

Abstract
Zero-valent iron nanoparticles (nZVI) treated by reduced sulfur compounds (i.e., sulfidated nZVI, S-nZVI) have attracted increased attention as promising materials for environmental remediation. While the preparation of S-nZVI and its reactions with various groundwater contaminants such as trichloroethylene (TCE) were already a subject of several studies, nanoparticle synthesis procedures investigated so far were suited mainly for laboratory-scale preparation with only a limited possibility of easy and cost-effective large-scale production and FeS shell property control. This study presents a novel approach for synthesizing S-nZVI using commercially available nZVI particles that are treated with sodium sulfide in a concentrated slurry. This leads to S-nZVI particles that do not contain hazardous boron residues and can be easily prepared off-site. The resulting S-nZVI exhibits a core–shell structure where zero-valent iron is the dominant phase in the core, while the shell contains mostly amorphous iron sulfides. The average FeS shell thickness can be controlled by the applied sulfide concentration. Up to a 12-fold increase in the TCE removal and a 7-fold increase in the electron efficiency were observed upon amending nZVI with sulfide. Although the FeS shell thickness correlated with surface-area-normalized TCE removal rates, sulfidation negatively impacted the particle surface area, resulting in an optimal FeS shell thickness of approximately 7.3 nm. This corresponded to a particle S/Fe mass ratio of 0.0195. At all sulfide doses, the TCE degradation products were only fully dechlorinated hydrocarbons. Moreover, a nearly 100% chlorine balance was found at the end of the experiments, further confirming complete TCE degradation and the absence of chlorinated transformation products. The newly synthesized S-nZVI particles thus represent a promising remedial agent applicable at sites contaminated with TCE.
Keywords: zero-valent iron, sulfidation, trichloroethylene, dechlorination, nanoparticles, selectivity
1. Introduction
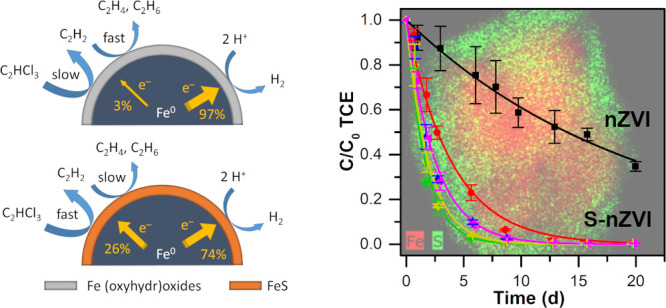
Nanoscale zero-valent iron (nZVI) presents an attractive and, according to current knowledge, environmentally benign reduction agent for the treatment of contaminated groundwater.1 Although many promising laboratory and field studies have been conducted in the past two decades, the field-scale application of this technology is limited mainly due to the low selectivity of nZVI due to reactions with natural reducible species (e.g., dissolved O2, NO3–, or water).2−5 For example, the selectivity of nZVI toward trichloroethylene (TCE) was reported to be as low as 3%, while the remaining 97% of available electrons was lost by iron corrosion in water.2 Fast particle corrosion leads to a rapid loss of the reductive capacity and the formation of an iron (oxyhydr)oxide passivation layer.2−4
Different strategies to increase the long-term performance of nZVI were investigated. For instance, nZVI has been immobilized onto various solid porous materials6−9 and coated with organic polymers10−12 to limit particle exposure to water. These materials, however, exhibited a lowered reduction capacity and/or increased contaminant sorption to the detriment of chemical reduction that could possibly result in a decreased rate of contaminant (bio)degradation and contaminant transport to other locations, causing secondary contamination.1 The doping of nZVI with a catalytic noble metal (e.g., Pd, Pt, or Ni) to increase the particle reactivity has also been widely studied.13−15 However, the superior reactivity of these materials has been found to be rapidly inhibited by natural water constituents, such as humic acids, and fast iron corrosion.16,17 Another problem of bimetallic systems can be the leaching of hazardous catalytic metals, which may generate a secondary pollution to the treated water.17
Over the past few years, great interest has been devoted to the chemical modification of iron materials, including nZVI, by reduced sulfur compounds (sulfidation) for environmental applications. Previous studies have shown that sulfidation can remarkably increase the reactivity of nZVI with various contaminants and, at the same time, significantly increase its selectivity.18−23 Treating nZVI with reduced sulfur species represents a technologically simple, cheap, and environmentally acceptable approach for increasing the performance of nZVI in contaminant removal.
Sulfidated nZVI (S-nZVI) particles are usually prepared via one-step or two-step solution synthesis methods (also referred to as aqueous–aqueous and aqueous–solid synthesis) that lead to different particle morphologies.24,25 A one-step synthesis method consists of adding dropwise a Fe3+ or Fe2+ solution into the mixed solution of a strong reductant (typically NaBH4) and sulfidation agent, e.g., dithionite or Na2S.19,26,27 This process leads to the parallel formation of Fe0 and FeS, resulting in a mixed-phase material.28 In the two-step method, the nZVI particles are synthesized first (typically using dissolved Fe3+ or Fe2+ and NaBH4), then recovered, and finally transferred into a solution of the sulfidation agent (or directly sulfidated in the nZVI synthesis mixture upon the reduction of Fe3+ or Fe2+ by NaBH4). During this process, nZVI reacts with water, generating Fe2+ ions, which are then precipitated with S2– ions originating from the sulfidation agent to form a FeS layer on the surface of the nZVI particles,19,22,29 thus giving rise to particles with a core–shell structure. A modified two-step synthesis includes the stoichiometric addition of Fe2+ in the sulfidation step.30 Other methods of S-ZVI synthesis were investigated, such as iron (oxyhydr)oxide reduction using gaseous SO231 and solid-phase synthesis by ball-milling microscale ZVI with elemental sulfur.32 Of the synthetic methods mentioned, the two-step aqueous–solid synthesis is of particular interest since it enables the production of nanoscale particles with a uniform particle-size distribution that can be easily separated from the suspension.23 Moreover, this approach allows for the sulfidation of nZVI materials synthesized using other methods than reduction by borohydride.
Several studies investigated the performance of S-nZVI particles prepared by the two-step aqueous–solid approach, all of them employing nZVI prepared by sodium borohydride reduction (nZVIBH) as a precursor material.19−23,29,33 While nZVIBH is a standard in laboratory testing, it is not well suited for large-scale production for several reasons: (i) its industrial scale-up is challenging due to the many synthesis steps involved and the large volume of wastewater produced,34 (ii) its production is cost-prohibitive (over $200 kg–1),34 and (iii) its slurry contains residual boron-containing species, which are hazardous for the environment.35,36 So far, aqueous–solid S-nZVI synthesis has been conducted at iron concentrations in slurries ranging from 0.2 to 20 g L–1,19−23,29,33 which are well-suited for laboratory batch experiments and/or direct application of a diluted slurry into the ground. Nevertheless, more concentrated slurries would be advantageous for off-site preparation to minimize shipping costs.
Herein, we describe a novel approach to synthesize a concentrated S-nZVI slurry with a high potential for production upscaling. The presented approach consists of the aqueous sulfidation of a commercially available nZVI prepared by the thermal reduction of iron (oxyhydr)oxide precursors in a H2 atmosphere (S-nZVIH2). nZVIH2 can be manufactured on a large scale at lower costs compared with nZVIBH (market price of $112–$147 kg–1)37,38 and without boron-containing residues. Sodium sulfide was chosen as the sulfidation agent since it contains sulfur in the most reduced form, and therefore, a larger amount of zero-valent iron is likely to be maintained during sulfidation compared with other sulfur-containing chemicals, e.g., dithionite.21 The specific goals of this study were to (i) prepare well-characterized S-nZVI particles with a controlled FeS shell thickness and (ii) demonstrate their high reactivity and increased selectivity toward TCE, a frequent groundwater contaminant, with an emphasis on kinetics and products of TCE reduction and iron corrosion, and the role of the sulfur to iron ratio and FeS shell thickness in TCE removal.
2. Materials and methods
2.1. Chemicals
The commercially available NANOFER 25P nZVI particles, supplied by NANO IRON, s.r.o. (Czech Republic), were used for all experiments.39 nZVI particles were stored in an airtight container under an inert atmosphere inside an Ar-filled glovebox prior to use. Sodium sulfide nonahydrate (≥98.0%, Alfa Aesar, Germany) was used as a sulfidation agent. Deoxygenated deionized (DO/DI) water prepared by purging Milli-Q purified water (Millipore Corp., Bedford, MA, USA) with N2 for 30 min was used in all experiments, including material synthesis. Details regarding the other chemicals used are provided in Text S1 in the Supporting Information (SI).
2.2. Sulfidation of nZVI
Sulfidated nZVI particles were prepared by treating nZVI particles in aqueous solutions of sodium sulfide in an anaerobic Ar-filled glovebox. Solutions of an appropriate concentration of Na2S were freshly prepared using DO/DI water (Table S1 in the SI). Aliquots of individual solutions (16 mL) were added to 4 g of NANOFER 25P nZVI particles in 42 mL glass vials to yield a nZVI concentration of 242 g L–1 (20% w/w) and nominal S/Fe mass ratios of 0.0025, 0.01, 0.05, 0.1, and 0.25. After the addition of sodium sulfide solutions, the mixtures were immediately dispersed using a T25 ULTRA-TURRAX disperser (IKA, Staufen, Germany) at 11 000 rpm for 2 min to yield a concentrated slurry. The vials were then sealed and allowed to react for 60 min on an horizontal shaker (160 rpm) outside the anaerobic glovebox. The concentration of the residual dissolved sulfide after S-nZVI synthesis was determined using iodometric titration,40 after separation of the particles with a magnet and filtration using a 0.1 μm PTFE syringe filter. A bare nZVI control sample was prepared by the same procedure using pure DO/DI water without the addition of sodium sulfide.
2.3. Material Characterization
All nZVI particles were characterized with a transmission electron microscope (TEM) JEOL 2100 at an electron acceleration voltage of 200 kV, which was equipped with an X-MaxN 80T SDD EDS detector (Oxford Instruments). High-resolution TEM (HR-TEM) images of individual particles were recorded with a spherical aberration corrected Titan G2 TEM (FEI), operated at 80 kV. Images were taken with a BM UltraScan CCD camera (Gatan). Energy-dispersive spectrometry (EDS) was performed in scanning (STEM) mode to obtain elemental maps with the Super-X system with four silicon drift detectors (Bruker). STEM images were taken with a HAADF detector 3000 (Fischione). The elemental mappings were performed on at least four representative images from different locations of the sample grid. Particle-size and shell-thickness distributions were evaluated using GIMP software (100 measurements were performed for each distribution).
The phase composition of nZVI samples was determined by powder X-ray diffraction (XRD) with an EMPYREAN diffractometer (PANalytical, B.V.) operating in a Bragg–Brentano geometry. The diffractometer was equipped in an iron-filtered Co Kα radiation source, programmable divergence and diffracted beam antiscatter slits, and a PIXcel detector. The patterns were measured in a 2θ range of 5 to 105°, and the data were processed using HighScorePlus software in conjunction with PDF-4+ and ICSD databases.
The speciation of S on the surface of the particles was evaluated by X-ray photoelectron spectroscopy (XPS) with a PHI 5000 VersaProbe II XPS system (Physical Electronics, Inc., Chanhassen, MN, USA) using monochromatic Al Kα radiation (15 kV, 50 W) and a photon energy of 1486.7 eV. The survey spectra were acquired with a pass energy of 187.850 eV and an electronvolt step of 0.8 eV, while, for the high-resolution spectra, a pass energy of 23.500 eV and an electronvolt step of 0.2 eV were used. Dual beam charge compensation was used for all measurements. All spectra were acquired in a vacuum of 1.4 × 10–7 Pa at room temperature (20 °C). The analyzed area on each sample was 100 μm in diameter. The spectra were evaluated with MultiPak (ULVAC-PHI, Inc.) software. All binding energy values were referenced to the aliphatic carbon peak C1s at 284.80 eV.
Iron speciation was analyzed using low-temperature (T = 150 K) 57Fe Mössbauer spectrometer (MS2007) equipped with a 50 mCi 57Co(Rh) source operating in a constant acceleration mode.41,42 MossWinn software43 was used for fitting the Mössbauer spectra. The values of the isomer shift were referred to an α-Fe foil at room temperature.
The size of the particles in suspensions diluted with Milli-Q water (final Fe conc. 1 g L–1) was measured using a DC24000UHR disc centrifuge (CPS Instruments, Inc., Prairieville, LA, USA) operated at a speed of 3780 rpm. The density gradient was prepared using sucrose solutions (8–24%) gravimetrically prepared by dissolving sucrose powder in DI water. The injected amount was 1.6 mL for each sucrose concentration. To protect the density gradient against water evaporation, a thin layer of dodecane was created by injecting 0.5 mL of it on top of the gradient. After the injection of sucrose solutions, an equilibration period of 20 min was applied to obtain a stable gradient. The injected sample volume was 0.1 mL. Each sample injection was preceded by injecting 0.1 mL of a PVC calibrant (particle diameter 1400 nm).
The particle hydrodynamic diameter and zeta-potential were determined with a ZetaSizer Nano instrument (Malvern Instruments, Malvern, United Kingdom). Nanoparticle suspensions were diluted to a concentration of 242 mg L–1 in 100 mM NaHCO3 (pH 8.7) and immediately analyzed in triplicate at a scattering angle of 173° with an acquisition time of 60 s.
The Brunauer–Emmett–Teller (BET) surface area and porosity were determined by nitrogen adsorption at 77.4 K up to the nitrogen saturation pressure using an Autosorb-iQ-C analyzer (Quantachrome).
Details regarding the sample preparation for individual characterizations are provided in Text S2 in the SI.
2.4. Geochemical Modeling
The precipitation of possible FeSx mineral phases during the sulfidation treatment was modeled on the basis of the evolution of the measured pH, redox potential, and dissolved sulfide and iron species concentrations in the slurry. The evolution of the pH and redox potential relative to the standard hydrogen electrode (Eh) was monitored for 26 h in a freshly prepared suspension of S-nZVI at a nominal S/Fe mass ratio of 0.05 (Fe concentration of 242 g L–1 and Na2S initial concentration of 0.039 mol L–1, with a volume of 500 mL) on an orbital shaker (230 rpm). At 15 min, 1 h, 3 h, and 26 h, the concentrations of the residual dissolved sulfide and iron were determined after the separation of the particles with a magnet and filtration using a 0.1 μm PTFE syringe filter. Further details regarding the analytical methods are provided in the Text S3 in the SI.
The Eh–pH diagram was constructed with Geochemist’s Workbench Professional 7.0 using the database minteq.dat. The same program was used to calculate saturation indices.
2.5. TCE Dechlorination Experiments
The ability of S-nZVI particles to dechlorinate TCE was tested in batch experiments conducted in 42 mL glass vials containing 20 mL of aqueous solution, with the remaining volume as headspace. The vials were capped with PTFE-lined Mininert valves (Sigma-Aldrich). The vials were spiked with 82 μL of freshly prepared S-nZVI suspensions to reach a nZVI concentration of 1 g L–1. Experiments were started by injecting 50 μL of a TCE stock solution in methanol (8 g L–1) to obtain an initial TCE concentration of 20 mg L–1. The reactors were placed on an horizontal shaker (125 rpm) at 22 ± 1 °C. Control experiments with bare nZVI, Na2S solutions only, and DO/DI water were performed in parallel. No attempt was made to adjust the pH. An aliquot (25–100 μL) of headspace gas was periodically withdrawn using a gastight syringe and analyzed using a 7890B gas chromatograph (GC) from Agilent Technologies, USA. The GC system was equipped with the following columns and detectors: (i) a 30 m Rtx-VMS capillary column (Restek, USA), followed by an electron capture detector (ECD) for the detection of TCE and dichloroethene isomers; (ii) a 30 m Rt-Q-BOND PLOT column (Restek, USA), connected to a valve coupled with (a) a 12 m MXT-Q BOND column (Restek, USA), followed by a flame ionization detector (FID) to detect ethene, ethane, acetylene, propane, and vinyl chloride and (b) a 30 m Carboxen-1010 PLOT column (Supelco, USA), coupled with a thermal conductivity detector (TCD) for H2 detection. Details regarding the GC analysis are provided in Text S3 in the SI. All experiments were run in four replicates for a period of 3 weeks. The pressure build-up was measured manually using a frictionless glass syringe (Poulten & Graf Ltd., Germany). The measured amounts of TCE and its reaction products were corrected for overpressure and sampling-induced headspace losses. Internal standard (propane) was used to validate the measured headspace losses. The detailed correction procedure is described elsewhere.2 The differences between the measured decreases of the internal standard within the vials and the calculated decreases (based on pressure equalization) were negligible. The total concentrations of all analytes were computed from the detected headspace concentrations using the respective Henry’s Law constants.44 The concentration of the dissolved chloride (Cl–) after reactivity experiments was analyzed using a 930 IC Flex ion chromatography system (Metrohm, Switzerland). Further information is provided in Text S3.
2.6. Calculation of Reaction Rates and Electron Efficiencies
The observed pseudo-first-order reaction rate constants for TCE removal, kobs, were calculated using nonlinear regression based on a Levenberg–Marquardt least-squares algorithm with OriginPro 2017 (OriginLab, Northampton, MA, USA). The initial surface-area-normalized rate constant, kSA, was obtained from the pseudo-first-order TCE degradation rate constant, kTCE, which was observed in water without headspace, using eqs 1 and 2.45
| 1 |
| 2 |
where VW is the aqueous volume, Vg the headspace volume, kH the dimensionless Henry’s law volatility constant, C (mg L–1) the aqueous phase TCE concentration, αs (m2 g–1) the surface area of the particles, and ρm (g L–1) the particle dosage. The value of kH = 0.33 was calculated for TCE at 22 °C.44
The electron efficiency, εe, describing the selectivity of electrons from Fe0 for TCE reduction over iron corrosion (production of H2) was calculated using eq 3.46
| 3 |
where ni is the stoichiometry for product i (i.e., 8 for ethane, 6 for ethene, and 4 for acetylene), pi the molar amount of that product, and pH2 the molar amount of H2 produced during the TCE dechlorination.
3. Results and Discussion
3.1. Characteristics and Surface Morphology of Fresh Particles
S-nZVI particles were prepared via aqueous–solid synthesis in concentrated nZVI slurries (20% w/w) by treating nZVI particles in sodium sulfide solutions of different concentrations (Table 1). The amount of sulfur deposited on the nZVI surface was calculated based on the sulfide consumption (Table S1 in the SI) and ranged 2.2–62.8 mg of S per g of Fe. The resulting S/Fe ratio was positively associated with the initial sulfide dose, while the efficiency of sulfide incorporation decreased with increasing sulfide addition in the studied range (from ca. 95 to 25%). The inefficient incorporation of sulfur at higher loadings is consistent with the available literature.20
Table 1. Sulfur Content, Specific Surface Area (SSA), Porosity, and TCE Degradation Kinetics of S-nZVI Particles Used in the Present Study.
| TCE
removal |
|||||
|---|---|---|---|---|---|
| particle typea | particle S/Fe mass ratio | BET SSA (m2 g–1) | pore surface area (m2 g–1) | kobs (10–3 h–1) | kSA (10–3 L m–2 h–1) |
| nZVI | 0 | 45.6b | 33.9b | 2.06 ± 0.06 | 0.0617 |
| 0.25S-nZVI | 0.22/100 | 36.7 | 36.0 | 10.6 ± 0.6 | 0.394 |
| 1S-nZVI | 0.94/100 | 32.4 | 29.4 | 16.4 ± 1.5 | 0.690 |
| 5S-nZVI | 1.95/100 | 32.7 | 25.9 | 25.3 ± 2.8 | 1.06 |
| 10S-nZVI | 3.19/100 | 25.0 | 21.2 | 21.8 ± 1.8 | 1.19 |
| 25S-nZVI | 6.29/100 | 15.5 | 15.4 | 16.4 ± 0.5 | 1.45 |
Labeled according to % nominal mass S/Fe ratio used for the synthesis.
After conditioning in DO/DI water.
The XRD patterns of untreated and sulfidated nZVI particles (Figure 1A) identified α-Fe0 (>90%), the major crystalline phase, with characteristic 2θ peaks at 52.5° and 99.5°. Magnetite (Fe2+Fe3+2O4) and wüstite (FeO) were detected as remaining nonreduced phases.39 No sulfur-bearing phases were discerned in the pattern due to their low concentration and/or, more probably, their low degree of crystallinity.24 Several unidentified peaks were present in the XRD pattern of the S-nZVI with the highest sulfur loading.
Figure 1.

(A) XRD patterns for bare nZVI and S-nZVI particles of varying S/Fe mass ratios. (B) Size distributions of bare nZVI and S-nZVI of varying S/Fe mass ratios. (C) XPS S 2p narrow region spectra of sulfidated nZVI with a S/Fe mass ratio 0.0195. XPS peak assignments were based on values summarized by Descostes et al. and Mullet et al.50,51 (D) 57Fe Mössbauer spectrum of sulfidated nZVI at a S/Fe mass ratio of 0.0195 measured at 150 K.
The particle size distribution and agglomeration were analyzed using disc centrifugal sedimentation (Figure 1B). The detected size distribution profile by weight showed two maxima, one corresponding to individual particles (40–120 nm) and the other to particle agglomerates (900–1400 nm). The size of individual particles was similar in all synthesized materials (see below), while its relative contribution was higher for bare nZVI. Nevertheless, in both nZVI and all S-nZVI materials, the dominant particle diameter corresponded to agglomerates (by weight). The typical agglomerate size was similar among bare nZVI and S-nZVI with sulfur content up to S/Fe 0.0319 (900–1100 nm). At a S/Fe mass ratio of 0.0629, the particles formed larger agglomerates (1200–1400 nm). This larger agglomerate size likely stems from the tendency of excessive FeS/FeOHx flakes to form networks that bind individual particles together.19
Agglomerate hydrodynamic diameters obtained with dynamic light scattering were roughly double, which can be attributed to increased S-nZVI particle agglomeration in solutions of higher ionic strength, i.e., 100 mM NaHCO347 (Table S2). The zeta-potential of bare nZVI particles was −18.3 mV and gradually decreased to −28 mV with an increasing S/Fe ratio (Table S2). This trend was observed previously for nZVI particles treated with dithionite.47 The lower zeta-potential of S-nZVI particles infers on their better colloidal stability compared with bare nZVI at a slightly basic pH that frequently occurs in natural waters. Indeed, the better colloidal stability of S-nZVI particles during aging was found in our previous study.48 It is likely that, even though sulfidation may increase the agglomerate size of fresh particles due to FeS bridging during particle synthesis, sulfidation improves the stability of particle agglomerates in the long term.
The morphology of the nZVI and S-nZVI particles was studied using TEM. Nanoscale ZVI particles, which were conditioned in DO/DI water without the addition of sulfide, had an irregular spherical shape, with an average particle size between 40 and 120 nm and an average thickness of the iron (oxyhydr)oxide shell of 3.9 nm (Figures S1A, S2A, and S3A). A similar morphology was observed for S-nZVI particles with a S/Fe mass ratio of 0.0195 (Figure S1B). At the highest sulfide dose (S/Fe mass ratio of 0.0629), the S-nZVI particles formed larger agglomerates (Figure S1C), but the average size of individual particles did not change substantially (Figure S2). The average shell thickness of S-nZVI particle shells ranged 6.1–9.7 nm (Figure S3). This is comparable with a previous study where S-nZVI particles with a theoretical S/Fe mass ratio 0.13 developed a 5 nm thick FeS shell.49 Although the shell thickness was not perfectly uniform for all particles of the same type, its mean value was associated with an increasing sulfide dose.
The BET specific surface area (SSA) and pore characteristics of the nZVI and S-nZVI sulfidated with different sulfide levels are shown in Table 1. The SSA of original nZVI particles was approximately 20–25 m2 g–139,52 and increased after conditioning in DO/DI water for the same duration as used for sulfidation to 45.6 m2 g–1. This is likely due to the formation of a thin layer of iron corrosion products on the particles, which increase their surface roughness.53 S-nZVI particles exhibited lower specific surface areas (15.5–36.7 m2 g–1) and also lower pore surface areas (except the particles with the lowest degree of sulfidation). Pore size distribution peaked at 38 Å for all particles (Figure S4). A decreasing trend in SSA was observed for particles with an increasing sulfide dose, except for particles with S/Fe ratios of 0.0094 and 0.0195, which exhibited similar SSAs. The observed trend may be explained by one or a combination of several of the following aspects: (i) limited iron corrosion in an aqueous environment (Figures S1 D–F) and thus slower formation of iron (oxyhydr)oxide flakes in sulfidated nZVI; (ii) coverage of the nZVI surface by layered FeS (Figures 2A, 2B), leading to a decrease in the surface roughness, e.g., by pore clogging;54 and (iii) increased agglomeration of particles in the sulfide environment.19 Since the measured particle agglomerate diameter of the S-nZVI sulfidated up to S/Fe mass ratio of 0.0319 was similar to the particle agglomerate diameter of bare nZVI, the decreased surface area for lower and intermediate sulfide doses seems to be a result of the reduced iron corrosion and/or decreased surface roughness. In case of the S-nZVI sulfidated with the highest sulfide dose, an increase in the particle agglomerate size was observed (by both the disc centrifuge and TEM). For this material, the agglomeration is likely a major factor influencing the decrease in surface area.
Figure 2.

(A) TEM image of a S-nZVI particle with a S/Fe mass ratio 0.0195. (B) HRTEM (high-resolution transmission electron microscopy) detail on the sulfide shell structure of a S-nZVI particle with a S/Fe mass ratio of 0.0094. The lattice spacing in areas 1 and 2 was calculated by fast Fourier transformation. (C–H) Overlay of Fe-S and Fe-S-O high-resolution EDS mapping of nanoscale zero-valent iron particles sulfidated with the following S/Fe ratios: 0.0094 (C and F), 0.0195 (D and G), and 0.0629 (E and H). Arrows indicate the average thickness of the particle FeS shell.
The Fe/FeS core–shell structure, typical for S-nZVI prepared using the two-step synthesis approach at lower sulfur doses and/or short sulfidation duration,24 was clearly visible on high-resolution transmission electron microscopy (HRTEM) (Figure 2). The shell consisted mostly of amorphous FeS sheets, which contained several semiordered atomic layers (Figure 2B). The d-spacing between these layers was around 5.3–5.4 Å, slightly larger compared with the d (001) of crystalline mackinawite (5.03 Å). The characteristics of the sulfide shell agree with previous studies that identified amorphous iron sulfide sheets with a mackinawite-like structure as the primary FeS precipitates.27,49,55,56
An EDS analysis of bare nZVI particles showed major bands only for iron and oxygen (Figure S5A), while S-nZVI particles contained also sulfur and sodium, whose abundance increased at higher sulfide doses (Figures S5B and C). The elemental distribution in the synthesized material was further investigated using single-particle high-resolution EDS elemental mapping. This confirmed the presence of a 5–10 nm thick sulfidic shell (Figure 2 C–H). Elemental mapping also indicated that the sulfidic shell was associated with an oxygen-rich layer. Sodium was detected as well in the shell region in particles with a S/Fe mass ratio 0.0195 or higher. More HRTEM images are provided in the SI (Figures S6–S8).
Additional data on the abundance and speciation of sulfur were obtained by XPS. The survey scans quantification showed that both sulfur and sodium abundance correlated to the applied sulfide dose (Table S3). The S 2p narrow spectra were fitted with S 2p3/2 and S 2p1/2 spin–orbit doublets separated by 1.18 eV with an intensity ratio of 2:1. Sulfur in the surface layer is predominantly present as sulfide (S2–), with a lower contribution of disulfide (S22–). (Figure 1C). The sulfur speciation was consistent in all samples of varying sulfide doses, with only slight differences in the relative abundance of S2– and S22– species. These findings are in agreement with the proposed structure of sulfidated nZVI having poorly ordered FeS as the dominant phases formed upon sulfidation.24
To get a deeper insight into the chemical nature of iron species upon sulfidation, the low-temperature 57Fe Mössbauer spectroscopy on S-nZVI with a S/Fe ratio of 0.0195 was employed (Figure 1D). The Mössbauer spectrum was deconvoluted into three sextet spectral components and one singlet component, with the derived values of the hyperfine Mössbauer parameters listed in Table S4. A dominant sextet spectral component represented zero-valent iron, which is present in the core of the synthesized nanoparticles. On the basis of the hyperfine Mössbauer parameters, the other two sextet components represented magnetite, a product of zero-valeniron oxidation43 and remaining nonreduced phase. The last singlet component in the spectrum reflected the presence of low-spin Fe(II) occupying the tetrahedral sites in a FeS system, which can be described as mackinawite-like.51,57,58
The formation of a FeS phase upon sulfidation (at a S/Fe mass ratio of 0.0195) was further confirmed by geochemical modeling. Before the nZVI addition, the Na2S solution pH was 12.99 and Eh was −430 mV. After the addition of nZVI, the pH changed only slightly to 12.9 and then decreased to 12.6 after 26 h (Figure S9). Eh dropped to −830 mV and then slightly increased to −820 mV at the end of the experiment. Respective calculated saturation indices for amorphous FeS (ppt), mackinawite (FeS), and pyrite (FeS2) changed from 4.18, 4.91, and 2.27 at the beginning of sulfidation to 3.69, 4.41, and 1.42 at the end (Table S5). The Eh–pH diagrams for the Fe-S-H2O system are shown in Figure 3.
Figure 3.

Eh–pH diagrams for the Fe-S-H2O system at 25 °C with amorphous (A) FeS and (B) mackinawite phases shown. The total Fe is 6 × 10–5 mol L–1, and the total S is 2 × 10–1 mol L–1. Blue and yellow fields represent dissolved species and mineral phases, respectively. Mackinawite is supressed in (A) and FeS (ppt) is supressed in (B). Hematite and pyrite are supressed in both diagrams. Dotted lines and numbers indicate speciation fields of S: 1 HSO4–, 2 SO42–, 3 rhombic S0, 4 H2S (aq), and 5 HS–. The position of the measured data is indicated by a rectangle at the bottom right.
Well-crystalline hematite and pyrite were supressed in the diagram, but magnetite, with mixed Fe valence, was retained because its formation has been reported in the literature.59 Sulfur speciation identified dissolved HS– ions as the dominant sulfur species, suggesting negligible H2S volatilization. The measured data are in the field of solid amorphous FeS (ppt) and crystalline mackinawite, suggesting favorable conditions for the formation of 1:1 ferrous sulfide with the composition of mackinawite. While the formation of fresh crystalline mackinawite is favored over amorphous FeS (ppt) thermodynamically (i.e., mackinawite has a higher saturation index), available laboratory data from FeS precipitation experiments and field data from a reducing lake and estuarine and marine sediments indicate that the formation of crystalline mackinawite is inhibited, and it is formed only later by the recrystallization of amorphous and poorly crystalline FeS phases.49,55,56,60,61 In this study, crystalline mackinawite has been observed in the XRD patterns of S-nZVI particles with high sulfur doses after the reactivity experiments (see section 3.4), corroborating the gradual crystallization of iron sulfides in time.
3.2. S-nZVI Reactivity and Selectivity toward TCE Removal
All S-nZVI particles degraded TCE much faster compared with the untreated nZVI (Figure 4A). Nearly 100% TCE dechlorination was achieved for all sulfur-treated materials within the 3-week experiment, while the untreated nZVI degraded only about 60% of TCE during the same period. As the S/Fe mass ratio increased from 0 to 0.0195 (mole S/Fe ratio of 0.034), the TCE removal rate increased (Table 1). The S/Fe mass ratios greater than 0.0195 resulted in decreased TCE removal rates. The existence of an optimal S/Fe ratio for TCE removal was previously observed.22,49 The kinetics of TCE reduction was well described by a first-order rate law.20 The observed pseudo-first-order rate constant (kobs) of the best performing S-nZVI was approximately 12-fold higher than for the untreated nZVI. Similarly, an 8- to 45-fold increase in the TCE removal rate has been reported for various nZVI systems after sulfidation.20,22,23,26,32,62 The surface-area-normalized reaction constants (kSA) for TCE degradation were positively associated with the S/Fe ratio (Table 1), while the surface area decreased with an increasing sulfide dose. Thus, the optimal S/Fe ratio represents the best compromise between sulfur coverage and surface area. The FeS shell thickness correlated with kSA values (Figure S10), but as the sulfidation negatively impacted the particle surface area, the kobs reached an optimal value at a shell thickness of about 7.3 nm.
Figure 4.

TCE dechlorination by pristine nZVI and sulfide-treated nZVI with varying S/Fe ratios: (A) TCE removal, (B) hydrogen production, (C) distribution of primary products after 1 day of reaction, and (D) schematic of the proposed mechanism of TCE dechlorination and hydrogen evolution. The reactions were carried out at an initial TCE concentration of 20 mg L–1 and a particle dose of 1 g L–1 with respect to the iron content without pH adjustment. Whiskers indicate the standard deviation (SD).
Sulfidation had a considerably different effect on the hydrogen production at various S/Fe ratios. At the lowest sulfur dosing (S/Fe mass ratio of 0.0022), hydrogen evolved faster compared with pristine nZVI (Figure 4B). Nonetheless, at a higher sulfur dosing, the hydrogen evolution was suppressed, as evidenced by earlier findings.18,20,22,23 This is likely a result of the complete particle surface coverage with a protective FeS film. Several mechanisms of hydrogen evolution suppression by a FeS shell have been described: (i) FeS is more hydrophobic than iron (oxyhydr)oxides and thus interacts less with water;62 (ii) a FeS shell increases the particle open circuit potential, which slows down the reaction with H+;20 and, concurrently, (iii) the particle coverage with FeS blocks the sites for hydrogen adsorption.63,64 Interestingly, the hydrogen production rate exhibited a minimum at intermediate sulfur loadings (S/Fe mass ratios of 0.0094–0.0195), while at S/Fe mass ratios of 0.0319 and 0.0629 more hydrogen was produced. This could be due to the disintegration of a thick FeS layer and the formation of secondary precipitates.24 This corroborates previous findings that sulfur dosing and sulfidation time influence the iron core corrosion rate.20,24
Electron efficiencies were calculated based on the detected levels of C2 hydrocarbons (ethane, ethane, and acetylene) that constitute the main TCE dechlorination products.18,20,23 Higher electron efficiencies were observed for S-nZVI particles in the course of the TCE dechlorination reaction compared with pristine nZVI (Table S6). While bare nZVI exhibited a 3.4% electron efficiency within 3 days of the reaction, the efficiency of S-nZVI particles with S/Fe ratios of 0.0094 and 0.0195 reached 26.4 and 22.9%, respectively, i.e., a 7-fold increase in selectivity. A substantial increase in the nZVI electron selectivity toward TCE removal after sulfidation was observed previously, especially at high TCE/Fe ratios.18,20,32,62 Electron efficiencies varied during the experiment due to (i) a gradual cease of hydrogen evolution by S-nZVI particles after several days of reaction18 and (ii) the exhaustion of TCE present in the reaction mixture. The highest difference in electron efficiencies throughout the experiment was observed for S-nZVI particles with the lowest sulfur dosing due to increased hydrogen production after 3 days of reaction.
A small residual amount of aqueous sulfide was present during the TCE degradation experiments discussed above, which originates from the incomplete sulfur incorporation into the shell of ZVI nanoparticles during sulfidation. In order to provide a conservative estimate of the potential TCE reduction by the residual dissolved sulfide, nominal concentrations of Na2S added to the best performing S-nZVI particles were evaluated for TCE removal (Figure S11). No decrease in the TCE concentration was detected within 3 weeks, confirming that dissolved sulfide does not degrade TCE.22
3.3. Distribution of TCE Dechlorination Products and Reaction Mechanism
Ethene, ethane, and acetylene were the main products of TCE dechlorination (Figure S12), while no less-chlorinated intermediates (i.e., dichloroethene isomers and vinyl chloride) were detected, which agrees with previous reports on TCE dechlorination using S-nZVI systems.18,20,23,32 This product pattern is consistent with the reductive β-elimination pathway.18,20,23,65,66 Elevated amounts of acetylene were produced by S-nZVI (Figure 4C). Acetylene was further hydrogenated to ethane and ethene over the course of the experiments (Figures S12C and S13). This change in the product distribution results from slower acetylene hydrogenation on the FeS surface compared with bare ZVI and/or its slower generation by bare ZVI.20,23,32 The observed carbon balance for all materials was within the range of 52.6–71.6% (Table S7). The relatively poor carbon balance stems from the formation of heavier hydrocarbons as a result of acetylene coupling reactions on ZVI or iron sulfide-based materials.23,65,67,68
Due to the incomplete carbon balance, the chlorine balance was calculated as an independent measure to check for possible TCE losses (Figure S14). The amount of total chlorine present in the reactors either incorporated in the TCE molecules or in a dissolved ionic form (Cl–) at the end of the experiment was remarkably close to the initial amount injected as TCE for all the tested materials. At the end of all S-nZVI experiments, chlorine was almost entirely present as Cl– ions (>95%), while TCE was almost completely degraded. The residual presence of low levels of TCE at the end of the experiment is due to TCE partitioning to the headspace where degradation does not occur. In the samples of bare nZVI, 58% of the chlorine was converted to Cl–, while 36% of the chlorine was still present as residual TCE. The almost 100% chlorine balance further confirms that no significant amounts of chlorinated transformation products were formed during the reaction of TCE with the tested materials, and losses due to leakage and sorption to particles were negligible.
The mechanisms driving the increased reactivity and selectivity of TCE removal using S-nZVI materials have been investigated in several studies. A schematic of the proposed mechanism is shown in Figure 4D. Iron sulfide has a lower resistance for electron transfer compared with iron (oxyhydr)oxides that occur on the nZVI surface,18,20,22,62 which promotes electron-mediated reactions such as β-elimination. Moreover, a FeS shell is more hydrophobic,62 thus it favors the sorption of organic contaminants and, at the same time, hinders interaction with hydrogen. The reaction with hydrogen is further reduced by blocking H adsorption sites with sulfur.63,64 These features of the FeS layer result in substantially higher electron efficiencies.18,20,32 The increased surface availability of electrons to the detriment of hydrogen atoms triggers elevated acetylene formation.20,62
3.4. Characteristics of nZVI Particles after Reactivity Experiments with TCE
The different effect of sulfidation on iron corrosion at various S/Fe ratios was detected through the XRD patterns of reacted particles. While the bare nZVI particles contained a crystalline fraction of α-Fe0 as low as 29 wt % after the 3-week experiment, the particles with S/Fe mass ratios of 0.0094–0.0629 contained much higher crystalline fractions of α-Fe0 (>71 wt %) (Figure 5A). Conversely, particles with the lowest sulfur dosing (S/Fe mass ratios of 0.0022) contained a lower crystalline fraction of α-Fe0 (16 wt %). These findings agree with the hydrogen evolution rates (see section 3.2). The higher stability of S-nZVI particles during aging was also observed in our previous study.48 The iron corrosion products detected using XRD were typically amakinite (Fe(OH)2), magnetite, and fougèrite ([Fe2+4Fe3+2(OH)12][CO3]·3H2O), a green rust mineral. In the two S-nZVI particle types with the highest sulfur doses, a broad 2θ peak at 19° corresponding to crystalline mackinawite was detected. Freshly prepared amorphous iron sulfide gradually converts to more crystalline phases,27,56 which explains why this phase was not discerned in the XRD pattern of freshly prepared particles.
Figure 5.

Characteristics of nZVI particles after the TCE degradation experiment: (A) XRD patterns for bare nZVI and S-nZVI particles of varying S/Fe mass ratios and (B) XPS S 2p narrow region spectrum of S-nZVI particles with a S/Fe ratio of 0.0195. Peak assignments were based on values summarized by Descostes et al. and Mullet et al.50,51
The slow oxidation of S-nZVI with S/Fe mass ratios of ≥ 0.0094 is corroborated by TEM images of particles after reactivity experiments. Distinct sheets of iron (oxyhydr)oxides, which result from iron oxidation, are apparent in the TEM micrographs of bare nZVI after the reaction (Figures S1D), while their prevalence for S-nZVI particles is lower (Figures S1E and F, respectively). Altogether, S-nZVI particles with S/Fe mass ratios in the range of 0.0094–0.0629 showed substantially higher longevity compared with bare nZVI.
The S speciation in the FeS shell was practically unaffected by aging. The XPS S 2p spectra show only a slight shift toward more oxidized forms between freshly synthesized and reacted particles (i.e., a decrease in the S2– and an increase in the S0 content) (Figure 5B). This agrees with a recent study where the FeS shell remained intact even after 180 days of particle aging in artificial groundwater,49 while partial disruption of FeS shell after 60 days of aging was observed in our previous study.48 Apparently, FeS shell is resistant to chemical oxidation but is prone to mechanical damage.22
4. Conclusions
This study investigates a new synthesis approach to produce sulfidated nZVI particles with controlled reactivity and selectivity toward TCE removal. Commercially available nZVI prepared by the thermal reduction of iron (oxyhydr)oxide precursors in a H2 atmosphere treated in a concentrated slurry with Na2S solutions resulted in S-nZVI particles with a core–shell character encompassing a metallic iron core and a laminar shell layer of a poorly ordered FeS phase with a mackinawite-like structure. The thickness of the FeS shell was not perfectly uniform but increased on average with increasing sulfur loadings. The average FeS shell thickness correlated with surface-area-normalized TCE removal constants. However, due to the negative effect of sulfidation on the surface area, the optimal average thickness of the FeS shell characterized by the highest TCE removal rate per mass of particles was found to be about 7.3 nm. An increased TCE removal rate by a factor of up to a 12 and a 7-fold increase in the electron efficiency compared with bare nZVI particles were observed at this FeS shell thickness, corresponding to the S/Fe mass ratio of 0.0195 (mole ratio 0.034). The XRD patterns and TEM images of fresh and reacted particles showed an increase in the stability of S-nZVI toward corrosion in water, leading to an increased selectivity of TCE removal, except for very low sulfur loadings. S-nZVI was found to degrade TCE by the β-elimination pathway, with ethene, ethane, and acetylene being the dominant reaction products. The less-chlorinated intermediates (i.e., dichloroethene isomers and vinyl chloride) were not detected. The proposed S-nZVI synthesis is more cost-efficient at a large-scale compared with the conventional synthesis using borohydride as the reducing agent. While the mere production costs of nZVIBH are estimated to be over $200 kg–1, the market price of nZVIH2 is only about $130 kg–1. Moreover, concentrated slurries (20% w/w) suitable for off-site production and without hazardous boron species can be prepared. S-nZVI particles synthesized using the proposed approach thus represent a very promising remedy for TCE groundwater pollution.
Acknowledgments
This project was supported by the bilateral Czech Science Foundation (GACR)/Austrian Science Fund (FWF) project ModelFace (Czech Grant No. 17-33779L, Austrian Grant No. I 3065-N34); by the Operational Program Research, Development and Education, European Regional Development Fund, Project No. CZ.02.1.01/0.0/0.0/16_019/0000754 of the Ministry of Education, Youth and Sports of the Czech Republic; and by the Technology Agency of the Czech Republic, Competence Centers (Project TE01020218). We thank Josef Kašlík, Martin Petr, Ondřej Tomanec, Josef Kopp, and Jiří Mikšíček for performing material characterization analyses and Ivo Medřík for technical assistance. Special thanks are given to Vesna Micic Batka and Wolfgang Obermaier for their valuable assistance in GC method development.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsami.0c08626.
Additional details on chemicals and materials, sample preparation, and analytical methods; concentrations of dissolved sulfide before and after sulfidation; DLS hydrodynamic diameter and zeta-potential; TEM images of samples; particle size and shell thickness distributions; pore surface area and pore size distributions; EDS analysis and STEM-XEDS elemental mappings; XPS surface element quantifications; Mössbauer hyperfine parameters; pH and Eh evolution; FeSx saturation indices; relationship between TCE removal kinetic constants and average S-nZVI particle FeS shell thickness; electron efficiencies; TCE evolution in control samples; evolution of TCE major dechlorination products and their distribution; and carbon and chlorine balance (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Stefaniuk M.; Oleszczuk P.; Ok Y. S. Review on Nano Zerovalent Iron (NZVI): From Synthesis to Environmental Applications. Chem. Eng. J. 2016, 287, 618–632. 10.1016/j.cej.2015.11.046. [DOI] [Google Scholar]
- Schöftner P.; Waldner G.; Lottermoser W.; Stöger-Pollach M.; Freitag P.; Reichenauer T. G. Electron Efficiency of NZVI Does Not Change with Variation of Environmental Parameters. Sci. Total Environ. 2015, 535, 69–78. 10.1016/j.scitotenv.2015.05.033. [DOI] [PubMed] [Google Scholar]
- Reinsch B. C.; Forsberg B.; Penn R. L.; Kim C. S.; Lowry G. V. Chemical Transformations during Aging of Zerovalent Iron Nanoparticles in the Presence of Common Groundwater Dissolved Constituents. Environ. Sci. Technol. 2010, 44 (9), 3455–3461. 10.1021/es902924h. [DOI] [PubMed] [Google Scholar]
- Liu H.; Wang Q.; Wang C.; Li X. Electron Efficiency of Zero-Valent Iron for Groundwater Remediation and Wastewater Treatment. Chem. Eng. J. 2013, 215–216, 90–95. 10.1016/j.cej.2012.11.010. [DOI] [Google Scholar]
- Liu Y.; Lowry G. V. Effect of Particle Age (Fe 0 Content) and Solution PH On NZVI Reactivity: H 2 Evolution and TCE Dechlorination. Environ. Sci. Technol. 2006, 40 (19), 6085–6090. 10.1021/es060685o. [DOI] [PubMed] [Google Scholar]
- Mackenzie K.; Bleyl S.; Georgi A.; Kopinke F.-D. Carbo-Iron – An Fe/AC Composite – As Alternative to Nano-Iron for Groundwater Treatment. Water Res. 2012, 46 (12), 3817–3826. 10.1016/j.watres.2012.04.013. [DOI] [PubMed] [Google Scholar]
- Petala E.; Dimos K.; Douvalis A.; Bakas T.; Tucek J.; Zbořil R.; Karakassides M. A. Nanoscale Zero-Valent Iron Supported on Mesoporous Silica: Characterization and Reactivity for Cr(VI) Removal from Aqueous Solution. J. Hazard. Mater. 2013, 261, 295–306. 10.1016/j.jhazmat.2013.07.046. [DOI] [PubMed] [Google Scholar]
- Zhan J.; Zheng T.; Piringer G.; Day C.; McPherson G. L.; Lu Y.; Papadopoulos K.; John V. T. Transport Characteristics of Nanoscale Functional Zerovalent Iron/Silica Composites for in Situ Remediation of Trichloroethylene. Environ. Sci. Technol. 2008, 42 (23), 8871–8876. 10.1021/es800387p. [DOI] [PubMed] [Google Scholar]
- Kim S. A.; Kamala-Kannan S.; Lee K.-J.; Park Y.-J.; Shea P. J.; Lee W.-H.; Kim H.-M.; Oh B.-T. Removal of Pb(II) from Aqueous Solution by a Zeolite–nanoscale Zero-Valent Iron Composite. Chem. Eng. J. 2013, 217, 54–60. 10.1016/j.cej.2012.11.097. [DOI] [Google Scholar]
- Zhang M.; He F.; Zhao D.; Hao X. Transport of Stabilized Iron Nanoparticles in Porous Media: Effects of Surface and Solution Chemistry and Role of Adsorption. J. Hazard. Mater. 2017, 322, 284–291. 10.1016/j.jhazmat.2015.12.071. [DOI] [PubMed] [Google Scholar]
- He F.; Zhao D.; Paul C. Field Assessment of Carboxymethyl Cellulose Stabilized Iron Nanoparticles for in Situ Destruction of Chlorinated Solvents in Source Zones. Water Res. 2010, 44 (7), 2360–2370. 10.1016/j.watres.2009.12.041. [DOI] [PubMed] [Google Scholar]
- He F.; Zhao D. Manipulating the Size and Dispersibility of Zerovalent Iron Nanoparticles by Use of Carboxymethyl Cellulose Stabilizers. Environ. Sci. Technol. 2007, 41 (17), 6216–6221. 10.1021/es0705543. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Wang C.-B.; Lien H.-L. Treatment of Chlorinated Organic Contaminants with Nanoscale Bimetallic Particles. Catal. Today 1998, 40 (4), 387–395. 10.1016/S0920-5861(98)00067-4. [DOI] [Google Scholar]
- Elliott D. W.; Zhang W. Field Assessment of Nanoscale Bimetallic Particles for Groundwater Treatment. Environ. Sci. Technol. 2001, 35 (24), 4922–4926. 10.1021/es0108584. [DOI] [PubMed] [Google Scholar]
- Marková Z.; Šišková K. M.; Filip J.; Čuda J.; Kolář M.; Šafářová K.; Medřík I.; Zbořil R. Air Stable Magnetic Bimetallic Fe–Ag Nanoparticles for Advanced Antimicrobial Treatment and Phosphorus Removal. Environ. Sci. Technol. 2013, 47 (10), 5285–5293. 10.1021/es304693g. [DOI] [PubMed] [Google Scholar]
- Yan W.; Herzing A. A.; Li X.; Kiely C. J.; Zhang W. Structural Evolution of Pd-Doped Nanoscale Zero-Valent Iron (NZVI) in Aqueous Media and Implications for Particle Aging and Reactivity. Environ. Sci. Technol. 2010, 44 (11), 4288–4294. 10.1021/es100051q. [DOI] [PubMed] [Google Scholar]
- Liu W.-J.; Qian T.-T.; Jiang H. Bimetallic Fe Nanoparticles: Recent Advances in Synthesis and Application in Catalytic Elimination of Environmental Pollutants. Chem. Eng. J. 2014, 236, 448–463. 10.1016/j.cej.2013.10.062. [DOI] [Google Scholar]
- He F.; Li Z.; Shi S.; Xu W.; Sheng H.; Gu Y.; Jiang Y.; Xi B. Dechlorination of Excess Trichloroethene by Bimetallic and Sulfidated Nanoscale Zero-Valent Iron. Environ. Sci. Technol. 2018, 52 (15), 8627–8637. 10.1021/acs.est.8b01735. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee S.; Ghoshal S. Optimal Design of Sulfidated Nanoscale Zerovalent Iron for Enhanced Trichloroethene Degradation. Environ. Sci. Technol. 2018, 52 (19), 11078–11086. 10.1021/acs.est.8b02399. [DOI] [PubMed] [Google Scholar]
- Xu J.; Cao Z.; Zhou H.; Lou Z.; Wang Y.; Xu X.; Lowry G. V. Sulfur Dose and Sulfidation Time Affect Reactivity and Selectivity of Post-Sulfidized Nanoscale Zerovalent Iron. Environ. Sci. Technol. 2019, 53 (22), 13344–13352. 10.1021/acs.est.9b04210. [DOI] [PubMed] [Google Scholar]
- Fan D.; O’Brien Johnson G.; Tratnyek P. G.; Johnson R. L. Sulfidation of Nano Zerovalent Iron (NZVI) for Improved Selectivity During In-Situ Chemical Reduction (ISCR). Environ. Sci. Technol. 2016, 50 (17), 9558–9565. 10.1021/acs.est.6b02170. [DOI] [PubMed] [Google Scholar]
- Rajajayavel S. R. C.; Ghoshal S. Enhanced Reductive Dechlorination of Trichloroethylene by Sulfidated Nanoscale Zerovalent Iron. Water Res. 2015, 78, 144–153. 10.1016/j.watres.2015.04.009. [DOI] [PubMed] [Google Scholar]
- Han Y.; Yan W. Reductive Dechlorination of Trichloroethene by Zero-Valent Iron Nanoparticles: Reactivity Enhancement through Sulfidation Treatment. Environ. Sci. Technol. 2016, 50 (23), 12992–13001. 10.1021/acs.est.6b03997. [DOI] [PubMed] [Google Scholar]
- Fan D.; Lan Y.; Tratnyek P. G.; Johnson R. L.; Filip J.; O’Carroll D. M.; Nunez Garcia A.; Agrawal A. Sulfidation of Iron-Based Materials: A Review of Processes and Implications for Water Treatment and Remediation. Environ. Sci. Technol. 2017, 51, 13070. 10.1021/acs.est.7b04177. [DOI] [PubMed] [Google Scholar]
- Li J.; Zhang X.; Sun Y.; Liang L.; Pan B.; Zhang W.; Guan X. Advances in Sulfidation of Zerovalent Iron for Water Decontamination. Environ. Sci. Technol. 2017, 51 (23), 13533–13544. 10.1021/acs.est.7b02695. [DOI] [PubMed] [Google Scholar]
- Kim E.-J.; Kim J.-H.; Azad A.-M.; Chang Y.-S. Facile Synthesis and Characterization of Fe/FeS Nanoparticles for Environmental Applications. ACS Appl. Mater. Interfaces 2011, 3 (5), 1457–1462. 10.1021/am200016v. [DOI] [PubMed] [Google Scholar]
- Mangayayam M.; Dideriksen K.; Ceccato M.; Tobler D. J. The Structure of Sulfidized Zero-Valent Iron by One-Pot Synthesis: Impact on Contaminant Selectivity and Long-Term Performance. Environ. Sci. Technol. 2019, 53 (8), 4389–4396. 10.1021/acs.est.8b06480. [DOI] [PubMed] [Google Scholar]
- Su Y.; Jassby D.; Song S.; Zhou X.; Zhao H.; Filip J.; Petala E.; Zhang Y. Enhanced Oxidative and Adsorptive Removal of Diclofenac in Heterogeneous Fenton-like Reaction with Sulfide Modified Nanoscale Zerovalent Iron. Environ. Sci. Technol. 2018, 52 (11), 6466–6475. 10.1021/acs.est.8b00231. [DOI] [PubMed] [Google Scholar]
- Fan D.; Anitori R. P.; Tebo B. M.; Tratnyek P. G.; Lezama Pacheco J. S.; Kukkadapu R. K.; Engelhard M. H.; Bowden M. E.; Kovarik L.; Arey B. W. Reductive Sequestration of Pertechnetate (99 TcO 4 – ) by Nano Zerovalent Iron (NZVI) Transformed by Abiotic Sulfide. Environ. Sci. Technol. 2013, 47 (10), 5302–5310. 10.1021/es304829z. [DOI] [PubMed] [Google Scholar]
- Du J.; Bao J.; Lu C.; Werner D. Reductive Sequestration of Chromate by Hierarchical FeS@Fe 0 Particles. Water Res. 2016, 102, 73–81. 10.1016/j.watres.2016.06.009. [DOI] [PubMed] [Google Scholar]
- Filip J.; Slunský J.; Nosek J.; Semerád J.; Kašlík J.; Oborná J.; Bachořík J.; Medřík I.. Sulfidized NZVI Particles for Groundwater Treatment: Synthesis, Complex Characterization and Laboratory-Scale Testing. In ACS National Meeting, San Francisco, CA, April 3–7, 2017; Abstract No. 307; American Chemical Society, 2017.
- Gu Y.; Wang B.; He F.; Bradley M. J.; Tratnyek P. G. Mechanochemically Sulfidated Microscale Zero Valent Iron: Pathways, Kinetics, Mechanism, and Efficiency of Trichloroethylene Dechlorination. Environ. Sci. Technol. 2017, 51 (21), 12653–12662. 10.1021/acs.est.7b03604. [DOI] [PubMed] [Google Scholar]
- Tang J.; Tang L.; Feng H.; Zeng G.; Dong H.; Zhang C.; Huang B.; Deng Y.; Wang J.; Zhou Y. PH-Dependent Degradation of p -Nitrophenol by Sulfidated Nanoscale Zerovalent Iron under Aerobic or Anoxic Conditions. J. Hazard. Mater. 2016, 320, 581–590. 10.1016/j.jhazmat.2016.07.042. [DOI] [PubMed] [Google Scholar]
- Li S.; Yan W.; Zhang W. Solvent-Free Production of Nanoscale Zero-Valent Iron (NZVI) with Precision Milling. Green Chem. 2009, 11 (10), 1618. 10.1039/b913056j. [DOI] [Google Scholar]
- Yan W.; Lien H.-L.; Koel B. E.; Zhang W. Iron Nanoparticles for Environmental Clean-up: Recent Developments and Future Outlook. Environ. Sci. Process. Impacts 2013, 15 (1), 63–77. 10.1039/C2EM30691C. [DOI] [PubMed] [Google Scholar]
- El-Temsah Y. S.; Oughton D. H.; Joner E. J. Effects of Nano-Sized Zero-Valent Iron on DDT Degradation and Residual Toxicity in Soil: A Column Experiment. Plant Soil 2013, 368 (1–2), 189–200. 10.1007/s11104-012-1509-8. [DOI] [Google Scholar]
- Cernik M.Nanoremediation in the EU – Impacts of NanoRem and Technology Combinations. In Practical Applications for Nanoremediation – Session 1. Nanorem, 2016. http://www.nanorem.eu/stream.aspx?p=/App_Data/docs/user14Gallery/1_Toolbox/8_Final_Conf_part2/NanoRem_TB_RemTech2016_Cernik_practical_applications_nanoremediation.pdf. [Google Scholar]
- Chmielewská E.Environmental Zeolites and Aqueous Media: Examples of Practical Solutions; Bentham Science Publishers, 2018. [Google Scholar]
- Kašlík J.; Kolařík J.; Filip J.; Medřík I.; Tomanec O.; Petr M.; Malina O.; Zbořil R.; Tratnyek P. G. Nanoarchitecture of Advanced Core-Shell Zero-Valent Iron Particles with Controlled Reactivity for Contaminant Removal. Chem. Eng. J. 2018, 354, 335–345. 10.1016/j.cej.2018.08.015. [DOI] [Google Scholar]
- American Public Health Association; A.W.W. Association; W.E. Federation. Standard Methods for the Examination of Water and Wastewater, 22nd ed.; American Public Health Association, American Water Works Association, Water Environment Federation: Washington, D.C., 2012. [Google Scholar]
- Pechousek J.; Jancik D.; Frydrych J.; Navarik J. N.; Novak P.. Setup of Mossbauer Spectrometers at RCPTM. In Mossbauer Spectroscopy in Materials Science – 2012; American Institute of Physics: Melville, NY, 2013; pp 186–194. [Google Scholar]
- Pechousek J.; Prochazka R.; Jancik D.; Frydrych J.; Mashlan M.. Universal LabVIEW-Powered Mössbauer Spectrometer Based on USB, PCI or PXI DevicesI. In International Conference on the Applications of the Mössbauer Effect (ICAME 2009); Muller H., Reissner M., Steiner W., Wiesinger G., Eds.; IOP Publishing Ltd: Bristol, UK, 2010. [Google Scholar]
- Klencsár Z.; Kuzmann E.; Vértes A. User-Friendly Software for Mössbauer Spectrum Analysis. J. Radioanal. Nucl. Chem. 1996, 210 (1), 105–118. 10.1007/BF02055410. [DOI] [Google Scholar]
- Sander R. Compilation of Henry’s Law Constants (Version 4.0) for Water as Solvent. Atmos. Chem. Phys. 2015, 15 (8), 4399–4981. 10.5194/acp-15-4399-2015. [DOI] [Google Scholar]
- Tratnyek P. G.; Scherer M. M.; Deng B.; Hu S. Effects of Natural Organic Matter, Anthropogenic Surfactants, and Model Quinones on the Reduction of Contaminants by Zero-Valent Iron. Water Res. 2001, 35 (18), 4435–4443. 10.1016/S0043-1354(01)00165-8. [DOI] [PubMed] [Google Scholar]
- Fan D.; O’Carroll D. M.; Elliott D. W.; Xiong Z.; Tratnyek P. G.; Johnson R. L.; Garcia A. N. Selectivity of Nano Zerovalent Iron in In Situ Chemical Reduction: Challenges and Improvements. Remediat. J. 2016, 26 (4), 27–40. 10.1002/rem.21481. [DOI] [Google Scholar]
- Kim E.-J.; Murugesan K.; Kim J.-H.; Tratnyek P. G.; Chang Y.-S. Remediation of Trichloroethylene by FeS-Coated Iron Nanoparticles in Simulated and Real Groundwater: Effects of Water Chemistry. Ind. Eng. Chem. Res. 2013, 52 (27), 9343–9350. 10.1021/ie400165a. [DOI] [Google Scholar]
- Semerád J.; Filip J.; Ševců A.; Brumovský M.; Nguyen N. H. A.; Mikšíček J.; Lederer T.; Filipová A.; Boháčková J.; Cajthaml T. Environmental Fate of Sulfidated NZVI Particles: The Interplay of Nanoparticle Corrosion and Toxicity during Aging. Environ. Sci.: Nano 2020, 7, 1794. 10.1039/D0EN00075B. [DOI] [Google Scholar]
- Mangayayam M. C.; Perez J. P. H.; Dideriksen K.; Freeman H. M.; Bovet N.; Benning L. G.; Tobler D. J. Structural Transformation of Sulfidized Zerovalent Iron and Its Impact on Long-Term Reactivity. Environ. Sci.: Nano 2019, 6 (11), 3422–3430. 10.1039/C9EN00876D. [DOI] [Google Scholar]
- Descostes M.; Mercier F.; Thromat N.; Beaucaire C.; Gautier-Soyer M. Use of XPS in the Determination of Chemical Environment and Oxidation State of Iron and Sulfur Samples: Constitution of a Data Basis in Binding Energies for Fe and S Reference Compounds and Applications to the Evidence of Surface Species of an Oxidized Py. Appl. Surf. Sci. 2000, 165 (4), 288–302. 10.1016/S0169-4332(00)00443-8. [DOI] [Google Scholar]
- Mullet M.; Boursiquot S.; Abdelmoula M.; Génin J.-M.; Ehrhardt J.-J. Surface Chemistry and Structural Properties of Mackinawite Prepared by Reaction of Sulfide Ions with Metallic Iron. Geochim. Cosmochim. Acta 2002, 66 (5), 829–836. 10.1016/S0016-7037(01)00805-5. [DOI] [Google Scholar]
- Klimkova S.; Cernik M.; Lacinova L.; Filip J.; Jancik D.; Zboril R. Zero-Valent Iron Nanoparticles in Treatment of Acid Mine Water from in Situ Uranium Leaching. Chemosphere 2011, 82 (8), 1178–1184. 10.1016/j.chemosphere.2010.11.075. [DOI] [PubMed] [Google Scholar]
- Filip J.; Karlický F.; Marušák Z.; Lazar P.; Černík M.; Otyepka M.; Zbořil R. Anaerobic Reaction of Nanoscale Zerovalent Iron with Water: Mechanism and Kinetics. J. Phys. Chem. C 2014, 118 (25), 13817–13825. 10.1021/jp501846f. [DOI] [Google Scholar]
- Hansson E. B.; Odziemkowski M. S.; Gillham R. W. Formation of Poorly Crystalline Iron Monosulfides: Surface Redox Reactions on High Purity Iron, Spectroelectrochemical Studies. Corros. Sci. 2006, 48 (11), 3767–3783. 10.1016/j.corsci.2006.03.010. [DOI] [Google Scholar]
- Rickard D.; Luther G. W. Chemistry of Iron Sulfides. Chem. Rev. 2007, 107 (2), 514–562. 10.1021/cr0503658. [DOI] [PubMed] [Google Scholar]
- Csákberényi-Malasics D.; Rodriguez-Blanco J. D.; Kis V. K.; Rečnik A.; Benning L. G.; Pósfai M. Structural Properties and Transformations of Precipitated FeS. Chem. Geol. 2012, 294–295, 249–258. 10.1016/j.chemgeo.2011.12.009. [DOI] [Google Scholar]
- Matsuo M.; Kawakami M.; Sugimori K. Mössbauer Spectroscopic Study on Chemical Changes of Iron Compounds with the Aid of Sulfate-Reducing Bacteria. Hyperfine Interact. 2000, 126 (1/4), 53–58. 10.1023/A:1012680325493. [DOI] [Google Scholar]
- Waanders F. B.; Silva L. F. O.; Saikia B. K. The Use of Mössbauer Spectroscopy in Environmental Research. Hyperfine Interact. 2017, 238 (1), 52. 10.1007/s10751-017-1423-9. [DOI] [Google Scholar]
- Baumgartner J.; Dey A.; Bomans P. H. H.; Le Coadou C.; Fratzl P.; Sommerdijk N. A. J. M.; Faivre D. Nucleation and Growth of Magnetite from Solution. Nat. Mater. 2013, 12 (4), 310–314. 10.1038/nmat3558. [DOI] [PubMed] [Google Scholar]
- Rickard D.; Morse J. W. Acid Volatile Sulfide (AVS). Mar. Chem. 2005, 97 (3–4), 141–197. 10.1016/j.marchem.2005.08.004. [DOI] [Google Scholar]
- Kraal P.; Burton E. D.; Bush R. T. Iron Monosulfide Accumulation and Pyrite Formation in Eutrophic Estuarine Sediments. Geochim. Cosmochim. Acta 2013, 122, 75–88. 10.1016/j.gca.2013.08.013. [DOI] [Google Scholar]
- Xu J.; Wang Y.; Weng C.; Bai W.; Jiao Y.; Kaegi R.; Lowry G. V. Reactivity, Selectivity, and Long-Term Performance of Sulfidized Nanoscale Zerovalent Iron with Different Properties. Environ. Sci. Technol. 2019, 53 (10), 5936–5945. 10.1021/acs.est.9b00511. [DOI] [PubMed] [Google Scholar]
- Bartholomew C. H.; Agrawal P. K.; Katzer J. R. Adv. Catal. 1982, 31, 135–242. 10.1016/S0360-0564(08)60454-X. [DOI] [Google Scholar]
- Burke M. L.; Madix R. J. Hydrogen on Pd(100)-S: The Effect of Sulfur on Precursor Mediated Adsorption and Desorption. Surf. Sci. 1990, 237 (1–3), 1–19. 10.1016/0039-6028(90)90515-A. [DOI] [Google Scholar]
- Arnold W. A.; Roberts A. L. Pathways and Kinetics of Chlorinated Ethylene and Chlorinated Acetylene Reaction with Fe(0) Particles. Environ. Sci. Technol. 2000, 34 (9), 1794–1805. 10.1021/es990884q. [DOI] [Google Scholar]
- Song H.; Carraway E. R. Catalytic Hydrodechlorination of Chlorinated Ethenes by Nanoscale Zero-Valent Iron. Appl. Catal., B 2008, 78 (1–2), 53–60. 10.1016/j.apcatb.2007.07.034. [DOI] [Google Scholar]
- Butler E. C.; Hayes K. F. Effects of Solution Composition and PH on the Reductive Dechlorination of Hexachloroethane by Iron Sulfide. Environ. Sci. Technol. 1998, 32 (9), 1276–1284. 10.1021/es9706864. [DOI] [Google Scholar]
- Butler E. C.; Hayes K. F. Factors Influencing Rates and Products in the Transformation of Trichloroethylene by Iron Sulfide and Iron Metal. Environ. Sci. Technol. 2001, 35 (19), 3884–3891. 10.1021/es010620f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.