

Abstract
We present a phylodynamic and phylogeographic analysis of this new severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) virus in this report. A tree of maximum credibility was constructed using the 72 entire genome sequences of this virus, from the three countries (China, Italy, and Spain) available as of 26 March 2020 on the GISAID reference frame. To schematize the current SARS‐CoV‐2 migration scenario between and within the three countries chosen, using the multitype bearth‐death model implemented in BEAST2. Bayesian phylogeographic reconstruction shows that SARS‐CoV‐2 has a rate of evolution of 2.11 × 10−3 per sites per year (95% highest posterior density: 1.56 × 10−3 to 3.89 × 10−3), and a geographic origin in Shanghai, where time until the most recent common ancestor (tMRCA) emerged, according to the analysis of the molecular clock, around 13 November 2019. While for Italy and Spain, there are two tMRCA for each country, which agree with the assumption of several introductions for these countries. That explains also this very short period of subepidermal circulation before the recent events. A total of 8 (median) migration events occurred during this short period, the largest proportion of which (6 events [75%]) occurred from Shanghai (China) to Spain and from Italy to Spain. Such events are marked by speeds of migration that are comparatively lower as compared with that from Shanghai to Italy. Shanghai's R0 and Italy's are closer to each other, though Spain's is slightly higher. All these results allow us to conclude the need for an automatic system of mixed, molecular and classical epidemiological surveillance, which could play a role in this global surveillance of public health and decision‐making.
Keywords: migration rate, multitype bearth‐death, number of introductions, SARS‐CoV‐2
Highlights
The multitype bearth‐death model implemented in BEAST2 is used.
The aim of this study was to estimate the migration rate of the SRAS‐CoV‐2 between and within the three countries chosen (CHINA, ITALY and SPAIN).
Identifying the R0 for each subpopulation.
Analyzing the number of transmission between these three geographic locations.
1. INTRODUCTION
The severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) pandemic (2019) has placed health systems around the world on high alert, many governments have declared a strengthening of measures at their borders to prevent this virus from spreading. 1 , 2 By the time China was the epicenter of this pandemic, a stringent framework for maintaining the distribution of these people had been placed in place, whether at local or international level, the virus had gained ground in many countries around the world, Partly because of its strong transmissibility, and also because a small fraction of infected persons experience little to no symptoms. 3 This global spread suggests a clear and actual understanding of this pathogen's dynamic properties, through modeling, adapting a phylogeographic method that allows for joint estimation of phylogeny and epidemiological parameters of interest. 4 By applying this discipline in this research, which is the most recent field of phylodynamics, 5 we can map the current SARS‐CoV‐2 migration scenario, between and within the three countries selected in this research (China [Shanghai], Italy, Spain), that we assume reflects what is happening around the world. By following an inference approach based on a multi‐type birth‐death model through a combined reconstruction of parameters of phylogeny and phylogeography, such as clusters; the routes of transmission and the routes of introduction of this Virus, which allow us to demonstrate the importance of international cooperation in matters of prevention and public health, and also highlights the importance of molecular surveillance in characterizing the spatio‐temporal links of dissemination of these epidemics.
2. MATERIALS AND METHODS
2.1. Data collection and phylogenetic analysis of SARS‐CoV‐2 genome
As of 26 March 2020, 72 genes of SARS‐CoV‐2 strains obtained from humans have been collected on GISAID (http://gisaid.org/). 6 Shanghai (n = 22 date of last sample 15 February 2020), Italy (n = 25 data from last sample 24 February 2020), Spain (n = 25 date of last sample 20 March 2020). These sequences were separated and selected sometimes manually, to maximize the length of the segments to be analyzed, which allows us to have a coverage of 29 778 pb of the complete genome of SARS‐CoV‐2 of the three countries in question, using the MIGA software. v10.0. 7 For the alignment of all the selected sequences we used CLOSTRALW2. To eliminate the recombination in the dataset, we used splitstreev4.15.1. 8 According to the Bayesian information criterion (BIC) method implemented in the jModelTest v2.1.10 software, 9 , 10 the most suitable nucleotide substitution model for this genomic data set is HKY + 4. Phylogenetic trees with maximum likelihood (ML) were constructed using the HASEGAWA‐KISHINO‐YANO nucleotide substitution model in phyML. 11 Bootstrap support values were calculated with 1000 trees, 12 and the trees were rooted at the peaks of the ML phylogenies perspectives. The Beast2 software, 13 also allows us to estimate the rate of evolution and the time until the most recent common ancestor (tMRCA) for the three locations Shanghai, italy and spain, using ML dating in the BEAST2 multitype bearth‐death package. 14
2.2. Reconstruction of time‐scaled phylogenies
Using Bayesian inference through a framework of Marcov Chain Monte Carlo (MCMC) implemented in BEAST2, we built the evolutionary history of SARS‐CoV‐2 across these three countries (Shanghai, Italy, and Spain). The BEAGLE Library program 15 has speeded up our calculations. To set the time scale prior for the dataset, we used a constrained evolution rate with a Log‐normal prior averaged at 10−3 by substitution per site per year. We performed phylogenetic Bayesian analyzes using a strict clock model. The MCMC chains were executed for 30 million steps with a sampling every 10 000 steps of the posterior distribution. Convergence was evaluated by calculating the effective size of the sample parameters using tracer v1.7.1. 16 All parameters have an effective sample size >200 indicates good mixing. The trees were summarized as tree with maximum credibility (MCC), using TREEANOTATEUR v1.8.4, after having eliminated the 10% as burn‐in, and then visualized in ICYTREE. 17
2.3. Reconstruction of special‐scaled phylogenies
Our data is divided into distinguished partitions called demes, interchangeable and which can be summed up in three geographic locations (Shanghai, Italy, and Spain), so in this dataset, we have strains from three different geographic locations. The Number of demes should be set to 3, which leaves the model estimating the reproduction number per type and the become uninfectious rate per type. This will allow us to see the differences in reproductive fitness and recovery speed between the three locations. To set the sampling proportions correctly by type, we are going to produce a tree that has the same proportion of samples for the whole period. To do this, we will set the first time interval values (minimum values) for the three geographic sites to the value 0, then we will also set the sampling change time parameter, which is the time slightly before the first sample, and the last sample at 0.23 follows our data. This allows us to estimate the following parameters:
-
(1)
R0.deme1, R0.deme2, and R0.deme3: These give the effective reproduction numbers for deme1 (Italy), deme2 (Shanghai), and deme3 (Spain), respectively.
-
(2)
becomeUninfectiousrate.deme1, becomeUninfectiousrate.deme2, and becomeUninfectiousrate.deme3: These are the recovery rates for an infected person in either location.
-
(3)
rateMatrix.deme1, rateMatrix.deme2, and rateMatrix.deme3: These give the migration rates (per lineage per year) between the three demes.
-
(4)
Tree.t:3deme.count between the three partitions: These give the number of ancestral migrations between the three countries on the inferred tree, going from the past to the present.
3. RESULTS
For the SARS‐CoV‐2 genomic dataset from the three geographic locations (Shanghai, Italy, and Spain) the most suitable model was HKY + 4 according to the BIC method. On all of our selected sequences, We found no statistically significant evidence of recombination (P = .89), according to the pairwise homoplasy index. Phylogenetic analyzes of the SARS‐CoV‐2 dataset, using the Bayesian multi‐type birth‐death model and molecular clock calibration, showed an estimate of the rate of evolution of whole genome sequences SARS‐CoV‐2 at 2.11 × 10−3 substitutions per site per year (95% highest posterior density [HPD]: 1.56 × 10−3 to 3.89 × 10−3). Figure 1 shows the MCC tree with Bayesian phylogeographic reconstruction of SARS‐CoV‐2 isolates. Each tree leaf could be interpreted as an end of infection, and each tree branch could be interpreted as a transmission event. The probable origin of SARS‐CoV‐2 is Shanghai with a state posterior probability (spp) of 0.99 dating back to the tMRCA of the human epidemic until 13 November 2019 (95% HPD: 1‐24 November 2019), while for Italy and Spain, there are two tMRCA for each country: Italy has two introduction of this virus, dated 9 and 23 November 2019, while for Spain we have a tMRCA as of 17 November 2019, and another tMRCA as of 27 November 2019, which is consistent with the hypothesis of several introductions for these two countries. 17 , 18 This also explains this very short period of sub‐epidermal circulation before the most recent events. According to our phylogeographic reconstruction, Shanghai is considered to be the source of this pandemic, with a first diffusion towards Italy with a spp of 0.99, followed by the emergence of two distinct lines, one with a further and rapid spread to Spain, with a spp of 0.85, and the second following a more complex scheme: from Shanghai to Italy, Spain (spp = 0.68). The multi‐type bearth‐death model also allowed us to estimate a total of eight migration events (medians) during this short four‐month period covered by this sample, the largest proportion of which were 6 events (75%) is produced from Shanghai to Spain, and from Italy to Spain (see Figure 2(B) and Table 1). In fact, the speed at which a line migrates from Shanghai to Spain m Shanghai to spain = 0.594, (95% HPD: 0.0216‐1.635) and m Italy to Spain = 0.207, (95% HPD: 0.014‐0.618), are relatively lower if compared with that of m Shanghai to Italy = 1.413 (95% HPD: 1.122 × 10−3 to 3.716) this explains that Italy has other routes of introduction apart from Shanghai and which are not figured in this study. The number of migration events within a country is greater than that between countries; because there are simply more events inside than between regions. In this analysis we find CItaly = 26 (95% HPD: 25‐27), CShanghai = 26 (95% HPD: 24‐27), s and CSpain = 20 (95% HPD: 18‐21), this is very reasonable because intra connectivity‐population is easier than interpopulation connectivity. Given the genetic information of these 72 sequences sampled across the three countries (Italy, Shanghai, and Spain) as well as the sampling dates and the subpopulation from which each sample was obtained, the multitype Birth‐death model shows the typical dynamics of SARS‐CoV‐2, the number of basic reproductive numbers R0 for each subpopulation. The Italy R0 = 1.032 (95% HPD: 0.969‐1.111) and the Shanghai R0 = 1.011 (95% HPD: 0.917‐1.124), are closest to each other and their most posterior density intervals raised to 95% including threshold one. So still remain very close to the epidemic threshold which validates the hypothesis of the Chinese introduction of this epidemic to Italy. While Spain R0 = 1.656 (95% HPD: 0.823‐3.015) is slightly higher, but its highest posterior density intervals at 95% also including the threshold one, which is in agreement with the two introductions shown on the tree (see Figure 2A and Table 1).
Figure 1.
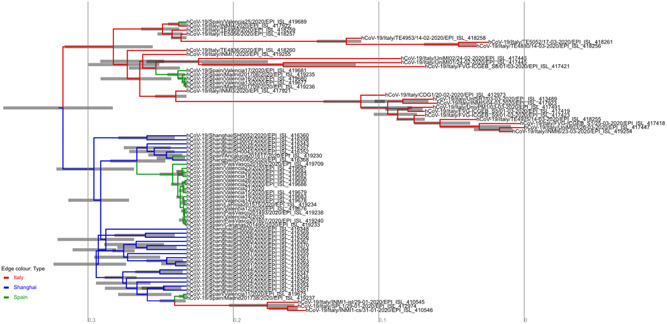
Bayesian maximum clade credibility trees assuming strict molecular clock, generated from the posterior distribution of 10% burn‐in. Branch lengths are shown in months according to the scale bar at the bottom of each panel. Tip branches are colored to represent the country of sampling: red = Italy, green = Spain, and blue = Shanghai (China). The full consensus tree annotated by the locations at coalescence nodes and showing node height uncertainty, with the width of the edges representing 99% how certain we can be of the location estimate at each point on the tree
Figure 2.
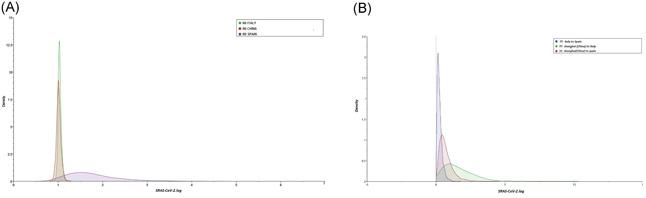
A, Compare the estimated R0 marginal posteriors between China, Italy, and Spain. B, Compare the inferred migration rates between China, Italy, and Spain
Table 1.
Epidemiological parameters estimated by multitype birth‐death analysis
| Parameter | Median | 95% HPD lower | 95% HPD upper |
|---|---|---|---|
| R0 Italy | 1.032 | 0.969 | 1.111 |
| R0 Shanghai | 1.011 | 0.917 | 1.124 |
| R0 Spain | 1.656 | 0.823 | 3.0155 |
| δItaly | 173.738 | 77.686 | 298.531 |
| δShanghai | 173.738 | 77.686 | 298.531 |
| δSpain | 173.738 | 77.686 | 298.531 |
| CShanghai to Italy | 2 | 0 | 3 |
| CShanghai to Spain | 3 | 2 | 4 |
| CItaly to Spain | 3 | 2 | 4 |
| CItaly | 26 | 25 | 27 |
| CShanghai | 26 | 24 | 27 |
| CSpain | 20 | 18 | 21 |
| mItaly to Spain | 0.207 | 0.014 | 0.618 |
| mShanghai to Italy | 1.413 | 1.122 × 10−3 | 3.716 |
| mShanghai to spain | 0.594 | 0.022 | 1.635 |
Note: Posterior parameter estimates of SRAS‐COV‐2 analysis. Median posterior estimates and 95% HPD intervals, the rate to become noninfectious δ, and the migration rates mij and estimated numbers of migration events Cij from subpopulation i to j for i, j∈{Italy, Shanghai, Spain}.
Abbreviations: HPD, highest posterior density; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2.
4. DISCUSSION
The main objective of this study is to schematize the current scenario of SARS‐CoV‐2 migration between and within the three countries of Shanghai, Italy, and Spain, as well as to identify and evaluate transmission routes and routes of introduction of this virus. These pathways which allowed the epidemic to progress rapidly from the initial epidemic in Shanghai to the pandemic which now affects almost all of the world. 19 , 20 , 21 To do this, we reported the phylodynamic and phylogeographic results, proposing the implementation of molecular modeling based on the multi‐type birth‐death model implemented in Beast2, using heterochromatic genomic data retrieved from the GISAID repository. The analysis confirmed the hypothesis that there are multiple sources of introduction in these two countries (Italy and Spain). These multiple sources of introduction in these two countries, give us a real picture on the degree of interconnection of the different sensitive human subpopulations, in the current time. It has allowed the virus to exploit, and spread so rapidly, multiple routes of introduction and transmission. This also indicates the need to set up collaboration mechanisms and coordination activities involving all countries at the planetary level to achieve the fight against epidemics, based on the results of our work. By creating a strong link between traditional epidemiology which aims to investigate the sources of transmission on the basis of traditional surveillance systems which follow the trajectory of the epidemic and genomic analyzes complete and conform, an independent source of information which is not subject to the biases associated to traditional epidemiological data. 22 , 23 We can end up with timely analyzes that are accompanied by different types of persuasive and reliable data. This will allow us to set up an automated mixed system, molecular and classical epidemiological surveillance which can play a role in this global public health and decision‐making surveillance.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
BN conceived of the presented idea. BS, BA, and BN collected data and prepared the datasets. BN, BS, and BA participated to phylogenetic analyses. All authors contributed to manuscript revision, read, and approved the submitted version.
Nabil B, Sabrina B, Abdelhakim B. Transmission route and introduction of pandemic SARS‐CoV‐2 between China, Italy, and Spain. J Med Virol. 2021;93:564–568. 10.1002/jmv.26333
REFERENCES
- 1. Novel coronavirus, Shanghai (2019‐nCoV). Border advisory (5). 2020. https://www.health.govt.nz/our-work/diseases-and-conditions/covid-19-novel-coronavirus
- 2. Wilson ME. What goes on board aircraft? passengers include Aedes, Anopheles, 2019‐nCoV, dengue, Salmonella, Zika, et al. Travel Med Infect Dis. 2020;33:101572. 10.1016/j.tmaid.2020.101572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Skums P, Kirpich A, Icer Baykal P, Zelikovsky A, Chowell G. Global transmission network of SARS‐CoV‐2: from outbreak to pandemic. medRxiv. 10.1101/2020.03.22.20041145 [DOI]
- 4. Kühnert D, Stadler T, Vaughan TG, Drummond AJ. Phylodynamics with migration: a computational framework to quantify population structure from genomic data. Mol Biol Evol. 2016;33(8):2102‐2116. 10.1093/molbev/msw064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Grenfell BT, Pybus OG, Gog JR, et al. Unifying the epidemiological and evolutionary dynamics of pathogens. Science. 2004;303(5656):327‐332. [DOI] [PubMed] [Google Scholar]
- 6. Elbe S, Buckland‐Merrett G. Data, disease and diplomacy: GISAID's innovative contribution to global health. Global Challenges. 2017;1:33‐46. 10.1002/gch2.1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35(6):1547‐1549. 10.1093/molbev/msy096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;23:254‐267. 10.1093/molbev/msj030 [DOI] [PubMed] [Google Scholar]
- 9. Posada D. jModelTest: phylogenetic model averaging. Mol Biol Evol. 2008;25:1253‐1256. [DOI] [PubMed] [Google Scholar]
- 10. Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696‐704. [DOI] [PubMed] [Google Scholar]
- 11. Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximumlikelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59(307):321‐321. 10.1093/sysbio/syq010 [DOI] [PubMed] [Google Scholar]
- 12. Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783‐791. 10.1111/j.1558-5646.1985.tb00420.x [DOI] [PubMed] [Google Scholar]
- 13. Bouckaert R, Vaughan TG, Barido‐Sottani J, et al. BEAST 2.5: an advanced software platform for Bayesian evolutionary analysis. PLoS Comput Biol. 2019;15(4):e1006650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Joëlle Barido‐Sottani Veronika, Bošková Louis, du Plessis Denise, et al. Taming the BEAST—A community teaching material resource for BEAST 2. Syst Biol. 2018;67(1):170‐174. 10.1093/sysbio/syx060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Suchard MA, Rambaut A. Many‐core algorithms for statistical phylogenetics. Bioinformatics. 2009;25:1370‐1376. 10.1093/bioinformatics/btp244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA. Posterior summarisation in Bayesian phylogenetics using Tracer 1.7. Syst Biol. 2018;67:901‐904. 10.1093/sysbio/syy032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zehender G, Lai A, Bergna A, et al. Genomic characterization and phylogenetic analysis of SARS‐COV‐2 in Italy. J Med Virol. 2020:1‐4. 10.1002/jmv.257944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Spiteri G, Fielding J, Diercke M, et al. Review of “First cases of coronavirus disease 2019 (COVID‐19) in the WHO European region, 24 January to 21 February 2020.” Toronto, ON: Queen's Printer for Ontario. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Giovanetti M, Angeletti S, Benvenuto D, Ciccozzi M. A doubt of multiple introduction of sars‐cov‐2 in italy: a preliminary overview. J Med Virol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Giovanetti M, Benvenuto D, Angeletti S, Ciccozzi M. The first two cases of 2019‐ncov in italy: where they come from? J Med Virol. 2020;92:518‐521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bedford T, Neher R, Hadfield J, Hodcroft E, Ilcisin M, Muller N, Genomic analysis of ncov spread. situation report 2020‐01‐23. Tech. rep., 2020.
- 22. Gwinn M, MacCannell D, Armstrong GL. Next‐generation sequencing of infectious pathogens. JAMA. 2019;321(9):893‐894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Armstrong GL, MacCannell DR, Taylor J, et al. Pathogen genomics in public health. N Engl J Med. 2019;381(26):2569‐2580. [DOI] [PMC free article] [PubMed] [Google Scholar]