

Abstract
Vaccination has been one of the most effective health intervention mechanisms to reduce morbidity and mortality associated with infectious diseases. Vaccines stimulate the body's protective immune responses through controlled exposure to modified versions of pathogens that establish immunological memory. However, only a few diseases have effective vaccines. The biological effects of nonthermal plasma on cells suggest that plasma could play an important role in improving efficacy of existing vaccines and overcoming some of the limitations and challenges with current vaccination strategies. This review summarizes the opportunities for nonthermal plasma for immunization and therapeutic purposes.
Keywords: adjuvant, COVID‐19, immunity, immunotherapy, nonthermal plasma, vaccine
Nonthermal plasma enhances innate and adaptive immune responses to infected cells. Nonthermal plasma induces oxidative stress in infected cells, increasing their immunogenicity and promoting recruitment of antigen presenting cells for phagocytic uptake. Antigen presenting cells then process pathogen antigens from the dead cell and migrate to the lymph nodes where they present the antigens to immune cells that mount an adaptive immune response and facilitate clearance of the infection.
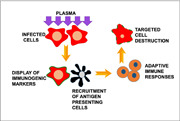
1. INTRODUCTION
Vaccination, also known as immunization, helps an individual's immune system to develop protection against diseases caused by infectious agents. Successful vaccination shields the individual from infection by the pathogen or facilitates its elimination from the body to mitigate disease. Therefore, vaccines serve as a relatively inexpensive means of improving the overall health of populations at risk for infection. The efficacy of a vaccine in an at‐risk population is dependent on the number of individuals in the population who have been vaccinated. Population‐level protection against the spread of a pathogen by sufficiently high levels of vaccination is referred to as herd immunity.
Early trials of vaccination in the 15th century paved the way for a system of worldwide infectious disease control that has reduced the morbidity and mortality associated with many pathogens that have been the cause of global infectious disease outbreaks, such as the 1918 flu pandemic.[ 1 , 2 ] Successful vaccination campaigns led to the complete eradication of smallpox in 1975, and they are the basis of contemporary efforts to eradicate polio. The current COVID‐19 pandemic has imposed a new urgency on researchers and industry to rapidly develop new strategies for vaccination that can be mass produced in a very short period of time. Advances in the knowledge of the biology of vaccine effects, along with the expansion of laboratory tools to study newly developed vaccines, have opened new avenues to be explored for developing “therapeutic” vaccines for treatment of chronic diseases, that is, they do not prevent disease but serve to reduce the severity of disease.
2. INFECTION AND IMMUNITY
In humans, a disease commonly occurs after infection with pathogens due to a breach in the protective physical or chemical barriers and/or subsequent failure of the innate and adaptive immune responses responsible for controlling the infection. Infected individuals may then transmit the pathogen among other members of the community, promoting local, regional, national, and even international dissemination of the pathogen (Figure 1). In contrast, a competent immune system controls and clears the pathogen before the appearance of disease symptoms or, alternatively, reduces the severity and long‐term sequelae of the infection. Several infections, such as smallpox, result in life‐long immunity (in those who survive) against re‐infection after a single exposure to the virus due to the development of specific immune memory against the pathogen.[ 3 , 4 , 5 ] This vaccine‐like effect of an infection is the factual basis for supporting the intentional exposure of children to patients who have the disease as a form of protection. In contrast, other pathogen‐associated diseases, such as malaria, require repeated exposures (with or without disease symptoms) to provide some level of long‐term immunity.[ 6 , 7 ] This protection is often lost if the individual leaves the endemic area for an extended period of time.
Figure 1.
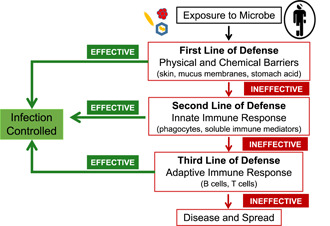
Three lines of defense protect against infection by pathogenic microbes. Pathogens can be blocked or inactivated upon the initial exposure by physical and chemical barriers that protect potential ports of entry on and in the body. If a pathogen gains entry into the body, the innate and adaptive immune systems can mount general and specific (respectively) responses to the pathogen. Failures of all three lines of defense can lead to disease in infected individuals and the transmission of the pathogen into the population
Many microbes do not confer long‐term immunity, even after a full‐blown disease. In addition, they continue to adapt to their environment in the host by acquiring mutations that allow them to become more virulent and possibly lethal without medical intervention. Some may even acquire mutations that facilitate cross‐species spread, as is the case with the pathogens responsible for COVID‐19, the severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) and the influenza virus.[ 8 , 9 ] Furthermore, as a part of their evolution, pathogens often employ methods of evading or suppressing immune responses. This has been a great barrier in containing the COVID‐19 pandemic, as the SARS‐COV‐2 virus overwhelms the antiviral response of the innate immune system.[ 10 ] The continuous evolution of this and other pathogens serves as a sustained challenge for the immune system, including any previously established immunological memory, which may or may not be sufficient to control the disease. Pathogens, like the human immunodeficiency virus type 1 (HIV‐1), almost always succeed in causing disease by disabling immune cells, evading the immune system, and acquiring mutations that provide resistance to antiviral drugs.[ 11 , 12 , 13 , 14 ] Control of pathogens that have such mechanisms to evade the host immune system necessitates a pharmacological intervention to prevent the disease. Therefore, each pathogen may present a unique set of challenges to the immune system that must be specifically addressed.
3. VACCINATION AND IMMUNOLOGICAL MEMORY
Vaccines work by introducing a specific and unique component of a pathogen, referred to as an antigen, that trains the host immune system without causing the symptoms associated with the infection. In other words, vaccines train the immune system to recognize a pathogen by mimicking the pathogen and enabling a specific and protective immune response. Ideally, the antigen promotes the generation of a strong immediate immune response, accompanied by memory responses specific to the antigen. Upon exposure to that pathogen at some time in the future, long‐lived memory cells awaken and recognize the antigen against which they were generated and mount a pathogen‐specific response, resulting in protection against the infection and associated disease.[ 15 , 16 ]
The effective development of long‐term memory cell responses depends greatly on the vaccine's ability to interact with and stimulate cells of the innate immune system, specifically antigen‐presenting cells (APC), at the site of inoculation (Figure 2). APCs act as the detectors of danger in the body and surveyors for foreign substances in the tissues. If APCs encounter foreign, nonself‐antigens, they engulf (phagocytose) them, travel to lymphoid organs, and present them on their surface. APCs display those antigens to highly specialized cells of the immune system (T and B cells) and trigger them to produce a specific immune response against the recognized antigens. Each T and B cell is designed to respond to a single antigen only, thus providing antigen specificity. This initial response takes up to 14 days to develop. A subset of these T and B cells, in turn, differentiates into long‐lived memory cells that are able to respond almost immediately upon re‐exposure to that antigen, bypassing the need for a de novo response.[ 15 , 16 , 17 , 18 , 19 , 20 ] It is, therefore, necessary for vaccine formulations to be designed with different combinations of antigens to promote protective immunity through strong initial interactions with APCs.
Figure 2.
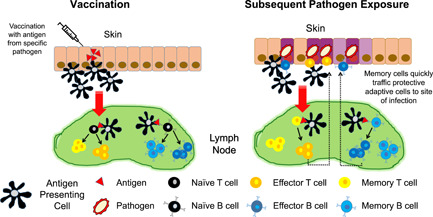
Vaccination results in the generation of immunological memory. Antigens in vaccine formulations are picked up by antigen‐presenting cells (APCs) at or near the site of inoculation. These APCs travel to lymph nodes and present the antigen to T cells that are specific for that antigen. The engaged T cells become activated and differentiate into effector T cells that travel back to the site of infection and attack the pathogen and/or the diseased cells. Some T cells establish memory and offer protection against subsequent exposures to that pathogen. Additionally, B‐cell activation results in production of antigen‐specific antibodies that may also persist to neutralize the antigen upon re‐exposure
4. VACCINE ANTIGENS
In general, antigens for vaccines may contain the entire live but attenuated pathogens or killed whole pathogens. Other vaccine formulations, called subunit vaccines, contain one or more antigenic proteins, peptides, or nucleic acids derived from the pathogen.
Live attenuated vaccines are composed of weakened forms of pathogenic microbes; therefore, they have all necessary components to stimulate an immune response. Administration of such vaccines may produce a mild, asymptomatic infection; however, they are not virulent enough to cause disease. Instead, they help to establish durable immunity. Pathogens are attenuated via the introduction of mutations in their genome to disrupt genes encoding virulence factors or metabolic pathways and/or promote a survival advantage for the less pathogenic strains over the more pathogenic forms.[ 21 , 22 ] However, these vaccines have a minute but real risk of the microorganisms reverting to a more virulent form, and they are generally not considered safe for the immunocompromised individual.[ 23 , 24 , 25 , 26 , 27 ]
Inactivated vaccines contain a heat‐ or chemically killed avirulent whole pathogen. An inactivated pathogen cannot replicate or produce proteins and toxins that contribute to pathogenesis; therefore, it does not cause disease. These vaccines tend to be weak immunogens and require the assistance of adjuvants to initiate the innate immune responses. Adjuvants work by triggering immune pathways distinct from those stimulated by the antigen, thereby altering the magnitude and quality of the adaptive responses to ensure maximum protection.[ 28 ]
Whole pathogen live attenuated and inactivated formulations allow for a number of antigens to be presented to APCs, with a greater potential for a multipronged robust immune response. In contrast, subunit vaccines only contain one or a few selected protein antigens that are potent stimulators of immunity.[ 29 , 30 , 31 , 32 , 33 , 34 ] As an alternative to direct delivery of antigens as vaccines, antigens can be encoded into nucleic acids. In this approach, DNA or messenger RNA encoding one or more antigens is injected as a part of a vaccine preparation. Once it is taken up by cells around the injection site, using host cellular machinery, there is a local antigen expression and presentation to APCs.[ 35 , 36 ]
Irrespective of the formulation of vaccine antigens, vaccines are designed to produce robust protective immune responses in a majority of healthy individuals.
5. VACCINE FORMULATIONS AND DELIVERY
Pathogen‐specific protection is influenced by the route of delivery, the correct formulation of vaccine antigens used, and controlled re‐exposure to antigens in the form of boosters (Table 1). Methods of delivery as well as formulations of vaccines have been optimized on the basis of the region where the initial contact between the pathogen and the immune system is expected to occur. In addition, there is a recipient‐dependent variation in the immune responses to vaccines. Generally, vaccine formulations are more efficient in healthy people than in individuals with compromised immune systems. Age or other pre‐existing conditions at the time of vaccination can influence the response to a delivered antigen.[ 38 , 39 , 40 , 41 , 42 ] Most vaccines are delivered on specific schedules during childhood to protect against bacterial and viral diseases that are more common in that age group. Other vaccines are available for adults as well as children for population‐level control of seasonal infections that change from year to year. The efficiency of some vaccines may be improved by incorporation of adjuvants (from adjuvare, to help) in vaccine formulations. These include aluminum salts, bacterial lipopolysaccharide (LPS), or cytosine phosphoguanine (CpG) oligodeoxynucleotides that are unmethylated DNA.[ 37 , 43 ] Although aluminum salts have been used for over 80 years and are the most commonly used adjuvants with confirmed safety and high efficacy for different vaccines, their specific mechanism(s) of action are still unclear.[ 44 , 45 ] Other adjuvants, like the synthetic CpG, have been more recently developed with more well‐known interactions to likewise promote a strong immune response.[ 46 , 47 ] Overall, adjuvants help to maximize APC engagement and function, more optimally stimulate adaptive immune responses, and establish immunological memory.
Table 1.
WHO‐recommended vaccines
| Antigen | Route of delivery | Formulation | Booster | Adjuvant |
|---|---|---|---|---|
| Recommended for all children | ||||
| BCG (tuberculosis) | Percutaneous | Live attenuated | No | None |
| Hepatitis B | Intramuscular | Protein subunit | No | Aluminum salts, LPS |
| Polio | Oral, intramuscular | Live attenuated, protein subunit | Yes, during travel | Aluminum salts |
| Diphtheria, tetanus, and pertussis | Intramuscular | Protein subunit | Yes, 3 | CpG |
| Haemophilus influenza type B | Intramuscular | Live attenuated | Yes, 1 | Polysaccharides and carrier proteins |
| Pneumococcal | Intramuscular | Protein subunit | Yes, 1 | Aluminum salts |
| Rotavirus | Oral | Live attenuated | No | Toxoid |
| Measles | Intramuscular | Live attenuated | Yes, 1 | None |
| Rubella | Intramuscular | Live attenuated | No | None |
| Human papillomavirus | Intramuscular | Protein subunit | No | Aluminum salts |
| Recommended for children residing in certain regions | ||||
| Japanese encephalitis | Intramuscular | Live attenuated | No | Aluminum salts |
| Yellow fever | Subcutaneous or intramuscular | Live attenuated | No | Aluminum salts |
| Tick‐borne encephalitis | Intramuscular | Whole inactivated | Yes, every 3 years | Aluminum salts |
| Recommended for children in some high‐risk populations | ||||
| Typhoid | Oral, intramuscular | Live attenuated | Yes, every 3 years | Aluminum salts, polysaccharides, carrier proteins |
| Cholera | Oral | Killed whole cell | Yes, every 2 years | Toxoid |
| Meningococcal | Intramuscular | Protein subunit | Yes, after 1 year | Toxoid |
| Hepatitis A | Intramuscular | Live attenuated, inactivated | None | Aluminum salts |
| Rabies | Intramuscular, intradermal | Live attenuated | None | Aluminum salts |
| Dengue | Intramuscular | Virus backbone vector | None | None |
| Recommended for children in certain immunization programs | ||||
| Mumps | Intramuscular | Live attenuated | None | None |
| Seasonal influenza | Intramuscular | Inactivated | Yes, annually | Vary based on formulation |
| Intranasal | Live attenuated | |||
| Varicella | Intramuscular | Live attenuated | None | CpG |
Note: Vaccines may be composed of whole organisms (live attenuated or inactivated) or a component derived from the pathogen (protein subunit, nucleic acid). Most vaccines are delivered via an injection intramuscularly or intradermally. Vaccines for diseases that initiate in the gastrointestinal system may be given orally (polio); those against respiratory diseases may be dispensed intranasally (influenza). The recommended programs are based on the routine vaccinations provided by the World Health Organization (WHO).[ 37 ]
Abbreviations: CpG, cytosine phosphoguanine; LPS, lipopolysaccharide.
A few vaccines have been suboptimal in establishing a sustained adaptive immune response that ensures a long‐term protection. A primary immunization, followed by a second booster dose (or even third, as with DPT) or multiple immunizations over a span of years, is necessary for sustained protection in such cases (e.g., tetanus).[ 38 , 39 , 40 , 48 ] Boosters also help address the limitations of conventional, mass‐produced vaccines.
5.1. Immunotherapy as vaccination
Successful vaccination has increased the life expectancy of people worldwide; however, in cases where there are no vaccines available, pathogens that lead to a chronic disease state result in severe morbidity and mortality. The persistence of a chronic disease is often a result of (a) immune defects in the individual as a consequence of the primary disease (ability of pathogen to suppress the immune function of the individual to avoid eradication) or (b) the evolution of the pathogen within the host to achieve survival benefits. Therefore, recent efforts have shifted toward examination of therapeutic approaches to overcome or counteract these immune defects. One such strategy is vaccination as a form of immunotherapy, because it is designed to specifically stimulate immune responses against disease antigens derived from people living with the disease.[ 49 , 50 ] This involves isolation of cells from the affected individual, their manipulation ex vivo to make the cells more immunogenic, and then reintroduction back into the patient to elicit optimal APC responses and subsequent protective adaptive immune responses (Figure 3). Here again, a personalized therapeutic approach may be more successful at combating each patient's individual disease, because fresh immune responses would be targeted against the pathogen variant found in the patient's body.
Figure 3.
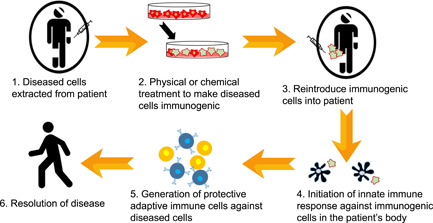
Ex vivo approaches to vaccination. Cells extracted from patients with chronic disease would be exposed to physical or chemical inactivators ex vivo. Those inactivated cells would be reintroduced into the patient, leading to host immune activation and re‐education against the specific antigenic form found in their body
Where classical vaccines work to establish an immune response based on a few preselected antigens, the use of whole cells that have been exposed to the pathogen in vivo and obtained from the patient for immunotherapy is considered optimal for chronic diseases, because it expands the selection of target antigens and is not limited by manufacturing issues.[ 49 , 50 , 51 , 52 ] Therefore, ex vivo immunotherapeutic vaccine approaches serve a more personalized as well as precision vaccination strategy to address the individual variation in disease‐associated antigens as well as immune defects specific to an individual. The key to the success of the ex vivo immunotherapeutic vaccination approach is the increase in immunogenicity of antigen‐experienced cells to promote the APC function. A recently identified mode of drastically increasing the immunogenic potential of diseased cells is a cellular process known as immunogenic cell death (ICD).
5.2. Inducers of ICD
ICD, largely characterized in cancer cells, is a type of cell death that is characterized by release or display of damage‐associated molecular patterns (DAMPs) that increase the cells' visibility to APCs. ICD, also referred to as immunogenic apoptosis, is distinct from nonimmunogenic apoptosis due to the specific spectrum of DAMPs released or displayed by the slowly dying cell.[ 53 , 54 ] DAMPs are described as “find me” molecules that are prochemotactic (which allow recruitment of APCs to the site of antigen introduction) or “eat me” molecules that are prophagocytic (promote recognition and uptake of the dying cell by phagocytes). Various chemical and physical ICD inducers have been identified, each of which induces a different subset of the characteristic ICD markers (Table 2). Molecules associated with ICD may function as adjuvants in vaccine immunotherapy due to the downstream effects caused by immunogenic apoptosis.
Table 2.
Examples of different ICD inducers and corresponding ICD markers demonstrated in vivo
| ICD inducer | ICD markers | In vivo disease model | Study | |
|---|---|---|---|---|
| Chemical methods | Mitoxantrone | Calreticulin, HMGB1 release | CT26 colon carcinoma | [55] |
| Cisplatin | CXCL10 release | Head and neck squamous cell carcinoma | [56] | |
| Doxorubicin | Calreticulin, ATP release, HMGB1 release | Triple‐negative breast cancer | [57] | |
| Viral vector | Oncolytic adenovirus | Calreticulin, ATP release, HMGB1 release | Mesothelioma | [58] |
| Physical methods | High hydrostatic pressure | Calreticulin, HSP70, HSP90 | Non‐small‐cell lung cancer | [59] |
| Photodynamic therapy | Calreticulin, HSP90, ATP release | CT26 colon carcinoma | [60] | |
| Radiation therapy | Calreticulin, ATP release, HMGB1 release | Prostate carcinoma | [61] | |
| Nonthermal plasma | Calreticulin, ATP release | CT26 colon carcinoma | [62] |
Note: Chemical methods of inducing ICD include chemotherapeutic drugs that are often administered to cancer patients, whereas physical methods are commonly drug‐free approaches directly applied to tumor cells.
Abbreviation: ICD, immunogenic cell death.
6. NONTHERMAL PLASMA (NTP) AS AN IMMUNOGENIC CELL DEATH INDUCER
Cancer therapeutics research continues to pursue methods of ICD induction that are characterized by optimal effectiveness with negligible or limited in vivo toxicity. Several chemotherapy drugs are now identified as ICD inducers when used in regimens not classically used in clinical practice, for example, oxaliplatin and mitoxantrone.[ 23 , 63 ] Physical methods such as radiation, photodynamic therapy, pulsed electric fields, and high hydrostatic pressure are also modalities under investigation as ICD inducers.[ 55 , 64 , 65 , 66 , 67 ] However, toxicity remains a major challenge to overcome with many of these treatments.
Although the mechanism of ICD induction associated with the different physical and chemical agents is not fully elucidated, a common theme is the induction of the oxidative and endoplasmic reticulum (ER) stress in exposed cells.[ 54 , 60 , 68 , 69 , 70 , 71 , 72 , 73 ] NTP has gained increasing attention over the last decade as being a highly controllable mechanism of delivering oxidative species to targets. The pioneering research by Lin et al. in 2017 first described NTP as a tool to enhance immunogenicity of cancer cells through the induction of the ER stress. It is being studied extensively in cancer research for its ability to selectively destroy tumor cells safely (in vivo).
NTP, the fourth state of matter, is an ionized gas composed of charged ions, electric fields, low amounts of UV light, and various reactive oxygen and nitrogen species (RONS). It is a high‐energy state that was originally utilized for sterilization due to its antimicrobial effects.[ 74 , 75 , 76 , 77 ] Initially, NTP‐mediated protection from disease was investigated in the context of disease prevention by inactivating the pathogen on surfaces or in aerosols. The concept of the antiviral effects of NTP is gaining particular attention these days due to the search for new solutions against COVID‐19. Its efficacy has been successfully demonstrated against viruses that cause foodborne illness such as norovirus as well as viruses that cause respiratory illness, including influenza, adenovirus, and respiratory syncytial virus, where NTP prevented viral replication and transmission.[ 78 , 79 , 80 , 81 , 82 , 83 , 84 , 85 ]
As NTP became widely recognized as a safe method for sterilization of various consumable goods and products in industry, the effects of NTP on mammalian cells started receiving attention. Shortly thereafter, the field of plasma medicine emerged, focusing largely on the antitumor potential associated with the NTP application.[ 86 ] During the generation of NTP, several different RONS are created, with hydroxyl ions, peroxides, superoxides, nitrates, nitrites, and peroxynitrites being the most biologically active.[ 86 , 87 , 88 , 89 , 90 ] When delivered to cells, these species induce oxidative stress, followed by the increased release of several DAMPs, including ATP and HMGB1. The plasma‐induced ER stress also causes the translocation of ER‐resident proteins like calreticulin (CRT) to the cell surface.[ 90 , 91 ] Together, these DAMPs act as multiple prophagocytic and prochemotactic ICD signals for APCs, an initiating step for all immune responses in the body (Figure 4). NTP may be applied directly to cells or indirectly via plasma‐exposed liquid or medium (PAL, PAM, or PALM) to obtain these immunogenic effects. Recent studies have also demonstrated success in causing ICD induction in tumors in many different animal models after direct or indirect exposure to NTP. Both modalities have also passed the “gold standard” test essential for any bona fide ICD inducer—the vaccination/challenge model. NTP has thus been characterized as a potent ICD inducer.
Figure 4.
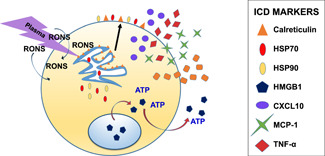
Treatment with NTP enhances immunogenicity of an exposed cell. NTP produces RONS that are biologically active. In response to these molecules, resident ER proteins such as calreticulin will become externalized and act as prophagocytic markers. The cell will also release specific proteins or molecules that act as “find me” signals, many of which are cytokines but also include ATP that has been translocated from the mitochondria. These will summon other immune cells to the site of stress and facilitate an immune response. CXCL, chemokine; ER, endoplasmic reticulum; HMGB1, high‐mobility group box1; HSP, heat shock protein; ICD, immunogenic cell death; MCP, monocyte chemoattractant protein; NTP, nonthermal plasma; RONS, reactive oxygen and nitrogen species; TNF, tumor necrosis factor
7. NTP ENHANCES IMMUNOGENICITY
The potential for using NTP to augment immunogenicity is evident in studies where either direct dielectric barrier discharge or plasma‐activated liquid applied to CT26 colon cancer cells or PDA6606 pancreatic cancer cells resulted in (a) increases in ICD markers CRT, HSP70, and HMGB1, as well as (b) the release of key proinflammatory cytokines that promote the APC function, such as MCP‐1, TNF‐α, and IFN‐γ. These results were corroborated by in vivo exposure of tumors where enhanced immunogenic phenotype of cancer cells was followed by an increased infiltration of macrophages into tumors. In addition, more tumor‐specific T cells were present in the spleens, indicating that local treatment of tumors produces a systemic protective response. T‐cell activation was validated ex vivo via co‐cultures of T cells isolated from these animals with NTP‐exposed CT26 cells.[ 62 , 92 ] The oxidative ER stress produced by the short‐term exposure of tumors to plasma RONS triggered the entire cascade of immune responses necessary to combat the disease. This would imply that a similar protective effect could be achieved by vaccination against cancer.
8. NTP FOR VACCINATION
The efficacy of NTP for vaccination against cancer was first tested in the CT26 colon cancer model.[ 62 ] An in vitro “cancer vaccine” was created by exposing CT26 cells to NTP and injected into mice after confirming the presence of DAMPs on these cells. A period of 1 week was allowed for the generation of de novo immune responses against this vaccine. The same mice were then challenged with an injection of live CT26 cells at a different site, and the tumor size was monitored for several days to assess the inhibition of cancer cell proliferation and tumor growth. If the initial vaccine was successful, the challenge should be protective against the development of new tumors at the challenge site. Immunization with NTP‐treated CT26 cells resulted in lower tumor volumes than that observed in control mice that did not receive the NTP‐CT26 vaccine, suggesting partial protection. Furthermore, 30% of the treated mice did not develop tumors and thus were considered fully protected.[ 62 ] This model of a whole‐cell vaccine suggests that plasma is sufficient to trigger events that are required for protective immune responses against a disease and may, in fact, result in NTP‐mediated autologous immunization.
The utility of NTP in combination with a peptide vaccine was also investigated in a mouse model of tumor growth and treatment.[ 62 ] To explore the benefits of the combination therapy, subcutaneous CT26 tumors in mice were treated with NTP in combination with the Ad5‐GUCY2C‐S1 vaccine, a vaccine already in clinical trials, targeting the GUCY2C tumor antigen of CT26 cells. Several weeks after treatment, antigen‐specific T cell activation was analyzed by IFN‐γ production. Comparisons between T‐cells isolated from mice treated with NTP alone and those that received the Ad5 vaccine alone revealed that levels of antigen‐specific cell activation were comparable. However, in mice that received both NTP and the Ad5 vaccine, activation of T cells targeting the GUCY2C tumor antigen was much more enhanced.[ 62 ] In addition, an expanded population of T cells targeting another tumor antigen, against which animals were not vaccinated (AH‐1), was also found, suggesting the generation of new antigens by NTP.
The implications of this observation would signify a much broader effect of plasma, relevant to modulation of the course of diseases. NTP could be useful as an immunomodulating tool in immunotherapeutic strategies that involve ex vivo exposure of cells and tissues, whereby cells not only emit DAMPs, but also display new antigens so that the de novo immune responses overcome the issue of the depleted immune function. Furthermore, proinflammatory cytokines and chemokines that attract and activate APCs are released in response to the NTP exposure. Successful vaccination strategies rely on differentiation of APCs promoted by antigens. Previous methods have relied on other maturation stimulants such as LPS along with the antigen. NTP application has been shown to alter the APC function directly in many studies. Kaushik et al.[ 93 ] demonstrated that the NTP exposure of monocytes caused them to differentiate into proinflammatory (M1) macrophages with antitumor and antimetastatic effects against cancer cells. Recruitment of APCs to the local site of NTP application has also been reported in other studies.[ 94 ] Yet, other studies document enhanced functional capacity of APCs.[ 95 , 96 , 97 ]
Furthermore, NTP may also have the potential to stimulate robust adaptive immune responses directly. In studies of NTP‐treated mouse melanoma cells, the upregulation of surface MHC I was observed in addition to increases in cell surface CRT.[ 98 ] This observation suggests an enhanced antigenicity of these cells, because an increase in surface MHC I potentially allows for more stable targeting by cytotoxic T lymphocytes (CTLs), which may thus mediate more efficient eradication upon encounter with these immune cells. This critical measure of immune protection after vaccination is particularly important in diseases in which progression is enabled by the downregulation of MHC I, including HIV‐1 infection. These results demonstrate the potential of NTP as not only a tool for enhancing immunogenicity of cells in chronic diseases, but also one that can reverse the immune dysfunctions that enable infections to persist.
Therefore, NTP fulfills many requirements for a good vaccine, at least in cancer model systems. It promotes antigenicity, stimulates innate immune responses (APC maturation and function directly, enhances APC function indirectly via DAMPs), modulates adaptive immune responses, and boosts the effects of other vaccines. These observations merit examination of NTP for vaccination (preventative and therapeutic) for infectious diseases.
Although several investigators report the direct antimicrobial effects of NTP, there is no evidence in the literature of immune protection conferred by plasma during an infectious disease. There is, however, evidence of NTP‐enhanced effects of vaccination against HIV‐1, a global public health challenge. A DNA vaccine targeting an HIV‐1 protein was administered to mice intradermally, followed by local NTP application at the site of injection.[ 99 ] The vaccine was selected, because it only produces weak immune responses. An enhanced cellular and humoral (antibody) immune response to the expressed antigen was reported in groups where vaccine and NTP were dispensed in conjunction as compared with animals that received the vaccine alone.[ 99 ] The authors propose that this adaptive immune response amplification may be a result of improved vaccine delivery and/or better local APC responses. Other studies suggested that NTP improves vaccine efficacy of inactivated whole‐pathogen vaccines. Vaccination with NTP‐inactivated H9N2 avian influenza and Newcastle viruses yielded higher antibody titers in chickens as compared with formaldehyde‐inactivated virus, presumably because NTP preserved the antigenic structure of the disabled virus better than formaldehyde.[ 88 ] The structural changes in the virus were dose‐dependent and correlated with the different plasma RONS.[ 88 ] This study highlights the ability of NTP to enhance adaptive immune responses after vaccination with NTP‐inactivated pathogens, besides aiding in vaccine delivery and promoting various APC functions at the site of vaccination. More detailed studies on other models of infection are needed to better elucidate additional immunological outcomes of plasma‐facilitated vaccinations.
Although not everything is known about the mechanisms that underlie NTP‐mediated enhancement of immunogenicity (besides induction of ICD), it is clear that NTP has the potential to serve as a multifaceted tool in vaccine development.
9. CONCLUSIONS
NTP has the potential to fulfill many requirements for an effective and efficient vaccination strategy through direct and indirect stimulation of both innate and adaptive immune responses. This conclusion is derived from the following effects of NTP:
NTP has demonstrated utility in enhancing immunogenicity of cells exposed to plasma RONS, which promotes APC recruitment, maturation, and cytokine production.
The direct exposure of APCs to NTP has also been shown to augment their migration, phagocytosis, and cytokine/chemokine production.
Increased recruitment of APCs is observed in tissues exposed to NTP.
Downstream proliferation and activation of T cells have been documented after the NTP exposure.
A direct stimulation of T cell function has been reported after the NTP exposure.
As a highly adaptable tool that continues to be extensively investigated in various disease models, NTP has the potential to synergize with existing vaccination strategies, including those involving ex vivo immunomodulation of immune cells. NTP also has the potential to be used as a partner in combinatorial vaccination strategies, as it was shown to promote immunogenicity of vaccines that were suboptimal when administered alone. The full potential of NTP to enhance immunogenicity needs to be explored further by assessment of the diversity of immunogenic antigens that are displayed post‐NTP exposure and how this NTP‐augmented antigen presentation impacts not just CTL responses but also B cell responses and the generation of antibodies. NTP could also become a novel tool in the fight against emerging infectious diseases, like COVID‐19, that even challenge the immune system of healthy individuals due to the host's immunological naivety of the pathogen. The most direct approach for NTP against SARS‐CoV‐2 would be inactivation of the virus in aerosols to interrupt transmission. However, virus particles inactivated by NTP can potentially be used for preventative vaccination to induce stronger and longer protection, and also facilitate delivery of SARS‐CoV‐2. The extensive range of vaccine forms, adjuvants, and dosing strategies offer novel opportunities for NTP to be developed as an emerging technology to be used in vaccinations against other infectious disease pathogens as well as cancers. The promotion of APC function, immune cell recruitment, T cell‐activation, and humoral responses suggests the potential for enhancing immunogenicity in the context of vaccination and chronic infectious diseases.
Mohamed H, Esposito RA, Kutzler MA, Wigdahl B, Krebs FC, Miller V. Nonthermal plasma as part of a novel strategy for vaccination. Plasma Process Polym. 2020;17:e2000051. 10.1002/ppap.202000051
REFERENCES
- 1. Short K. R., Kedzierska K., van de Sandt C. E., Front. Cell. Infect. Microbiol. 2018, 8, 343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Murray C. J., Lopez A. D., Chin B., Feehan D., Hill K. H., Lancet 2006, 368, 2211. [DOI] [PubMed] [Google Scholar]
- 3. Loggen H. G., Baerends‐Verburg J. L., Kreeftenberg J. G., J. Med. Primatol. 1983, 12, 192. [PubMed] [Google Scholar]
- 4. Kunasekaran M. P., Chen X., Costantino V., Chughtai A. A., MacIntyre C. R., Mil. Med. 2019, 184, e668. [DOI] [PubMed] [Google Scholar]
- 5. Kennedy R. B., Poland G. A., Ovsyannikova I. G., Oberg A. L., Asmann Y. W., Grill D. E., Vierkant R. A., Jacobson R. M., Vaccine 2016, 34, 3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jones T. R., Baird J. K., Basri H., Purnomo, Danudirgo E. W., Trop. Geogr. Med. 1991, 43, 1. [PubMed] [Google Scholar]
- 7. Sherwood J. A., Oster C. N., Adoyo‐Adoyo M., Beier J. C., Gachihi G. S., Nyakundi P. M., Ballou W. R., Brandling‐Bennett A. D., Schwartz I. K., Were J. B., Trans. R. Soc. Trop. Med. Hyg. 1991, 85, 336. [DOI] [PubMed] [Google Scholar]
- 8. Parrish C. R., Holmes E. C., Morens D. M., Park E. C., Burke D. S., Calisher C. H., Laughlin C. A., Saif L. J., Daszak P., Microbiol. Mol. Biol. Rev. 2008, 72, 457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang T., Wu Q., Zhang Z., Curr. Biol. 2020, 30, 1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Prompetchara E., Ketloy C., Palaga T., Asian Pac. J. Allergy. Immunol. 2020, 38, 1. [DOI] [PubMed] [Google Scholar]
- 11. Dampier W., Sullivan N. T., Chung C. H., Mell J. C., Nonnemacher M. R., Wigdahl B., Sci. Rep. 2017, 7, 14413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nagaraja P., Alexander H. K., Bonhoeffer S., Dixit N. M., Epidemics 2016, 14, 11. [DOI] [PubMed] [Google Scholar]
- 13. Schweighardt B., Wrin T., Meiklejohn D. A., Spotts G., Petropoulos C. J., Nixon D. F., Hecht F. M., J. Acquired Immune Defic. Syndr. 2010, 53, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Spector C., Mele A. R., Wigdahl B., Nonnemacher M. R., Med. Microbiol. Immunol. 2019, 208, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vasconcelos J. R., Dominguez M. R., Araujo A. F., Ersching J., Tararam C. A., Bruna‐Romero O., Rodrigues M. M., Front. Immunol. 2012, 3, 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Todryk S. M., Vaccines 2018, 6, 84. [Google Scholar]
- 17. Obaid A., Naz A., Ikram A., Awan F. M., Raza A., Ahmad J., Ali A., Sci. Rep. 2018, 8, 8874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ly A., Hansen D. S., Front. Immunol. 2019, 10, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Martin M. D., Badovinac V. P., Immunol. Res. 2014, 59, 35. [DOI] [PubMed] [Google Scholar]
- 20. Bannard O., Kraman M., Fearon D., Eur. J. Immunol. 2009, 39, 2083. [DOI] [PubMed] [Google Scholar]
- 21. Tennant S. M., Levine M. M., Vaccine 2015, 33, C36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sridhar S., Brokstad K. A., Cox R. J., Vaccines 2015, 3, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Workenhe S. T., Pol J. G., Lichty B. D., Cummings D. T., Mossman K. L., Cancer Immunol. Res. 2013, 1, 309. [DOI] [PubMed] [Google Scholar]
- 24. Ansaldi F., Trucchi C., Alicino C., Paganino C., Orsi A., Icardi G., Adv. Ther. 2016, 33, 1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Flasche S., Jit M., Rodriguez‐Barraquer I., Coudeville L., Recker M., Koelle K., Milne G., Hladish T. J., Perkins T. A., Cummings D. A., Dorigatti I., Laydon D. J., Espana G., Kelso J., Longini I., Lourenco J., Pearson C. A., Reiner R. C., Mier Y. T.‐R. L., Vannice K., Ferguson N., PLOS Med. 2016, 13, e1002181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Halasa N., Englund J. A., Nachman S., Weinberg G. A., Huber V. C., Allison K., Dubovsky F., Yi T., McCullers J. A., Flynn P. M., Vaccine 2011, 29, 4110. [DOI] [PubMed] [Google Scholar]
- 27. Gershon A. A., Steinberg S. P., Gelb L., Pediatrics 1986, 78, 757. [PubMed] [Google Scholar]
- 28. Coffman R. L., Sher A., Seder R. A., Immunity 2010, 33, 492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tamura S., Funato H., Nagamine T., Aizawa C., Kurata T., Vaccine 1989, 7, 503. [DOI] [PubMed] [Google Scholar]
- 30. Hashigucci K., Tamura S., Kurata T., Kamiya H., Ishidate T., Kansenshogaku Zasshi 1997, 71, 153. [DOI] [PubMed] [Google Scholar]
- 31. Shepardson K. M., Schwarz B., Larson K., Morton R. V., Avera J., McCoy K., Caffrey A., Harmsen A., Douglas T., Rynda‐Apple A., mBio 2017, 8. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32. Matsuo K., Yoshikawa T., Asanuma H., Iwasaki T., Hagiwara Y., Chen Z., Kadowaki S. E., Tsujimoto H., Kurata T., Tamura S. I., Vaccine 2000, 18, 2713. [DOI] [PubMed] [Google Scholar]
- 33. Tamura S., Yamanaka A., Shimohara M., Tomita T., Komase K., Tsuda Y., Suzuki Y., Nagamine T., Kawahara K., Danbara H., Vaccine 1994, 12, 419. [DOI] [PubMed] [Google Scholar]
- 34. de Pablo‐Maiso L., Domenech A., Echeverria I., Gomez‐Arrebola C., de Andres D., Rosati S., Gomez‐Lucia E., Reina R., Viruses 2018, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sardesai N. Y., Weiner D. B., Curr. Opin. Immunol. 2011, 23, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tateishi K., Fujihashi K., Yamamoto N., Hasegawa H., Ainai A., Sato K., Iho S., Yamamoto S., Maeyama J. I., Odagiri T., Asanuma H., Vaccine 2019, 37, 5382. [DOI] [PubMed] [Google Scholar]
- 37. World Health Organization (W.H.O.) . Table 1: Summary of WHO Position Papers—Recommendations for Routine Immunization, https://www.who.int/immunization/policy/Immunization_routine_table1.pdf?ua=1 (accessed: April 2019).
- 38. Eibl M. M., Mannhalter J. W., Zlabinger G., N. Engl. J. Med. 1984, 310, 198. [DOI] [PubMed] [Google Scholar]
- 39. Kanra G., Ceyhan M., Topal B., Bogaerts H., Vandevoorde D., Pediatr. Infect. Dis. J. 1992, 11, 598. [DOI] [PubMed] [Google Scholar]
- 40. Pichichero M. E., Badgett J. T., Rodgers G. C. Jr., McLinn S., Trevino‐Scatterday B., Nelson J. D., Pediatr. Infect. Dis. J. 1987, 6, 352. [DOI] [PubMed] [Google Scholar]
- 41. Ullberg‐Olsson K., Eriksson E., Eur. Surg. Res. 1975, 7, 249. [DOI] [PubMed] [Google Scholar]
- 42. Pichichero M. E., Passador S., Clin. Infect. Dis. 1997, 25, 1378. [DOI] [PubMed] [Google Scholar]
- 43. Shi S., Zhu H., Xia X., Liang Z., Ma X., Sun B., Vaccine 2019, 37, 3167. [DOI] [PubMed] [Google Scholar]
- 44. Marrack P., McKee A. S., Munks M. W., Nat. Rev. Immunol. 2009, 9, 287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Knudsen N. P., Olsen A., Buonsanti C., Follmann F., Zhang Y., Coler R. N., Fox C. B., Meinke A., D'Oro U., Casini D., Bonci A., Billeskov R., De Gregorio E., Rappuoli R., Harandi A. M., Andersen P., Agger E. M., Sci. Rep. 2016, 6, 19570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bode C., Zhao G., Steinhagen F., Kinjo T., Klinman D. M., Expert Rev. Vaccines 2011, 10, 499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Thompson B. S., Chilton P. M., Ward J. R., Evans J. T., Mitchell T. C., J. Leukocyte Biol. 2005, 78, 1273. [DOI] [PubMed] [Google Scholar]
- 48. Pichichero M. E., Blatter M. M., Kennedy W. A., Hedrick J., Descamps D., Friedland L. R., Pediatrics 2006, 117, 1084. [DOI] [PubMed] [Google Scholar]
- 49. Marin‐Acevedo J. A., Soyano A. E., Dholaria B., Knutson K. L., Lou Y., J. Hematol. Oncol. 2018, 11, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Aliru M. L., Schoenhals J. E., Venkatesulu B. P., Anderson C. C., Barsoumian H. B., Younes A. I., Ls K. M., Soeung M., Aziz K. E., Welsh J. W., Krishnan S., Immunotherapy 2018, 10, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ali M., Clemens J., Front. Public Health 2019, 7, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Keenan B. P., Jaffee E. M., Semin. Oncol. 2012, 39, 276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zitvogel L., Kepp O., Senovilla L., Menger L., Chaput N., Kroemer G., Clin. Cancer Res. 2010, 16, 3100. [DOI] [PubMed] [Google Scholar]
- 54. Garg A. D., Galluzzi L., Apetoh L., Baert T., Birge R. B., Bravo‐San Pedro J. M., Breckpot K., Brough D., Chaurio R., Cirone M., Coosemans A., Coulie P. G., De Ruysscher D., Dini L., de Witte P., Dudek‐Peric A. M., Faggioni A., Fucikova J., Gaipl U. S., Golab J., Gougeon M. L., Hamblin M. R., Hemminki A., Herrmann M., Hodge J. W., Kepp O., Kroemer G., Krysko D. V., Land W. G., Madeo F., Manfredi A. A., Mattarollo S. R., Maueroder C., Merendino N., Multhoff G., Pabst T., Ricci J. E., Riganti C., Romano E., Rufo N., Smyth M. J., Sonnemann J., Spisek R., Stagg J., Vacchelli E., Vandenabeele P., Vandenberk L., Van den Eynde B. J., Van Gool S., Velotti F., Zitvogel L., Agostinis P., Front. Immunol. 2015, 6, 588.26635802 [Google Scholar]
- 55. Tanaka M., Kataoka H., Yano S., Sawada T., Akashi H., Inoue M., Suzuki S., Inagaki Y., Hayashi N., Nishie H., Shimura T., Mizoshita T., Mori Y., Kubota E., Tanida S., Takahashi S., Joh T., Oncotarget 2016, 7, 47242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Economopoulou P., Koutsodontis G., Strati A., Kirodimos E., Giotakis E., Maragoudakis P., Prikas C., Papadimitriou N., Perisanidis C., Gagari E., Kotsantis I., Vagia E., Anastasiou M., Gkotzamanidou M., Kavourakis G., Lianidou E., Psyrri A., Oral Oncol. 2019, 94, 93. [DOI] [PubMed] [Google Scholar]
- 57. Lu J., Liu X., Liao Y. P., Wang X., Ahmed A., Jiang W., Ji Y., Meng H., Nel A. E., ACS Nano 2018, 12, 11041. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 58. Di Somma S., Iannuzzi C. A., Passaro C., Forte I. M., Iannone R., Gigantino V., Indovina P., Botti G., Giordano A., Formisano P., Portella G., Malfitano A. M., Pentimalli F., Front. Oncol. 2019, 9, 564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hradilova N., Sadilkova L., Palata O., Mysikova D., Mrazkova H., Lischke R., Spisek R., Adkins I., PLOS One 2017, 12, e0171539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Garg A. D., Agostinis P., Photochem. Photobiol. Sci. 2014, 13, 474. [DOI] [PubMed] [Google Scholar]
- 61. Gameiro S. R., Jammeh M. L., Wattenberg M. M., Tsang K. Y., Ferrone S., Hodge J. W., Oncotarget 2014, 5, 403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lin A. G., Xiang B., Merlino D. J., Baybutt T. R., Sahu J., Fridman A., Snook A. E., Miller V., Oncoimmunology 2018, 7, e1484978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Terenzi A., Pirker C., Keppler B. K., Berger W., J. Inorg. Biochem. 2016, 165, 71. [DOI] [PubMed] [Google Scholar]
- 64. Fucikova J., Moserova I., Truxova I., Hermanova I., Vancurova I., Partlova S., Fialova A., Sojka L., Cartron P. F., Houska M., Rob L., Bartunkova J., Spisek R., Int. J. Cancer 2014, 135, 1165. [DOI] [PubMed] [Google Scholar]
- 65. Galluzzi L., Kepp O., Kroemer G., Oncoimmunology 2013, 2, e26536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Galluzzi L., Kepp O., Kroemer G., EMBO J. 2012, 31, 1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Turubanova V. D., Balalaeva I. V., Mishchenko T. A., Catanzaro E., Alzeibak R., Peskova N. N., Efimova I., Bachert C., Mitroshina E. V., Krysko O., Vedunova M. V., Krysko D. V., J. Immunother. Cancer 2019, 7, 350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sun C., Wang H., Mao S., Liu J., Li S., Wang J., Immunol. Lett. 2015, 164, 65. [DOI] [PubMed] [Google Scholar]
- 69. Chen C., Ni X., Jia S., Liang Y., Wu X., Kong D., Ding D., Adv. Mater. 2019, 31, e1904914. [DOI] [PubMed] [Google Scholar]
- 70. Sagar V., Vatapalli R., Lysy B., Pamarthy S., Anker J. F., Rodriguez Y., Han H., Unno K., Stadler W. M., Catalona W. J., Hussain M., Gill P. S., Abdulkadir S. A., Cell Death Dis. 2019, 10, 801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kepp O., Menger L., Vacchelli E., Locher C., Adjemian S., Yamazaki T., Martins I., Sukkurwala A. Q., Michaud M., Senovilla L., Galluzzi L., Kroemer G., Zitvogel L., Cytokine Growth Factor Rev. 2013, 24, 311. [DOI] [PubMed] [Google Scholar]
- 72. Moserova I., Truxova I., Garg A. D., Tomala J., Agostinis P., Cartron P. F., Vosahlikova S., Kovar M., Spisek R., Fucikova J., Oncoimmunology 2017, 6, e1258505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tanaka H., Mizuno M., Katsumata Y., Ishikawa K., Kondo H., Hashizume H., Okazaki Y., Toyokuni S., Nakamura K., Yoshikawa N., Kajiyama H., Kikkawa F., Hori M., Sci. Rep. 2019, 9, 13657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sohbatzadeh F., Hosseinzadeh Colagar A., Mirzanejhad S., Mahmodi S., Appl. Biochem. Biotechnol. 2010, 160, 1978. [DOI] [PubMed] [Google Scholar]
- 75. Fricke K., Koban I., Tresp H., Jablonowski L., Schroder K., Kramer A., Weltmann K. D., von Woedtke T., Kocher T., PLOS One 2012, 7, e42539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Park S. R., Lee H. W., Hong J. W., Lee H. J., Kim J. Y., Choi B. B., Kim G. C., Jeon Y. C., J. Nanobiotechnol. 2014, 12, 29. [Google Scholar]
- 77. Sun Y., Yu S., Sun P., Wu H., Zhu W., Liu W., Zhang J., Fang J., Li R., PLOS One 2012, 7, e40629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ahlfeld B., Li Y., Boulaaba A., Binder A., Schotte U., Zimmermann J. L., Morfill G., Klein G., mBio 2015, 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Aboubakr H. A., Sampedro Parra F., Collins J., Bruggeman P., Goyal S. M., Food Microbiol. 2020, 85, 103307. [DOI] [PubMed] [Google Scholar]
- 80. Venezia R. A., Orrico M., Houston E., Yin S. M., Naumova Y. Y., Infect. Control Hosp. Epidemiol. 2008, 29, 430. [DOI] [PubMed] [Google Scholar]
- 81. Terrier O., Essere B., Yver M., Barthelemy M., Bouscambert‐Duchamp M., Kurtz P., VanMechelen D., Morfin F., Billaud G., Ferraris O., Lina B., Rosa‐Calatrava M., Moules V., J. Clin. Virol. 2009, 45, 119. [DOI] [PubMed] [Google Scholar]
- 82. Marsit N. M., Sidney L. E., Branch M. J., Wilson S. L., Hopkinson A., Plasma Processes Polym. 2017, 14, 1600134. [Google Scholar]
- 83. Alshraiedeh N. H., Alkawareek M. Y., Gorman S. P., Graham W. G., Gilmore B. F., J. Appl. Microbiol. 2013, 115, 1420. [DOI] [PubMed] [Google Scholar]
- 84. Zimmermann J. L., Dumler K., Shimizu T., Morfill G. E., Wolf A., Boxhammer V., Schlegel J., Gansbacher B., Anton M., J. Phys. D: Appl. Phys. 2011, 44, 505201. [Google Scholar]
- 85. Sakudo A., Toyokawa Y., Imanishi Y., Murakami T., Mater. Sci. Eng., C 2017, 74, 131. [DOI] [PubMed] [Google Scholar]
- 86. Kalghatgi S., Kelly C. M., Cerchar E., Torabi B., Alekseev O., Fridman A., Friedman G., Azizkhan‐Clifford J., PLOS One 2011, 6, e16270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wende K., Williams P., Dalluge J., Van Gaens W., Aboubakr H., Bischof J., von Woedtke T., Goyal S. M., Weltmann K.‐D., Bogaerts A., Masur K., Bruggeman P. J., Biointerphases 2015, 10, 029518. [DOI] [PubMed] [Google Scholar]
- 88. Wang G., Zhu R., Yang L., Wang K., Zhang Q., Su X., Yang B., Zhang J., Fang J., Vaccine 2016, 34, 1126. [DOI] [PubMed] [Google Scholar]
- 89. Vandamme M., Robert E., Lerondel S., Sarron V., Ries D., Dozias S., Sobilo J., Gosset D., Kieda C., Legrain B., Pouvesle J. M., Pape A. L., Int. J. Cancer 2012, 130, 2185. [DOI] [PubMed] [Google Scholar]
- 90. Ruwan Kumara M. H., Piao M. J., Kang K. A., Ryu Y. S., Park J. E., Shilnikova K., Jo J. O., Mok Y. S., Shin J. H., Park Y., Kim S. B., Yoo S. J., Hyun J. W., Oncol. Rep. 2016, 36, 2268. [DOI] [PubMed] [Google Scholar]
- 91. Lin A., Truong B., Patel S., Kaushik N., Choi E. H., Fridman G., Fridman A., Miller V., Int. J. Mol. Sci. 2017, 18, 966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Freund E., Liedtke K. R., van der Linde J., Metelmann H. R., Heidecke C. D., Partecke L. I., Bekeschus S., Sci. Rep. 2019, 9, 634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kaushik N. K., Kaushik N., Adhikari M., Ghimire B., Linh N. N., Mishra Y. K., Lee S. J., Choi E. H., Cancers 2019, 11, 842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Ranieri P., Shrivastav R., Wang M., Lin A., Fridman G., Fridman A. A., Han L.‐H., Miller V., Plasma Med. 2017, 7, 283. [Google Scholar]
- 95. Miller V., Lin A., Fridman G., Dobrynin D., Fridman A., Plasma Processes Polym. 2014, 11, 1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Miller V., Lin A., Kako F., Gabunia K., Kelemen S., Brettschneider J., Fridman G., Fridman A., Autieri M., Phys. Plasmas 2015, 22, 122005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Kaushik N. K., Kaushik N., Min B., Choi K. H., Hong Y. J., Miller V., Fridman A., Choi E. H., J. Phys. D: Appl. Phys. 2016, 49, 084001. [Google Scholar]
- 98. Bekeschus S., Rodder K., Fregin B., Otto O., Lippert M., Weltmann K. D., Wende K., Schmidt A., Gandhirajan R. K., Oxid. Med. Cell Longevity 2017, 2017, 4396467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Connolly R. J., Chapman T., Hoff A. M., Kutzler M. A., Jaroszeski M. J., Ugen K. E., Hum. Vaccin. Immunother. 2012, 8, 1729. [DOI] [PMC free article] [PubMed] [Google Scholar]