation of extracellular bacteria (i.e., at 1.5 h post‐infection) (Fig 5B). Conceivably, IMP is entering the macrophage through ongoing fluid‐phase uptake which continues despite the presence of intracellular S. aureus. In macrophages, purine nucleosides do not freely permeate the membrane and there exist dedicated transport systems to acquire nucleosides from the extracellular environment 41, 42. Moreover, purines stimulate the pinocytic function of macrophages ostensibly contributing to increased uptake of these metabolites from the extracellular milieu 43. To test whether fluid‐phase uptake supports IMP‐dependent growth of intracellular purK‐deficient S. aureus, we employed the PIK‐III/Dynasore treatment regimen already shown to perturb fluid‐phase consumption by macrophages. Under these conditions, and in contrast to IMP alone, pre‐treatment of macrophages with PIK‐III/Dynasore ablated IMP‐dependent rescue of purK mutant growth within macrophages (Fig 5B). Importantly, purK S. aureus in the presence of IMP and PIK‐III/Dynasore grow to wild‐type levels indicating the inhibitors themselves do not adversely affect the growth of S. aureus lacking purK (Fig 5A). Having shown that primary BMDMs internalize fluorescent dextran from the extracellular milieu, we reasoned that growth of purK S. aureus inside these primary macrophages would also be supported by the addition of IMP. Here, eFluor‐proliferation assays were performed where macrophages were infected with the S. aureus purK mutant expressing GFP. After gentamicin treatment, infected macrophages were exposed to 50 μM IMP or vehicle control and the fraction of macrophages containing replicating bacteria was determined at 12 h post‐infection. This analysis revealed that extracellular addition of IMP increased the fraction of primary BMDMs containing replicating purK‐deficient S. aureus as compared to vehicle‐treated cells, indicated by the presence of proliferation dye‐negative bacteria (Fig 5C and D). Attempts to utilize PIK‐III and Dynasore, even at half the concentration employed for RAW macrophages, to block IMP uptake failed as the compounds were toxic to the primary phagocytes. Indeed, by 12 h post‐infection BMDMs exposed to PIK‐III/Dynasore were detached from the substratum. Unfortunately, prolonged treatment with PIK‐III and Dynasore is necessary as it has already been established that S. aureus demonstrates delayed growth within macrophages 13, 15. We also performed a similar experiment using primary human M‐CSF‐derived macrophages where we first established that M‐CSF‐derived macrophages when infected with S. aureus maintain the ability to ruffle akin to uninfected cells (Movies EV7 and EV8). We also analyzed whether IMP supplementation could rescue intracellular growth of purK‐deficient S. aureus in an experiment using primary human macrophages. Entirely consistent with our observations using murine BMDMs, we observed that IMP supplementation enhanced intracellular growth of purK S. aureus as compared to macrophages without IMP. Unfortunately, PIK‐III and Dynasore were again toxic to primary human macrophages precluding our ability to perform prolonged experiments with these cells. Nevertheless, these observations in BMDMs (and in primary human macrophages) reveal that the addition of IMP promotes the intracellular growth of S. aureus, and with all these data taken together, we provide compelling evidence that IMP‐dependent growth of purK S. aureus within macrophages requires ongoing fluid‐phase uptake by the infected phagocyte.
Figure 5. Inosine monophosphate dependent growth of a purK S. aureus in macrophages requires macrophage‐driven fluid‐phase uptake.
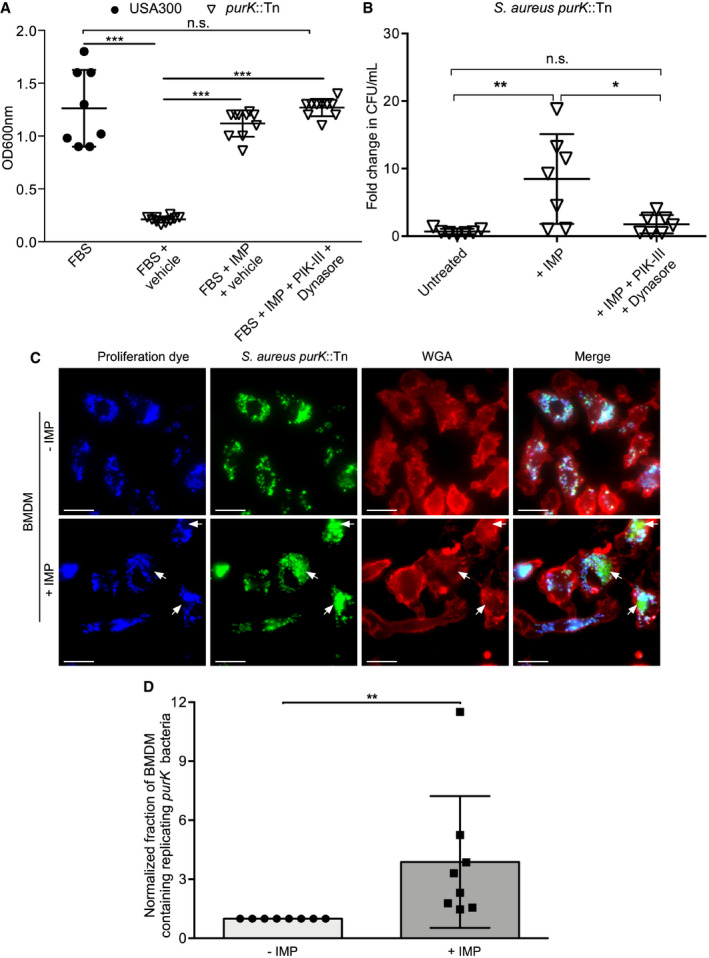
-
AGrowth of wild‐type S. aureus USA300 or a purK mutant. The data are the mean ± standard deviation of the end point optical density measured at 18 h at 600 nm for each strain in RPMI. Additives are indicated on the x‐axis and were either 5% (v/v) FBS, 50 μM IMP, 10 μM PIK‐III, and 100 μM Dynasore, or an equivalent volume of DMSO. Each symbol represents a biological replicate of S. aureus, and the data are derived from three independent experiments. Statistical significance was determined by a one‐way ANOVA with Holm–Sidak's multiple comparison test. n.s. indicates not significant, *** indicates P < 0.0001.
-
BThe graph depicts the fold change in bacterial burden for RAW 264.7 macrophages infected with purK S. aureus. Macrophages were left untreated or were exposed to 50 μM IMP or 50 μM IMP after pre‐treatment with PIK‐III and Dynasore which were maintained throughout the infection. The data are the mean ± standard deviation of the burden at 10 h post‐infection normalized to the burden at 1.5 h post‐infection prior to the addition of IMP. Each symbol represents a single biological replicate of S. aureus and derives from three independent experiments. Significance was determined by one‐way ANOVA with a Turkeys multiple comparison test. *P < 0.05, **P < 0.01.
-
C, DThe ability of purK S. aureus to grow in an IMP‐dependent manner within primary bone marrow‐derived macrophages is shown. (C) Fluorescence micrographs depicting purK replication analyzed by fluorescence proliferation assay where non‐replicating bacteria are both GFP and proliferation dye positive. In contrast, replicating bacteria (white arrows) are only GFP‐positive. The top row depicts purK‐infected BMDMs without IMP whereas the bottom row depicts macrophages carrying purK S. aureus in the presence of 50 μM IMP. Images were acquired at 12 h post‐infection and are representative of three independent experiments. Scale bars equal 10 μm. (D) The fraction of macrophages containing replicating bacteria was determined for purK‐infected BMDMs. Each data point represents a biological replicate (n = 8) and is derived from three independent experiments. The data shown are the mean ± standard deviation where the data were normalized to the no IMP condition. ** indicates P < 0.01 as determined by an unpaired two‐tailed t‐test with a Wilcoxon matched pairs test.
Vacuoles containing Salmonella enterica serovar Typhimurium also continue to interact with and receive material (e.g., dextran) from the endocytic pathway akin to S. aureus; however, it is unclear how this impacts nutrient availability for Salmonella 44. Previous work has demonstrated that intracellular Salmonella can access extracellular amino acids 45; however, this requires formation of salmonella‐induced filaments indicating the intracellular bacteria somehow alter amino acid and/or membrane trafficking within infected cells 45. In contrast, S. aureus does not perturb phagosomal maturation or, in so far as we can detect, redirect membrane trafficking within the cell 13, 17. We propose that the ability of S. aureus to reside in the macrophage phagolysosome and withstand killing without perturbing constitutive fluid‐phase uptake represents a strategy employed by this bacterium to allow the host cell to directly deliver nutrients to the pathogen within. Moreover, ongoing fluid‐phase uptake might represent a route by which novel therapeutics can be delivered to intracellular S. aureus.
Materials and Methods
Bacterial strains, plasmids, and bacterial culture conditions
The bacterial strains and plasmids used in this study are summarized in Table 1. S. aureus bacteria were routinely cultured in 4 ml of BD Bacto™ tryptic soy broth (TSB, Fischer scientific) in 20‐ml glass test tubes with constant shaking at 200 RPM at 37°C. When necessary, solid media were made by addition of Bacto agar at 1.5% (w/v). As needed, the following antibiotics were used for bacterial selection at the indicated final concentrations: erythromycin (3 μg/ml), lincomycin (20 μg/ml), and chloramphenicol (12 μg/ml). For every experiment, each biological replicate of S. aureus derives from an isolated colony of S. aureus, on an agar plate, that was used to inoculate individual bacterial culture.
Table 1.
Bacterial strains used in this study
| Strains | Description | Source |
|---|---|---|
| USA300 WT | USA300 LAC, cured of resistance plasmids | Lab stock |
| USA300 purK | USA300 with carrying the purK::ΦΝΣ allele that was transduced from the Nebraska transposon library into USA300 LAC | 48 |
| Plasmids | ||
| pCG44 | E. coli/S. aureus shuttle vector for constitutive expression of pHluorin in S. aureus; AmpR CmR | 49 |
| pCM29 | E. coli/S. aureus shuttle vector for constitutive expression of super folder GFP in S. aureus; AmpR CmR | 50 |
| pAH9 | Constitutive mCherry expression vector for S. aureus, EryR/LincR | 51 |
| PH‐Plcδ‐RFP | Lipid biosensor for PI(4,5)P2, encodes the PH domain of Plcδ fused to RFP, KanR | 52 |
CmR: chloramphenicol resistance; EryR: erythromycin resistance; LincR: lincomycin resistance; KanR: kanamycin resistance; AmpR: ampicillin resistance.
Mammalian tissue culture and transfection
RAW 264.7 (ATCC® TIB‐71™) immortalized murine macrophages were purchased directly from the ATCC® and were cultured in RPMI supplemented with 2 mM glutamine, 25 mM HEPES [4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid], and 5% (v/v) non‐heat‐inactivated fetal bovine serum (FBS, Wisent Inc.) without the use of antibiotics. RAW 264.7 macrophages were cultured in T25 vented tissue culture flasks at 37°C in the presence of 5% CO2. When RAW macrophages reached ~75% confluence, the cells were split using tissue culture scrapers as per the instructions provided by the ATCC® for this cell line.
To culture RAW macrophages for infection assays, 12‐well tissue culture plates were used. As needed, sterile 18‐mm glass coverslips (Electron Microscopy Sciences, No. 1) were also added to individual wells when microscopy was to be performed. To add macrophages, a uniform suspension of cells was generated by scraping RAW macrophages and diluting them to a density of ~4.0 × 105 cells per ml of RPMI with FBS. To a single well, 1 ml of this suspension (i.e., ~4.0 × 105) was added and the plate was incubated overnight (~16–18 h) for infection the next day. Typically, this yields ~6.0 × 105 macrophages at the time of infection.
Primary bone marrow‐derived macrophages (BMDMs) were derived from 6‐week‐old female BALB/c mice using established protocols 33, 46, 47. In brief, isolated precursor cells were diluted in RPMI supplemented with 10% (v/v) non‐heat‐inactivated FBS, 1× pen‐strep (Wisent), and recombinant murine M‐CSF (10 ng/ml; PeproTech®) such that 1.0 × 106 cells were added per well of a 12‐well tissue culture dish containing sterile coverslips. The culture medium was replaced on days 2 and 5; on day 5, antibiotics were omitted.
Primary human M‐CSF derived macrophages were cultured as previously described 13, 17, 33. In brief, blood was drawn from healthy volunteers in accordance with protocols approved by the University of Western Ontario Research Ethics Board. Peripheral blood monocytes were recovered and adhered to glass coverslips and differentiated using 10 ng/ml recombinant human M‐CSF (PeproTech) for 7 days prior to use.
To transfect eukaryotic expression vectors into RAW 264.7 macrophages, Fugene® HD transfection reagent (Promega) was used according to manufacturers’ instructions. In brief, 1 μg of plasmid DNA was combined with 3 μl of transfection reagent in a 100 μl volume of SF‐RPMI per well of a 12‐well tissue culture dish. Transfected cells were used for experimentation the following day after ~16‐ to 18‐h incubation.
Preparation of bacteria for phagocytosis
To infect macrophages with S. aureus, the bacteria were cultured overnight in TSB in the presence of antibiotics, as needed. The next day, 1 ml of the bacterial culture was pelleted and washed 1× in sterile saline (0.9% (v/v) NaCl). The resulting bacterial pellet was then re‐suspended in sterile saline supplemented with 10% (v/v) sterile heat‐inactivated human serum (Millipore Sigma) and incubated for 15 min at room temperature with constant mixing. Bacteria opsonized with human serum were then washed again once with 1 ml of sterile saline and re‐suspended in pre‐warmed serum‐free RPMI. The optical density at 600 nm (OD600 nm) of this bacterial suspension was determined, and the cells were diluted to an OD600 nm equal to 0.5 in SF‐RPMI or ~4.4 × 108 cfu/ml. This suspension was used to dilute the cells such that infections were set up at a multiplicity of infection (MOI) of 30 for 1 ml of SF‐RPMI per well of RAW macrophages unless otherwise indicated. For all experiments requiring BMDM infections, a MOI of 50 was employed.
In instances where bacterial fluorescence‐based proliferation assays were to be performed, bacteria that were cultured overnight were labeled with 1.25 μM eBioscience™ Cell Proliferation Dye eFluor™ 670 as previously described 17, 33 prior to opsonization of S. aureus with human serum. After eFluor™ 670 labeling, the dye was quenched by incubating the cells with 1 ml sterile TSB for 2 min prior to proceeding with opsonization as described above for bacteria from an overnight culture.
Macrophage infection assays
To infect macrophages, 1 ml of SF‐RPMI containing opsonized S. aureus was added to individual wells of a 12‐well tissue culture plate containing macrophages. To synchronize phagocytosis, the plates were immediately centrifuged at 277 × g for 2 min and then incubated at 37°C with 5% CO2 for 30 min. Next, the media was aspirated and replaced with SF‐RPMI supplemented with gentamicin (100 μg/ml) for 1 h. At this time, the medium was aspirated and each well washed twice with 1 ml of sterile 1× PBS. Wells containing infected macrophages were then either lysed with 0.5 ml of sterile 0.1% (v/v) Triton X‐100 diluted in PBS or were incubated further in RPMI containing 5% (v/v) FBS until the desired time point.
To determine the bacterial burden within infected macrophages, cells were lysed in 0.5 ml Triton X‐100 as described above and then subject to 10‐fold serial dilution in sterile saline before drop plating 10 μl samples onto agar plates. Lysates were generated at 1.5 h post‐infection to determine the number of gentamicin protected bacteria inside macrophages at the beginning of each experiment. Lysates were also generated at 10 h post‐infection, and the fold change in CFU/ml was determined by dividing the bacterial count for a given strain and/or biological replicate at 10 h post‐infection by the count for the same strain and/or replicate at 1.5 h post‐infection.
Treatment of macrophages with fluorescent tracers and pharmacological agents
To visualize fluid‐phase uptake and the delivery of macromolecules to intracellular compartments, macrophages were incubated with non‐fixable anionic Invitrogen™ fluorescein dextran (10,000 MW, Thermo Fisher Scientific) at 100 μg/ml after gentamicin treatment (i.e., at 1.5 h post‐infection). Cells were allowed to internalize dextran until 10 h post‐infection at which time they were washed twice with 1 ml sterile 1× PBS and then imaged by live‐cell fluorescence microscopy in SF‐RPMI. To better visualize fluorescein dextran within acidic compartments, macrophages were treated with 1 μM concanamycin A (Santa Cruz Biotechnology) for 30 min to inhibit v‐ATPase activity and unquench fluorescein fluorescence. To monitor degradation of macromolecules taken up by fluid‐phase uptake, macrophages were incubated with DQ™ green BSA (15 μg/ml) again at 1.5 h post‐infection after gentamicin treatment. At 10 h post‐infection, macrophages were washed and imaged as for dextran loaded cells; however, concanamycin A was not used to treat macrophages incubated with DQ™ green BSA.
To analyze the effect of internalizing gentamicin or vancomycin on S. aureus within macrophages, infected cells at 1.5 h post‐infection (after the initial 1 h gentamicin treatment) were either left untreated or were incubated with 300 μg/ml of gentamicin or vancomycin until 10 h post‐infection at which time cells were washed three times with 2 ml of sterile 1× PBS, then lysed with 500 μl of 0.1% (v/v) Triton X‐100 in 1× PBS, and plated for CFU determination.
To inhibit fluid‐phase uptake, macrophages were treated with 10 μM PIK‐III (Cayman Chemical), 100 μM Dynasore (Cayman Chemical), or an equal volume of DMSO (Sigma Millipore) as a vehicle control. Inhibitors were added at 1.5 h post‐infection after gentamicin treatment for infected and uninfected control cells and were added 30 min prior to additional compounds such as gentamicin (300 μg/ml) or inosine monophosphate (50 μM). In experiments where infections were not performed, cells were pre‐treated with PIK‐III and/or Dynasore for 30 min prior to incubation with 100 μg/ml fluorescein dextran for 2 h or Alexa Fluor® 647‐conjugated ChromPure mouse transferrin at 1 μg/ml (Jackson ImmunoResearch Laboratories Inc.) for 15–20 min. Dextran loaded cells were imaged live, whereas transferrin‐labeled cells were fixed with 4% (v/v) paraformaldehyde (PFA) for 20 min at room temperature prior to imaging.
In some instances, fluorescent wheat germ agglutinin (WGA) was used to demarcate the plasmalemma of macrophages and mark extracellular S. aureus. WGA conjugated to tetramethylrhodamine or Alex Fluor® 647 (Invitrogen™, Thermo Fisher Scientific) was used at a concentration of 1 μg/ml for 2 min prior to fixation with PFA as described above.
In vitro growth of S. aureus in the presence of PIK‐III and Dynasore
To analyze the impact of the PIK‐III and Dynasore on S. aureus growth wild‐type USA300 and purK bacteria were first streaked onto an agar plate is the presence of antibiotics, as necessary, and incubated overnight at 37°C. The following day, isolated colonies were re‐suspended in 200 μl of sterile saline (0.9% (w/v) NaCl); then, 5 μl of this bacterial suspension was used to inoculate 2 ml culture volumes of RPMI + 5% (v/v) FBS supplemented with DMSO or PIK‐III with Dynasore at 10 and 100 μM, respectively, in 13‐ml round bottom polypropylene snap cap tubes. To promote growth of purK‐deficient bacteria, inosine monophosphate (IMP) was also added at a final concentration of 50 μM. Cultures were incubated at 37°C with shaking at 200 rpm for 12 h at which point the optical density at 600 nm was determined. Alternatively, growth of wild‐type S. aureus USA300 was also monitored by growing the bacteria in a Bioscreen C™ automated microbiology growth curve analysis system (Growth Curves USA) in the same medium with vehicle and PIK‐III/Dyansore. Here, 250 μl aliquots of the inoculated medium were added to Bioscreen C™ honeycomb microplates (growth Curves USA) and incubated for 24 with continuous shaking with medium amplitude at 37°C.
Microscopy and image analysis
All DIC and fluorescence microscopy were performed on a Leica DMI6000 B inverted microscope equipped with 40× (NA 1.3), 63× (NA 1.4), and 100× (NA 1.4) oil immersion PL‐Apo objectives, a Leica 100 W Hg high pressure light source, and the Hamamatsu Orca Flash 4.0 and Photometrics Evolve 512 Delta EM‐CCD cameras. This microscope is also outfitted with an objective warmer and an enclosed heated stage insert with CO2 perfusion (Live Cell Instruments). For live‐cell imaging, coverslips carrying macrophages were placed in a magnetic imaging chamber and bathed in SF‐RPMI buffered with Na bicarbonate and 25 mM HEPES. All images were acquired using the Leica LASx software package. All image analysis and manipulation was performed using standard Image J tools (National Institutes of Health, Bethesda, MD). Typically, fields of view were imaged as a z series, and for presentation purposes, the sum of 5–10 consecutive z planes with a step size of ~0.2 μm was projected as a single image and then enhanced linearly for presentation purposes only. Quantitation of fluorescence intensity was only performed on individual z slices.
In instances where fluorescence intensity of FITC–dextran or DQ™ green BSA was quantified, all cells were imaged using identical acquisition parameters. Fluorescence intensity was measured using the ROI tool in Image J to encircle entire individual cells. A similar ROI was used to determine the background fluorescence where cells were not located for each field of view and value was subtracted from each single cell measurement for that field. All measurements were made on raw images prior to contrast enhancement. To analyze fluorescence intensity by line scan, a line was drawn across a single Z‐plane for the cell being analyzed using the line tool in Image J. Next, the plot profile feature was used under the analyze tab to measure the pixel intensity along this line for both the Alexa Fluor® 670 channel (i.e., transferrin) and TMR‐WGA channels. Next, the list tab was used to obtain the x‐axis (distance) and y‐axis (fluorescence intensity) values that were then plotted after background subtraction using GraphPad Prism® 6.0c software.
Ethics statement
Blood was obtained, with written permission, only from healthy adult volunteers, in compliance with protocol 109059 approved by the Office of Research Ethics at the University of Western Ontario. All animals were utilized in compliance with guidelines set out by the Canadian Council on Animal Care. All animal protocols (protocol 2017‐028) were reviewed and approved by the University of Western Ontario Animal Use Subcommittee, a subcommittee of the University Council on Animal Care.
Statistical analysis
All statistical analyses were performed using GraphPad Prism® 6.0c software.
Author contributions
RSF and DEH conceived of the experiments. RSF performed all experiments and analyzed the data. RSF and DEH co‐authored and edited the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7
Movie EV8
Review Process File
Acknowledgements
This study was supported by a Cystic Fibrosis Canada research grant awarded to D.E.H. We thank members of the Heinrichs laboratory for helpful discussions and comments on the manuscript. The synopsis image was created with BioRender.com.
EMBO Reports (2020) 21: e50348
Data availability
Primary datasets have not been deposited. The prism files containing the data presented in graphs are available upon request.
References
- 1. Lakhundi S, Zhang K (2018) Methicillin‐resistant Staphylococcus aureus : molecular characterization, evolution, and epidemiology. Clin Microbiol Rev 31: e00020–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Harkins CP, Pichon B, Doumith M, Parkhill J, Westh H, Tomasz A, de Lencastre H, Bentley SD, Kearns AM, Holden MTG (2017) Methicillin‐resistant Staphylococcus aureus emerged long before the introduction of methicillin into clinical practice. Genome Biol 18: 130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dordel J, Kim C, Chung M, Pardos de la Gándara M, Holden MTJ, Parkhill J, de Lencastre H, Bentley SD, Tomasz A (2014) Novel determinants of antibiotic resistance: identification of mutated loci in highly methicillin‐resistant subpopulations of methicillin‐resistant Staphylococcus aureus . MBio 5: e01000–e01013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kundsin RB, Walter CW, Morin P (1964) Staphylococcus aureus UC‐18: agent of nosocomial infections. Science 145: 1322–1323 [DOI] [PubMed] [Google Scholar]
- 5. Jones RN (2010) Microbial etiologies of hospital‐acquired bacterial pneumonia and ventilator‐associated bacterial pneumonia. Clin Infect Dis 51: S81–S87 [DOI] [PubMed] [Google Scholar]
- 6. Kallen AJ (2010) Health care‐associated invasive MRSA infections, 2005–2008. JAMA 304: 641 [DOI] [PubMed] [Google Scholar]
- 7. Herold BC, Immergluck LC, Maranan MC, Lauderdale DS, Gaskin RE, Boyle‐Vavra S, Leitch CD, Daum RS (1998) Community‐acquired methicillin‐resistant Staphylococcus aureus in children with no identified predisposing risk. JAMA 279: 593–598 [DOI] [PubMed] [Google Scholar]
- 8. DeLeo FR, Otto M, Kreiswirth BN, Chambers HF (2010) Community‐associated meticillin‐resistant Staphylococcus aureus . Lancet 375: 1557–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Flannagan R, Heit B, Heinrichs D (2015) Antimicrobial mechanisms of macrophages and the immune evasion strategies of Staphylococcus aureus . Pathogens 4: 826–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Richardson AR (2019) Virulence and metabolism. Microbiol Spectr 10.1128/microbiolspec.GPP3-0011-20 [DOI] [PubMed] [Google Scholar]
- 11. Foster TJ, Geoghegan JA, Ganesh VK, Höök M (2014) Adhesion, invasion and evasion: the many functions of the surface proteins of Staphylococcus aureus . Nat Rev Microbiol 12: 49–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fraunholz M, Sinha B (2012) Intracellular Staphylococcus aureus: live‐in and let die. Front Cell Infect Microbiol 2: 43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Flannagan RS, Heit B, Heinrichs DE (2016) Intracellular replication of Staphylococcus aureus in mature phagolysosomes in macrophages precedes host cell death, and bacterial escape and dissemination. Cell Microbiol 18: 514–535 [DOI] [PubMed] [Google Scholar]
- 14. Kobayashi SD, Braughton KR, Palazzolo‐Ballance AM, Kennedy AD, Sampaio E, Kristosturyan E, Whitney AR, Sturdevant DE, Dorward DW, Holland SM et al (2010) Rapid neutrophil destruction following phagocytosis of Staphylococcus aureus . J Innate Immun 2: 560–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Surewaard BGJ, Deniset JF, Zemp FJ, Amrein M, Otto M, Conly J, Omri A, Yates RM, Kubes P (2016) Identification and treatment of the Staphylococcus aureus reservoir in vivo . J Exp Med 213: 1141–1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jorch SK, Surewaard BGJ, Hossain M, Peiseler M, Deppermann C, Deng J, Bogoslowski A, van der Wal F, Omri A, Hickey MJ et al (2019) Peritoneal GATA6+ macrophages function as a portal for Staphylococcus aureus dissemination. J Clin Invest 129: 4643–4656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Flannagan RS, Kuiack RC, McGavin MJ, Heinrichs DE (2018) Staphylococcus aureus uses the GraXRS regulatory system to sense and adapt to the acidified phagolysosome in macrophages. MBio 9: e01143–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cunrath O, Bumann D (2019) Host resistance factor SLC11A1 restricts Salmonella growth through magnesium deprivation. Science 366: 995–999 [DOI] [PubMed] [Google Scholar]
- 19. Wang S, Tsun Z‐Y, Wolfson RL, Shen K, Wyant GA, Plovanich ME, Yuan ED, Jones TD, Chantranupong L, Comb W et al (2015) Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science 347: 188–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schwake M, Schröder B, Saftig P (2013) Lysosomal membrane proteins and their central role in physiology. Traffic 14: 739–748 [DOI] [PubMed] [Google Scholar]
- 21. Perland E, Hellsten SV, Lekholm E, Eriksson MM, Arapi V, Fredriksson R (2017) The novel membrane‐bound proteins MFSD1 and MFSD3 are putative SLC transporters affected by altered nutrient intake. J Mol Neurosci 61: 199–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chapel A, Kieffer‐Jaquinod S, Sagné C, Verdon Q, Ivaldi C, Mellal M, Thirion J, Jadot M, Bruley C, Garin J et al (2013) An extended proteome map of the lysosomal membrane reveals novel potential transporters. Mol Cell Proteomics 12: 1572–1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wyant GA, Abu‐Remaileh M, Wolfson RL, Chen WW, Freinkman E, Danai LV, Vander Heiden MG, Sabatini DM (2017) mTORC1 activator SLC38A9 is required to efflux essential amino acids from lysosomes and use protein as a nutrient. Cell 171: 642–654.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Canton J, Schlam D, Breuer C, Gütschow M, Glogauer M, Grinstein S (2016) Calcium‐sensing receptors signal constitutive macropinocytosis and facilitate the uptake of NOD2 ligands in macrophages. Nat Commun 7: 11284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bohdanowicz M, Schlam D, Hermansson M, Rizzuti D, Fairn GD, Ueyama T, Somerharju P, Du G, Grinstein S (2013) Phosphatidic acid is required for the constitutive ruffling and macropinocytosis of phagocytes. Mol Biol Cell 24: 1700–1712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Flannagan RS, Harrison RE, Yip CM, Jaqaman K, Grinstein S (2010) Dynamic macrophage “probing” is required for the efficient capture of phagocytic targets. J Cell Biol 191: 1205–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yoshida S, Pacitto R, Inoki K, Swanson J (2018) Macropinocytosis, mTORC1 and cellular growth control. Cell Mol Life Sci 75: 1227–1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Racoosin EL, Swanson JA (1993) Macropinosome maturation and fusion with tubular lysosomes in macrophages. J Cell Biol 121: 1011–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Commisso C, Davidson SM, Soydaner‐Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin JA, Thompson CB et al (2013) Macropinocytosis of protein is an amino acid supply route in Ras‐transformed cells. Nature 497: 633–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Futter CE, Pearse A, Hewlett LJ, Hopkins CR (1996) Multivesicular endosomes containing internalized EGF‐EGF receptor complexes mature and then fuse directly with lysosomes. J Cell Biol 132: 1011–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ohkuma S, Poole B (1978) Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proc Natl Acad Sci USA 75: 3327–3331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Frost LS, Dhingra A, Reyes‐Reveles J, Boesze‐Battaglia K (2017) The use of DQ‐BSA to monitor the turnover of autophagy‐associated cargo. Methods Enzymol 587: 43–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Flannagan RS, Heinrichs DE (2018) A fluorescence based‐proliferation assay for the identification of replicating bacteria within host cells. Front Microbiol 9: 3084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T (2006) Dynasore, a cell‐permeable inhibitor of dynamin. Dev Cell 10: 839–850 [DOI] [PubMed] [Google Scholar]
- 35. Dowdle WE, Nyfeler B, Nagel J, Elling RA, Liu S, Triantafellow E, Menon S, Wang Z, Honda A, Pardee G et al (2014) Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo . Nat Cell Biol 16: 1069–1079 [DOI] [PubMed] [Google Scholar]
- 36. Ferguson SM, De Camilli P (2012) Dynamin, a membrane‐remodelling GTPase. Nat Rev Mol Cell Biol 13: 75–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Drevets DA, Canono BP, Leenen PJM, Campbell PA (1994) Gentamicin kills intracellular Listeria monocytogenes . Infect Immun 62: 2222–2228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lehar SM, Pillow T, Xu M, Staben L, Kajihara KK, Vandlen R, DePalatis L, Raab H, Hazenbos WL, Hiroshi Morisaki J et al (2015) Novel antibody–antibiotic conjugate eliminates intracellular S. aureus . Nature 527: 323–328 [DOI] [PubMed] [Google Scholar]
- 39. Schnaith A, Kashkar H, Leggio SA, Addicks K, Krönke M, Krut O (2007) Staphylococcus aureus subvert autophagy for induction of caspase‐independent host cell death. J Biol Chem 282: 2695–2706 [DOI] [PubMed] [Google Scholar]
- 40. Brugarolas P, Duguid EM, Zhang W, Poor CB, He C (2011) Structural and biochemical characterization of N 5 ‐carboxyaminoimidazole ribonucleotide synthetase and N 5 ‐carboxyaminoimidazole ribonucleotide mutase from Staphylococcus aureus . Acta Crystallogr Sect D Biol Crystallogr 67: 707–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wei C, Pohorille A (2013) Permeation of aldopentoses and nucleosides through fatty acid and phospholipid membranes: implications to the origins of life. Astrobiology 13: 177–188 [DOI] [PubMed] [Google Scholar]
- 42. Soler C, García‐Manteiga J, Valdés R, Xaus J, Comalada M, Casado FJ, Pastor‐Anglada M, Celada A, Felipe A (2001) Macrophages require different nucleoside transport systems for proliferation and activation. FASEB J 15: 1979–1988 [DOI] [PubMed] [Google Scholar]
- 43. Gordon S, Cohn ZA (1973) The macrophage. Int Rev Cytol 36: 171–214 [DOI] [PubMed] [Google Scholar]
- 44. Drecktrah D, Knodler LA, Howe D, Steele‐Mortimer O (2007) Salmonella trafficking is defined by continuous dynamic interactions with the endolysosomal system. Traffic 8: 212–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Popp J, Noster J, Busch K, Kehl A, Zur Hellen G, Hensel M (2015) Role of host cell‐derived amino acids in nutrition of intracellular Salmonella enterica . Infect Immun 83: 4466–4475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Weischenfeldt J, Porse B (2008) Bone marrow‐derived macrophages (BMM): isolation and applications. CSH Protoc 2008: pdb.prot5080 [DOI] [PubMed] [Google Scholar]
- 47. Davis BK (2013) Isolation, culture, and functional evaluation of bone marrow‐derived macrophages. Methods Mol Biol 1031: 27–35. [DOI] [PubMed] [Google Scholar]
- 48. Goncheva MI, Flannagan RS, Heinrichs DE (2020) De novo purine biosynthesis is required for intracellular growth of Staphylococcus aureus and for the hypervirulence phenotype of a purR mutant. Infect Immun 88: e00104–e00120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gries CM, Sadykov MR, Bulock LL, Chaudhari SS, Thomas VC, Bose JL, Bayles KW (2016) Potassium uptake modulates Staphylococcus aureus metabolism. mSphere 1: e00125–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pang YY, Schwartz J, Thoendel M, Ackermann LW, Horswill AR, Nauseef WM (2010) agr‐dependent interactions of Staphylococcus aureus USA300 with human polymorphonuclear neutrophils. J Innate Immun 2: 546–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Boles BR, Horswill AR (2008) agr‐mediated dispersal of Staphylococcus aureus biofilms. PLoS Pathog 4: e1000052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Várnai P, Balla T (1998) Visualization of phosphoinositides that bind pleckstrin homology domains: calcium‐ and agonist‐induced dynamic changes and relationship to myo‐[3H]inositol‐labeled phosphoinositide pools. J Cell Biol 143: 501–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7
Movie EV8
Review Process File
Data Availability Statement
Primary datasets have not been deposited. The prism files containing the data presented in graphs are available upon request.