

We are confronted with the most dramatic pandemic world‐wide in the past 100 years. We are armed “to‐the‐teeth” compared to 1918, we know the agent, the genomic sequence, the bodily entry, the proliferation rate, the damage pathogenesis and the very nature of our enemy. We can identify its bodily presence and our resistance to it in terms of neutralizing antibody production. Nonetheless, the disease has laid lame the great nations of the current world and crippled the less fortunate countries. The primary disease features are not the kidney. However, the entry point has much to do with renal and cardiovascular disease. The kidney is a common target of corona‐virus (SARS‐CoV2) disease; the longer‐term consequences could be as well.
Corona‐virus disease 2019 (Covid‐19) is a global pandemic of the severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2). Corona viruses are RNA viruses. Corona‐virus attention was drawn through the severe acute respiratory syndrome (SARS), a viral respiratory disease of zoonotic origin caused by SARS‐CoV1. The Middle East respiratory syndrome (MERS), also known as camel flu, is a viral respiratory infection caused by a similar MERS‐coronavirus (MERS‐CoV). SARS and MERS thankfully did not result in a pandemic, while SARS‐CoV‐2 unfortunately did. Thanks to molecular biology, much is known about these pathogens and because of indefatigable work by basic scientists and clinicians, our knowledge of the disease entities is also quite complete. This state‐of‐affairs is not new. Over 100 years ago, we faced a quite similar pandemic, the outcome of which is still not clear. At least in the realm of literature, that epidemic yielded a Nobel prize in literature (Sinclair Lewis). Can the history of Arrowsmith help us further? 1
Corona viruses are known since 1930 and were first identified in fowl. The name is garnered from the roughly spherical‐shaped particles that exhibit bulbous surface projections, particularly the spike proteins. Human corona viruses infect the epithelial cells of the respiratory tract. Drosten et al identified a novel corona virus in patients with SARS (SARS‐Co‐V). 2 Their findings and those of others showed that novel corona viruses cause SARS. Fortunately, the SARS scare abated, albeit with serious reservations. 3 Next came MERS. Similarly, MERS also failed to cause a pandemic. Obviously, Covid‐19 disease is something quite different. 4 , 5 Nonetheless, basic research revealed some amazing findings, primarily appreciated in oriental countries. Imai, Kuba and Penniger et al pointed out that SARS had something to do with a cell‐surface receptor termed angiotensin‐converting enzyme 2 (ACE2). 6 ACE2 also serves as the entry point into cells for other corona viruses, including HCoV‐NL63, SARS‐CoV and SARS‐CoV‐2. The human version of the enzyme is often referred to as hACE2.
Angiotensin‐converting enzyme 2 (ACE2) is an enzyme attached to the cell membranes of cells in the lungs, arteries, heart, kidney and intestines. 7 ACE2 is novel human zinc metalloprotease that has considerable homology to human angiotensin‐converting enzyme (ACE). Expression in Chinese hamster ovary cells of a soluble, truncated form of ACE2, lacking the transmembrane and cytosolic domains, produced a glycoprotein of 120 kDa, which was able to cleave angiotensin I and angiotensin II but not bradykinin or Hip‐His‐Leu. ACE2 functions exclusively as a carboxypeptidase; the activity is not inhibited by classical ACE inhibitors such as captopril, lisinopril or enalaprilat. The ACE2 gene contains 18 exons, of which several have considerable size similarity with the first 17 exons of ACE. Reportedly, ACE2 can lower blood pressure by catalyzing the hydrolysis of angiotensin II (a vasoconstrictor peptide) into angiotensin (1‐9) 8 and angiotensin (1‐7) (a vasodilator peptide). 9 ACE2 is said to counter the activity of the related angiotensin‐converting enzyme (ACE) by reducing the amount of angiotensin‐II and increasing Ang (1‐7) making it a promising drug target for treating cardiovascular diseases. ACE2 is claimed to be an essential regulator of cardiac function. The conclusion is based on an ACE2 gene‐deleted model. 10 Nonetheless, a second ACE2 gene‐deleted model led to far more modest conclusions. 11 Any cardiovascular conclusions regarding ACE2 in humans remain to be proved.
Lv et al 12 extracted the nucleotide sequences of ACE and ACE2 genes from genomic assemblies of 36 representative vertebrates. They observed that most of the phylogenetic positions were consistent with the species tree; however, certain differences appeared in coelacanths and frogs, which could suggest a very slow evolutionary rate in the initial evolution of ACE and ACE2 in vertebrates. Our associates inspected the evolution of renin‐angiotensin‐aldosterone system components throughout evolution. 13 We found that important parts of the system began to appear with primitive chordates and tunicates and that all major components were present at the divergence of bony fish, with the exception of the Mas (putative angiotensin 1‐7) receptor. The Mas receptor first appears after the bony‐fish/tetrapod divergence. This phase of evolutionary innovation happened about 400 million years ago (Figure 1). Briefly, the renin‐angiotensin system shows up in bony fish; aldosterone is older still. ACE and ACE2 are absent in Drosophila, although an ancestral homolog (ACER) is present. We suggested that ACE could be the product of a gene duplication involving ACE2. In any event, ACE2 has been around longer than ACE or angiotensinogen (Figure 2). The timeframe is important since Darwin recognized that earlier functions could be adapted by later traits. We could conclude that the original functions of ACE2 are probably not related to cardiovascular regulation as we understand it today, but briefly the idea is that ACE2 produces the “good” (Ang 1‐7) while ACE produces the “bad” (Ang II). As a result (authors’ tongues in cheek), in the pandemic, the renin‐angiotensin‐system and its cardiovascular ramifications have received perhaps more attention than they deserved.
Figure 1.
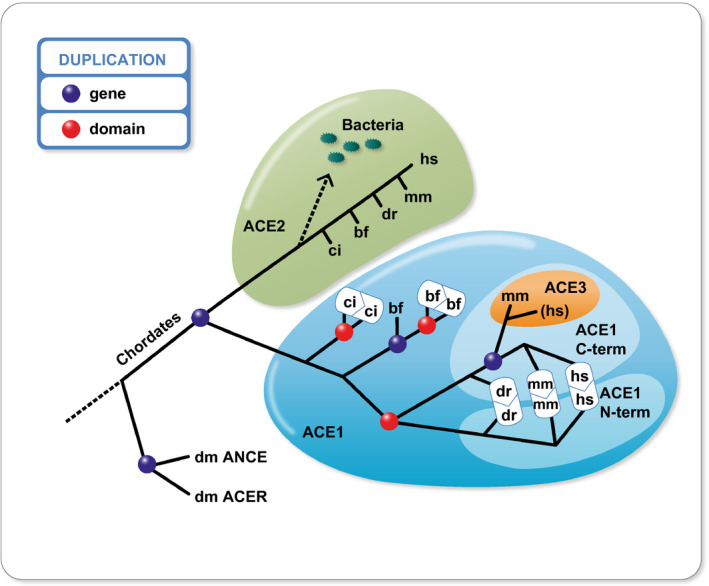
Evolution of the “anginotensin converting enzyme” (ACE) family. Abbreviations are as follows: Drosophila melanogaster = dm; Ciona intestinalis = ci; Branchiostoma floridae = bf; Danio rerio = dr; Mus musculus = mm; Homo sapiens = hs. The ACE family originated before the divergence of chordates from arthropods. 13 Gene duplications (blue spheres) have expanded this family leading to the existence of ACE1 and ACE2 in chordates. Multiple events of domain duplication (red spheres) have happened in the ACE1 subfamily, an important one leading to the vertebrate ACE1, which contains an N‐terminal and a C‐terminal‐catalytic domain. ACE3 is a single‐domain ACE, which stems from duplication of the mammalian C‐terminal domain of the ACE1. This sequence seems to have evolved into a pseudogene in humans. Vertebrate ACE2 orthologs are present in many bacterial species
Figure 2.
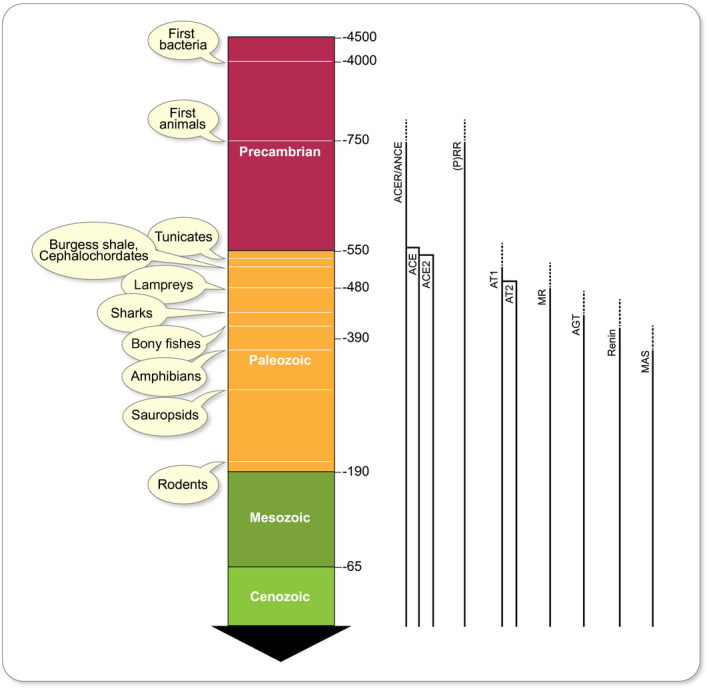
Time‐line emergence of the renin‐angiotensin‐aldosterone system (RAAS). 13 ACER and ANCE are Drosophila homolog variants. (P)RR is the (pro)renin receptor, an ATPase H+‐transporting lysosomal accessory protein. At left are geological eras and a timeline (scale in millions of years). At right are solid lines indicating appearance time. Hatched lines are uncertainty. While most genes appeared in the early Paleozoic, others might have emerged earlier in the Precambrian era and were adapted later for their use as part of the RAAS. ACE is one such example and might have evolved from an initial developmental function to physiological actions on volume regulation in vertebrates. The fact that the ACE family appear before angiotensin II receptors (AT1 and AT2), as well as the later appearance of angiotensinogen, renin and the MAS receptor gives us insight into long‐term evolution
Aside from putative cardiovascular regulation, Hashimoto et al suggested ACE2 as a key regulator of dietary amino acid homeostasis, innate immunity, gut microbial ecology and transmissible susceptibility to colitis. Their results provided a molecular explanation for how amino acid malnutrition could cause intestinal inflammation and diarrhoea. 14 ACE2 has a first cousin (homolog) that resides in the kidney. This homolog, collectrin, is an amino‐acid transporter and plays an essential role in amino‐acid reabsorption in gut and kidney, such as in Hartnup's disease, a tryptophan malady. 15 A cryo‐electron microscopy structures of full‐length human ACE2 in the presence of the neutral amino acid transporter collectrin (also known as B0AT1), with or without the receptor SARS‐CoV2 binding domain (RBD), of the surface spike glycoprotein (S protein) of SARS‐CoV‐2, both at an overall resolution of 2.9 angstroms, with a local resolution of 3.5 angstroms at the ACE2‐RBD interface has been elucidated in detail. 16 The amazing thing about this research is the vast detail we already know about Sars‐CoV‐2, the receptor binding domain and many interactions. Nonetheless, we are left with a series of interesting, albeit diverse, findings. We are confronted with an aggressive SARS virus (SARS‐CoV2), we confront a potentially lethal disease with short‐ and long‐term consequences, we deal with a putative “cardiovascular” receptor that far earlier probably had much different and unknown functions and moreover this receptor is somehow related to gastrointestinal and renal transport. ACE2 and ACE are both expressed in glomeruli. 17 We are also aware that Covid‐19 seems to involve the kidney directly and leads to acute kidney injury (AKI) and all the sequels to that conditions that are known. 18
ACE2 alone does not allow SARS‐CoV2 entry. Cell entry of corona viruses depends on binding of the viral spike (S) proteins to cellular receptors and on S protein priming by host cell proteases. SARS‐CoV‐2 uses the SARS‐CoV receptor ACE2 for entry and the serine protease, TMPRSS2, for S‐protein priming. Sera from convalescent SARS patients cross‐neutralized SARS‐2‐S‐driven entry. 19 Viral entry also depends on TMPRSS2 protease activity, an androgen‐dependent enzyme that might in part explain why men do more poorly with Covid‐19 disease than women. 20 Recently, Xu et al used state‐of‐the‐art single‐cell RNA sequencing techniques to analyse the transcriptomics of 15 human kidney samples. They found that ACE2 and TMPRSS2 are significantly co‐expressed in podocytes and proximal convoluted tubules, providing even more plausibility for direct viral involvement of the kidneys (https://www.preprints.org/manuscript/202002.0331/v1). Aside from “kidney cells”, the kidney is endowed with blood vessels. These structures, aside from expressing ACE2 could also be involved in Covid‐19 disease. Varga et al published endothelial infection by the virus. 21 Thus, no vascular tissue is safe. All these findings have necessarily markedly increased anxiety in patients ingesting renin‐angiotensin‐aldosterone system‐inhibiting drugs. 22 , 23 These anxieties, in spite of some retractions, can be laid to rest. Cardiovascular patients should NOT discontinue their medications because of the SARS‐Co2 risk.
The clinical pandemic moves so fast that no review can keep up. We present a tabular view of what is currently known (Table 1). New York is a hotspot and is monitored by a host of outstanding institutions. Early reports from these institutions showed that severe kidney disease is a hallmark of severe COVID disease. This interpretation is in stark contrast to early reports from China. Of 3235 patients hospitalized within the Mt Sinai hospital network between late February and the 15th of April, 43% developed an AKI. In ICU patients, this figure increased even further to 68%. This state‐of‐affairs resulted in a necessity for kidney replacement therapy in 8.4% of all patients and 34% of ICU patients. These frightening numbers have been confirmed by other groups both (Table 1).
Table 1.
A brief overview of Covid‐19 patients, acute kidney injury (AKI) and renal‐replacement therapies (RRT)
| Authors | Cohort | Median age | Study site, time period | AKI frequency | RRT frequency |
|---|---|---|---|---|---|
| Guan et al 45 |
N = 1099 Hospitalized N = 55 ICU |
47 |
552 hospitals, China, December 11, 2019 to January 29, 2020 |
Overall: 0.5% | Overall: 0.8% |
| Cheng et al 46 |
N = 701 Hospitalized N = 73 ICU |
63 |
Tongji hospital, Wuhan, January 14 to February 11 2020 |
Overall: 5.1% | Not reported |
| Mohamed et al |
N = 575 Hospitalized N = 105 ICU |
65 |
Ochsner medical center, New Orleans March 1 to March 30, 2020 |
Overall: 28% ICU: 61% |
Overall: 15.5% ICU: 44.5% |
| Cummings et al 47 | N = 257 ICU only | 62 | New York, March 2 to April 2, 2020 | Not reported | ICU: 31% |
| Hirsch et al |
N = 5449 Hospitalized N = 1395 ICU |
64 |
Northwell Health, NY March 1 to April 5 |
Overall: 37% Mechanically ventilated (N = 1190): 87.3% |
Overall: 5.2% Mechanically ventilated: 23.2% a |
| Chan et al |
N = 3235 Hospitalized N = 815 ICU |
66.5 |
Mt Sinai, NY February 27 to April 15,2020 |
Overall, 43% ICU: 68% |
Overall: 8.65% ICU: 34% |
| ICNARC database (report from July 3, 2020) | N = 9768 ICU only | 60 | NHS adult ICUs, England, Wales, Northern Ireland, March 1 to July 2, 2020 | Not reported | ICU: 26.6% |
Percentage of total intensive care unit (ICU) patients was not reported.
Transplanted kidney patients also have an increase risk from Covid‐19. The Montefiore Medical Center in New York reported 26 transplant patients with Covid‐19. These patients had ancillary risk factors. 24 Accordingly, a very high early mortality among kidney‐transplant recipients with Covid‐19—28% at 3 weeks was found. Two‐thirds of the patients died. The effect of Covid‐19 on renal transplant programs has been appreciated and discussed in detail. 25
That Covid‐19 patients develop acute kidney injury (AKI) would not be a surprise. However, underlying mechanisms might be. Investigators quantified the SARS‐CoV‐2 viral load in autopsy tissue samples obtained from 22 patients who had died from Covid‐19. Seventeen patients (77%) had more than two coexisting conditions. A greater number of coexisting conditions was associated with SARS‐CoV‐2 tropism for the kidneys in these patients, even in patients without a history of chronic kidney disease. The highest levels of SARS‐CoV‐2 copies per cell were detected in the respiratory tract, and lower levels were detected in the kidneys, liver, heart, brain and blood. 26 The findings indicate a broad organotropism of SARS‐CoV‐2. The kidney of this respiratory pandemic remained a focal point (article labelled as a “preprint”).
Nonetheless, all organ systems are targets in Covid‐19 disease. Autopsy studies, like the above, could help us further. Progressive respiratory failure, not renal failure, is the primary cause of death in the coronavirus disease 2019 (Covid‐19) pandemic. Pathologists recently examined 7 lungs obtained during autopsy from patients who died from Covid‐19 and compared them with 7 lungs obtained during autopsy from patients who died from acute respiratory distress syndrome (ARDS) secondary to influenza A(H1N1) infection and 10 age‐matched, uninfected control lungs. Vascular angiogenesis distinguished the pulmonary pathobiology of Covid‐19 from that of equally severe influenza virus infection. The pathology in the kidney might be quite similar. 27
Chinese investigators found that 251 of the 333 Covid‐19 patients (75.4%) had abnormal urine dipstick tests or AKI. Of 198 patients with renal involvement for the median duration of 12 days, 118 (59.6%) experienced remission of pneumonia during this period, and 111 of 162 (68.5%) patients experienced remission of proteinuria. 28 Among 35 patients who developed AKI, 16 (45.7%) experienced complete recovery of kidney function. The authors suspected that most AKI cases were intrinsic AKI. Patients with renal involvement had higher overall mortality compared with those without renal involvement (28 of 251 [11.2%] vs 1 of 82 [1.2%] respectively). Stepwise multivariate binary logistic regression analyses showed that severity of pneumonia was the risk factor most commonly associated with lower odds of proteinuric or haematuric remission and recovery from AKI. However, Hirsch et al were able to confirm these findings. 29 They published urine “dipstick” data from 542 AKI patients. Again, a striking 74% of patients showed traces of protein in their urine. Proteinuria of 1+ was found in 31.9% and proteinuria of 2+ and greater was found in 42.1%. Similar results were obtained for haematuria, 75.8% of patients had any blood detectable in their urine, in 46.1% Haematuria of 2+ or greater was observed. The dramatic involvement of AKI in Covid‐19 patients will not be reiterated here. The capabilities to serve AKI patients with Covid‐19 have become a major challenge in many countries.
Ultrastructural studies could help further. Farkash et al reported that viral particles in the renal tubular epithelium that were morphologically identical to SARS‐CoV‐2, and with viral arrays and other features of virus assembly, provide evidence of a productive direct infection of the kidney by SARS‐CoV‐2. 30 Their finding could offer confirmatory evidence that direct renal infection occurs in the setting of AKI in COVID‐19. The authors describe tubular isometric vacuolization observed with light microscopy, which correlated with double‐membrane vesicles containing vacuoles observed with electronic microscopy, may be a useful histological marker for active SARS‐CoV‐2 infection in kidney biopsy or autopsy specimens.
African Americans are particularly struck by Covid‐19. 31 Since APOL1 variants are a putative risk factor, Wu et al investigated this notion. 32 The authors found that collapsing glomerulopathy in black patients with COVID‐19 was associated with high‐risk APOL1 variants. They observed no direct viral infection in the kidneys, suggesting a possible alternative mechanism: a "two‐hit" combination of genetic predisposition and cytokine‐mediated host response to SARS‐CoV‐2 infection. Given this entity's resemblance with HIV‐associated nephropathy, the authors proposed the term COVID‐19‐associated nephropathy to describe the condition.
COVID‐19 is recent and, therefore, case records are common and valuable. A recent report by Sise et al is an example. 33 The 68‐year‐old patient suddenly developed fever to 39.9°C and cough. Because of a normal oxygen saturation value, he is sent home but returns with further symptoms, including dyspnoea and hypoxemia. Numerous risk factors were present such as hypertension, hyperlipidaemia, coronary artery disease, obstructive sleep apnoea and obesity. Among other serious problems, AKI developed. Renal replacement therapy became indicated; however, we learn that limitations on machinery and available dialysis replacement fluids are an issue in the most advanced hospital in the United States. These states‐of‐affairs confront nephrologists and intensivists in the COVID‐19 pandemic, in less fortunate clinical settings, all the more so.
Since COVID‐19 has afflicted many patients, AKI patient series have been collected. Daoud et al 34 have published a meta‐analysis. They found that the available limited published data to indicate that severe AKI in patients with COVID‐19 is an ominous clinical predictor and is associated with high mortality (no surprise here). Chen et al 35 have also trodden this territory. They were at least able to determine an incidence AKI rate of 10% in COVID‐19 patients, albeit the range in their series was huge (so as not to be helpful). The literature to date cannot help us further. We cannot determine a specific form of COVID‐19 AKI or chronic nephropathy. The clinical picture is likely to be diverse.
Jhaveri et al have recently described a carefully examined index patient. 36 A 69‐year‐old woman developed fever, cough and dyspnoea. Her physicians considered the diagnosis of COVID‐10 disease that was confirmed. Respiratory failure ensued; the renal findings suggested primary involvement and a renal biopsy was performed. The biopsy revealed thrombotic micro‐angiopathy. The glomerular picture was one of “collapsing” glomerular nephropathy. Treatment with anakinra and tocilizumab did not help the patient further. However, co‐agulopathy with Covid‐19 disease is widely reported. 37
AKI has many causes and a direct involvement of virus is difficult to discern. Even electron microscopy can lead to non‐specific findings. Apparently characteristic 60‐ to 140‐nm round particles surrounded by a corona of 9‐12 nm distinctive spikes have been identified and the photomicrographs appear convincing. Cassol et al have recently counselled for caution in interpreting these findings (Preprint https://doi.org/10.34067/KID.0002692020). This challenging task has been addressed earlier. 38 In situ hybridization could be a way out of this conundrum. Thus, pathogenic mechanisms aside from general processes have not helped us further. African Americans are far more likely to succumb to Covid‐19 than similarly aged, stratified, Caucasian Americans, even within the same health‐care system. 31 An autopsy series from New Orleans has recently been published. 39 Important findings include the presence of thrombosis and micro‐angiopathy in the small vessels and capillaries of the lungs, with associated haemorrhage that significantly contributed to death. Features of diffuse alveolar damage, including hyaline membranes, were present, even in patients who had not been ventilated. Cardiac findings included individual cell necrosis without lymphocytic myocarditis. The findings revolved around (thrombotic) micro‐angiopathy. The kidneys received scant mention in this report, even though they receive 20% of the cardiac output. The recently appreciated multisystem inflammatory syndrome (MISC) in children fortunately did not feature a major renal component (around 10%) in terms of target‐organ damage. 40 , 41
The question remains whether or not Covid‐19 renal disease is specific? 42 As a marker of illness, urinalysis and proteinuria have withstood the test of time. If the renal tropism hypothesis is true, 26 we would expect a regimented pattern in terms of AKI injuries. This state‐of‐affairs has not proved to be the case to our knowledge. 41 Possibly, the AKI observed in Covid‐19 disease is multifactorial and rests on numerous pathogenic mechanisms. Finally, are disease‐specific treatment modalities for those requiring renal replacement therapies indicated? Strategies have been recently reviewed by experts. 43 The same considerations for any AKI patients remain in place.
An appropriate stance is as follows: “When the going gets tough, the tough get going”. The basic science advancement in the authors’ view just in the last 8 months has been phenomenal. In contrast to the AIDS epidemic (largely ignored for decades), SARS‐Co‐V2 received immediate attention, in terms of genomic sequencing, assay development, sharing of diagnostic data, vaccine development and other international cooperation. The vascular community must remain at the forefront. Cardiac involvement (probably similar mechanisms) is common. There has never‐to‐fore been a more compelling argument for networking and collaboration than this one. Mechanistically, the data require a firm foundation. Our Covid‐19 renal‐pathology group in Berlin at the Charité is aiming to achieve that end. We, (Covid‐19 Charité investigators) are conducting a prospective investigation that includes incorporation of recent “biomarkers” for AKI in a patient population likely to need them. 44 Thus, clinical and pathological evaluations are necessary. Particularly, the lapse in anatomical pathology (the lapse in autopsies) is to be underscored here.
CONFLICT OF INTEREST
There are none.
REFERENCES
- 1. Eisenman DJ. Rereading Arrowsmith in the COVID‐19 Pandemic. JAMA. 2020; 324(4), 319‐320. [DOI] [PubMed] [Google Scholar]
- 2. Drosten C, Gunther S, Preiser W, et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348:1967‐1976. [DOI] [PubMed] [Google Scholar]
- 3. Peiris JS, Yuen KY, Osterhaus AD, Stohr K. The severe acute respiratory syndrome. N Engl J Med. 2003;349:2431‐2441. [DOI] [PubMed] [Google Scholar]
- 4. Berlin DA, Gulick RM, Martinez FJ. Severe Covid‐19. N Engl J Med. 2020. [DOI] [PubMed] [Google Scholar]
- 5. Gandhi RT, Lynch JB, Del Rio C. Mild or Moderate Covid‐19. N Engl J Med. 2020. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 6. Imai Y, Kuba K, Penninger JM. The discovery of angiotensin‐converting enzyme 2 and its role in acute lung injury in mice. Exp Physiol. 2008;93:543‐548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin‐converting enzyme. Cloning and functional expression as a captopril‐insensitive carboxypeptidase. J Biol Chem. 2000;275:33238‐33243. [DOI] [PubMed] [Google Scholar]
- 8. Donoghue M, Hsieh F, Baronas E, et al. A novel angiotensin‐converting enzyme‐related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ Res. 2000;87:E1‐E9. [DOI] [PubMed] [Google Scholar]
- 9. Eriksson U, Danilczyk U, Penninger JM. Just the beginning: novel functions for angiotensin‐converting enzymes. Curr Biol. 2002;12:R745‐R752. [DOI] [PubMed] [Google Scholar]
- 10. Crackower MA, Sarao R, Oudit GY, et al. Angiotensin‐converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417:822‐828. [DOI] [PubMed] [Google Scholar]
- 11. Gurley SB, Allred A, Le TH, et al. Altered blood pressure responses and normal cardiac phenotype in ACE2‐null mice. J Clin Invest. 2006;116:2218‐2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lv Y, Li Y, Yi Y, Zhang L, Shi Q, Yang J. A genomic survey of angiotensin‐converting enzymes provides novel insights into their molecular evolution in vertebrates. Molecules. 2018;23:2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fournier D, Luft FC, Bader M, Ganten D, Andrade‐Navarro MA. Emergence and evolution of the renin‐angiotensin‐aldosterone system. J Mol Med (Berl). 2012;90:495‐508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hashimoto T, Perlot T, Rehman A, et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature. 2012;487:477‐481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kuba K, Imai Y, Ohto‐Nakanishi T, Penninger JM. Trilogy of ACE2: a peptidase in the renin‐angiotensin system, a SARS receptor, and a partner for amino acid transporters. Pharmacol Ther. 2010;128:119‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of SARS‐CoV‐2 by full‐length human ACE2. Science. 2020;367:1444‐1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ye M, Wysocki J, William J, Soler MJ, Cokic I, Batlle D. Glomerular localization and expression of Angiotensin‐converting enzyme 2 and Angiotensin‐converting enzyme: implications for albuminuria in diabetes. J Am Soc Nephrol. 2006;17:3067‐3075. [DOI] [PubMed] [Google Scholar]
- 18. Soleimani M. Acute kidney injury in SARS‐CoV‐2 infection: direct effect of virus on kidney proximal tubule cells. Int J Mol Sci. 2020;21:3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hoffmann M, Kleine‐Weber H, Schroeder S, et al. SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271‐280.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Albini A, Di Guardo G, Noonan DM, Lombardo M. The SARS‐CoV‐2 receptor, ACE‐2, is expressed on many different cell types: implications for ACE‐inhibitor‐ and angiotensin II receptor blocker‐based cardiovascular therapies. Intern Emerg Med. 2020. 10.1007/s11739-020-02364-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Varga Z, Flammer AJ, Steiger P, et al. Endothelial cell infection and endotheliitis in COVID‐19. Lancet. 2020;395:1417‐1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. de Abajo FJ, Rodriguez‐Martin S, Lerma V, et al.; group M‐ACs . Use of renin‐angiotensin‐aldosterone system inhibitors and risk of COVID‐19 requiring admission to hospital: a case‐population study. Lancet. 2020;395:1705‐1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Reynolds HR, Adhikari S, Pulgarin C, et al. Renin‐angiotensin‐aldosterone system inhibitors and risk of Covid‐19. N Engl J Med. 2020;382:2441‐2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Akalin E, Azzi Y, Bartash R, et al. Covid‐19 and kidney transplantation. N Engl J Med. 2020;382:2475‐2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Martino F, Plebani M, Ronco C. Kidney transplant programmes during the COVID‐19 pandemic. Lancet Respir Med. 2020;8:e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Puelles VG, Lutgehetmann M, Lindenmeyer MT, et al. Multiorgan and renal tropism of SARS‐CoV‐2. N Engl J Med. 2020. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ackermann M, Verleden SE, Kuehnel M, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid‐19. N Engl J Med. 2020;383:120‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pei G, Zhang Z, Peng J, et al. Renal involvement and early prognosis in patients with COVID‐19 pneumonia. J Am Soc Nephrol. 2020;31:1157‐1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hirsch JS, Ng JH, Ross DW, et al. Northwell C‐RC and Northwell Nephrology C‐RC. Acute kidney injury in patients hospitalized with COVID‐19. Kidney Int. 2020;98:209‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Farkash EA, Wilson AM, Jentzen JM. Ultrastructural evidence for direct renal infection with SARS‐CoV‐2. J Am Soc Nephrol. 2020.31 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Price‐Haywood EG, Burton J, Fort D, Seoane L. Hospitalization and Mortality among Black Patients and White Patients with Covid‐19. N Engl J Med. 2020;382:2534‐2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wu H, Larsen CP, Hernandez‐Arroyo CF, et al. AKI and collapsing glomerulopathy associated with COVID‐19 and APOL 1 high‐risk genotype. J Am Soc Nephrol. 2020.31(7) [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sise ME, Baggett MV, Shepard JO, Stevens JS, Rhee EP. Case 17–2020: a 68‐year‐old man with Covid‐19 and acute kidney Injury. N Engl J Med. 2020;382:2147‐2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ali H, Daoud A, Mohamed MM, et al. Survival rate in acute kidney injury superimposed COVID‐19 patients: a systematic review and meta‐analysis. Ren Fail. 2020;42:393‐397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen YT, Shao SC, Hsu CK, Wu IW, Hung MJ, Chen YC. Incidence of acute kidney injury in COVID‐19 infection: a systematic review and meta‐analysis. Crit Care. 2020;24:346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jhaveri KD, Meir LR, Flores Chang BS, et al. Thrombotic microangiopathy in a patient with COVID‐19. Kidney Int. 2020. 10.1016/j.kint.2020.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fan BE, Chong VCL, Chan SSW, et al. Hematologic parameters in patients with COVID‐19 infection. Am J Hematol. 2020;95:E131‐E134. [DOI] [PubMed] [Google Scholar]
- 38. Goldsmith CS, Miller SE, Martines RB, Bullock HA, Zaki SR. Electron microscopy of SARS‐CoV‐2: a challenging task. Lancet. 2020;395:e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fox SE, Akmatbekov A, Harbert JL, Li G, Quincy Brown J, Vander Heide RS. Pulmonary and cardiac pathology in African American patients with COVID‐19: an autopsy series from New Orleans. Lancet Respir Med. 2020.8(7):681–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dufort EM, Koumans EH, Chow EJ, et al.; New York S, Centers for Disease C and Prevention . Multisystem inflammatory syndrome in children investigation T. multisystem inflammatory syndrome in children in New York State. N Engl J Med. 2020. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Feldstein LR, Rose EB, Horwitz SM, et al. Multisystem inflammatory syndrome in U.S. children and adolescents. N Engl J Med. 2020. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gross O, Moerer O, Weber M, Huber TB, Scheithauer S. COVID‐19‐associated nephritis: early warning for disease severity and complications? Lancet. 2020;395:e87‐e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ronco C, Reis T, Husain‐Syed F. Management of acute kidney injury in patients with COVID‐19. Lancet Respir Med. 2020;8:738‐742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Luft FC. Biomarkers and predicting acute kidney injury. Acta Physiol (Oxf). 2020:e13479. [DOI] [PubMed] [Google Scholar]
- 45. Guan W‐J, Ni Z‐Y, Hu Y, et al.; China Medical Treatment Expert Group for C. Clinical characteristics of coronavirus disease in China. N Engl J Med. 2019;2020(382):1708‐1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cheng Y, Luo R, Wang K, et al. Kidney disease is associated with in‐hospital death of patients with COVID‐19. Kidney Int. 2020;97:829‐838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cummings MJ, Baldwin MR, Abrams D, et al. Epidemiology, clinical course, and outcomes of critically ill adults with COVID‐19 in New York City: a prospective cohort study. Lancet. 2020;395:1763‐1770. [DOI] [PMC free article] [PubMed] [Google Scholar]