

Abstract
D614G spike glycoprotein (sgp) mutation in rapidly spreading severe acute respiratory syndrome coronavirus‐2 (SARS‐COV‐2) is associated with enhanced fitness and higher transmissibility in new cases of COVID‐19 but the underlying mechanism is unknown. Here, using atomistic simulation, a plausible mechanism has been delineated. In G614 sgp but not wild type, increased D(G)614‐T859 Cα‐distance within 65 ns is interpreted as S1/S2 protomer dissociation. Overall, ACE2‐binding, post‐fusion core, open‐state and sub‐optimal antibody‐binding conformations were preferentially sampled by the G614 mutant, but not wild type. Furthermore, in the wild type, only one of the three sgp chains has optimal communication route between residue 614 and the receptor‐binding domain (RBD); whereas, two of the three chains communicated directly in G614 mutant. These data provide evidence that D614G sgp mutant is more available for receptor binding, cellular invasion and reduced antibody interaction; thus, providing framework for enhanced fitness and higher transmissibility in D614G SARS‐COV‐2 mutant.
Keywords: COVID‐19, molecular dynamics simulation, mutation, SARS‐COV‐2, spike glycoprotein
The causative agent of COVID‐19; SARS‐COV‐2 has acquired a rare mutation (G23403A) in the region encoding spike glycoprotein (D614G) resulting in enhanced fitness and higher transmissibility. Here, MD simulation has revealed evidence of S1/S2 protomer disaggregation, preferential sampling of post‐fusion, and receptor‐binding (ACE2) conformations and enhanced communication between residue 614 and receptor‐binding domain in D6 mutant; thus, providing structural framework for improved cellular invasion and higher fitness in D614G mutant but not wild type SARS‐COV‐2.
1. BACKGROUND
COVID‐19 is global pandemic caused by severe acute respiratory syndrome coronavirus‐2 (SARS‐COV‐2). This disease was first reported in patients presenting pneumonia‐like symptom in Wuhan province of China but has now spread to other countries, infecting more than 5 million and with case‐to‐fatality ratio of ~1.38%. 1 The race to find cure had relied on the genome sequence and with the release of Wuhan reference strain, diagnostic kits, candidate drugs and vaccine development had commenced. 2 Vaccine development may be the hope as most of the re‐purposed drug candidates have not produced the desired outcome in clinical settings. Most of the promising vaccine candidates under investigation recognize and bind the spike glycoproteins. 3 , 4 Therefore, SARS‐COV‐2 genetic drift, especially in the region encoding spike glycoprotein has implications on its virulence, and more importantly, the potency or applicability of target vaccines in clinical settings. A major finding from a recent study is guanine‐to‐adenine transition in position 23,403 in the Wuhan reference strain leading to aspartate‐to‐glycine mutation in spike glycoprotein residue 614 (D614G). Although, the G614 is thought to originate from China or Europe, its intercontinental spread and prevalence in sequenced samples in other continents would suggest strongly that the mutation confers a selective advantage and enhanced fitness over D614. 5
Among the mechanisms put forward by Korber et al. (2020) 5 to explain the enhanced fitness; perhaps, the most compelling came from the resolved structure of the spike glycoprotein itself. D614 carboxylic functional group located on the S1 protomer, proximal to the N‐terminal domain (NTD) and receptor binding domain (RBD) forms hydrogen bond with hydroxyl group of T859 of viral membrane bound S2 protomer; thus making D614‐T859 interaction important in spike protein oligomeric state formation. 6 The loss of this critical interaction in G614 is thought to promote viral glycoprotein shedding. In order to test this hypothesis, following two independent 150 ns, atomistic simulations, the minimal distance between D(G)614 and T859 were monitored with time along the trajectories. Indeed, both were on the average of 1.5 nm apart at the beginning of the simulation, at around 65 ns, G614‐T859 inter‐residue distance began to increase to 2.0 nm while D614‐T859 decreased to approximately 0.5 nm which was maintained throughout the simulation. Notably, at 75 ns, the G614‐T859 inter‐residue distance further increased to ~2.5 nm and was maintained till about 135 ns but moved back to its initial state of 1.0 nm at the end of the simulation; thus, providing the first evidence in support of S1 shedding from viral‐membrane‐bound S2 following D614G mutation (Figure 1a). Theoretically, increased shedding, interpreted as increased S1/S2 unit distance in this study, will ultimately facilitate viral attachment to host cells, hence, enhanced fitness, and increased transmissibility.
FIGURE 1.
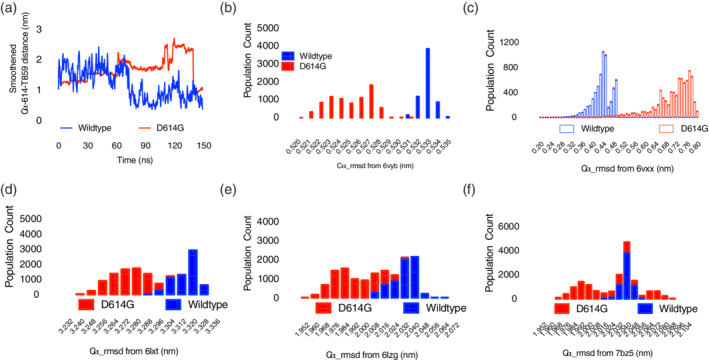
Sampled population and dynamics of wild type and D614G SARS‐COV‐2 spike glycoprotein . (a) Distance‐time plot of Ca‐distance between residue 614 and T859 during atomistic simulation. (b–f) Population count‐rmsd histogram plots; see the x‐axes for the proteins used in comparison (unless otherwise stated, each plot represents the mean of 2 independent simulations) [Color figure can be viewed at wileyonlinelibrary.com]
Another plausible mechanism proposed is its influence on RBD conformation. RBD is required for the receptor angiotensin converting enzyme‐2 (ACE2) recognition and binding. 7 Indeed, such binding is known to be preceded by closed‐to‐open state conformational change in the RBD. How the mutation in position 614 would influence this transitional event is yet to be investigated. To provide further insight, root‐mean‐square deviation (rmsd) tool and population distribution plot were used to investigate the conformations sampled by the two biosystems in comparison with the open (PDB ID: 6vyb) 2 and closed (PDB ID: 6vxx) states. 2 The results showed that the open state was preferentially sampled by G614 biosystem (Figure 1b) while the D614 biosystem sampled the closed state conformation (Figure 1c). Generally, while these data do not explicitly give information about the communication between 614 residue and RBD, it, however, provide a proof that events at 614 influence conformations around the RBD. To further establish these findings, the conformations sampled were also compared with that of S2‐post‐fusion core (PDB ID: 6lxt). 8 Recall that once S1 unit disaggregates (shedding), from the spike glycoprotein trimer, and attaches through ACE2 to host cells, the S2 unit re‐assembles into a unit leading to the virus‐host membrane fusion; the entire fusion process, relies on conformational changes within the heptad repeats (HR1 and HR2; fusion core) of the S2 unit. The conformations sampled by S2 unit in the G614 biosystem were predominantly those of the post‐fusion core (Figure 1d), while the wild type sampled the pre‐fusion core conformation (data not shown); and clearly, the conformations of the RBD of G614 biosystem were those previously characterized to bind ACE2 (PDB ID: 6lzg, Figure 1e) but not the wild type. 7 These data strongly lend credence to the fitness and selectivity of G614‐clade above D614‐clade.
Another key consideration in this study was to evaluate how well antibody will bind the conformations sampled by both biosystems. Given that ACE2 and antibody binding regions and RBD are overlapped in most cases, it may be taken for granted that since G614 biosystem preferentially sampled ACE2 binding conformation, same may apply for antibody‐binding conformation, and its transmissibility success will be lessened due to immune response. The results obtained from comparison with antibody (B38, PDB ID: 7bz5) 9 ‐bound conformation revealed a rather unusual pattern. G614 biosystems sampled wide range of conformations; those close to the antibody binding conformation (rmsd <2.008 nm) and those non‐recognizable by the antibody (> 2.056 nm) while the wild type sampled a very narrow range of conformations (rmsd between 2.016 ~ 2.056 nm) (Figure 1f).
To answer the most important question of how a mutation at a remote site (614) dictates the conformational events at the RBD, network of communities and communication analyses were performed on the trajectories. Community clusters formed by residues is a direct reflection of internal motions and functional dynamics of protein complexes. 10 In the wild type biosystem, one defining feature is the presence of a large community at the N‐terminal domain (NTD, blue edge‐node; (Figure 2a) while every other regions present a near continuous homogenous community interspaced by small communities. G614 mutant presents a rather unique pattern, revealing three (3) large communities around the NTD, RDB and the region bordering G614 residue towards the viral membrane‐binding region while the rest (Figure 2b); just like the wild type is a near continuous stretch of community.
FIGURE 2.
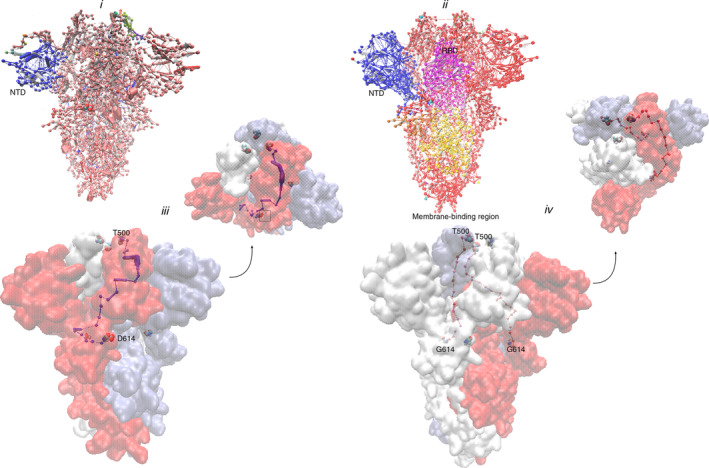
Protein network and communication analyses. (a) Communities evolved by the wild type (b) and D614G mutant during simulation. (c) Surface representation of spike glycoprotein trimer showing the optimal communication path between residue 614 and T500 (RBD) in wild type and D614G mutant (d) [Color figure can be viewed at wileyonlinelibrary.com]
The coronavirus spike glycoprotein NTD confers receptor specificity 11 ; the seeming communication with the RBD (G614) may represent key adaptive mechanism for RBD optimization for receptor binding and the diffusion of such information toward the viral membrane may provide the necessary signal for host invasion; thus, providing a good evidence that G614 clade may be more clinically successful than the wild type.
Communication between residue 614 and RBD (T500) was also established; and surprisingly, in the wild type, such optimal communication route could only be found on only one chain (chain B); on the other hand, in G614 biosystem, two of the three chains (chains B and C) have optimal communication route between residue 614 and T500; further providing evidence that 614 mutations have positive implication on the receptor recognition, binding and overall fitness of G614 clade SARS‐COV‐2.
2. METHODS
The spike glycoprotein trimer coordinates used for atomistic simulation had been previously reported. 2 The structure is essentially the NTD, RBD, and S2 (1 ~ 1,230) domain; since the fibritin C superfamily region (1,231 ~ 1,270) required for membrane attachment was not captured in the starting structure, it was therefore treated as the soluble fraction without a membrane. Broken chains and incomplete residue re‐modeling and glycan removal were performed using DeepView suite. D614G mutant was generated using mutagenesis plugin in PyMol.
TIP3P‐solvated, and NaCl‐neutralized biosystems were prepared using HTMD platform with charmm36 forcefield parameters. 12 Equilibration (80 ns) and production simulation (150 ns) were subsequently performed 13 using ACEMD3 software 14 . Prior to analysis, all biosystems were checked for convergence (data not shown). RMSD from selected structures (see text) were performed using GROMACS in‐built gmx rms tool, protein community and network analyses were performed using networkTools in VMD software (see supplementary methods for details). 15 All line graphs were plotted as mean of two independent simulations using GraphPad prism (ver. 7.0).
Supporting information
Appendix S1. Supporting Information
Omotuyi IO, Nash O, Ajiboye OB, et al. Atomistic simulation reveals structural mechanisms underlying D614G spike glycoprotein‐enhanced fitness in SARS‐COV‐2. J Comput Chem. 2020;41:2158–2161. 10.1002/jcc.26383
REFERENCES
- 1. Bedford J., Enria D., Giesecke J., Heymann D. L., Ihekweazu C., Kobinger G., Lane H. C., Memish Z., Oh M. D., Sall A. A., Schuchat A., Ungchusak K., Wieler L. H., Strategic W. H. O., H. Lancet 2020, 395, 1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Walls A. C., Park Y. J., Tortorici M. A., Wall A., McGuire A. T., Veesler D., Cell 2020, 181, 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ou X., Liu Y., Lei X., Li P., Mi D., Ren L., Guo L., Guo R., Chen T., Hu J., Xiang Z., Mu Z., Chen X., Chen J., Hu K., Jin Q., Wang J., Qian Z., Nat Commun 2020, 11, 1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yuan M., Wu N. C., Zhu X., Lee C. D., So R. T. Y., Lv H., Mok C. K. P., Wilson I. A., Science 2020, 368, 630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Korber B., Fischer W., Gnanakaran S., Yoon H., Theiler J., Abfalterer W., Foley B., Giorgi E., Bhattacharya T., Parker M., Partridge D., Evans C., de Silva T., LaBranche C., Montefiori D., bioRxiv 2020, 2020.2004.2029.069054. [Google Scholar]
- 6. Wrapp D., Wang N., Corbett K. S., Goldsmith J. A., Hsieh C. L., Abiona O., Graham B. S., McLellan J. S., Science 2020, 367, 1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang Q., Zhang Y., Wu L., Niu S., Song C., Zhang Z., Lu G., Qiao C., Hu Y., Yuen K. Y., Wang Q., Zhou H., Yan J., Qi J., Cell 2020, 181, 894 e899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xia S., Liu M., Wang C., Xu W., Lan Q., Feng S., Qi F., Bao L., Du L., Liu S., Qin C., Sun F., Shi Z., Zhu Y., Jiang S., Lu L., Cell Res 2020, 30, 343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wu Y., Wang F., Shen C., Peng W., Li D., Zhao C., Li Z., Li S., Bi Y., Yang Y., Gong Y., Xiao H., Fan Z., Tan S., Wu G., Tan W., Lu X., Fan C., Wang Q., Liu Y., Qi J., Gao G. F., Gao F., Liu L., medRxiv 2020, 2020.2005.2001.20077743. [Google Scholar]
- 10. Blacklock K., Verkhivker G. M., PLoS One 2013, 8, e71936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tsai J. C., Zelus B. D., Holmes K. V., Weiss S. R., J Virol 2003, 77, 841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huang J., MacKerell A. D. Jr., J Comput Chem 2013, 34, 2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Olaposi O. I., Oyekanmi N., Ojo A. A., Eniafe G. O., Drug Res (Stuttg) 2019, 69, 643. [DOI] [PubMed] [Google Scholar]
- 14. Harvey M. J., Giupponi G., Fabritiis G. D., J Chem Theory Comput 2009, 5, 1632. [DOI] [PubMed] [Google Scholar]
- 15. Omotuyi O. I., Nagai J., Ueda H., Sci Rep 2015, 5, 13343. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting Information