

Abstract
Cervical cancer (CC) is one of the most prevalent types of cancer affecting females worldwide. However, the molecular mechanisms underlying the development and progression of CC remains to be elucidated. Taking the high incidence and mortality rates amongst women into consideration, the identification of novel biomarkers to prevent CC is of great significance and required to improve diagnosis. Using three raw microarray datasets from the Gene Expression Omnibus database, 188 differentially expressed genes (DEGs) were identified. Gene Ontology and pathway analyses were performed on the DEGs. Through protein-protein interaction network construction and module analysis, eight hub genes [cell division cycle 6, cyclin-dependent kinase 1 (CDK1), cell division control protein 45, budding uninhibited by benzimidazoles 1 (BUB1), DNA topoisomerase II α (TOP2A) and minichromosome maintenance complex component 4, CCNB2 and CCNB1] were identified, but only TOP2A was considered a prognostic factor in survival analysis. There were strong positive correlations between TOP2A and BUB1 (P<0.0001, rs=0.635), CDK1 (P<0.0001, rs=0.511), centromere protein F (CENPF) (P<0.0001, rs=0.677), Rac GTPase activating protein 1 (RACGAP1) (P<0.0001, rs=0.612), F-box protein 5 (FBXO5) (P<0.0001, rs=0.585) and BUB1 mitotic checkpoint serine/threonine kinase B (BUB1B) (P<0.0001, rs=0.584). Additionally, BUB1, CDK1, CENPF, RACGAP1, FBXO5 and BUB1B are all potentially suitable candidate targets for the diagnosis and treatment of CC. In conclusion, the present study identified TOP2A as a potential tumor oncogene and a biomarker for the prognosis of CC.
Keywords: TOP2A, cervical cancer, bioinformatics analysis, differentially expressed genes, protein-protein interactions
Introduction
Cervical cancer (CC) is the fourth most common type of female malignancy with a high mortality rate worldwide, and almost all cases of CC are caused by human papillomavirus (HPV) infection (1). In China, HPV infection is the most common etiological factor underlying CC (2). Our previous study showed that the prevalence of HPV in the Jiangxi Province is 22.49% (3). However, although HPV infection is a prerequisite of CC, it is not sufficient to induce CC alone (4). The specific molecular mechanisms between the persistent high-risk HPV infection and the pathological process of CC are still controversial (5). Increasing evidence has shown that the abnormal expression of multiple genes are involved in the pathogenesis of CC (6). As tumorigenesis is a complex pathological process involving various genetic and epigenetic events, such as the inactivation of suppressor genes and/or the overexpression of oncogenes (7), the identification of dysregulated genes in cancer-associated pathways may highlight the molecular mechanisms underlying tumorigenesis, assisting in the development of novel strategies for treatment of CC. Therefore, it is important to elucidate the potential molecular mechanisms underlying CC, to provide novel therapeutic targets and prognostic biomarkers of CC (8,9).
To improve our understanding of the molecular mechanisms underlying CC, bioinformatics analysis has been widely used to identify differentially expressed genes (DEGs), functional pathways and hub genes involved in the carcinogenesis and progression of CC. Although several genes have been identified in predicting the clinical outcome of CC, there have been inconsistencies between previous studies (10,11), which may be due to small sample sizes, heterogeneous histological subtypes, different detection platforms and various data processing methods. In the present study, three mRNA microarray datasets were downloaded from the Gene Expression Omnibus (GEO) database to acquire DEGs between CC and normal tissues. Subsequently, Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis, protein-protein interaction (PPI) network analysis and the Cytoscape plugin, CytoHubba, were used to identify hub genes in CC. Furthermore, Kaplan-Meier plotter was used on the cBioPortal website and the DNA topoisomerase II α (TOP2A) gene was identified as an independent prognostic biomarker of disease-free survival in CC. A total of 188 DEGs and 8 hub genes were identified, which may be candidate biomarkers for CC.
Materials and methods
Microarray data information
GSE63514(12), GSE9750(13) and GSE7410(14) were obtained from the NCBI GEO database (ncbi.nlm.nih.gov/geo) (15,16). The GSE63514 dataset used the GPL570 [HG-U133 Plus 2] Affymetrix Human Genome U133 Plus 2.0 Array platform, which contained 28 CC tissue samples and 24 normal samples. The GSE9750 dataset used the GPL96 Affymetrix Human Genome U133A Array platform and included 33 CC tissue samples that were primarily marked by HPV16 or HPV18 and 21 normal cervical samples. GSE7410 contained 40 CC and 5 normal cervical samples, and used the GPL1708 Agilent-012391 Whole Human Genome Oligo Microarray G4112A platform.
Screening of DEGs in CC
The DEGs between CC and normal cervical samples were screened using GEO2R (ncbi.nlm.nih.gov/geo/geo2r). The adjusted P-values (adj.P) and Benjamini and Hochberg false discovery rates were used to provide a balance between the discovery of statistically significant genes and limit false-positives. Probe sets without corresponding gene symbols or genes with >1 probe set were removed or averaged, respectively. Only genes with a log fold-change >1 and adj.P-value of <0.01 were regarded as statistically significant DEGs.
Functional annotation and pathway enrichment analysis
The screened DEGs were submitted to the Database for Annotation, Visualization and Integrated Discovery (DAVID; david.abcc.ncifcrf.gov/; version 6.7) (17), an online tool for functional annotation. KEGG is a database for understanding higher-level functions and biological information generated by high throughput experimental technologies (18). GO analysis is a useful bioinformatics tool for annotating genes and analyzing their biological processes (19). The significant enrichment analysis of DEGs was evaluated using GO and KEGG, with P<0.05 as the cutoff. If there were >5 terms enriched in the category, the top 10 were selected based on the P-value.
PPI network construction and module analysis
The identified DEGs were imported into the Search Tool for the Retrieval of Interacting Genes (STRING; string-db.org; version 10.0) (20) database to evaluate the functional interactions among them. Subsequently, Cytoscape (cytoscape.org/; version 3.6.1) was used to screen the modules from the PPI network (21), as a molecular interaction network tool. The plugins Cytoscape and CytoHubba were used to predict and explore the important nodes and subnetworks in the network with 12 topological algorithms, which included degree, edge percolated component, maximum neighborhood component and density of maximum neighborhood component (22). CytoHubba was used to rank nodes in a network by their network features and select the overlapping top 15 hub genes by 6 ranked methods. Finally, all identified hub genes in this module were mapped to DAVID to perform KEGG and GO enrichment analysis.
Key gene selection and analysis
Overall and disease-free survival analysis of hub genes using Kaplan-Meier survival curves, as well as correlation analysis of TOP2A and co-expression genes, were performed in cBioPortal (cbioportal.org; version 2.4.3) (23,24). The expression profile of TOP2A was analyzed and displayed using the online database UALCAN (ualcan.path.uab.edu). The relationship between TOP2A and expression patterns and HPV viral infection status were analyzed using the 201291_s_at and 201292_probes in the Zhai Cervix dataset available from Oncomine (oncomine.com) (13,14,25). The heat maps of TOP2A gene expression in clinical CC samples vs. normal tissues are respectively: Cervical Squamous Cell Carcinoma vs. Normal Biewenga Cervix, Gynecol Oncol, 2008(14); Cancer Type: CC Bittner Multi-cancer, unpublished data, 2006; Cervical Squamous Cell Carcinoma vs. Normal Scotto Cervix 2, Genes Chromosomes Cancer, 2008(15); and Cervical Squamous Cell Carcinoma Epithelia vs. Normal Zhai Cervix, Cancer Res, 2007(25).
Statistical analysis
All statistical analyses were performed using the aforementioned bioinformatics tools. Adj.P were corrected for in multiple comparisons using the Benjamini and Hochberg's false discovery rate. Co-expression and networks were calculated according to the cBioPortal online instructions. A log-rank test was performed to identify the significance of the Spearman's Correlation coefficient between the mRNA expression z-Scores (RNASeq V2 RSEM). Spearman's Correlation coefficient >0.5 and P<0.05 was considered to indicate a statistically significant difference.
Results
Identification of DEGs
Following the standardization of the microarray results, 3,816 genes in the GSE63514 dataset, 2,625 genes in the GSE9750 dataset and 2,093 genes in the GSE7410 dataset were identified as DEGs. The 188 overlapping DEGs amongst the three datasets were determined using a Venn diagram (Fig. 1), and consisted of 56 downregulated genes and 132 upregulated genes between normal and cancerous tissues.
Figure 1.
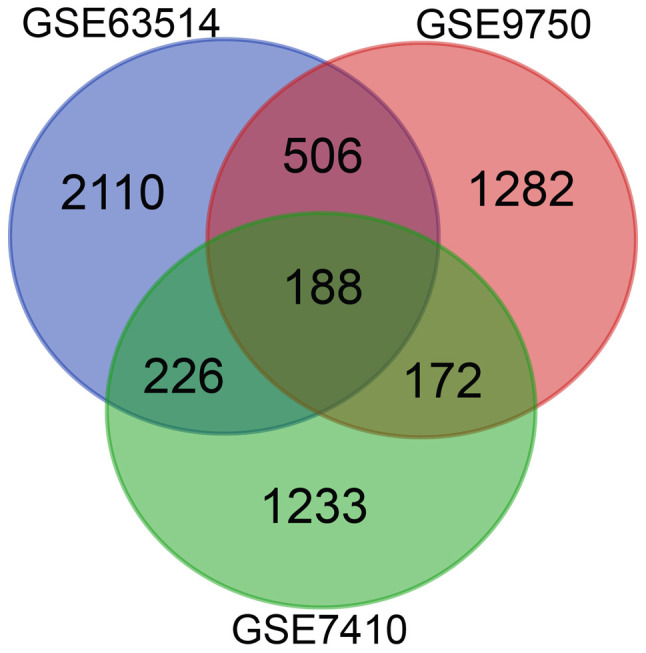
Differentially expressed genes were selected for using a fold-change of >2 and an adjusted P-value of <0.01 among the mRNA expression profiling datasets GSE63514, GSE9750 and GSE7410. An overlap of 188 genes was observed among the three datasets.
GO and KEGG enrichment analysis of DEGs
To determine the biological significance of the 188 DEGs, functional and pathway enrichment analyses were performed using DAVID. As shown by GO analysis, the DEGs were significantly enriched in cell cycle, cell cycle phase, mitotic cell cycle, cell cycle process and M phase (Fig. 2A). In terms of cellular components, the DEGs were primarily enriched in microtubule cytoskeleton, spindle, chromosome, nuclear lumen and chromosomal part (Fig. 2B). The molecular function terms were primarily enriched in ATP binding, adenyl ribonucleotide binding, adenyl nucleotide binding, purine nucleoside binding and nucleoside binding (Fig. 2C). These significantly enriched terms may assist in improving our understanding of the roles of the DEGs in the development and progression of CC. In the KEGG analysis, the DEGs were primarily enriched in cell cycle, DNA replication and mismatch repair (Fig. 2D).
Figure 2.

Top 10 significant changes in the Gene Ontology and KEGG significant enrichment analyses of differentially expressed mRNAs. (A) Biological process. (B) Cellular components. (C) Molecular function. (D) KEGG. KEGG, Kyoto Encyclopedia of Genes and Genomes.
PPI network construction and interaction network analysis of the hub genes
The screened DEGs were uploaded to the STRING website to reconstruct the PPI network. The resulting network contained 188 nodes and 2,854 edges (Fig. 3A). Subsequently, hub genes were filtered using CytoHubba. Cell division cycle 6 (CDC6), cyclin-dependent kinase 1 (CDK1), cell division control protein 45 (CDC45), budding uninhibited by benzimidazoles 1 (BUB1), TOP2A, minichromosome maintenance complex component 4 (MCM4), CCNB2 and CCNB1 were identified as the 8 hub genes, all of which were imported into STRING to reconstruct the PPI network of 8 nodes and 28 edges (Fig. 3B). A network of the hub genes and their co-expression genes was then analyzed using the cBioPortal online platform, which shows the gene-gene interaction network among the hub genes and the most frequently altered neighboring co-expression genes (Fig. 3C). These results suggest that the above hub genes may serve a critical role in the development of CC.
Figure 3.

PPI network and the hub gene module of DEGs. (A) PPI network of DEGs was constructed with Cytoscape. Upregulated genes are marked in light red and downregulated genes in light blue. (B) Hub gene module obtained from the PPI network based on STRING with 8 nodes and 28 edges. (C) Hub genes and their co-expression genes were analyzed using cBioPortal. Nodes with bold black outlines represent the hub genes. Nodes with thin black outline represent the co-expression genes. PPI, protein-protein interaction; DEGs, differentially expressed genes; STRING, Search Tool for the Retrieval of Interacting Genes; CDC6, cell division cycle 6; CDK1, cyclin-dependent kinase 1; CDC45, cell division control protein 45; BUB1, budding uninhibited by benzimidazoles 1; MCM4, minichromosome maintenance complex component 4; TOP2A, DNA topoisomerase II α.
TOP2A mRNA expression and gene expression correlation analysis
cBioPortal was used to perform the survival analysis of these hub genes to evaluate their effects on CC. Briefly, only TOP2A was clearly associated with the prognosis of patients (Fig. 4A). Patients whose tissues exhibited upregulated expression levels of TOP2A had significantly shorter disease/progression-free survival times compared to those with lower expression levels (P<0.01). The expression profile of TOP2A in human tissues was displayed using UALCAN. It was found that the TOP2A mRNA expression in bladder, breast, cervical, esophageal, liver, kidney and lung cancer, as well as glioblastoma multiforme, was higher compared with the respective normal tissues (Fig. 4B). Oncomine analysis of cancer vs. normal tissue confirmed that TOP2A expression was significantly upregulated in CC (Fig. 4C). In the Zhai Cervix dataset, higher TOP2A mRNA expression levels were associated with cancer type and human papillomavirus (HPV) infection status (Fig. 5A-D).
Figure 4.

Expression analysis of the TOP2A gene. (A) Disease-free survival analyses of hub genes were performed in the cBioPortal online platform. (B) Expression profiles for TOP2A in human cancer analyzed using UALCAN. (C) Oncomine analysis of TOP2A in cancer vs. normal tissue. TOP2A, DNA topoisomerase II α; TCGA, The Cancer Genome Atlas.
Figure 5.

Association between the expression of TOP2A, cancer type and HPV Infection Status in the 201291_s_at and 201292_at probes in the Zhai Cervix dataset. (A and B) TOP2A mRNA expression in CC between normal CC and precursor tissues. 0, no value (3); 1, cervix squamous epithelium (10); 2, cervical squamous cell carcinoma (21); 3, high grade cervical squamous intraepithelial neoplasia (7). (C and D) TOP2A mRNA expression and HPV Infection Status. 0, no value (3); 1, HPV negative (10); 2, HPV type 16 positive (14); 3, HPV type 18 positive (weak) (1); 4, HPV type 18 positive (4); 5, HPV type 18 and 45 positive (1); 6, HPV type 33 positive (1); 7, HPV type 33, 52 and 58 positive (4); 8, HPV type 56 positive (1); 9, HPV type 58 positive (1); 10, HPV type 59 positive (1). TOP2A, DNA topoisomerase II α; CC, cervical cancer; HPV, human papillomavirus. The numbers in the brackets refers to the number of samples.
TOP2A gene expression correlation analysis results from the cBioPortal online platform showed that, of the hub genes, BUB1 (P<0.0001, rs=0.635) and CDK1 (P<0.0001, rs=0.511) had a close association with TOP2A (Fig. 6A and B). In addition, strong positive correlations were observed between TOP2A mRNA expression levels and centromere protein F (CENPF; P<0.0001, rs=0.677), Rac GTPase activating protein 1 (RACGAP1; P<0.0001, rs=0.612), F-box protein 5 (FBXO5; P<0.0001, rs=0.585) and BUB1 mitotic checkpoint serine/threonine kinase B (P<0.0001, rs=0.584; Fig. 6C-F).
Figure 6.

Gene expression correlation analysis for BUB1, CDK1, CENPF, RACGAP1, FBXO5 and BUB1B with the TOP2A gene using cBioPortal. The scatter plot shows the Spearman's correlation of TOP2A expression with the expression of (A) BUB1, (B) CDK1, (C) CENPF, (D) RACGAP1, (E) FBXO5 and (F) BUB1B. BUB1, budding uninhibited by benzimidazoles 1; CDK1, cyclin-dependent kinase 1; CENPF, centromere protein F; RACGAP1, Rac GTPase activating protein 1; FBXO5, F-box protein 5; BUB1B, BUB1 mitotic checkpoint serine/threonine kinase B; TOP2A, DNA topoisomerase II α.
Discussion
In the present study the key words ‘cervical cancer’ were used to search the GEO database and screen out the datasets which contained Homo sapiens mRNA expression profiles of both CC tissues and normal tissues. A total of three datasets (GSE63514, GSE9750 and GSE7410) were selected for further investigation. Among these, GSE63514 and GSE9750 have been analyzed together (26,27). Other studies have also mined the GSE63514(28) and GSE9750(29) datasets; only the GSE7410 dataset had not yet been thoroughly examined previously, to the best of our knowledge. Therefore, the GEO2R online tool was used to perform the analyses of the three datasets to determine any potentially relevant factors.
In the present study, 188 DEGs were identified in CC, including 132 upregulated and 56 downregulated genes. As shown by the results of KEGG and GO enrichment analysis, the 188 DEGs were primarily enriched in cell cycle. Previous studies have reported that the abnormal regulation of cell cycle process serves a vital role in the tumorigenesis and progression of several types of cancer (30-32). In addition, with regard to GO terms, the DEGs were primarily enriched in cell cycle, mitotic cell cycle, microtubule cytoskeleton, spindle, ATP binding and adenyl ribonucleotide binding, whereas changes in the KEGG pathway were largely enriched in cell cycle, DNA replication and mismatch repair. Previous studies have shown the antitumor effect of disrupting the microtubule cytoskeleton (33,34). Furthermore, HPV infection facilitating DNA replication and stimulating DNA damage response are potential important causes of a range of diseases, including CC (35). Therefore, these potential mechanisms support the results of the present study. The results of the present study may improve our understanding of the underlying mechanisms by which the identified DEGs may promote development or progression of CC. The other enriched functions and pathways may also be involved in CC carcinogenesis, and thus should be further studied.
Cytoscape is an important open source bioinformatics software platform for visualizing molecular interaction networks. The CytoHubba plugin was used to screen out core genes for subsequent prognosis analysis similar to previous studies (36,37). These hub genes may serve an important role in the development of CC. Through the PPI network, CDC6, CDK1, CDC45, BUB1, TOP2A, MCM4, CCNB2 and CCNB1 were further selected as hub genes and these hub gens were considered important. Subsequently, the hub genes were chosen for further survival analysis. Only TOP2A was associated with patient prognosis, and TOP2A dysregulation was significantly associated with reduced disease-free survival, but not overall survival (P<0.01). TOP2A is an essential nuclear enzyme for chromosome condensation, chromatid separation and the relief of supercoiled DNA during mitosis, and it is crucial for the segregation of daughter chromosomes at the end of cell division (38). Upregulated expression of TOP2A is significantly associated with increased CC cell division (39) and reduced survival periods (40-43). In addition, TOP2A may be used as an immunohistochemical biomarker for CC (39). Several studies have confirmed the use of TOP2A as a sensitive biomarker for the screening of carcinoma tissues (39,44,45). TOP2A was also frequently overexpressed in several types of cancer, including lung (46), prostate (47), breast (42,48), hepatic (49) and ovarian cancer (50), and glioma (51). Upregulated expression of TOP2A is significantly associated with the progression from cervical intraepithelial neoplasia grade 2 to more advanced cervical lesion division (38), and the Oncomine analysis performed in the present study confirmed these results. In addition, due to HPV infection being the primary etiological factor of CC, the data in the Zhai Cervix dataset suggested that high-risk HPV infection resulted in increased TOP2A expression, compared with no HPV infection. Santin et al (52) showed that TOP2A was coordinately dysregulated in primary HPV16 and HPV18-infected stage-IB-IIA CC, potentially representing a common signaling pathway initiated by HPV transformation. Thus, the encoding enzyme gene is regarded as the target of several anticancer agents (53-55) and a variety of mutations in this gene have been associated with the development of drug resistance (56-58).
To examine the underlying molecular mechanisms in CC, gene expression correlation analysis was performed in cBioPortal. CENPF, RACGAP1 FBXO5 and BUB1B had a strong positive correlation with TOP2A expression, as well as the hub genes BUB1 and CDK1 in CC. Nevertheless, previous studies have not shown interactions between TOP2A and co-expression genes BUB1, CDK1, CENPF, RACGAP1, FBXO5 and BUB1B in CC. However, Guo et al (59) suggested that BUB1, CDK1, RACGAP1 and TOP2A may be involved in the tumorigenesis of adrenocortical carcinoma. Li et al (60) showed that lower expression levels of TOP2A and CENPF were associated with improved overall survival in patients with bladder cancer. BUB1 is a mitotic checkpoint serine/threonine kinase that is crucial for physiological chromosomal segregation and is correlated with cancer stem cell potential; it may also be a target for anti-breast cancer stem cell therapies (61). CDK1 is a member of the Ser/Thr protein kinase family, which is associated with proliferation and has been demonstrated to eb a prognostic factor in the development of ovarian cancer (62,63). Schwermer et al (64) confirmed a pivotal role of the CDK1/CCNB1 complex in tumor cell survival. CENPF is a centromere-kinetochore complex and chromosomal segregation-associated protein during mitosis (65). Preclinical analysis of mouse models showed that FOXM1 and CENPF were master drug treatment-responsive genes in prostate cancer (65). RACGAP1 serves a key role in metastasis, regulating cell morphology, motility and establishment of cell polarity (66). Furthermore, RACGAP1 has been shown to function as a potential biomarker, due to its significant association with poor disease-free and overall survival in colorectal cancer (67). FBXO5, also known as EMI1, is an inhibitor of the anaphase promoting complex/cyclosome (APC/C) and is upregulated in HPV16 E7 oncoprotein-expressing mitotic cells, where it interferes with the degradation of APC/C substrates in CC (68). BUB1B encoded a kinase involved in spindle checkpoint function, and also serves a role in inhibiting the APC/C; the spindle checkpoint function has been shown to be impaired in several types of cancer (69-71). Further studies are required to confirm whether TOP2A also participates in any biological processes associated with cancer development or progression, due to its strong positive correlation with FBXO5 and BUB1B.
The present study has some limitations. Firstly, although upregulated TOP2A mRNA expression levels was a biomarker for the prognosis of CC, all the data in the present study are based on online databases; further studies with larger sample sizes are required to validate these results and to determine potential targets for diagnosis and treatment of CC. Gene Set Enrichment Analysis was not performed, which is a more powerful strategy for functional and pathway enrichment analysis, to assess the significant enrichment analysis of DEGs; thus, future studies are required to identify the significant molecular signaling pathways in CC. Finally, due to the lack of sufficient clinical sample information, multivariate Cox regression analysis could not be performed to further elucidate the significance of TOP2A. In future studies, the underlying mechanisms of TOP2A in CC should be clarified.
In conclusion, TOP2A may be used a biomarker which predicts poor prognosis and may serve as a therapeutic target for treatment of CC. Further studies are required to explore and demonstrate the potential use of the identified hub genes for the diagnosis, prognosis and treatment of CC.
Acknowledgements
The authors are grateful for the help provided by Dr Rui Hou (Harry Perkins Institute of Medical Research, The Western University of Australia).
Funding
This study was partially supported by the National Natural Science Foundation of China (grant no. 81702580) and the Natural Science Foundation of Jiangxi Province (grant nos. 2017ACB21066 and 20171BBG70051).
Availability of data and materials
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.
Authors' contributions
QZ, HL and TZ designed the study. QZ, LZ, SH, XX, and TZ analyzed the data and prepared the figures. QZ and YL revised the manuscript. HL, YL and JL conceived the study and interpreted the data. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Small W Jr, Bacon MA, Bajaj A, Chuang LT, Fisher BJ, Harkenrider MM, Jhingran A, Kitchener HC, Mileshkin LR, Viswanathan AN, Gaffney DK. Cervical cancer: A global health crisis. Cancer. 2017;123:2404–2412. doi: 10.1002/cncr.30667. [DOI] [PubMed] [Google Scholar]
- 2.Soto D, Song C, McLaughlin-Drubin ME. Epigenetic alterations in human papillomavirus-associated cancers. Viruses. 2017;9(248) doi: 10.3390/v9090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhong TY, Zhou JC, Hu R, Fan XN, Xie XY, Liu ZX, Lin M, Chen YG, Hu XM, Wang WH, et al. Prevalence of human papillomavirus infection among 71,435 women in Jiangxi Province, China. J Infect Public Health. 2017;10:783–788. doi: 10.1016/j.jiph.2017.01.011. [DOI] [PubMed] [Google Scholar]
- 4.Woodman CB, Collins SI, Young LS. The natural history of cervical HPV infection: Unresolved issues. Nat Rev Cancer. 2007;7:11–22. doi: 10.1038/nrc2050. [DOI] [PubMed] [Google Scholar]
- 5.Integrated genomic and molecular characterization of cervical cancer. Nature. 2017;543:378–384. doi: 10.1038/nature21386. Cancer Genome Atlas Research Network; Albert Einstein College of Medicine; Analytical Biological Services; Barretos Cancer Hospital; Baylor College of Medicine; Beckman Research Institute of City of Hope; Buck Institute for Research on Aging; Canada's Michael Smith Genome Sciences Centre; Harvard Medical School; Helen F. Graham Cancer Center & Research Institute at Christiana Care Health Services; HudsonAlpha Institute for Biotechnology et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He H, Liu X, Liu Y, Zhang M, Lai Y, Hao Y, Wang Q, Shi D, Wang N, Luo XG, et al. Human papillomavirus E6/E7 and long noncoding RNA TMPOP2 mutually upregulated gene expression in cervical cancer cells. J Virol. 2019;93:e01808–e01818. doi: 10.1128/JVI.01808-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 8.Zhang J, Yao T, Lin Z, Gao Y. Aberrant methylation of MEG3 functions as a potential plasma-based biomarker for cervical cancer. Sci Rep. 2017;7(6271) doi: 10.1038/s41598-017-06502-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen AH, Qin YE, Tang WF, Tao J, Song HM, Zuo M. MiR-34a and miR-206 act as novel prognostic and therapy biomarkers in cervical cancer. Cancer Cell Int. 2017;17(63) doi: 10.1186/s12935-017-0431-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yi YX, Fang Yan, Wu KJ, Liu YY, Zhang W. Comprehensive gene and pathway analysis of cervical cancer progression. Oncol Lett. 2020;19:3316–3332. doi: 10.3892/ol.2020.11439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang HJ, Xue JM, Li J, Wan LH, Zhu YX. Identification of key genes and pathways of diagnosis and prognosis in cervical cancer by bioinformatics analysis. Mol Genet Genomic Med. 2020;8(e1200) doi: 10.1002/mgg3.1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.den Boon JA, Pyeon D, Wang SS, Horswill M, Schiffman M, Sherman M, Zuna RE, Wang Z, Hewitt SM, Pearson R, et al. Molecular transitions from papillomavirus infection to cervical precancer and cancer: Role of stromal estrogen receptor signaling. Proc Natl Acad Sci USA. 2015;112:E3255–E3264. doi: 10.1073/pnas.1509322112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scotto L, Narayan G, Nandula SV, Arias-Pulido H, Subramaniyam S, Schneider A, Kaufmann AM, Wright JD, Pothuri B, Mansukhani M, Murty VV. Identification of copy number gain and overexpressed genes on chromosome arm 20q by an integrative genomic approach in cervical cancer: Potential role in progression. Genes Chromosomes Cancer. 2008;47:755–765. doi: 10.1002/gcc.20577. [DOI] [PubMed] [Google Scholar]
- 14.Biewenga P, Buist MR, Moerland PD, Ver Loren van Themaat E, van Kampen AH, ten Kate FJ, Baas F. Gene expression in early stage cervical cancer. Gynecol Oncol. 2008;108:520–526. doi: 10.1016/j.ygyno.2007.11.024. [DOI] [PubMed] [Google Scholar]
- 15.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, et al. NCBI GEO: Archive for functional genomics data sets-update. Nucleic Acids Res. 2013;41 (Database Issue):D991–D995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang DW, Sherman BT, Tan Q, Collins JR, Alvord WG, Roayaei J, Stephens R, Baseler MW, Lane HC, Lempicki RA. The DAVID Gene Functional Classification Tool: A novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007;8(R183) doi: 10.1186/gb-2007-8-9-r183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanehisa M. The KEGG database. Novartis Found Symp. 2002;247:91–101. discussion 101-103, 119-128, 244-152. [PubMed] [Google Scholar]
- 19.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: Tool for the unification of biology The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C, Jensen LJ. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41 (Database Issue):D808–D815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics. 2011;27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, Lin CY. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;8 (Suppl 4)(S11) doi: 10.1186/1752-0509-8-S4-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(pl1) doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhai Y, Kuick R, Nan B, Ota I, Weiss SJ, Trimble CL, Fearon ER, Cho KR. Gene expression analysis of preinvasive and invasive cervical squamous cell carcinomas identifies HOXC10 as a key mediator of invasion. Cancer Res. 2007;67:10163–10172. doi: 10.1158/0008-5472.CAN-07-2056. [DOI] [PubMed] [Google Scholar]
- 26.Medi K, Esra G, Kazim YA. Novel genomic biomarker candidates for cervical cancer as identified by differential co-expression network analysis. OMICS. 2019;23:261–273. doi: 10.1089/omi.2019.0025. [DOI] [PubMed] [Google Scholar]
- 27.Dai FF, Chen GT, Wang YQ, Zhang L, Long YM, Yuan MQ, Yang DY, Liu SY, Cheng YX, Zhang LP. Identification of candidate biomarkers correlated with the diagnosis and prognosis of cervical cancer via integrated bioinformatics analysis. Onco Targets Ther. 2019;12:4517–4532. doi: 10.2147/OTT.S199615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu JH, Nie SP, Gao M, Jiang Y, Wan YC, Ma XL, Zhou SL, Cheng WJ. Identification of EPHX2 and RMI2 as two novel key genes in cervical squamous cell carcinoma by an integrated bioinformatic analysis. J Cell Physiol. 2019;234:21260–21273. doi: 10.1002/jcp.28731. [DOI] [PubMed] [Google Scholar]
- 29.van Dam PA, Rolfo C, Ruiz R, Pauwels P, Van Berckelaer C, Trinh XB, Ferri Gandia J, Bogers JP, Van Laere S. Potential new biomarkers for squamous carcinoma of the uterine cervix. ESMO Open. 2018;3(e000352) doi: 10.1136/esmoopen-2018-000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iqbal J, Ejaz SA, Miliutina M, Langer P, Saeed A. Anti-proliferative effects of chromones: Potent derivatives affecting cell growth and apoptosis in breast, bone-marrow and cervical cancer cells. Med Chem. 2019;15:883–891. doi: 10.2174/1573406415666190621155843. [DOI] [PubMed] [Google Scholar]
- 31.Zhao H, Li S, Wang G, Zhao W, Zhang D, Wang F, Li W, Sun L. Study of the mechanism by which dinaciclib induces apoptosis and cell cycle arrest of lymphoma Raji cells through a CDK1-involved pathway. Cancer Med. 2019;8:4348–4358. doi: 10.1002/cam4.2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang WH, Forde D, Lai AG. Dual prognostic role of 2-oxoglutarate-dependent oxygenases in ten cancer types: Implications for cell cycle regulation and cell adhesion maintenance. Cancer Commun (Lond) 2019;39(23) doi: 10.1186/s40880-019-0369-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee JW, Park S, Kim SY, Um SH, Moon EY. Curcumin hampers the antitumor effect of vinblastine via the inhibition of microtubule dynamics and mitochondrial membrane potential in HeLa cervical cancer cells. Phytomedicine. 2016;23:705–713. doi: 10.1016/j.phymed.2016.03.011. [DOI] [PubMed] [Google Scholar]
- 34.Saha SK, Khuda-Bukhsh AR. Berberine alters epigenetic modifications, disrupts microtubule network, and modulates HPV-18 E6-E7 oncoproteins by targeting p53 in cervical cancer cell HeLa: A mechanistic study including molecular docking. Eur J Pharmacol. 2014;744:132–146. doi: 10.1016/j.ejphar.2014.09.048. [DOI] [PubMed] [Google Scholar]
- 35.Das D, Bristol ML, Smith NW, James CD, Wang X, Pichierri P, Morgan IM. Werner Helicase control of human papillomavirus 16 E1-E2 DNA replication is regulated by SIRT1 deacetylation. mBio. 2019;10(e00263-19) doi: 10.1128/mBio.00263-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lou W, Ding B, Xu L, Fan W. Construction of potential glioblastoma multiforme-related miRNA-mRNA regulatory network. Front Mol Neurosci. 2019;12(66) doi: 10.3389/fnmol.2019.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li L, Lei Q, Zhang S, Kong L, Qin B. Screening and identification of key biomarkers in hepatocellular carcinoma: Evidence from bioinformatic analysis. Oncol Rep. 2017;38:2607–2618. doi: 10.3892/or.2017.5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brown CA, Bogers J, Sahebali S, Depuydt CE, De Prins F, Malinowski DP. Role of protein biomarkers in the detection of high-grade disease in cervical cancer screening programs. J Oncol. 2012;2012(289315) doi: 10.1155/2012/289315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peres AL, Paz E Silva KM, de Araújo RF, de Lima Filho JL, de Melo Júnior MR, Martins DB, de Pontes Filho NT. Immunocytochemical study of TOP2A and Ki-67 in cervical smears from women under routine gynecological care. J Biomed Sci. 2016;23(42) doi: 10.1186/s12929-016-0258-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smrkolj S, Erzen M, Rakar S. Prognostic significance of topoisomerase II alpha and collagen IV immunoexpression in cervical cancer. Eur J Gynaecol Oncol. 2010;31:380–385. [PubMed] [Google Scholar]
- 41.Korkolopoulou P, Vassilakopoulos TP. Topoisomerase IIalpha as a prognostic factor in mantle cell lymphoma. Leukemia. 2004;18:1347–1349. doi: 10.1038/sj.leu.2403413. [DOI] [PubMed] [Google Scholar]
- 42.Fritz P, Cabrera CM, Dippon J, Gerteis A, Simon W, Aulitzky WE, van der Kuip H. c-erbB2 and topoisomerase IIalpha protein expression independently predict poor survival in primary human breast cancer: A retrospective study. Breast Cancer Res. 2005;7:R374–R384. doi: 10.1186/bcr1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wong N, Yeo W, Wong WL, Wong NL, Chan KY, Mo FK, Koh J, Chan SL, Chan AT, Lai PB, et al. TOP2A overexpression in hepatocellular carcinoma correlates with early age onset, shorter patients survival and chemoresistance. Int J Cancer. 2009;124:644–652. doi: 10.1002/ijc.23968. [DOI] [PubMed] [Google Scholar]
- 44.Del Pino M, Svanholm-Barrie C, Torné A, Marimon L, Gaber J, Sagasta A, Persing DH, Ordi J. mRNA biomarker detection in liquid-based cytology: A new approach in the prevention of cervical cancer. Mod. Pathol. 2015;28:312–320. doi: 10.1038/modpathol.2014.106. [DOI] [PubMed] [Google Scholar]
- 45.Scapulatempo-Neto C, Veo C, Fregnani JHTG, Lorenzi A, Mafra A, Melani AGF, Loaiza EAA, Rosa LAR, de Oliveira CM, Levi JE, Longatto-Filho A. Characterization of topoisomerase II α and minichromosome maintenance protein 2 expression in anal carcinoma. Oncol Lett. 2017;13:1891–1898. doi: 10.3892/ol.2017.5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu D, Huang CL, Kameyama K, Hayashi E, Yamauchi A, Sumitomo S, Yokomise H. Topoisomerase IIalpha gene expression is regulated by the p53 tumor suppressor gene in nonsmall cell lung carcinoma patients. Cancer. 2002;94:2239–2247. doi: 10.1002/cncr.10450. [DOI] [PubMed] [Google Scholar]
- 47.Schaefer-Klein JL, Murphy SJ, Johnson SH, Vasmatzis G, Kovtun IV. Topoisomerase 2 alpha cooperates with androgen receptor to contribute to prostate cancer progression. PLoS One. 2015;10(e0142327) doi: 10.1371/journal.pone.0142327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hua W, Sa KD, Zhang X, Jia LT, Zhao J, Yang AG, Zhang R, Fan J, Bian K. MicroRNA-139 suppresses proliferation in luminal type breast cancer cells by targeting Topoisomerase II alpha. Biochem Biophys Res Commun. 2015;463:1077–1083. doi: 10.1016/j.bbrc.2015.06.061. [DOI] [PubMed] [Google Scholar]
- 49.Panvichian R, Tantiwetrueangdet A, Angkathunyakul N, Leelaudomlipi S. TOP2A amplification and overexpression in hepatocellular carcinoma tissues. Biomed Res Int. 2015;2015(381602) doi: 10.1155/2015/381602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Erriquez J, Becco P, Olivero M, Ponzone R, Maggiorotto F, Ferrero A, Scalzo MS, Canuto EM, Sapino A, Verdun di Cantogno L, et al. TOP2A gene copy gain predicts response of epithelial ovarian cancers to pegylated liposomal doxorubic in: TOP2A as marker of response to PLD in ovarian cancer. Gynecol Oncol. 2015;138:627–633. doi: 10.1016/j.ygyno.2015.06.025. [DOI] [PubMed] [Google Scholar]
- 51.Deguchi S, Katsushima K, Hatanaka A, Shinjo K, Ohka F, Wakabayashi T, Zong H, Natsume A, Kondo Y. Oncogenic effects of evolutionarily conserved noncoding RNA ECONEXIN on gliomagenesis. Oncogene. 2017;36:4629–4640. doi: 10.1038/onc.2017.88. [DOI] [PubMed] [Google Scholar]
- 52.Santin AD, Zhan F, Bignotti E, Siegel ER, Cané S, Bellone S, Palmieri M, Anfossi S, Thomas M, Burnett A, et al. Gene expression profiles of primary HPV16- and HPV18-infected early stage cervical cancers and normal cervical epithelium: Identification of novel candidate molecular markers for cervical cancer diagnosis and therapy. Virology. 2005;331:269–291. doi: 10.1016/j.virol.2004.09.045. [DOI] [PubMed] [Google Scholar]
- 53.Jain M, Zhang L, He M, Zhang YQ, Shen M, Kebebew E. TOP2A is overexpressed and is a therapeutic target for adrenocortical carcinoma. Endocr Relat Cancer. 2013;20:361–370. doi: 10.1530/ERC-12-0403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang H, Yang B, Geng T, Li B, Dai P, Chen C. Tissue-specific selection of optimal reference genes for expression analysis of anti-cancer drug-related genes in tumor samples using quantitative real-time RT-PCR. Exp Mol Pathol. 2015;98:375–381. doi: 10.1016/j.yexmp.2014.10.014. [DOI] [PubMed] [Google Scholar]
- 55.Khoo BL, Lee SC, Kumar P, Tan TZ, Warkiani ME, Ow SG, Nandi S, Lim CT, Thiery JP. Short-term expansion of breast circulating cancer cells predicts response to anti-cancer therapy. Oncotarget. 2015;6:15578–15593. doi: 10.18632/oncotarget.3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thougaard AV, Langer SW, Hainau B, Grauslund M, Juhl BR, Jensen PB, Sehested M. A murine experimental anthracycline extravasation model: Pathology and study of the involvement of topoisomerase II alpha and iron in the mechanism of tissue damage. Toxicology. 2010;269:67–72. doi: 10.1016/j.tox.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 57.Melaiu O, Cristaudo A, Melissari E, Di Russo M, Bonotti A, Bruno R, Foddis R, Gemignani F, Pellegrini S, Landi S. A review of transcriptome studies combined with data mining reveals novel potential markers of malignant pleural mesothelioma. Mutat Res. 2012;750:132–140. doi: 10.1016/j.mrrev.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 58.Meagher NS, Schuster K, Voss A, Budden T, Pang CNI, de Fazio A, Ramus SJ, Friedlander ML. Does the primary site really matter? Profiling mucinous ovarian cancers of uncertain primary origin (MO-CUP) to personalise treatment and inform the design of clinical trials. Gynecol Oncol. 2018;150:527–533. doi: 10.1016/j.ygyno.2018.07.013. [DOI] [PubMed] [Google Scholar]
- 59.Guo J, Gu Y, Ma X, Zhang L, Li H, Yan Z, Han Y, Xie L, Guo X. Identification of hub genes and pathways in adrenocortical carcinoma by integrated bioinformatic analysis. J Cell Mol Med. 2020;8:4428–4438. doi: 10.1111/jcmm.15102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li S, Liu X, Liu T, Meng X, Yin X, Fang C, Huang D, Cao Y, Weng H, Zeng X, Wang X. Identification of biomarkers correlated with the TNM staging and overall survival of patients with bladder cancer. Front Physiol. 2017;8(947) doi: 10.3389/fphys.2017.00947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Han JY, Han YK, Park GY, Kim SD, Kim JS, Jo WS, Lee CG. Corrigendum: Bub1 is required for maintaining cancer stem cells in breast cancer cell lines. Sci Rep. 2016;6(17984) doi: 10.1038/srep17984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xi Q, Huang M, Wang Y, Zhong J, Liu R, Xu G, Jiang L, Wang J, Fang Z, Yang S. The expression of CDK1 is associated with proliferation and can be a prognostic factor in epithelial ovarian cancer. Tumour Biol. 2015;36:4939–4948. doi: 10.1007/s13277-015-3141-8. [DOI] [PubMed] [Google Scholar]
- 63.Chen S, Chen X, Xiu YL, Sun KX, Zhao Y. MicroRNA-490-3P targets CDK1 and inhibits ovarian epithelial carcinoma tumorigenesis and progression. Cancer Lett. 2015;362:122–130. doi: 10.1016/j.canlet.2015.03.029. [DOI] [PubMed] [Google Scholar]
- 64.Schwermer M, Lee S, Köster J, van Maerken T, Stephan H, Eggert A, Morik K, Schulte JH, Schramm A. Sensitivity to cdk1-inhibition is modulated by p53 status in preclinical models of embryonal tumors. Oncotarget. 2015;6:15425–15435. doi: 10.18632/oncotarget.3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mitrofanova A, Aytes A, Zou M, Shen MM, Abate-Shen C, Califano A. Predicting drug response in human prostate cancer from preclinical analysis of in vivo mouse models. Cell Rep. 2015;12:2060–2071. doi: 10.1016/j.celrep.2015.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Grewal S, Carver JG, Ridley AJ, Mardon HJ. Implantation of the human embryo requires Rac1-dependent endometrial stromal cell migration. Proc Natl Acad Sci USA. 2008;105:16189–16194. doi: 10.1073/pnas.0806219105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Imaoka H, Toiyama Y, Saigusa S, Kawamura M, Kawamoto A, Okugawa Y, Hiro J, Tanaka K, Inoue Y, Mohri Y, Kusunoki M. RacGAP1 expression, increasing tumor malignant potential, as a predictive biomarker for lymph node metastasis and poor prognosis in colorectal cancer. Carcinogenesis. 2015;36:346–354. doi: 10.1093/carcin/bgu327. [DOI] [PubMed] [Google Scholar]
- 68.Yu Y, Munger K. Human papillomavirus type 16 E7 oncoprotein inhibits the anaphase promoting complex/cyclosome activity by dysregulating EMI1 expression in mitosis. Virology. 2013;446:251–259. doi: 10.1016/j.virol.2013.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yost S, de Wolf B, Hanks S, Zachariou A, Marcozzi C, Clarke M, de Voer R, Etemad B, Uijttewaal E, Ramsay E, et al. Biallelic TRIP13 mutations predispose to Wilms tumor and chromosome missegregation. Nat Genet. 2017;49:1148–1151. doi: 10.1038/ng.3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tan CL, Teissier S, Gunaratne J, Quek LS, Bellanger S. Stranglehold on the spindle assembly checkpoint: The human papillomavirus E2 protein provokes BUBR1-dependent aneuploidy. Cell Cycle. 2015;14:1459–1470. doi: 10.1080/15384101.2015.1021519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Suematsu T, Li Y, Kojima H, Nakajima K, Oshimura M, Inoue T. Deacetylation of the mitotic checkpoint protein BubR1 at lysine 250 by SIRT2 and subsequent effects on BubR1 degradation during the prometaphase/anaphase transition. Biochem Biophys Res Commun. 2014;453:588–594. doi: 10.1016/j.bbrc.2014.09.128. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.