

Abstract
Parkinson’s disease (PD), the second most common age-related progressive neurodegenerative disorder, is characterized by dopamine depletion and the loss of dopaminergic (DA) neurons with accompanying neuroinflammation. Zonisamide is an-anti-convulsant drug that has recently been shown to improve clinical symptoms of PD through its inhibition of monoamine oxidase B (MAO-B). However, zonisamide has additional targets, including voltage-gated sodium channels (Nav), which may contribute to its reported neuroprotective role in preclinical models of PD. Here, we report that Nav1.6 is highly expressed in microglia of post-mortem PD brain and of mice treated with the parkinsonism-inducing neurotoxin MPTP. Administration of zonisamide (20 mg/kg, i.p. every 4 h × 3) following a single injection of MPTP (12.5 mg/kg, s.c.) reduced microglial Nav 1.6 and microglial activation in the striatum, as indicated by Iba-1 staining and mRNA expression of F4/80. MPTP increased the levels of the pro-inflammatory cytokine TNF-α and gp91phox, and this was significantly reduced by zonisamide. Together, these findings suggest that zonisamide may reduce neuroinflammation through the down-regulation of microglial Nav 1.6. Thus, in addition to its effects on parkinsonian symptoms through inhibition of MAO-B, zonisamide may have disease modifying potential through the inhibition of Nav 1.6 and neuroinflammation.
Keywords: MPTP, Parkinson’s disease, Zonisamide, Voltage-gated sodium channels, Nav1.6, Microglia, Neuroinflammation, TNF-α, gp91phox
1. Introduction
Neuroinflammation and microglial activation contributes to the pathogenesis of a variety of neurodegenerative diseases, including Alzheimer’s disease (AD) and Parkinson’s disease (PD) (Bronzuoli et al., 2016; Heneka et al., 2015; Tansey and Goldberg, 2010; Wang et al., 2015). Microglia play an active role in neuroprotection and repair of neurons in the central nervous system following toxic insult and neuronal injury. However, persistent activation of microglia can mediate neuronal death and neurodegeneration by increasing the secretion of inflammatory molecules and cytokines, including tumor necrosis factor alpha (TNF-α) and reactive oxygen species (ROS) (Harrigan et al., 2008; Liu et al., 2010). Given the association between microglial activation and neurodegeneration, a significant amount of effort has been focused on identifying interventions that can dampen the neuroinflammatory response and slow the initiation or progression of neurodegeneration. Unfortunately, these interventions have not yet been successful in clinical trials.
Microglia express a number of ion channels, including Na+ channels that regulate various aspects of inflammatory process, providing a potential target for intervention (Black et al., 2009; Hossain et al., 2017; Pappalardo et al., 2016; Richardson and Hossain, 2013). Although generation of action potentials is the primary function of voltage-gated sodium channels (VGSC), several recent studies demonstrated that VGSC can regulate a number of cellular functions such as morphological transformation, migration, and phagocytosis of microglia when stimulated with lipopolysaccharide (LPS) (Black et al., 2009; Stevens et al., 2013), suggesting potential immunomodulatory properties of VGSC. We recently reported that Na+ influx through VGSC initiates activation of microglia and subsequently triggers an inflammatory pathway by accumulation of intracellular sodium [(Na+)i] after exposure to LPS (Hossain et al., 2013) and pyrethroid insecticides (Hossain et al., 2017), which appears to involve Nav 1.6. The Nav 1.6 isoform is one of most abundantly expressed isoforms within peripheral and central nervous system of the adult (Goldin, 2001; Krzemien et al., 2000; Tzoumaka et al., 2000) and is also found to be a predominant isoform in microglia (Black et al., 2009; Black and Waxman, 2012; Hossain et al., 2017; Hossain et al., 2013). Additional studies from Waxman’s group demonstrated that mice exhibit a significant upregulation of Nav 1.6 in activated microglia in an experimental inflammatory/demyelinating model of multiple sclerosis (Craner et al., 2005). Furthermore, they reported that the expression of Nav 1.6 expression increases with morphological transformation of microglia to an amoeboid like appearance. Together, these data suggest that microglial Nav 1.6 plays an important role in regulation of microglial inflammation and could serve as a potential therapeutic target in neurodegeneration.
Zonisamide is an anti-convulsant drug approved by the FDA for the treatment of epilepsy (Sonsalla et al., 2010). The primary mechanism by which zonisamide is thought to exert its anti-epileptic effect is through inhibition of the voltage-gated sodium and T-type calcium channels (Biton, 2007; Kito et al., 1996; Matar et al., 2009; Okada et al., 2002). Recently, zonisamide has been reported to improve symptoms in PD patients when used in combination with other anti-parkinsonian drugs, likely through its ability to inhibit monoamine oxidase B (MAO-B) (Murata et al., 2007). In preclinical models, zonisamide has been shown to provide neuroprotection against seizure (Mares, 2010; Ueda et al., 2005) and ischemia (Minato et al., 1997; Owen et al., 1997). Pre-treatment of mice with zonisamide attenuated the MPTP-induced reduction in striatal DA, DOPAC, and tyrosine hydroxylase (TH), which was likely the result of MAO-B inhibition and decreased MPP+ formation (Sonsalla et al., 2010). More recent studies demonstrate that zonisamide can be protective when given after MPTP administration (Choudhury et al., 2011). However, the mechanisms responsible for this effect have not been fully elucidated.
In the present study, we investigated the expression of microglial Nav1.6 in PD brain and MPTP mice. We further assessed the ability of zonisamide to prevent neuroinflammation in an acute mouse model of neuroinflammation produced by a single injection of MPTP (12.5 mg/kg, s.c.) that results in striatal injury and neuroinflammation within 12 h after MPTP administration (O’Callaghan et al., 1990). Our findings demonstrate that Nav1.6 expression is increased in microglia in PD brain and in mice treated with MPTP. Repeated zonisamide treatment administered post-MPTP significantly reduced this expression, which was accompanied by reduction of microglial activation and associated pro-inflammatory mediators. Thus, in addition to its effects on MAO-B, zonisamide may also have a neuroprotective role in PD by reducing microglial activation through down-regulation of Nav1.6.
2. Materials and methods
2.1. Case selection and neuropathological assessment
Specimens were obtained from the Emory Alzheimer Disease Research Center Neuropathology Core, Atlanta, GA. Tissues were fixed in 4% paraformaldehyde for 1 to 2 weeks before being transferred to cryopreservative. The neuropathologic diagnosis of PD was based on the presence of nigral degeneration and Lewy bodies. Control cases had no clinical history or neuropathologic diagnosis of neurologic disease. Case descriptions are provided in Supplemental Table 1.
2.2. Immunofluorescent staining for human tissues
Fixed, cryopreserved, free-floating, 50-μm-thick sections were prepared using a freezing sliding microtome. Basal ganglia samples underwent antigen retrieval using citrate/Tween-20 at 96 °C for 25 min followed by incubation for 60 min in glycine +10% donkey serum. Sections were incubated with mouse anti-Nav 1.6 (1:250 dilution, Cat #WH0006334M4, Sigma Aldrich, St. Louis, MO) and rabbit anti-Iba1 (1:250 dilution, Cat #019–19,741, Wako Pure Chemical Industries, Ltd., Osaka, Japan) overnight at 4 °C. Following incubation, sections were washed in PBS and TruBlack (Biotium) for one minute and then rinsed 3 times with PBS. Sections were then incubated with secondary antibodies conjugated with Alexa Fluor 594 or 488 for 1.5 h and then rinsed 4 times with PBS. Sections were mounted on glass slides and coverslipped using Fluormount G mounting media. The specificity of the Nav1.6 antibody was confirmed by omitting the primary antibody, which resulted in no detectable signal. Fluorescent images were captured on a Zeiss Axio Observer D1 microscope (Zeiss Inc., Germany) with an X-Cite series 120Q fluorescent illuminator, a ProgRes® MF camera (Jenoptik Optical Systems GmbH, Germany), and ProgRes®CapturePro 2.8 software (Jenoptik Inc., Easthampton, MA).
Optical intensity of fluorescence of Nav1.6 stain was semi-quantified in individual cells that were co-labeled with Iba1 (20–25 cells/section) using ImageJ software (NIH) and the data were presented as density/intensity changes in the expression of Nav 1.6.
2.3. Animals
Ten-week-old male C57BL/6 J mice were purchased from Jackson Laboratory (Bar Harbor, ME). They were housed 5 per cage in a temperature-controlled animal care facility under a 12 h light/dark cycle with ad libitum access to food and water. Experiments were performed in compliance with the NIH Guide for the Care and Use of Laboratory Animals and approved by the animal care and utilization committee of Rutgers-Robert Wood Johnson Medical School.
2.4. MPTP treatment
A total of 36 mice were randomly divided into 9 mice per group across four treatment groups (saline, MPTP + saline, zonisamide + saline, and MPTP + zonisamide). MPTP (Sigma, St. Louis, MO) and zonisamide (Sigma, St. Louis, MO) were freshly prepared in saline (0.9% NaCl, Hospira, Lake Forest, IL). Mice were given a single injection of MPTP (base free, 12.5 mg/kg s.c., Sigma-Aldrich, St. Luis, MO) and 90 min later mice were administered the first dose of zonisamide (20 mg/kg i.p.) or saline followed by an injection every 4 h for total of 3 injections. We selected a single dose of 12.5/kg of MPTP because this dosing regimen has been shown to result in striatal injury and neuroinflammation that occurs rapidly after administration (O’Callaghan et al., 1990). The timing of the zonisamide treatment was to ensure that it occurred after the peak of MPP+ levels in the brain, which peaks at 90 min following administration (J.P. O’Callaghan, personal communication), such that its ability to inhibit MAO-B would not confound the experiment (Sonsalla et al., 2010). Animals were euthanized with CO2 2.5 h after the last dose of zonisamide. Striata were rapidly dissected out, frozen in liquid nitrogen, and stored at −80 °C until processing for RNA isolation.
2.5. Immunofluorescent staining for mouse tissues
Mice were anesthetized with sodium pentobarbital (50 mg/kg) and transcardially perfused with phosphate buffered saline (PBS) followed by 4% paraformaldehyde in PBS (pH 7.4). Brains were removed and post-fixed in 4% paraformaldehyde at 4 °C for 7 days, and then transferred into 30% sucrose with 0.1% sodium azide in PBS. The entire striatum was cut into 30 μm coronal sections on a freezing, sliding microtome.
Immunofluorescent staining was performed as described previously with some modification (Alam et al., 2017). Briefly, free-floating sections were rinsed, then blocked with 10% goat serum in PBS and 0.3% Triton X-100 for 60 min at room temp. Sections were then incubated overnight at 4 °C with anti-Iba1 (1:250 dilution, Cat #019–19741, Wako Pure Chemical Industries, Ltd., Osaka, Japan), anti-Nav1.6 (1:250 dilution, Cat #WH0006334M4, Sigma-Aldrich), anti-TNF-α (1:500 dilution, cat #AB1793, Abcam, Cambridge, MA), anti-gp91phox (1:250 dilution, Cat # SC27635, Santa Cruz, CA) with 3% goat serum in PBS containing 0.3% Triton X-100. After being washed three times, sections were incubated with appropriate secondary antibody conjugated to Alexa Fluor (1:250 dilution, Life Technologies, Grand Island, NY) for 1 h at room temperature in the dark. Sections were then rinsed of excess secondary antibody, mounted onto slides, dried, and cover lipped with Prolong Gold containing 4′,6-diamidino-2-phenylindole (Life Technologies). Fluorescent images were captured as described above for the human samples. A negative control consisting of incubations without primary antibody was included to determine specific staining of Nav1.6. Semi-quantification of images for Nav1.6 were performed as described above.
2.6. RNA isolation and real-time quantitative PCR
RNA isolation and qPCR was performed as described previously (Fortin et al., 2013; Hossain et al., 2015). Briefly, RNA was isolated using RNeasy mini kits as per manufacturer’s instructions (QIAGEN, Valencia, CA). RNA concentration was then determined on a NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA) and cDNA was synthesized from 1 μg of total RNA with the First Strand Super Script cDNA synthesis kit (Invitrogen, San Diego, CA). Real-time qPCR was performed in a 384-well plate using the ABI 7900HT and SYBR Green (Applied Biosystems, Foster City, CA) detection system. Melt curves were obtained to confirm the quality of product for each primer set. Each primer set was checked by agarose gel electrophoresis to confirm a single PCR product of the expected size. The expression of GAPDH was used to normalize the amount of mRNA in each sample. Data were analyzed using the ΔΔCt method and results are presented as relative levels of mRNA. The primer sequences for all target mRNAs are designed with the Primer Blast program (NCBI) and provided in Supplemental Table 2.
2.7. Statistical analysis
Statistical analyses were performed using Prism 5.01 software (Graphpad software, San Diego, CA). Student’s t-test was used to determine differences between control and PD brain expression of Nav 1.6. All analyses for the animal treatments were performed using two-way ANOVA followed by Bonferroni’s post hoc multiple comparison test to compare the difference among groups. All values were expressed as mean ± SEM and the p value < 0.05 was considered statistically significant.
3. Results
3.1. Increased expression of Nav 1.6 in striatal microglia in Parkinson’s disease patients
Because increased markers of inflammation and microglial activation have been observed in the post mortem brains of PD patients (McGeer et al., 1988) and Nav 1.6 has been found to be a significant contributor to microglial activation in vitro (Black et al., 2009; Hossain et al., 2017; Hossain et al., 2013), we examined Nav 1.6 and microglial activation in the striatum of post mortem brains from PD patients by immunofluorescence staining (Fig. 1; Fig. S1). To this end, we used Iba1 to immunostain microglia in human PD brain and aged matched controls. We found that Nav1.6 is expressed at low levels in Iba1+ microglia in control human striatum and is mainly present in microglia demonstrating signs of activation. Activated microglia in the striatum of post-mortem PD patients showed strong expression of Nav 1.6 (bottom panel) when compared to age matched control brains (top panel). To confirm differences between control and PD, we quantified the fluorescent expression of Nav1.6 in Iba1-labeled cells and the data showed a significant increase in the expression of microglial Nav1.6 in PD brain when compared to control brain.
Fig. 1.
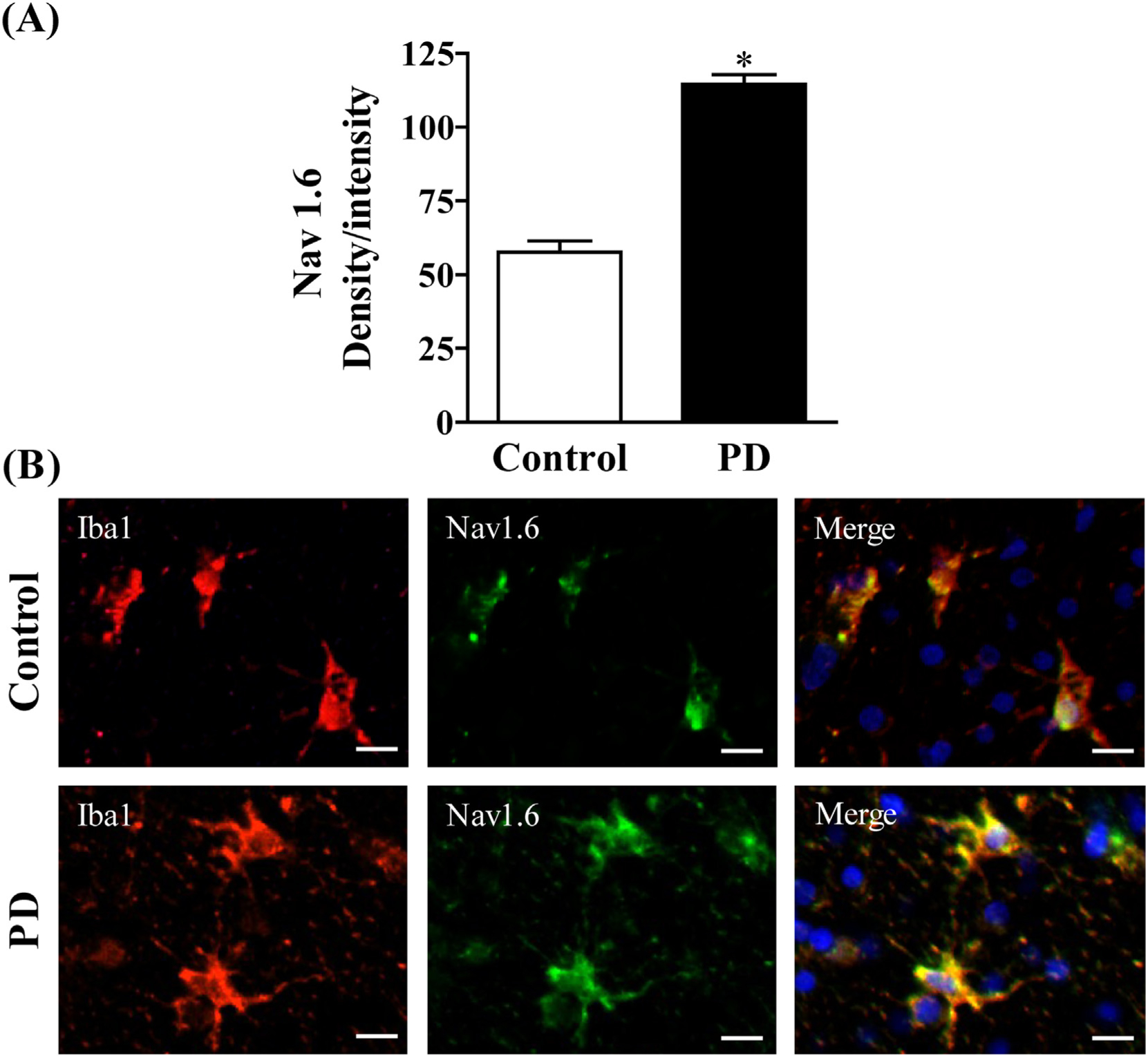
(A) Semi-quantification of Nav1.6 staining.(B) Expression of Nav1.6 in microglia of postmortem brain. Age matched control (Top panel) and PD (Bottom). Microglia were labeled with anti-Iba1 antibody (red) and expression of Nav1.6 was visualized by immunolabeling with anti-Nav1.6 antibody (green). Scale bar = 20 μM. Images were captured on a Zeiss Observer D1 microscope (Zeiss Inc., Thornwood, NY) with an X-Cite series 120Q fluorescent illuminator and a Jenoptik camera with ProgRes CapturePro 2.8 software (Jenoptik, Easthampton, MA). Optical density per intensity of fluorescence against Nav1.6 stain was semi-quantified in individual cells using ImageJ software (NIH). The values represent mean density/intensity ± SEM from 20 to 25 cells/section/brain. * indicates significant difference from control (p < 0.05) by Student’s t-test.
3.2. Increased expression of Nav 1.6 in striatal microglia of mice treated with MPTP and its reduction by zonisamide
To further study Nav 1.6 in activated microglia and neuroinflammation, we used a mouse model of PD by MPTP injection. C57BL/6 J mice were given a single injection of MPTP (12.5 mg/kg, s.c.) and sacrificed 12 h later. The expression of Nav 1.6 in striatal microglia was visualized by immunofluorescence staining together with Iba1 (Fig. 2) andMPTP increased the expression of Nav 1.6 in activated microglia in the striatum. Next, we examined the effect of zonisamide on MPTP-induced Nav 1.6. Repeated treatment of mice with zonisamide (20 mg/kg, i.p.), every 4 h for 3 injections, significantly attenuated MPTP-induced expression of microglial Nav 1.6. The quantified immunofluorescent data demonstrate that MPTP significantly increased expression of microglial Nav1.6 when compared to control, which was reduced by zonisamide treatment.
Fig. 2.
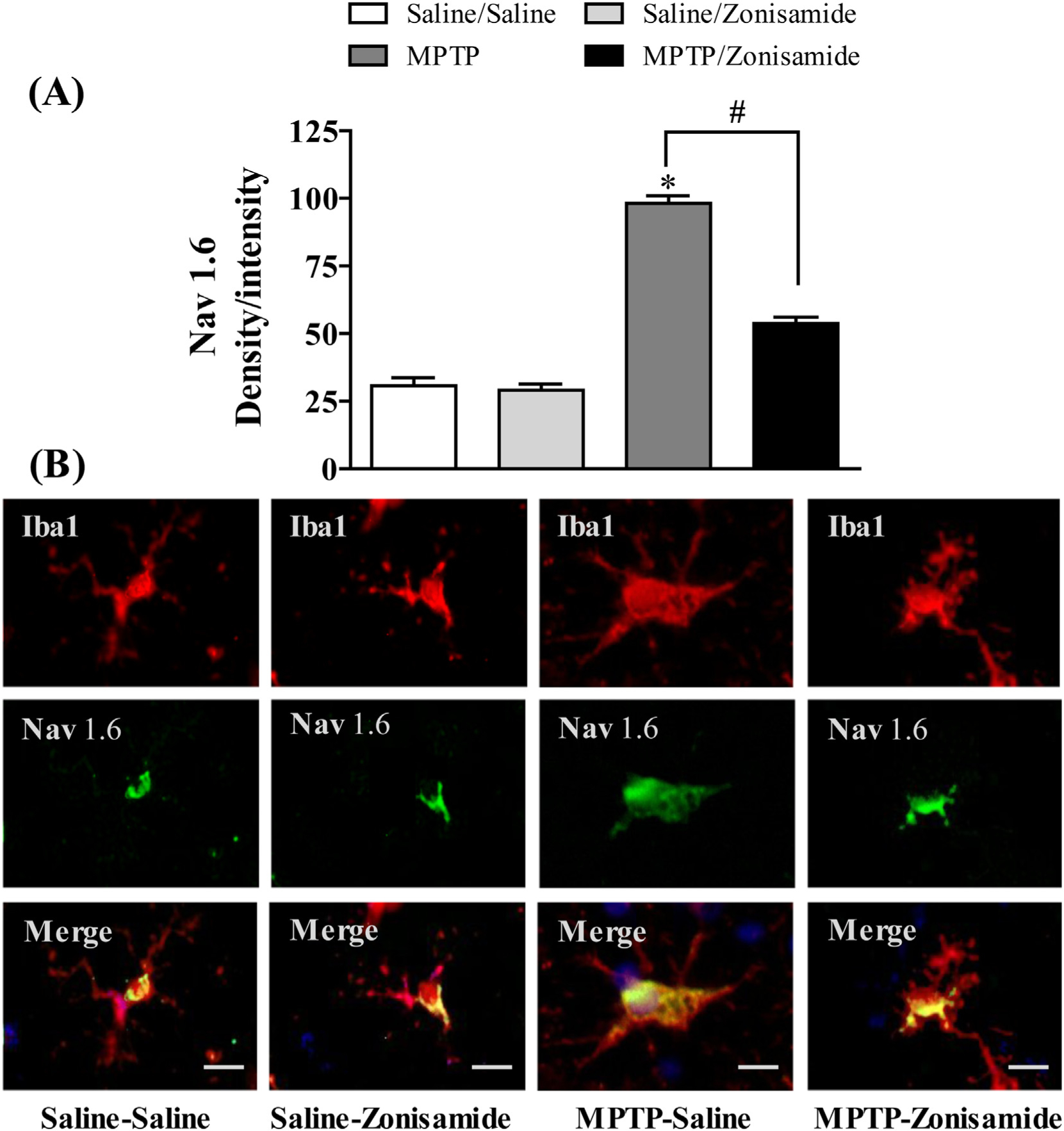
Zonisamide treatment reduces Nav 1.6 expression in striatal microglia following acute exposure to MPTP. (A) Semi-quantification for Nav1.6 stain. (B) Microglia were labeled with anti-Iba1 antibody (red) and expression of Nav1.6 was visualized by immunolabeling with anti-Nav1.6 antibody (green) Scale bar = 20 μM. Images were captured on a Zeiss Observer D1 microscope (Zeiss Inc., Thornwood, NY) with an X-Cite series 120Q fluorescent illuminator and a Jenoptik camera with ProgRes CapturePro 2.8 software (Jenoptik, Easthampton, MA). Optical density per intensity of fluorescence against Nav1.6 stain was semi-quantified in individual cells using ImageJ software (NIH). The values represent mean density/intensity ± SEM from 20 to 25 cells/section/animal. * indicates significant difference from control and # indicates significant differences from MPTP + Saline when compared with MPTP +Zonisamide (p < 0.05).
3.3. Repeated treatment with Zonisamide prevents microglial activation and decreases F4/80 mRNA expression in the striatum of MPTP treated mice
To evaluate the inflammatory response of microglia following a single MPTP exposure, striatal brain sections were examined with Iba1 immunofluorescence staining (Fig. 3A). Activation of microglia is accompanied by characteristic morphological changes, including be-coming more amoeboid. We revealed an amoeboid appearance of microglia with large soma in the striatum of MPTP treated mice compared to control, indicating activation of microglia by MPTP. Repeated zonsamide treatments attenuated MPTP-induced activation of microglia, as morphological evidence of activated microglia was reduced. Furthermore, MPTP increased mRNA expression of striatal F4/80 mRNA, a mouse-specific microglial marker, which was significantly reduced by Zonisamide treatment (Fig. 3B).
Fig. 3.
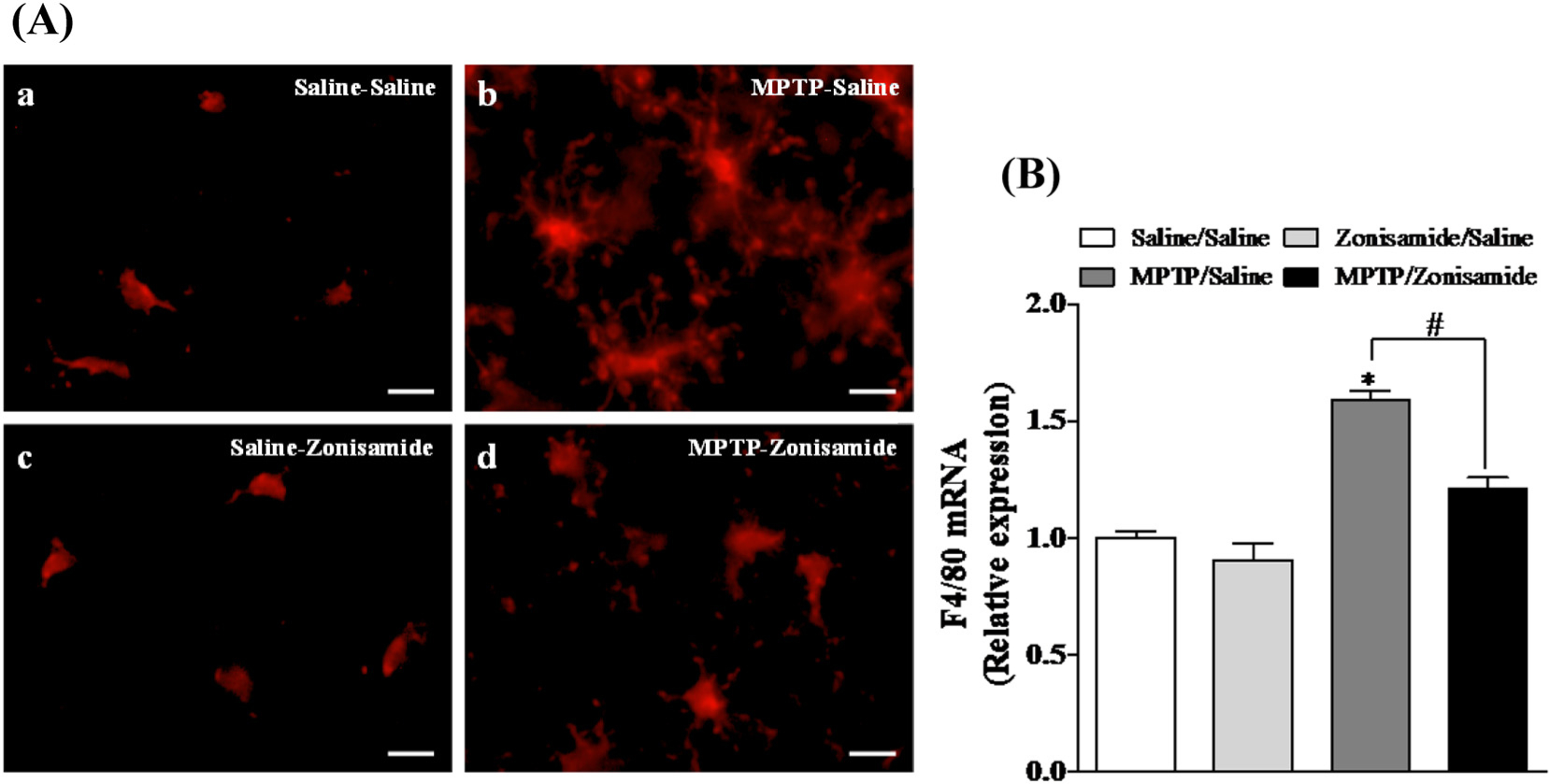
Zonisamide treatment reduces microglial activation and mRNA expression of strial F4/80 following a single exposure to MPTP. (A) Microglia were labeled with anti-Iba1 antibody (red) after 12 h MPTP (12.5 mg/kg, s.c.) exposure with or without zonisamide (20 mg/kg, i.p. every 4 h × 3). Microglia in resting condition in saline (A.a) and zonisamide control (A.b) and activated microglia in MPTP treated animals (A.c). Administration of zonisamide reduces MPTP-induced microglial activation (A.d). Scale bar = 20 μM. (B) mRNA expression of striatal F4/80. All values were expressed as mean ± SEM from 5 animals per group. * indicates significant difference from control and # indicates significant differences from MPTP + Saline when compared with MPTP + zonisamide (p < 0.05).
3.4. Zonisamide treatment suppresses gp91phox mRNA and protein expression in MPTP treated mice
The glycoprotein gp91phox is an important catalytic subunit of NADPH oxidase that influences dopaminergic neurodegeneration by generating ROS and cytokine secretion in brain (Lull and Block, 2010; Qin and Crews, 2012). Consistent with previous studies (Chung et al., 2017; Joglar et al., 2009), we found that a single exposure to MPTP significantly increased mRNA expression of gp91phox in the striatum (Fig. 4A). The induction of gp91phox mRNA was 1.5-fold higher in MPTP treated mice when compared to saline control. In addition, immunofluorescence staining results showed that gp91phox is predominantly expressed in striatal microglia and is also enhanced after MPTP treatment (Fig. 4B). Zonisamide significantly decreased gp91phox mRNA induction and reduced staining of microglial gp91phox following MPTP treatment.
Fig. 4.
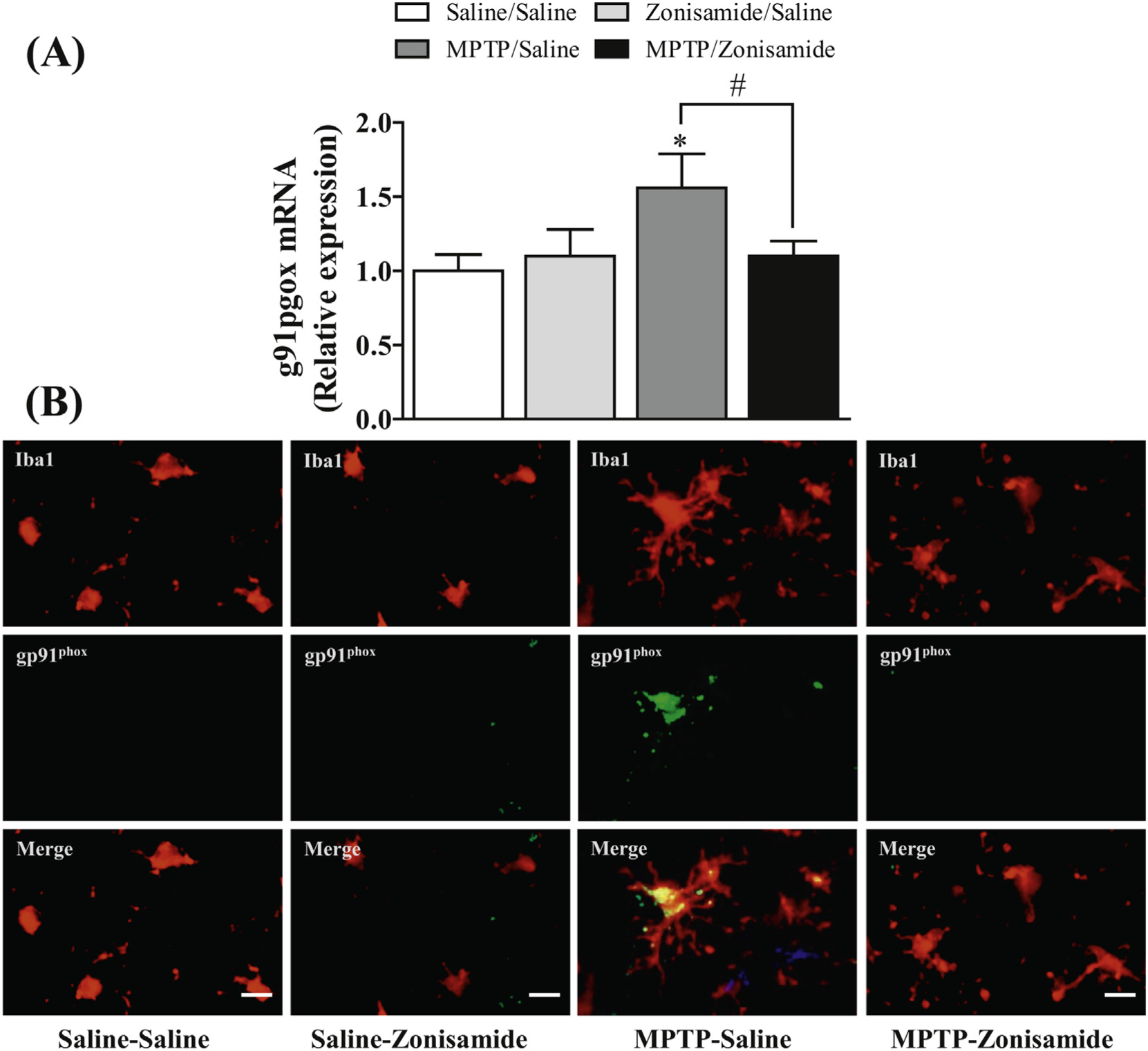
Zonisamide reduces the expression of gp91phox in microglia activated with MPTP. (A) mRNA expression of gp91phox in the striatum. All values were expressed as mean ± SEM from 5 animals per group. * indicates significant difference from control and # indicates significant differences from MPTP + Saline when compared with MPTP + zonisamide (p < 0.05). (B) immunolabeling of microglia and gp91phox in the striatum. Microglia were label with anti-Iba1 antibody (red) and expression of gp91phox was visualized by immunolabeling with anti-gp91phox antibody (green) at 12 h MPTP (12.5 mg/kg, s.c.) exposure with or without zonisamide (20 mg/kg, i.p. every 4 h × 3) administration. Scale bar = 20 μM.
3.5. Zonisamide treatment reduced the induction of TNF-α in MPTP treated mice
Activation of NADPH oxidase in microglia can increase the release of a number of cytokines and chemokines (Hossain et al., 2013; Liu et al., 2010; Turchan-Cholewo et al., 2009). Here, we assayed mRNA and protein expression of Tnf-α following MPTP exposure with or without zonisamide treatment. MPTP administration significantly increased striatal mRNA expression of Tnf-α (Fig. 5A) by about 25-fold in MPTP treated mice when compared to control. Because activated microglia are a significant source for releasing of Tnf-α, we examined immunofluorescence staining of Tnf-α in striatal microglia. We found that MPTP exposure resulted in increased expression of Tnf-α in activated microglia as compared to saline control (Fig. 5B). Mice treated with zonsimide significantly attenuated MPTP-induced up-regulation of mRNA expression of Tnf-α in the striatum and protein expression of Tnf-α in Iba1-stained microglia.
Fig. 5.
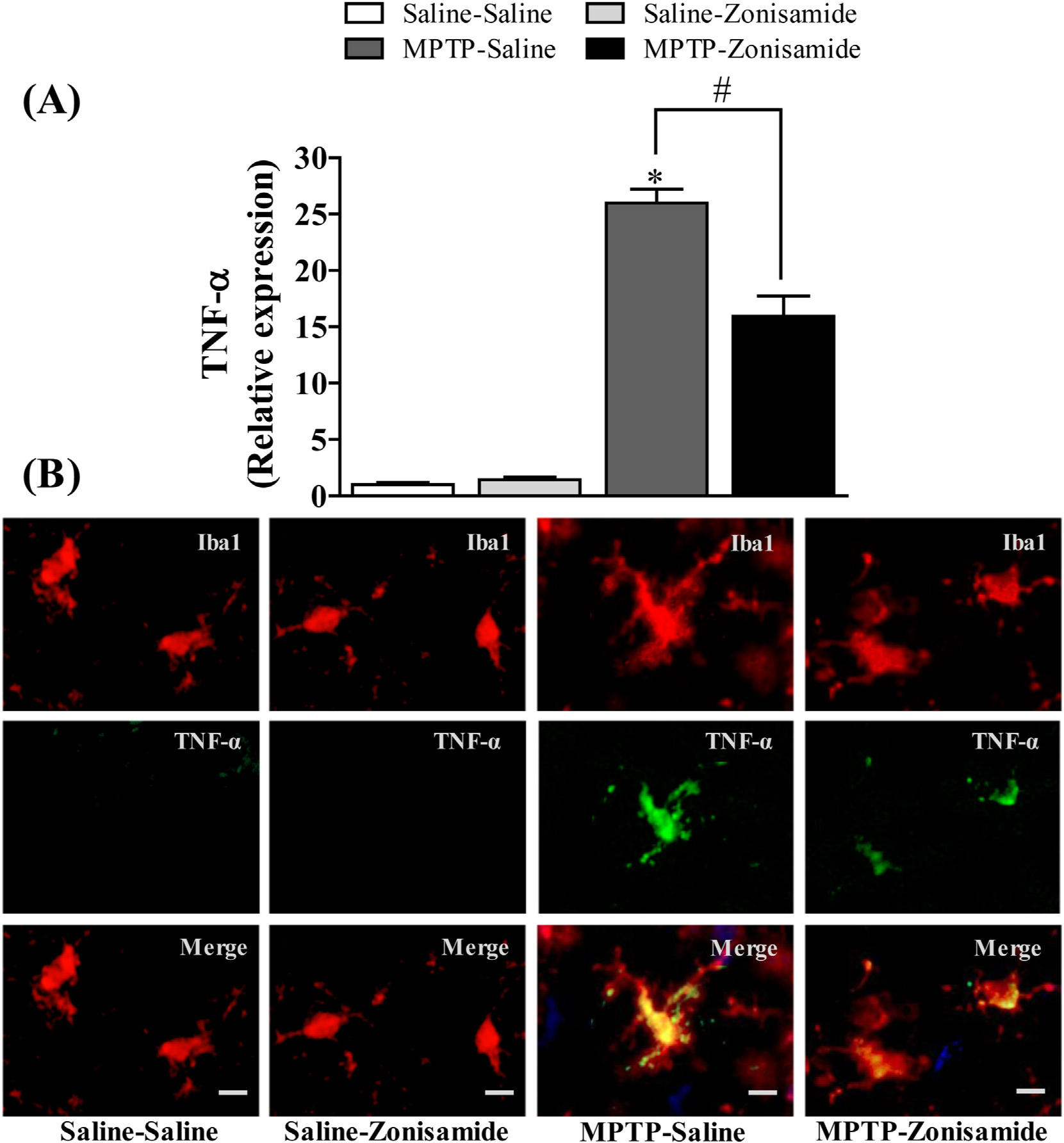
Zonisamide reduces the expression of TNF-α in microglia activated with MPTP. (A) mRNA expression of TNF-α. All values were expressed as mean ± SEM from 5 animals per group. * indicates significant difference from control and # indicates significant differences from MPTP + Saline when compared with MPTP + zonisamide (p < 0.05). (B) Immunofluorescent staining of microgila and TNF-α in the striatum. Microglia were labeled with anti-Iba1 antibody (red) and expression of TNF-α was visualized by with anti-TNF-α antibody (green) at 12 h following MPTP (12.5 mg/kg, s.c.) exposure with or without zonisamide (20 mg/kg, i.p. every 4 h × 3) administration. Scale bar = 20 μM.
4. Discussion
Chronic neuroinflammation is a common feature of many neurodegenerative diseases and activation of microglia have been proposed to play both causative and aggravating roles in the pathogenic process of DA neuron degeneration in PD (Hirsch et al., 2012; Lofrumento et al., 2011; Lull and Block, 2010). Post-mortem brains of human PD patients consistently display evidence of inflammation and microglial activation, first shown by McGeer and colleagues (McGeer et al., 1988). Additionally, increased expression of many inflammatory mediators, including TNF-α, inducible nitric oxide synthase (iNOS), and cyclooxygenase 2 (COX2) have been identified in activated microglia located in the SN of PD patients (Knott et al., 2000; Nagatsu et al., 2000; Richardson and Hossain, 2013). In experimental animal models of PD, sustained release of TNF-α and production of ROS by activated microglia leads to DA neuron degeneration, suggesting the regulation of microglial function could be a potential therapeutic target (Kempuraj et al., 2013; Lull and Block, 2010; Richardson and Hossain, 2013; Wang et al., 2015). However, to date, no neuroprotective drug has been successful in PD clinical trials. Here, we report that activated microglia in PD brain and in MPTP-treated mice display prominent expression of Nav 1.6. This expression in MPTP treated mice was reduced by treatment with zonisamide and accompanied by decreased microglial activation and expression of TNF-α and gp91phox. These data suggest that zonisamide may exert neuroprotective properties through the dampening of Nav 1.6 expression and decreased microglial activation.
The neuroprotective potential for zonisamide has been an active area of research in recent years (Rosler et al., 2010; Grover et al., 2012). In PD models, a number of mechanisms have been explored, including inhibition of MAO-B (Sonsalla et al., 2010), increases in antioxidant enzymes (Kawajiri et al., 2010), suppression of ER stress (Tsuji et al., 2015), alteration of proteasome function (Bentea et al., 2016), increased neurotrophin levels (Sano et al., 2015) and general disruption of the inflammatory response (Yokoyama et al., 2010). The role of inflammation is a particular attractive target, as many, if not all, of the other protective effects noted above can be attributed to a dampened inflammatory response. However, the mechanism responsible for zonisamide’s effects on neuroinflammation has not been established.
Microglia express a number of VGSC and blockade of these channels attenuates multiple functions of these immune cells (Black et al., 2009; Black and Waxman, 2012; Mantegazza et al., 2010). Experimental studies demonstrated that the VGSC contribute to activation of microglia, cytokine secretion and respiratory burst after LPS stimulation (Black et al., 2009; Black and Waxman, 2012; Hossain et al., 2017; Hossain et al., 2013). There are several VGSC isoforms present in microglia, however, Nav1.6 appears to play the predominant role in modulating microglial function (Black et al., 2009; Black and Waxman, 2012; Hossain et al., 2013). In addition, Nav1.6 is prominently expressed in both primary microglia (Hossain et al., 2017; Hossain et al., 2013) and in neurons. The channel mediates a persistent sodium current that is not normally seen with other sodium channel isoforms (Tan and Soderlund, 2010). Here, Nav1.6 was found to be present in murine and human microglia. The expression was particularly strong within activated microglia of post-mortem PD brains and in MTPT-treated mice. This observation is similar to that reported in experimental autoimmune encephalomyelitis and an inflammatory model of multiple sclerosis (Black and Waxman, 2012; Craner et al., 2005). When MPTP-treated mice were administered zonisamide, the increase in Nav1.6 was reduced and was accompanied by morphological changes of microglia consistent with reduction of activation and by reduced expression of F4/80, a mouse-specific microglial marker. These findings are consistent with data generated from microglia derived from med mice, which lack functional Nav1.6 (Kohrman et al., 1996), and show a reduced response to inflammatory signaling following stimulation with ATP (Black and Waxman, 2012; Craner et al., 2005). Together, these data provide support for the hypothesis that the sodium channel Nav 1.6 is a key participant in molecular pathways associated with microglial activation.
NADPH oxidase is a prime generator of microglial ROS both in vitro (Liu et al., 2010) and in vivo (Qin and Crews, 2012). Microglial NADPH oxidase is upregulated in post-mortem PD brain as shown by increased immunostaining for gp91phox, indicating a potential role for microglial NADPH oxidase in neurodegeneration in PD (Wu et al., 2003). Accumulating evidence indicates that the NADPH oxidase pathway influences dopaminergic neurodegeneration following exposure to LPS and MPTP. Studies using neuron-glia cultures from NADPH oxidase-deficient mice reported that NADPH oxidase initiates an intracellular ROS signaling pathways that can activate microglia and release inflammatory cytokines (Forman and Torres, 2002; Qin et al., 2004). Further, mice lacking the catalytic subunit gp91phox exhibit reduced microglial activation and neurodegeneration (Cengiz et al., 2011; Qin and Crews, 2012; Qin et al., 2004). Here, a single exposure to MPTP increased the mRNA expression of gp91phox and TNF-α and staining for these molecules in activated microglia in the striatum, which was significantly attenuated when mice were treated with zonisamide. These data clearly demonstrate the ability of zonisamide to reduce pro-inflammatory processes and provide a plausible mechanism for its reported neuroprotective properties.
Although the exact mechanism by which Nav 1.6 regulates microglial pro-inflammatory processes remains to be precisely elucidated (Black and Waxman, 2012), we recently reported that stimulation of microglial cells with pyrethroid pesticides or LPS causes a rapid influx of Na+ through VGSC (Hossain et al., 2017; Hossain et al., 2013). Following this influx, a series of pro-inflammatory signaling events occurred, including activation of NADPH oxidase, release of TNF-α and production of ROS (Hossain et al., 2013). Blockade of VGSC by tetrodotoxin (TTX) effectively blocked or reduced these pro-inflammatory processes. During these initial experiments, we discovered that there is a compensatory increase in the Na+/H+ exchanger (NHE1) that appears to function as a means to counteract the Na+ influx that results from LPS treatment (Hossain et al., 2013). Indeed, combination of TTX and the NHE1 inhibitor caripropride reduced intracellular Na+ levels and pro-inflammatory cytokine release more potently than either alone. These data suggest that intracellular Na+ homeostasis is an important component of microglial activation and maintenance of pro-inflammatory cytokine secretion and ROS formation. However, additional study is required to dissect the molecular pathways in microglial activation regulated by VGSC.
5. Conclusion
Because microglial activation and neuroinflammation contribute to the initiation and progression of neurodegeneration in PD, several drugs targeting neuroinflammation have been reported to attenuate the loss of DA neurons and the behavioral deficits seen in animal models of PD. Unfortunately, most have failed in clinical trials. Therefore, there is a significant need to find therapeutics that target the disease process. This study provides evidence that Nav1.6 highly expressed in activated murine and human microglia within the basal ganglia and that zonisamide treatment reduces this expression along with that of pro-inflammatory cytokines and NADPH oxidase. Taken in concert with data from the literature and these data demonstrating that zonisamide can reduce inflammation in the MPTP model, it appears that Nav1.6 may play an integral role in the molecular pathway(s) responsible for microglial activation and neuroinflammation in PD. Finally, these data suggest that targeting Nav1.6 with zonisamide may provide novel pathway for therapeutic intervention for reducing neuroinflammation in PD, while also improving PD symptoms through its ability to inhibition MAO-B.
Supplementary Material
Funding
This work was supported in part by the following NIH Grants: R01ES021800, R01NS088627, P30ES005022, U01NS079249, P30NS055077 and P50AG025688. Additional support was provided by the Michael J Fox Foundation and private support from the Glenn and Karen Leppo Parkinson’s Research Fund, the Richard Nicely Parkinson’s Research Fund and the Alan and Janice Woll Parkinson’s Research Fund. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or any other funding source.
Footnotes
Conflict of interest
The authors declare that they have no competing financial interests or other conflicts of interest.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.expneurol.2018.07.005.
References
- Alam G, Edler M, Burchfield S, Richardson JR, 2017. Single low doses of MPTP decrease tyrosine hydroxylase expression in the absence of overt neuron loss. Neurotoxicology 60, 99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentea E, Van Liefferinge J, Verbruggen L, Martens K, Kobayashi S, Deneyer L, Demuyser T, Albertini G, Maes K, Sato H, Smolers I, Lewerenze J, Massie A, 2017. Zonisamide attenuates lactacystin-induced parkinsonism in mice without affecting system Xc-. Exp Neurol 290, 15–28. [DOI] [PubMed] [Google Scholar]
- Biton V, 2007. Clinical pharmacology and mechanism of action of zonisamide. Clin. Neuropharmacol 30, 230–240. [DOI] [PubMed] [Google Scholar]
- Black JA, Waxman SG, 2012. Sodium channels and microglial function. Exp. Neurol 234, 302–315. [DOI] [PubMed] [Google Scholar]
- Black JA, Liu S, Waxman SG, 2009. Sodium channel activity modulates multiple functions in microglia. Glia 57, 1072–1081. [DOI] [PubMed] [Google Scholar]
- Bronzuoli MR, Iacomino A, Steardo L, Scuderi C, 2016. Targeting neuroinflammation in Alzheimer’s disease. J. Inflamm. Res 9, 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cengiz P, Kleman N, Uluc K, Kendigelen P, Hagemann T, Akture E, Messing A, Ferrazzano P, Sun D, 2011. Inhibition of Na+/H+ exchanger isoform 1 is neuroprotective in neonatal hypoxic ischemic brain injury. Antioxid. Redox Signal 14, 1803–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury ME, Moritoyo T, Kubo M, Kyaw WT, Yabe H, Nishikawa N, Nagai M, Matsuda S, Nomoto M, 2011. Zonisamide-induced long-lasting recovery of dopaminergic neurons from MPTP-toxicity. Brain Res. 1384, 170–178. [DOI] [PubMed] [Google Scholar]
- Chung YC, Baek JY, Kim SR, Ko HW, Bok E, Shin WH, Won SY, Jin BK, 2017. Capsaicin prevents degeneration of dopamine neurons by inhibiting glial activation and oxidative stress in the MPTP model of Parkinson’s disease. Exp. Mol. Med 49, e298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craner MJ, Damarjian TG, Liu S, Hains BC, Lo AC, Black JA, Newcombe J, Cuzner ML, Waxman SG, 2005. Sodium channels contribute to microglia/macrophage activation and function in EAE and MS. Glia 49, 220–229. [DOI] [PubMed] [Google Scholar]
- Forman HJ, Torres M, 2002. Reactive oxygen species and cell signaling: respiratory burst in macrophage signaling. Am. J. Respir. Crit. Care Med 166, S4–S8. [DOI] [PubMed] [Google Scholar]
- Fortin MC, Aleksunes LM, Richardson JR, 2013. Alteration of the expression of pesticide-metabolizing enzymes in pregnant mice: potential role in the increased vulnerability of the developing brain. Drug Metab. Dispos 41, 326–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin AL, 2001. Resurgence of sodium channel research. Annu. Rev. Physiol 63, 871–894. [DOI] [PubMed] [Google Scholar]
- Grover ND, Limaye RP, Gokhale DV, Patil TR, 2013. Zonisamide: a review of the clinical and experimental evidence for its use in Parkinson’s disease. Indian J Pharmacol 45, 547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrigan TJ, Abdullaev IF, Jourd’heuil D, Mongin AA, 2008. Activation of microglia with zymosan promotes excitatory amino acid release via volume-regulated anion channels: the role of NADPH oxidases. J. Neurochem 106, 2449–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM, Herrup K, Frautschy SA, Finsen B, Brown GC, Verkhratsky A, Yamanaka K, Koistinaho J, Latz E, Halle A, Petzold GC, Town T, Morgan D, Shinohara ML, Perry VH, Holmes C, Bazan NG, Brooks DJ, Hunot S, Joseph B, Deigendesch N, Garaschuk O, Boddeke E, Dinarello CA, Breitner JC, Cole GM, Golenbock DT, Kummer MP, 2015. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch EC, Vyas S, Hunot S, 2012. Neuroinflammation in Parkinson’s disease. Parkinsonism Relat. Disord 18 (Suppl 1), S210–S212. [DOI] [PubMed] [Google Scholar]
- Hossain MM, Sonsalla PK, Richardson JR, 2013. Coordinated role of voltage-gated sodium channels and the Na+/H+ exchanger in sustaining microglial activation during inflammation. Toxicol. Appl. Pharmacol 273, 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain MM, Dicicco-Bloom E, Richardson JR, 2015. Hippocampal ER stress and learning deficits following repeated pyrethroid exposure. Toxicol. Sci 143, 220–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain MM, Liu J, Richardson JR, 2017. Pyrethroid insecticides directly activate microglia through interaction with voltage-gated sodium channels. Toxicol. Sci 155, 112–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joglar B, Rodriguez-Pallares J, Rodriguez-Perez AI, Rey P, Guerra MJ, Labandeira-Garcia JL, 2009. The inflammatory response in the MPTP model of Parkinson’s disease is mediated by brain angiotensin: relevance to progression of the disease. J. Neurochem 109, 656–669. [DOI] [PubMed] [Google Scholar]
- Kawajiri S, Machida Y, Saiki S, Sato S, Hattori N, 2010. Zonisamide reduces cell death in SH-SY5Y cells via an anti-apoptotic effect and by upregulation MnSOD. Neurosci Lett 481, 88–91. [DOI] [PubMed] [Google Scholar]
- Khan MM, Kempuraj D, Thangavel R, Zaheer A, 2013. Protection of MPTP-induced neuroinflammation and neurodegeneration by Pycnogenol. Neurochem. Int 62, 379–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kito M, Maehara M, Watanabe K, 1996. Mechanisms of T-type calcium channel blockade by zonisamide. Seizure 5, 115–119. [DOI] [PubMed] [Google Scholar]
- Knott C, Stern G, Wilkin GP, 2000. Inflammatory regulators in Parkinson’s disease: iNOS, lipocortin-1, and cyclooxygenases-1 and -2. Mol. Cell. Neurosci 16, 724–739. [DOI] [PubMed] [Google Scholar]
- Kohrman DC, Harris JB, Meisler MH, 1996. Mutation detection in the med and medJ alleles of the sodium channel Scn8a. Unusual splicing due to a minor class AT-AC intron. J. Biol. Chem 271, 17576–17581. [DOI] [PubMed] [Google Scholar]
- Krzemien DM, Schaller KL, Levinson SR, Caldwell JH, 2000. Immunolocalization of sodium channel isoform NaCh6 in the nervous system. J. Comp. Neurol 420, 70–83. [PubMed] [Google Scholar]
- Liu Y, Kintner DB, Chanana V, Algharabli J, Chen X, Gao Y, Chen J, Ferrazzano P, Olson JK, Sun D, 2010. Activation of microglia depends on Na+/H+ exchange-mediated H+ homeostasis. J. Neurosci 30, 15210–15220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lofrumento DD, Saponaro C, Cianciulli A, De Nuccio F, Mitolo V, Nicolardi G, Panaro MA, 2011. MPTP-induced neuroinflammation increases the expression of pro-inflammatory cytokines and their receptors in mouse brain. Neuroimmunomodulation 18, 79–88. [DOI] [PubMed] [Google Scholar]
- Lull ME, Block ML, 2010. Microglial activation and chronic neurodegeneration. Neurotherapeutics 7, 354–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantegazza M, Curia G, Biagini G, Ragsdale DS, Avoli M, 2010. Voltage-gated sodium channels as therapeutic targets in epilepsy and other neurological disorders. Lancet Neurol. 9, 413–424. [DOI] [PubMed] [Google Scholar]
- Mares P, 2010. Zonisamide suppresses the tonic phase but not the clonic phase of generalized seizures in developing rats. Epilepsy Res. 92, 244–248. [DOI] [PubMed] [Google Scholar]
- Matar N, Jin W, Wrubel H, Hescheler J, Schneider T, Weiergraber M, 2009. Zonisamide block of cloned human T-type voltage-gated calcium channels. Epilepsy Res. 83, 224–234. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Boyes BE, McGeer EG, 1988. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38, 1285–1291. [DOI] [PubMed] [Google Scholar]
- Minato H, Kikuta C, Fujitani B, Masuda Y, 1997. Protective effect of zonisamide, an antiepileptic drug, against transient focal cerebral ischemia with middle cerebral artery occlusion-reperfusion in rats. Epilepsia 38, 975–980. [DOI] [PubMed] [Google Scholar]
- Murata M, Hasegawa K, Kanazawa I, Japan Zonisamide On PDSG, 2007. Zonisamide improves motor function in Parkinson disease: a randomized, double-blind study. Neurology 68, 45–50. [DOI] [PubMed] [Google Scholar]
- Nagatsu T, Mogi M, Ichinose H, Togari A, 2000. Cytokines in Parkinson’s disease. J. Neural Transm. Suppl 143–151. [PubMed] [Google Scholar]
- O’Callaghan JP, Miller DB, Reinhard JF Jr., 1990. Characterization of the origins of astrocyte response to injury using the dopaminergic neurotoxicant, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Brain Res. 521, 73–80. [DOI] [PubMed] [Google Scholar]
- Okada M, Zhu G, Yoshida S, Kanai K, Hirose S, Kaneko S, 2002. Exocytosis mechanism as a new targeting site for mechanisms of action of antiepileptic drugs. Life Sci. 72, 465–473. [DOI] [PubMed] [Google Scholar]
- Owen AJ, Ijaz S, Miyashita H, Wishart T, Howlett W, Shuaib A, 1997. Zonisamide as a neuroprotective agent in an adult gerbil model of global forebrain ischemia: a histological, in vivo microdialysis and behavioral study. Brain Res. 770, 115–122. [DOI] [PubMed] [Google Scholar]
- Pappalardo LW, Black JA, Waxman SG, 2016. Sodium channels in astroglia and microglia. Glia 64, 1628–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosler TW, Arias-Carrion O, Hoglinger GU, 2010. Zonisamide: aspects in neuroprotection. Exp Neurol 224, 336–339. [DOI] [PubMed] [Google Scholar]
- Sano H, Murata M, Nambu A, 2015. Zonisamide reduces nigrostriatal dopaminergic neurodegneration in a mouse genetic model of Parkinson’s disease. J Neurochem. 134, 371–381. [DOI] [PubMed] [Google Scholar]
- Qin L, Crews FT, 2012. NADPH oxidase and reactive oxygen species contribute to alcohol-induced microglial activation and neurodegeneration. J. Neuroinflammation 9, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Liu Y, Wang T, Wei SJ, Block ML, Wilson B, Liu B, Hong JS, 2004. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J. Biol. Chem 279, 1415–1421. [DOI] [PubMed] [Google Scholar]
- Richardson JR, Hossain MM, 2013. Microglial ion channels as potential targets for neuroprotection in Parkinson’s disease. Neural Plast. 2013, 587418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonsalla PK, Wong LY, Winnik B, Buckley B, 2010. The antiepileptic drug zonisamide inhibits MAO-B and attenuates MPTP toxicity in mice: clinical relevance. Exp. Neurol 221, 329–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens M, Timmermans S, Bottelbergs A, Hendriks JJ, Brone B, Baes M, Tytgat J, 2013. Block of a subset of sodium channels exacerbates experimental autoimmune encephalomyelitis. J. Neuroimmunol 261, 21–28. [DOI] [PubMed] [Google Scholar]
- Tan J, Soderlund DM, 2010. Divergent actions of the pyrethroid insecticides S-bioallethrin, tefluthrin, and deltamethrin on rat Na(v)1.6 sodium channels. Toxicol. Appl. Pharmacol 247, 229–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tansey MG, Goldberg MS, 2010. Neuroinflammation in Parkinson’s disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis 37, 510–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujii S, Ishisaka M, Shimazawa M, Hashizume T, Hara H, 2015. Zonisamide suppresses endoplasmic reticulum stress-induced neuronal cell damage in vitro and in vivo. Eur J Pharmacol 746, 301–307. [DOI] [PubMed] [Google Scholar]
- Turchan-Cholewo J, Dimayuga VM, Gupta S, Gorospe RM, Keller JN, Bruce-Keller AJ, 2009. NADPH oxidase drives cytokine and neurotoxin release from microglia and macrophages in response to HIV-Tat. Antioxid. Redox Signal 11, 193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzoumaka E, Tischler AC, Sangameswaran L, Eglen RM, Hunter JC, Novakovic SD, 2000. Differential distribution of the tetrodotoxin-sensitive rPN4/NaCh6/Scn8a sodium channel in the nervous system. J. Neurosci. Res 60, 37–44. [DOI] [PubMed] [Google Scholar]
- Ueda Y, Doi T, Tokumaru J, Nakajima A, Nagatomo K, 2005. In vivo evaluation of the effect of zonisamide on the hippocampal redox state during kainic acid-induced seizure status in rats. Neurochem. Res 30, 1117–1121. [DOI] [PubMed] [Google Scholar]
- Wang Q, Liu Y, Zhou J, 2015. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener 4, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DC, Teismann P, Tieu K, Vila M, Jackson-Lewis V, Ischiropoulos H, Przedborski S, 2003. NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. Proc. Natl. Acad. Sci. U. S. A 100, 6145–6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama H, Yano R, Kuroiwa H, Tsukada T, Uchida H, Kato H, Kasahara J, Araki T, 2010. Therapeutic effect of a novel anti-parkinsonian agent zonisamide against MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity in mice. Metab Brain Dis. 25, 305–313. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.