

Abstract
Cytokine storm syndrome (CSS) is a critical clinical condition induced by a cascade of cytokine activation, characterized by overwhelming systemic inflammation, hyperferritinaemia, haemodynamic instability and multiple organ failure (MOF). At the end of 2019, the disease caused by severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) emerged in Wuhan, China, and rapidly developed into a global pandemic. More and more evidence shows that there is a dramatic increase of inflammatory cytokines in patients with COVID‐19, suggesting the existence of cytokine storm in some critical illness patients. Here, we summarize the pathogenesis, clinical manifestation of CSS, and highlight the current understanding about the recognition and potential therapeutic options of CSS in COVID‐19.
Keywords: COVID‐19, cytokine storm syndrome, recognition, SARS‐CoV‐2, treatment

Introduction
Synonymous with cytokine release syndrome (CRS), cytokine storm syndrome (CSS) is a cytokine‐mediated systemic inflammatory response induced by a variety of initiating factors, resulting in a clinical presentation of unremitting high fever, lymphadenopathy, hepatosplenomegaly, cytopaenia, hyperferritinaemia and central nervous system (CNS) abnormalities, and, if untreated, progression to multiple organ failure (MOF) is almost inevitable [1, 2, 3, 4, 5, 6]. The hallmark of CSS is an unchecked feed‐forward activation and amplification of host immune, causing the massive release of a wide range of cytokines, such as interferon (IFN)‐γ, tumour necrosis factor (TNF), interleukin (IL)‐1, IL‐6 and IL‐18, which contributes to the formation of a cytokine storm [3, 6, 7, 8]. CSS was firstly reported in the early 1990s, as a systemic reaction when the anti‐T‐cell antibody muromonab‐CD3 (OKT3) was used for the immunosuppressive treatment of solid organ transplantation [9]. The triggers leading to CSS are heterogeneous and derive from infection [10, 11], malignant tumour [12], rheumatic disease [13, 14], iatrogenic injury [15] and the application of immunotherapeutic drugs [7, 16, 17], amongst which infection is the most common cause [1].
Coronaviruses (CoV) are enveloped, positive‐sense RNA viruses infecting multiple host species, including humans and several other vertebrates, which may cause cross‐species transmission [18]. Generally, coronaviruses infecting humans can be classified into low pathogenic CoVs and highly pathogenic CoVs. Low pathogenic CoVs infect upper airway and cause seasonal respiratory illness, whilst highly pathogenic CoVs, such as severe acute respiratory syndrome CoV (SARS‐CoV) and Middle East respiratory syndrome CoV (MERS‐CoV), infect the lower respiratory tract and may lead to acute lung injury (ALI), acute respiratory distress syndrome (ARDS) and even death [19, 20]. Since December 2019, the severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2; previously named 2019 novel coronavirus or 2019‐nCoV) disease (COVID‐19) had rapidly spread from Wuhan of China to over 200 countries and regions in the world, caused global pandemic as claimed by WHO [21, 22, 23]. The disease has affected over 6 500 000 people and killed more than 380 000 by 5 June 2020 [24]. Previous studies have shown that cytokine storms are associated with the deterioration of patients with SARS and MERS [10]. Similar to other highly pathogenic CoVs, there is growing evidence that cytokine storms caused by the excessive production of inflammatory factors may participate in the pathogenesis of patients with COVID‐19, which may be one of the key factors leading to the rapid worsening of the disease [25, 26].
Therefore, early identification and treatment of CSS may be crucial for improving the outcome of critically ill patients with COVID‐19. Here, we summarize the pathogenesis, clinical manifestations of CSS, and highlight the current perspective about the recognition and potential therapeutics for CSS in COVID‐19.
Pathogenesis of CSS
To date, the pathogenesis of CSS has not been fully elucidated. Previous studies have shown that the development of CSS involves the imbalance of proinflammatory and anti‐inflammatory mechanisms and the interaction of a variety of cells and cytokines, resulting in immune regulation disorder, causing a series of clinical manifestations [3, 8, 27]. The pathogenesis of CSS is summarized in Fig. 1.
Fig. 1.
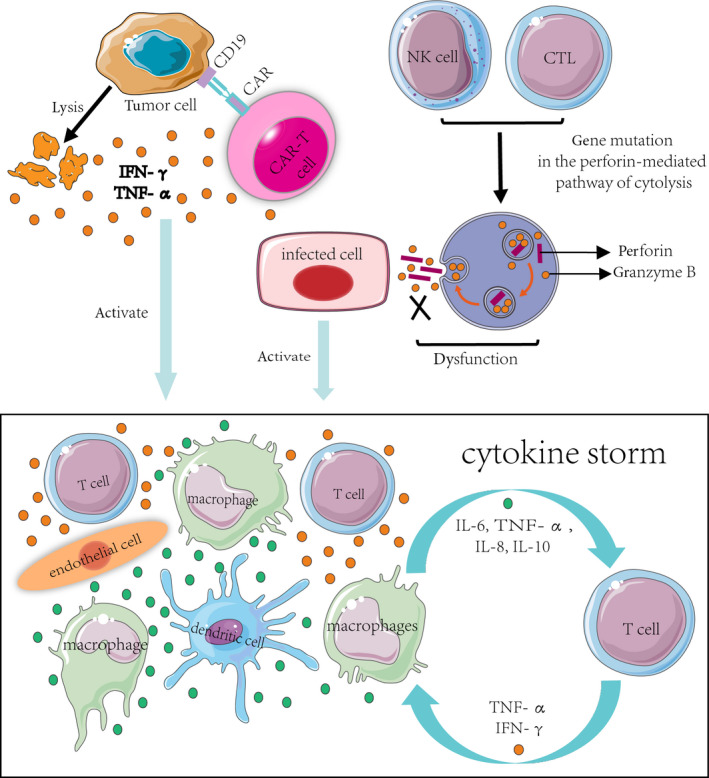
Proposed pathogenesis of CSS. In the setting of CAR T‐cell therapy, CAR T cells can recognize target cells (tumour cells) and induce the lysis of target cells, along with the activation of CAR T cells and T cell, causing a consecutive release of cytokines including IFN‐γ or TNF‐α. These cytokines trigger a cascade reaction by activation of innate immune cells including macrophages, DCs and endothelial cells with further cytokine releasing, which finally leads to a cytokine storm. In the setting of HLH, mutations in perforin coding gene or genes essential for perforin transport will cause a failure of normal cytolytic function and inability to clear the antigenic stimulus, leading to the persistent activation of macrophages and T cells by the infected cells, accompanied by excessive secretion of proinflammatory cytokines, which finally leads to a cytokine storm. Abbreviations: CSS: cytokine storm syndrome; CAR: chimeric antigen receptor; IFN‐γ: interferon‐gamma; TNF‐α: tumour necrosis factor‐alpha; IL: interleukin; CTL: cytotoxic T lymphocytes; NK cell: natural killer cell; and DC: dendritic cell.
Cytolytic cell dysfunction
CSS has been described in haemophagocytic lymphohistiocytosis (HLH), which can be classified into primary (pHLH) and secondary HLH (sHLH). pHLH, also referred to as familial HLH (fHLH), often occurs in infants, whilst sHLH is mainly seen in adults and can be triggered by malignancy, infectionand autoimmunity [28]. Macrophage activation syndrome (MAS) belongs to the spectrum of sHLH and complicates several rheumatic diseases, especially systemic juvenile idiopathic arthritis (sJIA) [29, 30, 31]. The most progress in the understanding of the mechanism of CSS has been made in pHLH, which is a result of mutations in genes involved in the perforin‐mediated pathway of cytolysis shared by natural killer cells (NK cells) and cytotoxic CD8+ T cells [32]. Normally, perforin in NK cells and cytotoxic CD8+ T cells can be packaged into cytolytic granules and then released into the immunologic synapse to form a pore between the lytic cell and the target cell (infected or tumour cells), allowing granzyme B, co‐packaged with perforin, to induce the apoptotic cell death of the target cell [8]. Therefore, mutations in perforin coding gene or genes essential for perforin transport will cause a failure of normal cytolytic function and inability to clear the antigenic stimulus, leading to the hyperactivation of macrophages and T helper 1 (Th1) cells accompanied by excessive secretion of proinflammatory cytokines, causing a self‐amplifying hyperinflammatory state known as the cytokine storm [30, 33]. In addition, some work also suggested that more than 40% of sHLH patients had heterozygous mutations in the fHLH‐associated genes [34]. Similarly, gene mutations in sHLH alter cytolytic function in cytotoxic CD8+ T cells and NK cells as well.
Activation of immune and nonimmune cells
Macrophages
Previous studies have shown that haemophagocytic macrophages were found to produce the proinflammatory cytokine TNF‐α and IL‐6 in the liver of MAS patients, suggesting that activated macrophages may participate in the pathogenesis of MAS [35]. As we know, activated macrophages can produce a variety of cytokines, especially TNF‐α and a variety of interleukins (ILs, e.g. IL‐6, IL‐1 βand IL‐18), which may trigger the cascade reaction of inflammatory factors, and finally form a cytokine storm [8]. Macrophages also play a crucial role in the CSS induced by chimeric antigen receptor (CAR) T‐cell therapy, which has demonstrated remarkable success in the treatment of CD19‐positive B‐cell malignant tumours. Van der Stegen and his colleagues showed that both toxicity and IL‐6 release could be ameliorated by prior macrophage depletion in a severe combined immunodeficiency (SCID)/beige mouse model of CSS induced by ErbB‐specific CAR T‐cell therapy [36]. Subsequently, Singh and his colleagues demonstrated that monocyte‐derived cells were responsible for IL‐6 secretion in response to CAR T‐cell activation and IL‐6 blockade did not affect the therapeutic effect of CAR T cell [37]. Similarly, using a murine model of CSS that developed within 2–3 days of CAR T‐cell infusion, Giavridis and his colleagues showed that the severity of CSS was not mediated by CAR T cell‐derived cytokines but by IL‐6, IL‐1 produced by recipient macrophages [38]. Further, Norelli et al found that human monocytes were the major source of IL‐1 and IL‐6 during CSS in the humanized mice with high leukaemia burden. Accordingly, they found that both monocyte depletion and blocking IL‐6 or IL‐1 receptor could avoid the occurrence of CSS [39]. All of these findings suggest that monocytes/macrophages play a crucial role in the development of CSS after CAR T‐cell therapy and enable new therapeutic interventions in this filed. Previous studies also demonstrated that macrophages infected by SARS and MERS‐CoV showed delayed but elevated levels of IFN and other proinflammatory cytokines, suggesting macrophages could play an important role in SARS and MERS pathogenesis [10].
Dendritic cells
The role of dendritic cells (DCs) in the pathogenesis of CSS is mainly mediated by their ability to present antigen to T cells [40]. Hermans et al demonstrated that cytotoxic T cell‐mediated clearance of antigen‐loaded DC may serve as a negative feedback mechanism to limit the activity of DC within the lymph node [41]. As we mentioned above, patients with pHLH or some patients with MAS have cytolytic cell dysfunction, which means cytotoxic T cells may fail to clear the antigen‐loaded DCs. This leads to persistent activation of DCs and antigen presentation to T cells [42], which in turn leads to the overproduction of proinflammatory cytokines.
Endothelial cells
Endothelial activation may also participate in the pathogenesis of CSS, which may contribute to haemodynamic instability, capillary leak and consumptive coagulopathy [16]. On the one hand, the excessive production of proinflammatory cytokines such as IL‐6 and IFN‐γ can induce endothelial activation in severe CSS, characterized by the release of stored von Willebrand factor and angiopoietin‐2. Angiopoietin‐2 can further amplify endothelial activation [43]. On the other hand, Amrom et al reported that vascular endothelial cell was also a key source of IL‐6 in a patient who died of CSS after CAR T‐cell therapy [44].
Excessive production of cytokines
Previous studies have shown that a variety of cytokines can be markedly increased in patients with CSS, which may vary according to the heterogeneity of the disease background (Table 1). In the setting of MAS, several studies suggested that IL‐1 receptor antagonists and IL‐6 antagonists were effective in patients with sJIA complicated with MAS [13, 45, 46, 47], but there is still no evidence that IL‐1β and IL‐6 are directly related to the pathogenesis of MAS. The increase of IL‐18 level is also related to the susceptibility of MAS in sJIA, but the specific mechanism is still unclear [48]. In the context of CAR T‐cell therapy, CSS is induced by the excessive release of IFN‐γ by activated T cells or tumour cells. IFN‐γ can then facilitate the activation of other immune cells, especially macrophages. The activated macrophages produce excessive amounts of additional cytokines such as IL‐6, TNF‐α, IL‐8 and IL‐10, which leads to other corresponding clinical manifestations [16, 49]. In the setting of influenza virus‐related CSS, Farrar and his colleagues found that patients infected with H5N1 had higher levels of monocyte chemoattractant protein 1 (MCP‐1), monokine induced by IFN‐gamma (MIG), interferon‐gamma‐induced protein‐10 (IP‐10) and IL‐8 than patients infected with seasonal H1N1 common influenza [11]. In HIN1‐related CSS, Kelvin and his colleagues demonstrated that patients with more severe infection had higher secretion of Th1 and Th17 cytokines, such as IL‐15, IL‐12p70 and IL‐6 [50]. Moreover, previous studies also confirmed that cytokines played an important role in the pathogenesis of severe CoV infection. The serum proinflammatory cytokines (IFN‐γ, transforming growth factor‐β (TGF‐β), IL‐1, IL‐6, IL‐12) in severe SARS patients were significantly higher than those with mild to moderate symptoms, and serum proinflammatory cytokines (IFN‐α, IL‐6, IL‐8) in severe MERS patients were significantly increased as well [10].
Table 1.
Major cytokines involved in CSS in the context of different diseases
| Disease background | Cytokines | References |
|---|---|---|
| MAS | IFN‐γ, TNF‐α, IL‐1β, IL‐2, sIL2Rα, IL‐6, IL‐10, IL‐12, IL‐18 | [51, 52, 53] |
| CAR T cell therapy | IFN‐γ, TNF‐α, IL‐1, IL‐2, sIL2Rα, IL‐6, IL‐8, IL‐10, IL‐12, MCP‐1, MIP‐1a, GM‐CSF | [16, 49] |
| H5N1 influenza | IFN‐γ, IL‐6, IL‐8, IL‐10, MCP‐1, MIG, IP‐10, | [11] |
| H1N1 influenza | IFN‐γ, TNF‐α, IL‐6, IL‐8, IL‐9, IL‐17, IL‐15, IL‐12p70 | [50] |
| SARS | IFN‐γ, IL‐1, IL‐6, IL‐12, TGF‐β, MCP‐1, MIG, IP‐10, IL‐8 | [10] |
| MERS | IFN‐α, IL‐1β, IL‐2, IL‐6, IL‐8, MCP‐1, MIP‐1a, CCL‐5 | [10] |
CAR T cell, chimeric antigen receptor T cell; CCL, chemokine (C‐C motif) ligand; CSS, cytokine storm syndrome; GM‐CSF, granulocyte‐macrophage colony‐stimulating factor; IFN‐γ, interferon‐γ; IL, interleukin; IP‐10, interferon‐gamma‐induced protein‐10; MAS, macrophage activation syndrome; MCP‐1, monocyte chemoattractant protein 1; MERS, Middle East respiratory syndrome; MIG, monokine induced by IFN‐gamma; MIP‐1a, macrophage inflammatory protein 1a; SARS, severe acute respiratory syndrome; sIL2Rα, soluble IL2 receptor α; TGF‐β, transforming growth factor‐β; TNF‐α, tumour necrosis factor‐α.
Possible mechanisms of CSS in COVID‐19
The cytokine storm in COVID‐19 may have some differences from the cytokine storms in other clinical settings. Remarkably, the autopsy findings revealed that the lymphoid tissues and organs had been destroyed in COVID‐19 patients [54, 55], which is very unusual from CSS in sepsis and CAR T‐cell therapy. Spleen atrophy and lymph node atrophy are observed in patients with COVID‐19 [54, 55], whilst in other CSS‐related diseases, lymphadenopathy and splenomegaly are more common [4]. However, the specific mechanisms for these differences remain unclear and need to be further clarified. Here, we summarize the evidence of CSS in COVID‐19 and the possible pathogenesis of cytokine storm induced by SARS‐CoV‐2.
The direct effect of SARS‐CoV‐2
Coronaviruses (CoVs) are enveloped, positive‐sense, single‐stranded RNA viruses, which have caused two large‐scale pandemics in the last two decades, SARS and MERS [10]. Spike (S) proteins of coronaviruses, including the SARS‐CoV, facilitate viral entry into their target cells via the interaction with functional cellular receptor identified as angiotensin‐converting enzyme 2 (ACE2) [56]. Generally, ACE2 was highly expressed in alveolar epithelial cells, vascular endothelial cells, intestinal epithelial cells, cardiomyocytes and renal proximal tubular cells [57]. Functionally, ACE2, belonging to the ACE family, inactivates angiotensin Ⅱ(AngⅡ) and generates angiotensin 1–7, a biologically active heptapeptide characterized by a potent vasodilator function [58]. It has been demonstrated that the binding of the coronavirus spike protein to ACE2 leads to the down‐regulation of ACE2, which in turn results in excessive production of vasoconstrictor AngⅡ and reduced production of vasodilator angiotensin 1–7. AngⅡ also plays the role as a proinflammatory cytokine via angiotensin receptor 1 (AT1R). The AngⅡ‐AT1R axis further activates NF‐κB and metalloprotease 17 (ADAM17), which stimulates the production of the mature form of epidermal growth factor receptor (EGFR) ligands and TNF‐α [59]. Additionally, the induction of ADAM17 also processes the membrane form of IL‐6Rα to the soluble form (sIL‐6Rα), followed by the gp130‐mediated activation of STAT3 via the sIL‐6Rα‐IL‐6 complex. The activation of both NF‐κB and STAT3, which in turn activate the IL‐6 amplifier (IL‐6 Amp), a mechanism for the hyperactivation of NF‐κB by STAT3, will lead to a hyperinflammatory state, resulting in increased pulmonary vascular permeability [60]. Apart from ACE2, the entry of CoVs also requires S protein priming by cellular proteases, and SARS‐CoV employs the cellular serine protease TMPRSS2 for S protein priming [61]. Recent studies also indicated that ACE2 was a functional receptor of SARS‐CoV‐2 [62, 63]. Additionally, Wrapp and his colleagues found that the receptor‐binding ability of SARS‐CoV‐2 was 10‐20 times stronger than that of SARS‐CoV [64]. Moreover, Hoffmann and his colleagues found that SARS‐CoV‐2 also used the serine protease TMPRSS2 for S protein priming [65]. Based on the similarity between SARS‐CoV‐2 and SARS‐CoV, a similar mechanism of hyperinflammatory state can be expected for SARS‐CoV‐2. Furthermore, using the (AT1R) blockers and TMPRSS2 inhibitors may be a promising treatment option for SARS‐CoV‐2 infection.
Immune dysfunction
A postmortem study in patients with COVID‐19 demonstrated that the pathological features of COVID‐19 greatly resembled those seen in SARS and MERS, with a histological examination showing a bilateral diffuse alveolar damage with cellular fibromyxoid exudates [66, 67]. Of note, using the method of flow cytometry, they found the number of peripheral CD4+ and CD8+ T cells substantially reduced, whilst the status of these cells was over‐activated. CD8+T cells were found to harbour high concentrations of cytotoxic granules, such as perforin and granulysin. Additionally, there was an increased concentration of highly proinflammatory Th17 cells [66]. In summary, the over‐activation of T cells, including the increase of proinflammatory Th17 and high cytotoxicity of CD8+ T cells, may partly account for the severe immune injury in this patient. Zhou’s group also demonstrated that CD4+ T lymphocytes are rapidly activated to become pathogenic T helper (Th) 1 cells and inflammatory CD14+CD16+ monocytes were found in peripheral blood after the SARS‐CoV‐2 infection [68]. In a retrospective study of 123 patients with COVID‐19, the percentage of NK cell reduced 34.31% and 47.62% in mild and severe groups respectively, suggesting a more obvious NK cell reduction in severe patients [69]. In addition, other autopsy findings of COVID‐19 patients also demonstrated the characteristic diffused alveolar damage, with major monocytes and macrophages infiltration rather than lymphocytes [54, 55], indicating that macrophages may also play an important role in cytokine storm induced by SARS‐CoV‐2.
Elevation of inflammatory cytokines
Previous studies showed that high serum levels of proinflammatory cytokines and chemokines were found in SARS and MERS patients, indicating the exaggerated cytokine and chemokine responses played an important role in the pathogenesis of CoV infection [10]. A retrospective study also found that compared with nonsevere patients, intensive care unit (ICU) patients showed higher plasma concentrations of IL‐2, IL‐7, IL‐10, granulocyte colony‐stimulating factor (GCSF), IP‐10, MCP‐1, MIP‐1aand TNF‐α, suggesting there might be a cytokine storm in the body of severe patients [26]. Zhou’s group revealed that a high percentage of granulocyte‐macrophage colony‐stimulating factor (GM‐CSF)+ and IL‐6 + expressions could be found in CD4 + T cells from COVID‐19 patients in both ICU and non‐ICU patients compared to healthy controls. CD14 + CD16+ inflammatory monocyte from COVID‐19 patients also showed the capability to secrete GM‐CSF. Importantly, the level of IL‐6 secreted from these inflammatory monocytes was significantly higher in ICU patients than non‐ICU patients, suggesting targeting GM‐CSF or IL‐6 may be effective in blocking inflammatory storms in COVID‐19 patients [68].
Finally, it can be speculated that the direct effect of the virus, immune response and massive release of inflammatory factors eventually contribute to a cytokine storm in the pathogenesis of COVID‐19 (Fig. 2). However, the number of related studies is still very small, and further research is needed to investigate the mechanism of cytokine storms in COVID‐19 patients.
Fig. 2.
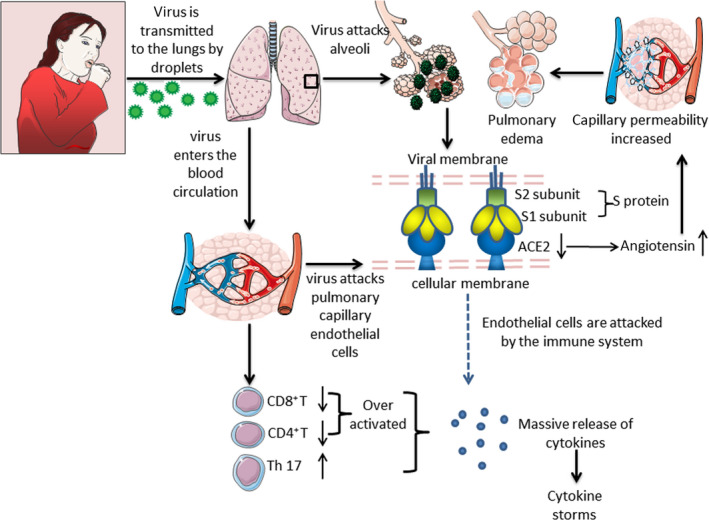
Possible mechanisms of cytokine storms in COVID‐19. SARS‐CoV‐2 transmitted by air droplets reaches the lungs. On the one hand, the S protein on the surface of the virus binds to the ACE2 receptor in alveolar epithelial cells, resulting in the down‐regulation of ACE2 expression and an increase of angiotensin level, which leads to increased pulmonary capillary permeability and pulmonary oedema. On the other hand, SARS‐CoV‐2 reaches the lungs again through blood circulation and interacts with ACE2 receptors on the surface of alveolar capillary endothelial cells, making the alveolar capillary endothelium the target of attack by the immune system, thus inducing a series of immune responses and aggravating lung injury. The imbalance of lymphocyte subsets characterized by the decrease of CD4+, CD8+ T cells, the increase of the number of proinflammatory Th17 cells and the increase of CD8+ cytotoxic particles, aggravated the disorder of host immune system. Inflammatory monocytes amplify cytokines production. In addition, many kinds of cytokines are released in patients with COVID‐19, which contribute to the formation of a cytokine storm. Abbreviations: ACE2: angiotensin‐converting enzyme 2 and NK cell: natural killer cell.
Clinical characteristics
Clinical manifestations of CSS
The clinical symptoms of patients with CSS vary from mild influenza‐like symptoms to severe ones that may have uncontrolled systemic inflammatory responses, disseminated intravascular coagulation (DIC), shock and MOF [4, 6]. The common clinical features of CSS include persistent fever, splenomegaly, hepatomegaly accompanied by liver dysfunction, lymphadenopathy, coagulation disorders, cytopaenia and symptoms of the central nervous system, mainly characterized by an acute or subacute (1–4 weeks) course of disease [5]. Respiratory symptoms are also common in patients with CSS, and mild cases can be characterized by cough and shortness of breath, whilst severe ones can progress to ALI and ARDS with radiological abnormalities. In addition, renal failure or cardiac insufficiency may also occur in CSS [6]. Common laboratory abnormalities in CSS patients include pancytopaenia (anaemia, leukopenia, thrombocytopaenia), elevated serum creatinine, liver enzymes and C‐reactive protein (CRP), as well as coagulation abnormalities. In HLH/MAS patients, hyperferritinaemia and hypertriglyceridemia may also occur, and haemophagocytes can be seen in bone marrow aspiration [31].
Clinical characteristics of severe patients with COVID‐19
According to a report of 72,314 cases from the Chinese Center for Disease Control and Prevention, most cases of COVID‐19 were classified as mild (81%), 14% were severe (dyspnoea, respiratory rate ≥ 30/min, blood oxygen saturation ≤ 93%, the partial pressure of arterial oxygen to fraction of inspired oxygen ratio < 300 and/or lung infiltrates> 50% within 24 to 48 h), and 5% were critical (respiratory failure, septic shock and/or multiple organ dysfunction or failure). The overall case‐fatality rate was 2.3% [70]. Severe patients who need critical care have tended to be older (median age ≈60 years), and 40% have had comorbidities, such as diabetes and cardiac disease [71]. Lymphocytopaenia is one of the most prominent characteristics of COVID‐19 [26, 71, 72, 73, 74], indicating that SARS‐CoV‐2 may attack the immune system. Moreover, a retrospective study of 452 patients with COVID‐19 showed that severe cases tend to have lower lymphocytes counts, higher leucocytes counts and neutrophil–lymphocyte ratio (NLR). It also demonstrated that the number of T cells was more hampered in severe cases and helper T cells were lower in the severe group, suggesting that the surveillance of NLR and lymphocyte subsets may contribute to the early screening of critical illness [75]. In addition, compared with nonsevere patients, severe patients may have significantly higher levels of inflammatory parameters, including CRP, ferritin and proinflammatory cytokines (such as IL‐2R, IL‐6, GM‐CSF, IP‐10, MCP‐1, MCP‐1α and TNF‐α) [26, 76], indicating the presence of a cytokine storm in severe patients. Moreover, patients with COVID‐19 have been found to have an elevated level of D‐dimer, and there is growing evidence that some severe patients can have myocardial dysfunction or myocarditis, which may be partly associated with the cytokine storm [77]. Liver involvement or renal injury may also present in severe patients with COVID‐19, which may also raise the possibility that this reflects the presence of a cytokine storm [26, 72, 73, 74]. Summary of clinical characteristics of severe patients with COVID‐19 in various studies is shown in Table 2.
Table 2.
Summary of clinical characteristics of severe patients with COVID‐19 in various studies
| Studies | Huang et al [26] | Wang et al [71] | Guan et al [72] | Cao et al [73] | Qi et al [74] | |
|---|---|---|---|---|---|---|
| Basic clinical features | Number of severe patients | 13 | 36 | 173 | 19 | 50 |
| Age, years | 49.0 (41.0–61.0) | 66 .0 (57.0–78.0) | 52.0 (40.0–65.0) | 63.7 | 71.5 (65.8, 77.0) | |
| Male (%) | 85 | 61.1 | 57.8 | 89.5 | 78.0 | |
| Comorbidities | Hypertension | 15.0 | 58.3 | 23.7 | 31.6 | 26.0 |
| Diabetes | 8.0 | 22.2 | 16.2 | 10.5 | 24.0 | |
| Cardiovascular disease | 23.0 | 25.0 | 2.3 | 26.3 | NR | |
| Common symptoms | Fever (%) | 100.0 | 100.0 | 91.3 | 94.7 | 78.0 |
| Headache (%) | 8.3 | 8.3 | 15.0 | 0 | NR | |
| Myalgia (%) | 33.3 | 33.3 | 17.3 | 31.6 | 74.0 | |
| Cough (%) | 76.0 | 58.3 | 70.5 | 36.8 | 68.0 | |
| Dyspnoea (%) | 92.0 | 63.9 | 37.6 | 36.8 | 34.0 | |
| Diarrhoea (%) | 0 | 16.7 | 5.8 | 0 | NR | |
| Laboratory examinations | Lymphocytopaenia (%) | 85 | NR | 95.5 | 84.2 | 92.0 |
| Thrombocytopaenia (%) | 8 | NR | 57.7 | 31.6 | 38.0 | |
| AST↑(%) | 62 | NR | 39.4 | 42.1 | NR | |
| Creatinine↑ (%) | 15 | NR | 4.3 | 15.8 | NR | |
| CK↑ (%) | 46 | NR | 19.0 | NR | 36.0 | |
| LDH↑ (%) | 92 | NR | 58.1 | NR | 36.0 | |
| Troponin↑ (%) | 31 | NR | NR | 47.4 | 14.3 | |
| CRP↑ (%) | NR | NR | 81.5 | 78.6 | 58.0 | |
| ESR↑ (%) | NR | NR | NR | 100 | NR | |
| Serum ferritin↑ (%) | NR | NR | NR | NR | NR | |
| D‐dimer↑ (%) | NR | NR | 59.6 | 63.2 | 26.0 | |
| Day from illness onset to dyspnoea (day) | 8.0 (6.0‐17.0) | 6.5 (3.0‐10.8) | 5.0 (3.0‐8.0) | NR | 3.0 (2.0‐5.0) | |
| Complications | ARDS (%) | 85 | 61.1 | 15.6 | NR | 54.0 |
| Acute cardiac injury* (%) | 31 | 22.2 | NR | NR | 6.0 | |
| Acute kidney injury (%) | 23 | 8.3 | 2.9 | NR | NR | |
| Co‐infection (%) | 31 | NR | NR | NR | NR | |
| Shock (%) | 23 | 30.6 | 6.4 | NR | 22.0 |
Acute cardiac injury* was defined if the serum levels of cardiac biomarkers (e.g. troponin I) were above the 99th percentile upper reference limit or new abnormalities were shown in electrocardiography and echocardiography; ARDS, acute respiratory distress syndrome; AST, aspartate aminotransferase; CK, creatinine kinase; CRP, C‐reactive protein; ESR, erythrocyte sedimentation rate; LDH: lactose dehydrogenase; NR, not reported.
Recognition of CSS in COVID‐19
There is no unified standard for the diagnosis of COVID‐19 associated with CSS, and further clinical and laboratory researches are needed. Here, we propose a basic principle for consideration of CSS in COVID‐19: (i) a sudden or rapid progression with multiple organ involvement (such as liver, cardiac or renal injury); (ii) the significant decline of peripheral blood lymphocyte counts; (iii) the significant elevation of systematic inflammatory indicators (such as CRP, serum ferritin, erythrocyte sedimentation rate); and (iv) the elevation of multiple cytokines, such as IL‐1β, IL‐2R, IL‐6, IFN‐γ, IP‐10, MCP‐1, TNF‐α and MIP1a. Clinicians should keep highly alert on the possibility of CSS under these circumstances. However, given that CSS is a highly heterogeneous disease and may present with unspecific syndromes, the diagnosis of CSS in COVID‐19 is very challenging and the development of a specific diagnostic test that helps to make the diagnosis of CSS earlier is a high priority for future research.
Potential therapeutic options for CSS in COVID‐19
Glucocorticoid
Glucocorticoid has powerful anti‐inflammatory properties and is an effective choice for the treatment of CSS [1]. Corticosteroids were widely used during the outbreaks of SARS and MERS. Several studies demonstrated that timely administration of corticosteroids often leads to improvement of radiographic outcome and oxygenation in SARS as a consequence of more effective control of immunopathological lung damage [78, 79]. However, a meta‐analysis of corticosteroid use in patients with SARS showed that in 29 studies of steroid use, 25 were inconclusive and four were classified as causing possible harm [80]. A retrospective observational study of critically ill patients with MERS reported that the administration of corticosteroids did not improve the 90‐day mortality (adjusted odds ratio 0·8, 95% CI 0.5–1.1; P = 0.12) [81]. Additionally, the early application of corticosteroids may delay viral clearance in SARS‐CoV [82] and MERS‐CoV [81] infections, and increase secondary infection rates and mortality in patients of influenza pneumonitis [83]. Therefore, glucocorticoids should not be used in theearly phases of disease unless there is a clear indication for their use [84].
For patients with COVID‐19, a newly published autopsy report demonstrated that pulmonary oedema and hyaline membrane formation were present in the lung of the patient, indicating timely and appropriate use of corticosteroids together with ventilator support may be considered for the severe patients to prevent ARDS development [66]. At present, several retrospective studies have reported that corticosteroids were used for the treatment of severe patients to reduce inflammatory‐induced lung injury [25, 26, 71]. However, there is still no evidence from randomized clinical trials to support the application of glucocorticoids in patients with COVID‐19.
In China, the newly updated version of the guidelines for the prevention, diagnosis and treatment of pneumonia caused by COVID‐19 pointed out that patients with progressive deterioration of oxygenation indicators, rapid imaging progress and excessive activation of the inflammatory response are recommended to use glucocorticoid within a short period time (3–5 day) and a recommended dose not exceeding the equivalent of methylprednisolone 1–2 mg kg‐1 d‐1 [54]. The efficacy and safety of glucocorticoids in COVID‐19 still needs to be elucidated in further high‐quality clinical trials.
Blood purification therapy
The application of blood purification technology is helpful to the removal of cytokines and may be beneficial to improve the clinical outcome of critically ill patients. Commonly used extracorporeal blood purification treatments in CSS include plasma exchange, blood/plasma filtration, adsorption, perfusion and continuous renal replacement therapy (CRRT).
Previous studies have suggested that therapeutic plasma exchange plays an important role in the treatment of severe HLH [85, 86]. Similarly, high‐volume haemofiltration has also been reported to improve organ function in critically ill children with HLH by reducing the levels of cytokines (such as TNF‐α and IL‐6) [87]. In addition, a positive therapeutic effect was also seen in septic patients due to its ability to successfully remove some inflammatory cytokines and improve the sequential organ failure assessment (SOFA) score after the application of high‐volume haemofiltration for 6 h [88]. Moreover, a randomized controlled study showed that compared with conventional‐dose CRRT, high‐dose CRRT treatment could reduce blood IL‐6 and IL‐8 levels in septic patients with AKI more significantly [89]. In terms of the proper time for initiation of RRT, a previous double‐blind randomized controlled study suggested that early initiation of RRT [started within 8 h at stage 2 of KDIGO (Kidney Disease: Improving Global Outcomes) guideline] could significantly reduce the 90‐day mortality of critically ill patients compared with delayed (started over 12 h of KDIGO stage 3) [90], whilst others reported that there was no significant difference in overall mortality at 90 days between an early strategy (within 12 h after documentation of failure‐stage acute kidney injury) for the initiation of RRT or a delayed strategy (48 h if renal recovery had not occurred) [91]. The discrepancies may partly due to the differences in inclusion criteria and dialysis techniques, and further multicentre trials of this intervention are warranted.
CytoSorb®, a recently developed, commercially available haemoadsorption device that utilizes extracorporeal blood purification, is designed to reduce systemic cytokine burden. Numerous case reports and case series have suggested improved clinical outcomes with CytoSorb in patients with septic shock [92]. Recently, CytoSorb® was also reported to be effective in a patient developed CSS after CAR T‐cell application [93]. Given that there is excessive production of various cytokines in patients with COVID‐19, CytoSorb® may become a potential therapeutic candidate in COVID‐19 patients complicated with CSS.
Based on the previous experience in the treatment of patients with SARS and MERS and collected clinical experience in treatment of critical ill patients in China [94, 95], the newly updated version of guidelines for the prevention, diagnosis, and treatment of pneumonia caused by COVID‐19 in China also recommended that COVID‐19 patients with high inflammatory response could consider to use extracorporeal blood purification technology for removal of cytokines and mitigate CSS [54].
Biological agents
IL‐1‐inhibiting agent
Dysfunction of the innate immune system involving IL‐1 is important to CSS pathogenesis. Anakinra is a recombinant, nonglycosylated form of human IL‐1Ra, which can block the biologic activity of both IL‐1α and IL‐1β by competitively inhibiting their binding to IL‐1R. In the treatment of MAS associated with sJIA and AOSD, numerous studies supported the application of anakinra [45, 46, 47]. In the setting of CSS induced by CAR T‐cell therapy, Norelli and his colleagues demonstrated that anakinra could abolish both CSS and neurotoxicity, resulting in substantially extended leukaemia‐free survival using a humanized mice with high leukaemia burden [39]. In terms of severe sepsis, a previous, randomized, multicentre phase III trial reported that anakinra failed to demonstrate a statistically significant reduction in 28‐day mortality [96]. However, a reanalysis of data from this phase III randomized trial found that interleukin‐1 receptor blockade was associated with significant improvement in survival of patients with hepatobiliary dysfunction and DIC [97]. Recent clinical studies showed that IL‐1β was also markedly elevated in patients with COVID‐19 [26]. Currently, there are several anakinra studies registered for COVID‐19 (NCT04357366, NCT04324021, NCT04339712, NCT04330638, NCT04341584, NCT02735707). Apart from anakinra, there are currently other anti‐IL‐1 agents available, such as canakinumab and rilonacept. The potential therapeutic effect of IL‐1 inhibition still needs to be investigated in these clinical trials.
IL‐6‐inhibiting agent
IL‐6 seems to play an important role in the pathophysiology of CSS since highly elevated IL‐6 levels are seen in patients with CSS [3, 8] and murine models of the disease [39]. Recent studies have also shown that there is a significant elevation of IL‐6 in COVID‐19, especially in critically ill patients [25, 26]. Furthermore, elevated levels of the IL‐6 in the blood have been reported to be predictive of a fatal outcome in patients with COVID‐19 [98]. Recently, Chen et al reported that detectable serum SARS‐CoV‐2 RNA (RNAaemia) in COVID‐19 patients was associated with elevated IL‐6 concentration and poor prognosis, suggesting IL‐6 could be a potential therapeutic target for severe patients in the hyperinflammatory state [99].
Tocilizumab, a recombinant human IL‐6 monoclonal antibody, specifically binds to soluble and membrane‐bound IL‐6 receptors (IL‐6R), thus blocking IL‐6 signalling and its mediated inflammatory response, which has been demonstrated to show outstanding efficacy in the rescue of CSS accompanied by T‐cell engaged therapy [49]. However, clinical experience with tocilizumab in viral disease is very limited. Meanwhile, the application of tocilizumab may also increase the risk of opportunistic infections which may be a barrier for the wide use of tocilizumab in the treatment of COVID‐19 [100]. In addition, it has been demonstrated that IL‐6 is necessary for resolution influenza virus infection by reducing virus‐induced death of neutrophils in the lung and by promoting neutrophil‐mediated viral clearance [101], so similar studies on COVID‐9 are urgently needed.
Currently, tocilizumab is suggested for the treatment of patients with extensive lung lesions and elevated IL‐6 levels in China [54], and the phase IV clinical trials of tocilizumab in COVID‐19 (registration number: ChiCTR2000029765) are undergoing. The efficacy and safety data needs to be verified in the future.
Janus kinase (JAK) inhibitor
Several cytokines signal through the JAK/STAT pathway, which is now recognized as a major target to inhibit the effect of a wide array of cytokines, including ILs (IL‐2, IL‐3, IL‐4, IL‐5, IL‐6, IL‐7, IL‐9, IL‐10, IL‐12, IL‐15, IL‐21, IL‐23), IFN‐(α, β, γ) and growth factors (GM‐CSF, TGF‐β). Thus, JAK inhibitors are increasingly used in the setting of inflammatory and autoimmune diseases [102, 103]. As we mentioned above, the receptors of SARS‐CoV‐2 might be ACE2, which is widely distributed in multiple cells, especially lung AT2 alveolar epithelial cells. Most viruses, including the SARS‐CoV‐2, could enter cells through receptor‐mediated endocytosis, and one of the known regulators of endocytosis is the AP2‐associated protein kinase 1 (AAK1). Baricitinib, a JAK inhibitor, may block AAK1, as well as cyclin G‐associated kinase (GAK), which also regulates viral endocytosis. Thus, baricitinib is proposed to have the ability to reduce both the viral entry and the inflammation, which is suggested as a possible candidate for the treatment of COVID‐19 [104]. However, JAK inhibitors also block INF‐a production, which is important in fighting virus, and may increase the risk of viral reactivation [105]. Additionally, baricitinib has known to cause lymphocytopaenia which may not be suitable for patients with COVID‐19 who often have low lymphocyte counts [106]. Currently, there are several baricitinib studies registered for COVID‐19, testing 2‐4 mg daily oral for 7–14 days (NCT04340232, NCT04346147, NCT04320277, NCT04321993, NCT04345289).
Recently, a multicentre, single‐blind, randomized controlled trial indicated that ruxolitinib (a JAK1/2 inhibitor) contributed to a numerically faster clinical improvement in patients with severe COVID‐19, with a significant reduction of the levels of various cytokines in the ruxolitinib group when compared to the control group [107]. Although the patients enrolled in this study were relatively small, it is also informative to future trials to test the efficacy of ruxolitinib in a larger population. Further data from clinical trials are urgently needed to evaluate the safety and efficacy of JAK inhibitors for the treatment of COVID‐19.
Chloroquine and hydroxychloroquine
Previous studies have reported the broad‐spectrum antiviral activity of chloroquine (CQ), a widely used anti‐malarial and autoimmune disease drug, including SARS‐CoV [108]. The potential antiviral activities of CQ have been attributed to multiple mechanisms, including increasing endosomal pH required for virus/cell fusion [109], as well as interfering with the glycosylation of ACE2 during the viral entry [110]. Moreover, CQ affects immune system activity by down‐regulating the production of cytokines (such as TNF‐α and IL‐6), and the expression of TNF‐α receptor, which might reduce damage due to the exaggerated inflammatory response induced by viral infection [108].
Recent publications have drawn attention to the use of CQ in the treatment of patients with COVID‐19. Wang et al. demonstrated that CQ was highly effective in control of SARS‐CoV‐2 infection at both viral entry and postentry stages in vitro [111]. Gao et al pointed that CQ phosphate had more benefits than control treatment in preventing the exacerbation of pneumonia, improving lung imaging findings, promoting a virus‐negative conversion and shortening the disease course in more than 100 patients with COVID‐19 [112]. However, the margin between the therapeutic and toxic dose is relatively narrow and the side effect of CQ poisoning may even lead to death [113]. The preliminary safety results from a parallel, double‐blinded, randomized, phase IIb clinical trial showed that the high dosage CQ group (12 g total dose over 10 days) presented more QTc > 500 ms and a trend towards higher lethality than the lower dosage (5 days of treatment, total dose 2.7 g), suggesting the higher CQ dosage should not be recommended for critically ill patients with COVID‐19 because of its potential safety hazards [114].
Besides, there is a growing trend of preference for hydroxychloroquine (HCQ), a less toxic derivative of CQ. Yao et al. reported that HCQ was more effective in vitro than chloroquine for both prophylaxis and treatment [115]. However, the results of clinical trials of HCQ remain controversial. An open‐label nonrandomized clinical trial of 20 patients with COVID‐19 in France treated with HCQ alone or in combination with azithromycin showed a significant reduction of the viral carriage 6 days after the inclusion when compared to controls [116], whilst other similar studies found no difference in the rate of virologic clearance and clinical outcomes [117, 118]. These inconsistent results may be partly due to the small sample size or the difference in the severity of the patient’s condition.
To date, evidence for the use of CQ and HCQ in COVID‐19 is still limited and inconclusive, which is mainly from in vitro and small‐scale, poorly controlled or uncontrolled clinical studies. Additionally, safety hazards should also be placed a premium for the use of CQ and HCQ in patients with COVID‐19. Therefore, safety data and results from high‐quality, well‐performed randomized clinical trials in patients with COVID‐19 are urgently needed to elucidate the true clinical application value of CQ and HCQ.
Prospect
COVID‐19 is the third highly pathogenic human coronavirus infectious disease after SARS and MERS. Accumulating evidence has shown that CSS might be one of the most important and deadly complications in severe patients with COVID‐19, whilst current knowledge about this is still very limited. Given the different manifestations of CSS in various clinical issues, it is of paramount importance to further recognize the nature of the initiation and progression of this systemic inflammatory process, which will be greatly helpful to curb this deadly clinical situation by reducing the mortality in the setting of COVID‐19 and other diseases.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Author Contribution
Yue‐ming Gao: Conceptualization (equal); Writing‐original draft (lead). Bi‐Cheng Liu: Conceptualization (equal); Writing‐review & editing (lead). Gang Xu: Writing‐original draft (supporting); Writing‐review & editing (supporting). Bin Wang: Conceptualization (supporting); Writing‐review & editing (supporting).
Acknowledgment
This work was financially supported by the key international cooperation program of China National Natural Science Foundation (81720108007, 81670696) and the key research project of the Ministry of China Science and Technology (2018YFC1314000) to Prof. Bi‐Cheng Liu as PI.
Gao Y‐M, Xu G, Wang B, Liu B‐C (Zhongda Hospital, Southeast University School of Medicine, Nanjing, China; Tongji Hospital, University of HuaZhong Science and Technology, Wuhan, China). Cytokine storm syndrome in coronavirus disease 2019: A narrative review (Review). J Intern Med, 2021; 289: 147–161. 10.1111/joim.13144
References
- 1. Behrens EM, Koretzky GA. Review: cytokine storm syndrome: looking toward the precision medicine era. Arthritis Rheumatol 2017; 69: 1135–43. [DOI] [PubMed] [Google Scholar]
- 2. Chen H, Wang F, Zhang P et al. Management of cytokine release syndrome related to CAR‐T cell therapy. Front Med 2019; 13: 610–7. [DOI] [PubMed] [Google Scholar]
- 3. Chousterman BG, Swirski FK, Weber GF. Cytokine storm and sepsis disease pathogenesis. Semin Immunopathol 2017; 39: 517–28. [DOI] [PubMed] [Google Scholar]
- 4. Cron RQ, Behrens EM. Cytokine storm syndrome. Switzerland: Springer Nature, 2019. [Google Scholar]
- 5. Murthy H, Iqbal M, Chavez JC, Kharfan‐Dabaja MA. Cytokine release syndrome: current perspectives. Immunotargets Ther 2019; 8: 43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shimabukuro‐Vornhagen A, Godel P, Subklewe M et al. Cytokine release syndrome. J Immunother Cancer 2018; 69: 56–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gauthier J, Turtle CJ. Insights into cytokine release syndrome and neurotoxicity after CD19‐specific CAR‐T cell therapy. Curr Res Transl Med 2018; 66: 50–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Crayne CB, Albeituni S, Nichols KE, Cron RQ. The immunology of macrophage activation syndrome. Front Immunol 2019; 10: 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chatenoud L, Ferran C, Reuter A et al. Systemic reaction to the anti‐T‐cell monoclonal antibody OKT3 in relation to serum levels of tumor necrosis factor and interferon‐gamma. N Engl J Med 1989; 321: 63. Corrected and republished from: N Engl J Med 1989; 320: 1420–1. [DOI] [PubMed] [Google Scholar]
- 10. Channappanavar R, Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin Immunopathol 2017; 39: 529–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. De Jong MD, Simmons CP, Thanh TT et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat Med 2006; 12: 1203–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Singh A, Dawman L, Seth R. Malignancy associated hemophagocytic lymphohistiocytosis in children. J Cancer Res Ther 2018; 14: 559–62. [DOI] [PubMed] [Google Scholar]
- 13. Schulert GS, Grom AA. Pathogenesis of macrophage activation syndrome and potential for cytokine‐directed therapies. Annu Rev Med 2015; 66: 145–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Henderson LA, Cron RQ. Macrophage activation syndrome and secondary hemophagocytic lymphohistiocytosis in childhood inflammatory disorders: diagnosis and management. Paediatr Drugs 2020; 22: 29–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nebelsiek T, Beiras‐Fernandez A, Kilger E, Möhnle P, Weis F. Routine use of corticosteroids to prevent inflammation response in cardiac surgery. Recent Pat Cardiovasc Drug Discov 2012; 7: 170–4. [DOI] [PubMed] [Google Scholar]
- 16. Hay KA, Hanafi LA, Li D et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor‐modified T‐cell therapy. Blood 2017; 130: 2295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Williams M, Khalid T, Hughes S, Bonney D, Wynn R. Rituximab‐induced cytokine storm in the absence of overt lymphoproliferative disease. J Pediatr Hematol Oncol 2016; 38: e29–e31. [DOI] [PubMed] [Google Scholar]
- 18. Peck KM, Burch CL, Heise MT, Baric RS. Coronavirus host range expansion and middle east respiratory syndrome coronavirus emergence: biochemical mechanisms and evolutionary perspectives. Annu Rev Virol 2015; 2: 95–117. [DOI] [PubMed] [Google Scholar]
- 19. Su S, Wong G, Shi W et al. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol 2016; 24: 490–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cui J, Li F, Shi ZL. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol 2019; 17: 181–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhou P, Yang XL, Wang XG et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020; 579: 270–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li Q, Guan X, Wu P et al. Early transmission dynamics in Wuhan, China, of novel coronavirus‐infected pneumonia. N Engl J Med 2020; 382: 1199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lai CC, Shih TP, Ko WC, Tang HJ, Hsueh PR. Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) and coronavirus disease‐2019 (COVID‐19): the epidemic and the challenges. Int J Antimicrob Agents 2020; 55: 105924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Coronavirus disease (COVID‐2019) situation reports . Available at: https://www.who.int/emergencies/diseases/novelcoronavirus‐2019/situation‐reports.
- 25. Chen N, Zhou M, Dong X et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet 2020; 395: 507–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huang C, Wang Y, Li X et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020; 395: 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. Into the eye of the cytokine storm. Microbiol Mol Biol Rev 2012; 76: 16–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Weitzman S. Approach to hemophagocytic syndromes. Hematology Am Soc Hematol Educ Program 2011; 178–83. [DOI] [PubMed] [Google Scholar]
- 29. Alongi A, Naddei R, De Miglio L, Natoli V, Ravelli A. Macrophage activation syndrome in pediatrics. Pediatr Allergy Immunol 2020; 31(Suppl 24): 13–5. [DOI] [PubMed] [Google Scholar]
- 30. Carter SJ, Tattersall RS, Ramanan AV. Macrophage activation syndrome in adults: recent advances in pathophysiology, diagnosis and treatment. Rheumatology (Oxford) 2019; 58: 5–17. [DOI] [PubMed] [Google Scholar]
- 31. Sen ES, Clarke SL, Ramanan AV. Macrophage activation syndrome. Indian J Pediatr 2016; 83: 248–53. [DOI] [PubMed] [Google Scholar]
- 32. Filipovich AH, Chandrakasan S. Pathogenesis of hemophagocytic lymphohistiocytosis. Hematol Oncol Clin North Am 2015; 29: 895–902. [DOI] [PubMed] [Google Scholar]
- 33. Brisse E, Wouters CH, Matthys P. Advances in the pathogenesis of primary and secondary haemophagocytic lymphohistiocytosis: differences and similarities. Br J Haematol 2016; 174: 203–17. [DOI] [PubMed] [Google Scholar]
- 34. Kaufman KM, Linghu B, Szustakowski JD et al. Whole‐exome sequencing reveals overlap between macrophage activation syndrome in systemic juvenile idiopathic arthritis and familial hemophagocytic lymphohistiocytosis. Arthritis Rheumatol 2014; 66: 3486–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Billiau AD, Roskams T, Van Damme‐Lombaerts R et al. Macrophage activation syndrome: characteristic findings on liver biopsy illustrating the key role of activated, IFN‐gamma‐producing lymphocytes and IL‐6‐ and TNF‐alpha‐producing macrophages. Blood 2005; 105: 1648–51. [DOI] [PubMed] [Google Scholar]
- 36. Van der Stegen SJ, Davies DM, Wilkie S et al. Preclinical in vivo modeling of cytokine release syndrome induced by ErbB‐retargeted human T cells: identifying a window of therapeutic opportunity? J Immunol 2013; 191: 4589–98. [DOI] [PubMed] [Google Scholar]
- 37. Singh N, Hofmann TJ, Gershenson Z et al. Monocyte lineage‐derived IL‐6 does not affect chimeric antigen receptor T‐cell function. Cytotherapy 2017; 19: 867–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. CAR T cell‐induced cytokine release syndrome is mediated by macrophages and abated by IL‐1 blockade. Nat Med 2018; 24: 731–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Norelli M, Camisa B, Barbiera G et al. Monocyte‐derived IL‐1 and IL‐6 are differentially required for cytokine‐release syndrome and neurotoxicity due to CAR T cells. Nat Med 2018; 24: 739–48. [DOI] [PubMed] [Google Scholar]
- 40. Steinman RM. Decisions about dendritic cells: past, present, and future. Annu Rev Immunol 2012; 30: 1–22. [DOI] [PubMed] [Google Scholar]
- 41. Hermans IF, Ritchie DS, Yang J, Roberts JM, Ronchese F. CD8+ T cell‐dependent elimination of dendritic cells in vivo limits the induction of antitumor immunity. J Immunol 2000; 164: 3095–101. [DOI] [PubMed] [Google Scholar]
- 42. Jenkins MR, Rudd‐Schmidt JA, Lopez JA et al. Failed CTL/NK cell killing and cytokine hypersecretion are directly linked through prolonged synapse time. J Exp Med 2015; 212: 307–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gust J, Hay KA, Hanafi LA et al. Endothelial activation and bloodbrain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR‐T cells. Cancer Discov 2017; 7: 1404–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Obstfeld AE, Frey NV, Mansfield K et al. Cytokine release syndrome associated with chimeric‐antigen receptor T‐cell therapy: clinicopathological insights. Blood 2017; 130: 2569–72. [DOI] [PubMed] [Google Scholar]
- 45. Durand M, Troyanov Y, Laflamme P et al. Macrophage activation syndrome treated with anakinra. J Rheumatol 2010; 37: 879–80. [DOI] [PubMed] [Google Scholar]
- 46. Miettunen PM, Narendran A, Jayanthan A, Behrens EM, Cron RQ. Successful treatment of severe paediatric rheumatic disease‐associated macrophage activation syndrome with interleukin‐1 inhibition following conventional immunosuppressive therapy: case series with 12 patients. Rheumatology (Oxford) 2011; 50: 417–9. [DOI] [PubMed] [Google Scholar]
- 47. Sönmez HE, Demir S, Bilginer Y, Özen S. Anakinra treatment in macrophage activation syndrome: a single center experience and systemic review of literature. Clin Rheumatol 2018; 37: 3329–35. [DOI] [PubMed] [Google Scholar]
- 48. Shimizu M, Nakagishi Y, Inoue N et al. Interleukin‐18 for predicting the development of macrophage activation syndrome in systemic juvenile idiopathic arthritis. Clin Immunol 2015; 160: 277–81. [DOI] [PubMed] [Google Scholar]
- 49. Hay KA. Cytokine release syndrome and neurotoxicity after CD19 chimeric antigen receptor‐modified (CAR‐) T cell therapy. Br J Haematol 2018; 183: 364–74. [DOI] [PubMed] [Google Scholar]
- 50. Bermejo‐Martin JF, Ortiz de Lejarazu R, Pumarola T et al. Th1 and Th17 hypercytokinemia as early host response signature in severe pandemic influenza. Crit Care 2009; 13: R201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fujiwara F, Hibi S, Imashuku S. Hypercytokinemia in hemophagocytic syndrome. Am J Pediatr Hematol Oncol 1993; 15: 92–8. [DOI] [PubMed] [Google Scholar]
- 52. Takada H, Nomura A, Ohga S, Hara T. Interleukin‐18 in hemophagocytic lymphohistiocytosis. Leuk Lymphoma 2001; 42: 21–8. [DOI] [PubMed] [Google Scholar]
- 53. Takada H, Ohga S, Mizuno Y, Nomura A, Hara T. Increased IL‐16 levels in hemophagocytic lymphohistiocytosis. J Pediatr Hematol Oncol 2004; 26: 567–73. [DOI] [PubMed] [Google Scholar]
- 54. National Health Commission . Diagnosis and treatment protocol for novel coronavirus pneumonia (Trial Version 7). Chin Med J (Engl) 2020; 133: 1087–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yao XH, Li TY et al. A pathological report of three COVID‐19 cases by minimally invasive autopsies. Chin. J. Pathol 2020; 49: E009. [DOI] [PubMed] [Google Scholar]
- 56. Li W, Moore MJ, Vasilieva N et al. Angiotensin‐converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003; 426: 450–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Xu H, Zhong L, Deng J et al. High expression of ACE2 receptor of 2019‐nCoV on the epithelial cells of oral mucosa. Int J Oral Sci 2020; 12: 8–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Santos RA, Simoes e Silva AC, Maric C et al. Angiotensin‐(1–7) is an endogenous ligand for the G protein‐coupled receptor Mas. Proc Natl Acad Sci U S A 2003; 100: 8258–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Eguchi S, Kawai T, Scalia R, Rizzo V. Understanding angiotensin II Type 1 receptor signaling in vascular pathophysiology. Hypertension 2018; 71: 804–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Murakami M, Kamimura D, Hirano T. Pleiotropy and specificity: insights from the interleukin 6 family of cytokines. Immunity 2019; 50: 812–31. [DOI] [PubMed] [Google Scholar]
- 61. Glowacka I, Bertram S, Muller MA et al. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J Virol 2011; 85: 4122–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS. Angiotensin‐converting enzyme 2 (ACE2) as a SARS‐CoV‐2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med 2020; 46: 586–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chen Y, Guo Y, Pan Y, Zhao ZJ. Structure analysis of the receptor binding of 2019‐nCoV. Biochem Biophys Res Commun 2020; 525: 135–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wrapp D, Wang N, Corbett KS et al. Cryo‐EM structure of the 2019‐nCoV spike in the prefusion conformation. Science 2020; 367: 1260–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hoffmann M, Kleine‐Weber H, Schroeder S et al. SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020; 181: 271–80.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Xu Z, Shi L, Wang Y et al. Pathological findings of COVID‐19 associated with acute respiratory distress syndrome. Lancet Respir Med 2020; 8: 420–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhang H, Zhou P, Wei Y et al. Histopathologic changes and SARS‐CoV‐2 immunostaining in the lung of a patient with COVID‐19. Annals Internal Med 2020; 172: 629–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhou Y, Fu B, Zheng X et al. Pathogenic T cells and inflammatory monocytes incite inflammatory storm in severe COVID‐19 patients. National Sci Rev 2020; 7: 998–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wan S, Yi Q, Fan S et al. Characteristics of lymphocyte subsets and cytokines in peripheral blood of 123 hospitalized patients with 2019 novel coronavirus pneumonia (NCP). medRxiv 2020. 10.1101/2020.02.10.20021832. [DOI] [Google Scholar]
- 70. Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID‐19) outbreak in China: summary of a report of 72 314 cases from the Chinese center for disease control and prevention. JAMA 2020; 323: 1239. [DOI] [PubMed] [Google Scholar]
- 71. Wang D, Hu B, Hu C et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus–infected Pneumonia in Wuhan, China. JAMA 2020; 323: 1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Guan WJ, Ni ZY, Hu Y et al. Clinical characteristics of 2019 novel coronavirus infection in China. medRxiv 2020. Available at doi: 10.1101/2020.02.06.20020974. [DOI] [Google Scholar]
- 73. Cao M, Zhang DD, Wang YH et al. Clinical features of patients infected with the 2019 novel coronavirus (COVID‐19) in Shanghai, China. medRxiv 2020. 10.1101/2020.03.04.20030395. [DOI] [Google Scholar]
- 74. Qi D, Yan XF, Tang XM et al. Epidemiological and clinical features of 2019‐nCoV acute respiratory disease cases in Chongqing municipality, China: a retrospective, descriptive, multiple‐center study. medRxiv 2020. 10.1101/2020.03.01.20029397. [DOI] [Google Scholar]
- 75. Qin C, Zhou L, Hu Z et al. Dysregulation of immune response in patients with COVID‐19 in Wuhan, China. Clin Infect Dis 2020. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Chen L, Liu HG, Liu W et al. Analysis of clinical features of 29 patients with 2019 novel coronavirus pneumonia. Zhonghua Jie He He Hu Xi Za Zhi 2020; 43: E005. [DOI] [PubMed] [Google Scholar]
- 77. Clerkin KJ, Fried JA, Raikhelkar J et al. Coronavirus disease 2019 (COVID‐19) and cardiovascular disease. Circulation 2020; 141: 1648–55. [DOI] [PubMed] [Google Scholar]
- 78. Ho JC, Ooi GC, Mok TY et al. High‐dose pulse versus nonpulse corticosteroid regimens in severe acute respiratory syndrome. Am J Respir Crit Care Med 2003; 168: 1449–56. [DOI] [PubMed] [Google Scholar]
- 79. Yam LY, Lau AC, Lai FY, Shung E, Chan J, Wong V. Corticosteroid treatment of severe acute respiratory syndrome in Hong Kong. J Infect 2007; 54: 28–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Stockman LJ, Bellamy R, Garner P. SARS: systematic review of treatment effects. PLoS Medicine 2006; 3: e343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Arabi YM, Mandourah Y, Al‐Hameed F et al. Corticosteroid therapy for critically ill patients with middle east respiratory syndrome. Am J Respir Crit Care Med 2018; 197: 757–67. [DOI] [PubMed] [Google Scholar]
- 82. Lee N, Allen Chan KC, Hui DS et al. Effects of early corticosteroid treatment on plasma SARS‐associated Coronavirus RNA concentrations in adult patients. J Clin Virol 2004; 31: 304–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ni YN, Chen G, Sun J, Liang BM, Liang ZA. The effect of corticosteroids on mortality of patients with influenza pneumonia: a systematic review and meta‐analysis. Crit Care 2019; 23: 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Russell CD, Millar JE, Baillie JK. Clinical evidence does not support corticosteroid treatment for 2019‐nCoV lung injury. Lancet 2020; 395: 473–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kinjo N, Hamada K, Hirayama C, Shimizu M. Role of plasma exchange, leukocytapheresis, and plasma diafiltration in management of refractory macrophage activation syndrome. J Clin Apher 2018; 33: 117–20. [DOI] [PubMed] [Google Scholar]
- 86. Demirkol D, Yildizdas D, Bayrakci B et al. Hyperferritinemia in the critically ill child with secondary hemophagocytic lymphohistiocytosis/sepsis/multiple organ dysfunction syndrome/macrophage activation syndrome: what is the treatment? Crit Care 2012; 16: R52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Cui Y, Zhang YC, Kang YL et al. High‐volume hemofiltration in critically ill patients with secondary hemophagocytic lymphohistiocytosis/macrophage activation syndrome: a prospective study in the PICU. Pediatr Crit Care Med 2016; 17: e437–e43. [DOI] [PubMed] [Google Scholar]
- 88. Ghani RA, Zainudin S, Ctkong N et al. Serum IL‐6 and IL‐1‐ra with sequential organ failure assessment scores in septic patients receiving high‐volume haemofiltration and continuous venovenous haemofiltration. Nephrology 2006; 11: 386–93. [DOI] [PubMed] [Google Scholar]
- 89. Park JT, Lee H, Kee YK et al. High‐dose versus conventional‐dose continuous venovenous hemodiafiltration and patient and kidney survival and cytokine removal in sepsis‐associated acute kidney injury: a randomized controlled trial. Am J Kidney Dis 2016; 68: 599–608. [DOI] [PubMed] [Google Scholar]
- 90. Zarbock A, Kellum JA, Schmidt C et al. Effect of early vs delayed Initiation of renal replacement therapy on mortality in critically ill patients with acute kidney injury: the ELAIN randomized clinical trial. JAMA 2016; 315: 2190–9. [DOI] [PubMed] [Google Scholar]
- 91. Barbar SD, Clere‐Jehl R, Bourredjem A et al. Timing of renal‐replacement therapy in patients with acute kidney injury and sepsis. N Engl J Med 2018; 379: 1431–42. [DOI] [PubMed] [Google Scholar]
- 92. Kogelmann K, Jarczak D, Scheller M, Drüner M. Hemoadsorption by CytoSorb in septic patients: a case series. Crit Care 2017; 21: 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Friesecke S, Stecher SS, Gross S, Felix SB, Nierhaus A. Extracorporeal cytokine elimination as rescue therapy in refractory septic shock: a prospective single‐center study. J Artif Organs 2017; 20: 252–9. [DOI] [PubMed] [Google Scholar]
- 94. Chu KH, Tsang WK, Tang CS et al. Acute renal impairment in coronavirus‐associated severe acute respiratory syndrome. Kidney Int 2005; 67: 698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Arabi YM, Arifi AA, Balkhy HH et al. Clinical course and outcomes of critically ill patients with Middle East respiratory syndrome coronavirus infection. Ann Intern Med 2014; 160: 389–97. [DOI] [PubMed] [Google Scholar]
- 96. Opal SM, Fisher CJ Jr, Dhainaut JF et al. Confirmatory interleukin‐1 receptor antagonist trial in severe sepsis: a phase III, randomized, double‐blind, placebo‐controlled, multicenter trial. The Interleukin‐1 Receptor Antagonist Sepsis Investigator Group. Crit Care Med 1997; 25: 1115–24. [DOI] [PubMed] [Google Scholar]
- 97. Shakoory B, Carcillo JA, Chatham WW et al. Interleukin‐1 receptor blockade is associated with reduced mortality in sepsis patients with features of macrophage activation syndrome: reanalysis of a prior phase III trial. Crit Care Med 2016; 44: 275–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Ruan Q, Yang K, Wang W, Jiang L, Song J. Clinical predictors of mortality due to COVID‐19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med 2020; 46: 846–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Chen X, Zhao B, Qu Y et al. Detectable serum SARS‐CoV‐2 viral load (RNAaemia) is closely correlated with drastically elevated interleukin 6 (IL‐6) level in critically ill COVID‐19 patients. Clin Infect Dis 2020. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Rutherford AI, Subessinghe S, Hyrich K, Galloway JB. Serious infection across biologic‐treated patients with rheumatoid arthritis: results from the British Society for Rheumatology Biologics Register for Rheumatoid Arthritis. Ann Rheum Dis 2018; 77: 905–10. [DOI] [PubMed] [Google Scholar]
- 101. Dienz O, Rud JG, Eaton SM et al. Essential role of IL‐6 in protection against H1N1 influenza virus by promoting neutrophil survival in the lung. Mucosal Immunol 2012; 5: 258–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Jamilloux Y, El Jammal T, Vuitton L, Gerfaud‐Valentin M, Kerever S, Sève P. JAK inhibitors for the treatment of autoimmune and inflammatory diseases. Autoimmun Rev 2019; 18: 102390. [DOI] [PubMed] [Google Scholar]
- 103. Tv A, Haikarainen T, Raivola J, Silvennoinen O. Selective JAKinibs: prospects in inflammatory and autoimmune diseases. BioDrugs 2019; 33: 15–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Richardson P, Griffin I, Tucker C et al. Baricitinib as potential treatment for 2019‐nCoV acute respiratory disease. Lancet 2020; 395: e30–e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Zhang W, Zhao Y, Zhang F et al. The use of anti‐inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID‐19): the perspectives of clinical immunologists from China. Clin Immunol 2020; 214: 108393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Praveen D, Puvvada RC, Vijey Aanandhi M. Janus kinase inhibitor baricitinib is not an ideal option for management of COVID‐19. Int J Antimicrobial Agents 2020; 55: 105967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Cao Y, Wei J, Zou L et al. Ruxolitinib in treatment of severe coronavirus disease 2019 (COVID‐19): a multicenter, single‐blind, randomized controlled trial. J Allergy Clin Immunol 2020; S0091–6749: 30738–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Savarino A, Boelaert JR, Cassone A, Majori G, Cauda R. Effects of chloroquine on viral infections: an old drug against today’s diseases? Lancet Infect Dis 2003; 3: 722–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Salata C, Calistri A, Parolin C, Baritussio A, Palù G. Antiviral activity of cationic amphiphilic drugs. Expert Rev Anti Infect Ther 2017; 15: 483–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Vincent MJ, Bergeron E, Benjannet S et al. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol J 2005; 2: 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019‐nCoV) in vitro. Cell Res 2020; 30: 269–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Gao J, Tian Z, Yang X. Breakthrough: chloroquine phosphate has shown apparent efficacy in treatment of COVID‐19 associated pneumonia in clinical studies. Biosci Trends 2020; 14: 72–3. [DOI] [PubMed] [Google Scholar]
- 113. Frisk‐Holmberg M, Bergqvist Y, Englund U. Chloroquine intoxication [letter]. Br J Clin Pharmacol 1983; 15: 502–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Borba MGS, Val FFA, Sampaio VS et al. Effect of high vs low doses of chloroquine diphosphate as adjunctive therapy for patients hospitalized with severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) infection: a randomized clinical trial. JAMA Netw Open 2020; 3: e208857. [DOI] [PubMed] [Google Scholar]
- 115. Yao X, Ye F, Zhang M et al. In vitro antiviral activity and projection of optimized dosing design of hydroxychloroquine for the treatment of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2). Clin Infect Dis 2020. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Gautret P, Lagier JC, Parola P et al. Hydroxychloroquine and azithromycin as a treatment of COVID‐19: results of an open‐label non‐randomized clinical trial. Int J Antimicrob Agents 2020. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Chen J, Liu D, Lui L et al. A pilot study of hydroxychloroquine in treatment of patients with common coronavirus disease‐19 (COVID‐19). J Zhejiang Univ (Med Sci) 2020; 49: 215–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Molina JM, Delaugerre C, Le Goff J et al. No evidence of rapid antiviral clearance or clinical benefit with the combination of hydroxychloroquine and azithromycin in patients with severe COVID‐19 infection. Médecine et Maladies Infectieuses 2020; 50: 384. [DOI] [PMC free article] [PubMed] [Google Scholar]