

Abstract
Imatinib and second generation BCR/ABL tyrosine kinase inhibitors (TKIs) serve now as standard therapies for patients with chronic myelogenous leukemia (CML); however, CML stem cells are intrinsically insensitive to the cell death-inducing effects of TKIs, allowing the persistence of a “reservoir” of BCR/ABL- expressing CML-initiating cells potentially responsible for disease relapse and progression. Although it is still controversial whether the “insensitivity” of CML stem cells to treatment with TKI is due to BCR/ABL-dependent or independent mechanisms, treatment with IM appears to suppress BCR/ABL-dependent signaling in CML stem cells with no adverse effects on their survival. Recent evidence indicates that BCR/ABL suppresses and treatment of CML cells with IM/TKIs induces autophagy, a genetically- regulated process of adaptation to metabolic stress which could allow tumor cells to become metabolically inert enabling their survival under conditions that may mimic growth factor/nutrients deprivation. Based on this hypothesis, TKI-induced autophagy may “antagonize” TKI-induced cell death and inhibition of autophagy may eliminate this survival mechanism by restoring “sensitivity” of CML stem cells to treatment with IM/TKI. Consistent with this, phenotypically and functionally defined CML- enriched stem cells insensitive to treatment with TKI are efficiently eliminated by the combination of TKI and chloroquine, an inhibitor of late stage autophagy. Thus, inhibition of autophagy may improve the potent and specific effects of TKIs by rendering CML stem cells sensitive to these targeted therapies.
Keywords: Oncogene, Leukemia, Cell Death, Therapy, Review
2. INTRODUCTION
CML is a malignancy arising from transformation of the hematopoietic stem cell which typically progresses in three distinct disease stages: an indolent chronic phase (CP) characterized by the accumulation of mature granulocytes and myeloid precursors in the bone marrow and the peripheral blood; an accelerated phase (AP) characterized by an increase in disease burden and in the frequency of myeloid precursors; and an acute phase called blast crisis (BC) marked by increasing numbers of differentiation-arrested blast cells of either myeloid or lymphoid lineage (1–3).The hallmark of all phases is the Philadelphia chromosome (Ph1) which results from a reciprocal translocation of chromosomes 9 and 22 and generates BCR/ABL fusion genes that encodes a p210 KD protein with constitutive tyrosine kinase activity (4). Transformation of hematopoietic stem cells by p210BCR/ABL requires its tyrosine kinase activity and depends on its ability to activate a multitude of intracellular signaling pathways which include the classical PI-3K/Akt and MAP kinase and the more recently described β- catenin/LEF-1 and Hedgehog pathways. Together, these pathways promote an expansion of the pool of committed myeloid progenitors and lead to their increased survival and proliferation and limited dependence on growth factors (5– 10).
The generation of the BCR/ABL kinase ATP- competitive inhibitor imatinib mesylate (IM) has revolutionized the therapy of CML, since this drug is highly effective in the CP of the disease (11). However, there are three major problems with IM-based therapy: i) the limited response of CML-BC or Ph1 B-acute lymphoblastic leukemia (ALL) patients to IM (12–14); ii) the development of resistance caused in approximately 40% of cases by mutations in the BCR/ABL kinase domain which impair the ability of IM to interact with the protein (15–17); and iii) the relative insensitivity of Ph1 stem cells to IM resulting in the persistence of residual leukemic cells (and possibly disease relapse) even in patients with complete cytogenetic response (CCyR) (18). For these reasons, more potent BCR/ABL inhibitors, also targeting IM-resistant mutants including BCR/ABLT315I, are being developed and tested (19–22). The insensitivity of primitive Ph1 stem cells (which overexpress p210BCR/ABL) to treatment with IM and second generation (nilotinib, dasatinib, bosutinib) TKI (18, 23–27) is specially concerning because even new TKI capable of blocking the activity of the T315I mutant which is not bound by IM and second generation TKI (i.e. AP24534; reference 22) may not be able to eradicate Ph1 stem cells from patients with the T315I mutation. The reasons for the insensitivity of CML stem cells to treatment with TKI are incompletely understood; several BCR/ABL-dependent and independent mechanisms including increased expression/activity of BCR/ABL, autocrine production of growth factors, down-regulation of drug transporters and persistent activation of proliferation/survival signaling pathways in spite of BCR/ABL inhibition have been proposed (28). Although it is still controversial whether TKI can suppress BCR/ABL activity in Ph1 stem cells (25,26,29), a recent study provides strong evidence in support of IM being able to inhibit the tyrosine kinase activity of BCR/ABL in CML stem cells (29); however, this study also showed that cytokines support allowed Ph1 stem cells to grow and survive like the normal counterpart (29), suggesting that CML stem cells may be “insensitive” to treatment with TKI via BCR/ABL-independent mechanisms or that the inhibition of BCR/ABL signaling in CML stem cells leads to “context-specific” metabolic changes that may antagonize its cell death-promoting effects. Thus, it is crucial to understand mechanistically why CML stem cells are insensitive to TKI and to develop novel therapeutic strategies that, in combination with TKI, might be effective in targeting the stem cell population.
3. REGULATION OF AUTOPHAGY BY BCR/ABL
3.1. Autophagy is an adaptation process that provides a survival mechanism for cancer cells
Macroautophagy (hereafter referred to as autophagy) is a degradative process in eukaryotic cells that results in the sequestration and the breakdown of intracellular organelles and proteins within specialized lysosomes called autolysosomes under homeostatic conditions or in response to stress (30, 31). This process allows cells to adapt to developmental changes and/or unfavorable environmental conditions (30, 31). Autophagy is a genetically controlled process which progresses through discrete steps which lead to the engulfment of long-lived proteins and whole organelles into multi- membraned vacuoles called autophagosomes (30,31). Autophagosomes then fuse with lysosomes to produce autolysosomes for final destruction and recycling (30,31). While in certain cellular contexts autophagy can serve as a cell death mechanism, named type II cell death (32, 33), it is becoming increasingly clear that this process can provide a cell survival mechanism which allows cells to adapt their metabolism to starvation caused by a decrease in metabolite concentrations or extracellular nutrients, typical consequences of loss of growth factor signaling (34–36). In this context, autophagy serves as an adaptation mechanism to evade programmed cell death. Consistent with a survival role played by autophagy, its genetic (knockdown of autophagy genes) or pharmacological (by use of drugs such as chloroquine, an inhibitor of autophagosome-lysosome fusion and lysosomal acidification (37)) inhibition results in cell death of growth factor-starved cells in which apoptosis has been genetically ablated (36,38). The pro- survival effect associated with induction of autophagy is not limited to growth factor-starved normal cells but was also observed in tumor cells: treatment of Myc-induced lymphomas with chemotherapy or drugs inducing p53 re- activation led to the appearance of morphological and biochemical markers of autophagy and pharmacological or genetic inhibition of autophagy enhanced the p53- or chemotherapy-dependent anti-tumor effects in vivo (37, 41). Together, these and other studies suggest that induction of autophagy provides a protective mechanism to tumor cells.
However, in tumors displaying defective apoptosis, inhibition of autophagy causes caspase- independent necrotic cell death, which, in turn, augments inflammation, leading to enhanced tumor burden (42). Autophagy can limit inflammation also in normal cells, thus counteracting tumorigenesis (42). Thus, the consequences of autophagy inhibition might be double- faced, as it can either promote or suppress tumorigenesis depending on the tumor type or inflammation status (37, 39, 42).
As shown by the study by Amaravadi et al. (39) in which mice with Myc-induced lymphomas were treated with alkylating drugs, the autophagic process can be also activated by chemotherapy and it provides a mechanism to chemotherapy-treated cells to evade cell death. Consistent with the findings of this study, autophagy inhibition sensitizes tumor cells to cell death induced by irradiation (43–45), alkylating agents (46), or arsenic trioxide (47), suggesting that cancer cells can react to chemotherapy by inducing autophagy as a self-defence mechanism (34). Such a mechanism might be particularly important in allowing persistence of drug-resistant dormant cancer cells (48, 49).
How an increase in intracellular lysosome- mediated catabolism results in reduced sensitivity to cell death is still unclear. Perhaps, clearance of mitochondria through autophagy (mitophagy) might sequester pro- apoptotic proteins (i.e. BH3-only proteins) and prevent the release into the cytosol of apoptosis activators such as cytochrome C (50). Alternatively, autophagy suppression may lead to reduced availability of metabolites for bioenergetic needs, thus impairing the survival capacity of cancer cells (see also below). Finally, as specific proteins can be targeted for degradation via the p62 cargo pathway (51), autophagy suppression could result in reduced degradation of cell death inducers.
3.2. BCR/ABL suppresses autophagy by a tyrosine kinase-dependent mechanism
The oncogenic p210BCR/ABL protein has the ability to activate several survival pathways which mimic those activated by growth factor signaling (8,52), allowing transformed cells to acquire resistance to apoptosis induced by growth factor deprivation (8, 52). Inhibition of tyrosine kinase activity by IM results in the induction of cell death which might be caused by a sudden decrease of intracellular survival signals and a relative increase in pro- apoptotic signals (53). We have recently shown that BCR/ABL expression also prevents IL-3-deprivation- induced autophagy and that pharmacological inhibition of BCR/ABL tyrosine kinase activity, in addition to markers of apoptosis, leads to the rapid appearance of morphological and biochemical changes consistent with autophagy (54). Mechanistically, induction of autophagy by treatment with TKIs is not due to inhibition of c-Abl tyrosine kinase activity since expression of IM-resistant T315I c-Abl did not rescue IM-induced autophagy in hematopoietic cells expressing native BCR/ABL. Induction of autophagy was also independent from apoptosis as inhibition of caspase activity or Bcl-2 overexpression did not prevent IM-induced autophagy in BCR/ABL-expressing myeloid progenitor cells. Induction of autophagy did not require expression of wild type p53 since it was also induced by TKIs of p53-null K562 cells (54).
Although the precise mechanisms responsible for the ability of BCR/ABL to suppress autophagy remain unknown, it is likely that the PI-3K/Akt/mTOR is involved since treatment of BCR/ABL-expressing cells with Akt or mTOR inhibitors induced autophagy (54, data not shown). Moreover, the autophagic process activated by Imatinib treatment of BCR/ABL-expressing cells was preceded by the induction of endoplasmic reticulum (ER) stress and relied on intracellular calcium as autophagy was blocked by intracellular calcium chelators (54). However, it is unclear whether inhibition of Akt or mTOR is functionally linked to the ER stress response in the induction of autophagy.
Regardless of the mechanisms involved in the BCR/ABL-dependent suppression of autophagy, treatment with pharmacological inhibitors of autophagosome- lysosome fusion (chloroquine or bafilomycin A) potentiates IM-induced cell death in CML cell lines and primary CML cells, including those carrying partially IM-resistant BCR/ABL mutants (52). Moreover, candidate CML stem cells, defined as colony forming CD34+CD38- cells and LTC-IC were also exquisitely susceptible to combination treatment (54). Genetic suppression of autophagy by knockdown of the autophagy genes Atg5 and Atg7 in K562 and primary CML cells also enhanced TKI-induced cell death, supporting the specificity of the effects induced by pharmacological inhibitors (54).
Together, these findings indicate that induction of autophagy provides a survival mechanism to IM-treated BCR/ABL-expressing cells, including the stem cell population, and suggest that inhibition of autophagy may improve the therapeutic efficacy of TKI in the treatment of CML by a more efficient elimination of stem cells. Based on this study, a randomized phase 2 clinical trial of IM versus hydrochloroquine (HCQ)/IM in CML patients with major cytogenetic response and residual disease by Q-PCR detection of bcr/abl transcripts has been approved and patients are presently being recruited in the UK at three centers (Glasgow, Liverpool and London) under the leadership of Dr Holyoake.
3.3. Is there a molecular basis for tki-induced autophagy versus apoptosis in cml stem cells?
The enhanced TKI-induced cell death associated with pharmacological inhibition of late stage autophagy (chloroquine treatment) in CML cells suggests that induction of autophagy by TKI has an “antagonistic effect” on TKI-induced cell death. This effect appears to be especially prominent in CML stem cells which were completely insensitive to apoptosis induction by TKI treatment unless autophagy was also suppressed (52). An explanation for these data is that TKI suppress BCR/ABL activity in CML stem cells but the consequence of this effect is a preferential induction of autophagy rather than apoptosis. Perhaps, inhibition of BCR/ABL tyrosine kinase activity in CML stem cells preferentially activates signal transduction pathway (s) leading to autophagy rather than apoptosis; although a number of pathways and genes regulating autophagy overlap with those involved in apoptosis (55), there are some (i.e. mTOR –dependent signaling) which appear to be more important for the regulation of autophagy than apoptosis (56), raising the possibility that TKI treatment of distinct CML progenitor/stem cell subsets elicits differences in the “intensity” of autophagy- versus apoptosis-regulatory signals.
One possibility is that IM treatment activates markers of autophagy (i.e., LC3-II expression and subcellular localization) rather than apoptosis (i.e., Bim levels) in stem cell-enriched (CD34+CD38-) versus progenitor-enriched (CD34+CD38+) CML subsets and this may reflect the preferential activation of signaling pathways which regulate autophagy versus apoptosis.
Although autophagy and apoptosis share many molecular regulators (i.e., the PI-3K/Akt pathway), another possibility is that different signal thresholds may be required in specific cell subsets (i.e. CD34+CD38- versus CD34+CD38+) to inhibit autophagy versus apoptosis (i.e. phosphorylation of Akt) and the consequence of suppressing Akt signaling (via IM treatment, or directly through an Akt inhibitor) may be the preferential induction of autophagy versus apoptosis (Figure 1).
Figure 1.
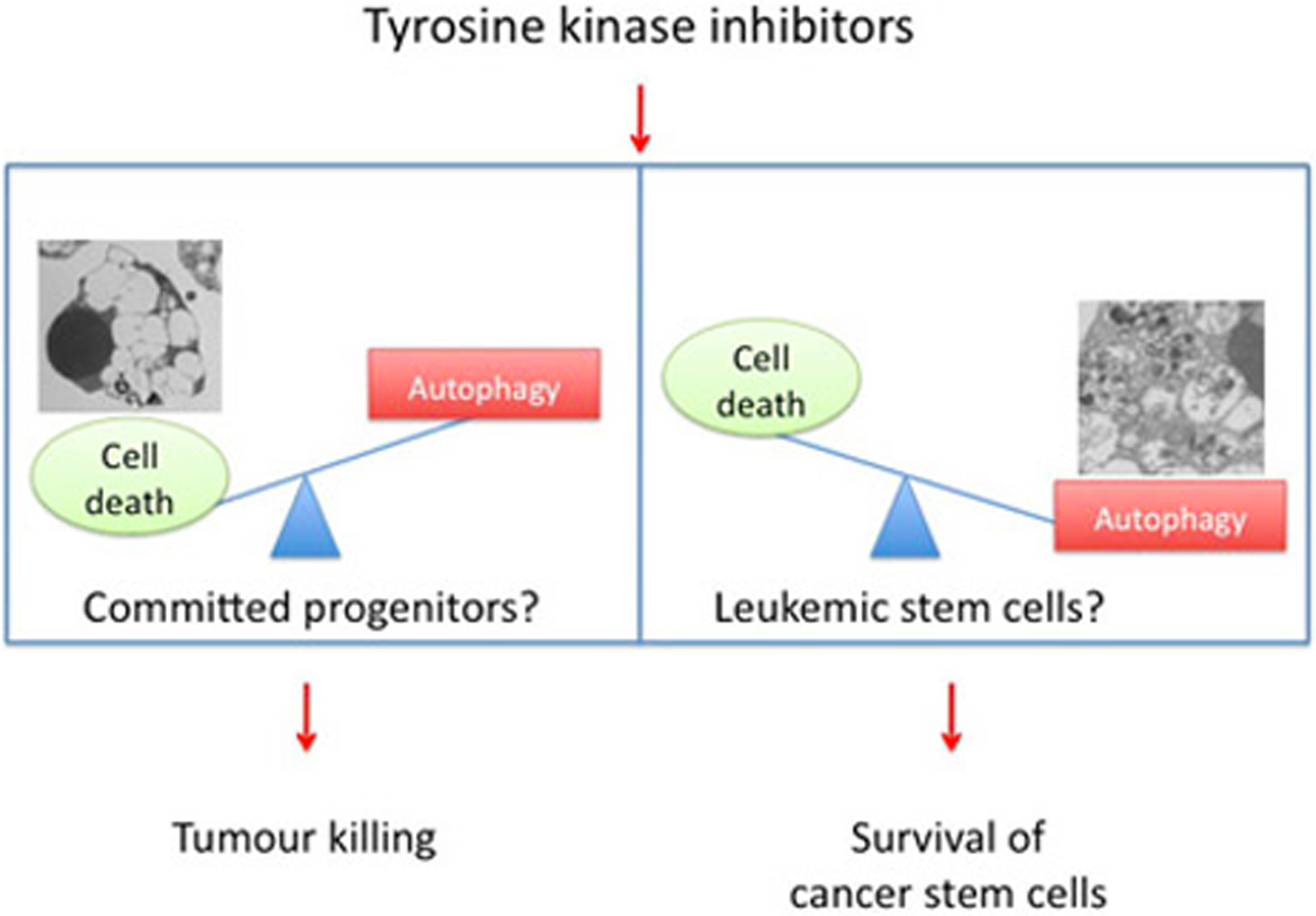
Treatment with TKI may elicit different responses in distinct CML progenitor subsets. The response to TKI may be different in distinct CML progenitor subsets. One possibility is that in committed progenitors TKI preferentially activate cell death rather than autophagy. By contrast, more primitive progenitors (stem cells) would be more resistant to cell death due to their propensity to activate preferentially the process of autophagy. As a result, the debulking effect of TKIs would be accompanied by survival of resistant leukemic stem cells. Survival of these autophagy-prone stem cells would be responsible for promoting disease progression.
Thus, there may be differences in the activation of autophagy- versus apoptosis- regulatory Akt-dependent signaling pathways in distinct progenitor subsets.
If such differences are not detected in spite of the fact that IM treatment induces autophagy but not apoptosis in the CD34+CD38- CML subset (54), alternative mechanisms (i.e., increased expression/activity of autophagy activators or apoptosis inhibitors) may explain the predominance of autophagy versus apoptosis in TKI- treated CML stem cells. Experimental approaches to identify such alternative mechanisms may include screening of oligonucleotide RNA arrays, proteomic analyses and screening of pathway-specific or comprehensive phospho antibody arrays using RNA/proteins of IM-treated CD34+CD38- versus CD34+CD38+ CML cells. It is also conceivable that CML stem cells are resistant to cell death following inhibition of BCR/ABL tyrosine kinase activity because of their quiescent state and limited bioenergetic needs. In this context, induction of autophagy would supply sufficient metabolites to provide energy to quiescent stem cells (Figure 2).
Figure 2.
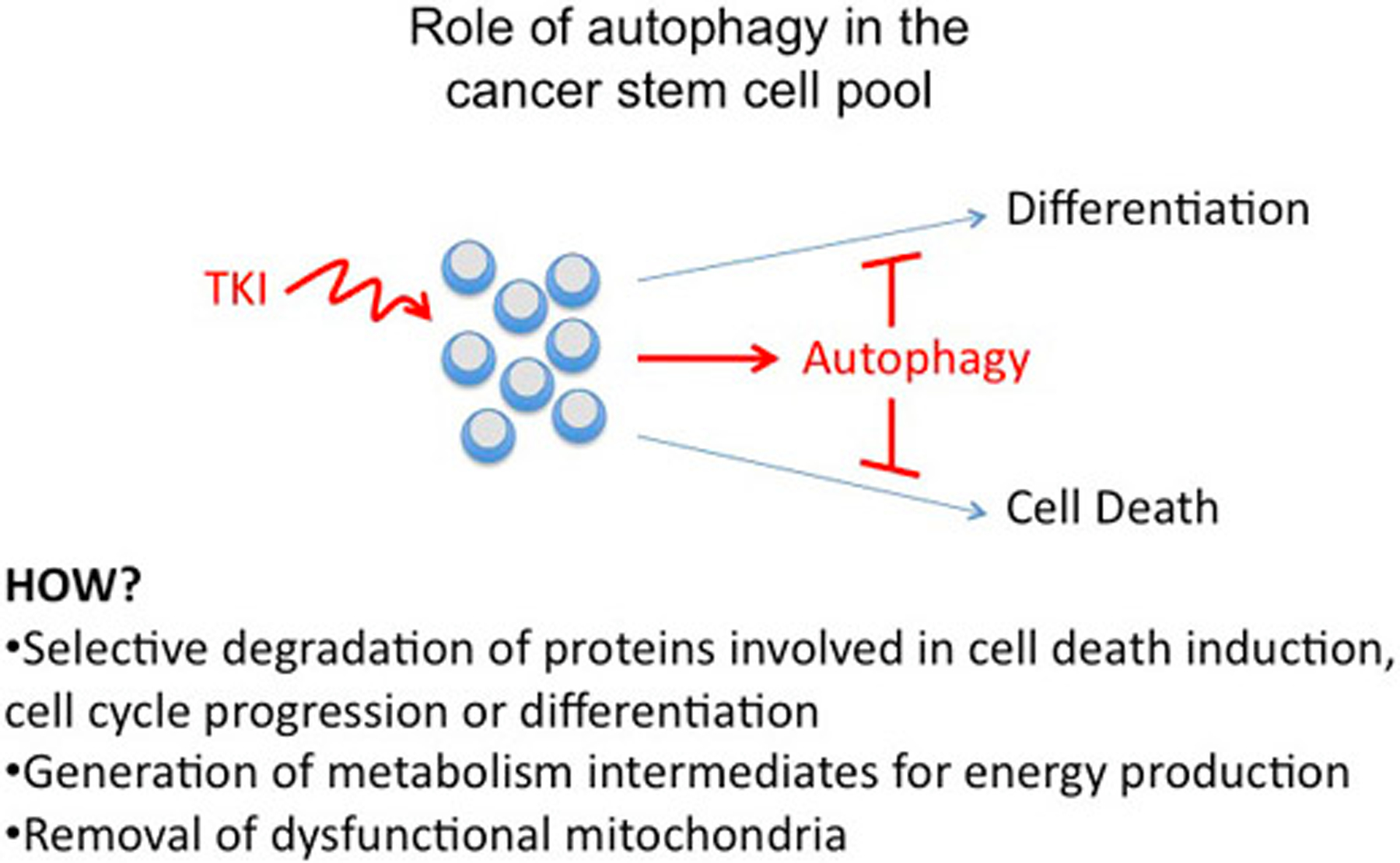
Role of autophagy in the cancer stem cell pool. Induction of autophagy by TKI treatment may maintain CML stem cells by preventing cell death, inhibiting differentiation, or blocking cell cycle progression. Metabolism intermediates generated by autophagy may be also needed for maintenance of stem cells.
Induction of autophagy in TKI-treated CML stem cells may, mechanistically, prevent apoptotic cell death by autophagosome sequestration of regulators (i.e. BH3-only proteins) of the intrinsic, mitochondrial apoptotic pathway (Figure 2). In IM-treated K562 cells, inhibition of autophagosome formation by RNAi of Atg5 or Atg7 expression enhanced IM-induced cell death as effectively as inhibition of “autophagy flux” by chloroquine or bafilomycin A1 treatment (54). CML-enriched stem cells were sensitized to TKI by treatment with chloroquine, but genetic approaches are necessary to determine whether inhibition of autophagosome formation also enhances TKI- induced cell death of CML stem cells. The identification of the stage of the autophagic process which “antagonizes” TKI-induced cell death is not only biologically relevant but has also translational implications; the insensitivity of CML stem cells to TKI-induced cell death could be manipulated by harnessing mitochondria-independent cell death pathways or by restoring the expression/activity of cell death mediators lost/inactivated via enhanced autophagic flux.
4. CONCLUSION
Although autophagy appears to be induced earlier than apoptosis in CML cells (54), it is also possible that signals leading to either process could be activated simultaneously. In this regard, failure of TKI to induce apoptosis of CML stem cells may depend on the predominant activation of autophagy rather than apoptosis. Reversal of this “antagonistic” effect of autophagy seems capable of “sensitizing” CML stem cells to the apoptosis-promoting effects of TKI, although in vivo studies have not been performed yet to assess the effects of autophagy inhibition on TKI-treated CML stem cells. Since failure to eliminate CML stem cells remains, probably, the major problem of TKI-based therapies (including second and third generation inhibitors targeting mutant BCR/ABL, including the T315I mutation), the ability of autophagy inhibition to “sensitize” CML stem cells to TKI would make unnecessary to target CML stem cells through BCR/ABL-independent, less specific approaches. At the same time, normal stem cells would be spared as pharmacological inhibition of autophagy alone has modest or no effects on normal or CML progenitors (54).
5. ACKNOWLEDGMENTS
This work was supported, in part, by National Cancer Institute grants CA 95111 and PO1 78890 (BC). PS is supported by the Samantha Dickson Brain Tumour Trust and the Wellcome Trust.
6. REFERENCES
- 1.Kantarjian HM, Keating MJ, Talpaz M, Walters RS, Smith TL, Cork A, McCredie KB, Freireich EJ: Chronic myelogenous leukemia in blast crisis. Analysis of 242 patients. Am J Med 83,445–454 (1987) [DOI] [PubMed] [Google Scholar]
- 2.Savage DG, Szydlo RM, Goldman JM. Clinical features at diagnosis in 430 patients with chronic myeloid leukaemia seen at a referral centre over a 16-year period: Br J Haematol 96,111–116 (1997) [DOI] [PubMed] [Google Scholar]
- 3.Spiers AS. Clinical manifestations of chronic granulocytic leukemia: Semin Oncol 22, 380–340 (1995) [PubMed] [Google Scholar]
- 4.Ben-Neriah Y, Daley GQ, Mes-Masson AM, Witte ON, Baltimore D. The chronic myelogenous leukemia-specific P210 protein is the product of the bcr/abl hybrid gene: Science 233, 212–214 (1986) [DOI] [PubMed] [Google Scholar]
- 5.Cortez D, Kadlec L, Pendergast AM: Structural and signaling requirements for BCR-ABL-mediated transformation and inhibition of apoptosis. Mol Cell Biol 15, 5531–5541 (1995) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goga A, McLaughlin J, Afar DE, Saffran DC, Witte ON: Alternative signals to RAS for hematopoietic transformation by the BCR-ABL oncogene. Cell 82, 981– 988 (1995) [DOI] [PubMed] [Google Scholar]
- 7.Gordon MY: Biological consequences of the BCR/ABL fusion gene in humans and mice. J Clin Pathol 52, 719–722 (1999) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Etten RA: Oncogenic signaling: new insights and controversies from chronic myeloid leukemia. J. Exp. Med 204, 461–465 (2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, Gotlib J, Li K, Manz MG, Keating A, Sawyers CL, Weissman IL: Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med 351, 657–667 (2004) [DOI] [PubMed] [Google Scholar]
- 10.Zhao C, Chen A,Jamieson CH, Fereshteh M, Abrahamsson A, Blum J, Kwon HY, Kim J, Chute JP, Rizzieri D, Munchhof M, VanArsdale T, Reya T: Hedgehog signaling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 458, 776–779 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deininger M, Buchdunger E, Druker BJ:The development of Imatinib as therapeutic agent for chronic myeloid leukemia. Blood 108, 2640–2649 (2005) [DOI] [PubMed] [Google Scholar]
- 12.Ottmann OG, Druker BJ, Sawyers CL, Goldman JM, Reiffers J, Silver RT, Tura S, Fisher T, Deininger MW, Schiffer CA, Baccarani M, Gratwohl A, Hochhaus A, Hoelzer D, Fernandes-Reese S, Gathmann I, Capdeville R, O’Brien SG: A phase 2 study of imatinib in patients with relapsed or refractory Philadelphia chromosome-positive acute lymphoid leukemias. Blood 100, 1965–1971 (2002) [DOI] [PubMed] [Google Scholar]
- 13.Sawyers CL, Hochhaus A, Feldman E, Goldman JM,Miller CB, Ottmann OG, Schiffer CA, Talpaz M, Guilhot F, Deininger MW, Fisher T, O’Brien SG, Stone RM, Gambacorti-Passerini CB, Russel NH, Reiffers JJ, Shea TC, Chapuis B, Coutre S, Tura S, Morra E, Larson RA, Saven A, Peschel C, Gratwohl A, Mandelli F, Ben-Am M, Gathmann I, Capdeville R, Paquette RL, Druker BJ: Imatinib induces hematologic and cytogenetic responses in patients with chronic myelogenous leukemia in myeloid blast crisis: results of a phase II study. Blood 99, 3530–3539 (2002) [DOI] [PubMed] [Google Scholar]
- 14.Hofmann WK, Jones LC, Lemp NA, de Vos S, Gschaidmeier H, Hoelzer D, Ottmann OG, Koeffler HP: Ph (+) acute lymphoblastic leukemia resistant to the tyrosine kinase inhibitor STI571 has a unique BCR-ABL gene mutation. Blood 99,1860–1862 (2002) [DOI] [PubMed] [Google Scholar]
- 15.Branford S, Rudzki Z, Walsh S, Parkinson I, Grigg A, Szer J, Taylor K, Hermann R, Seymour JF, Arthur C, Joske D, Lynch K, Hughes T: Detection of BCR-ABL mutations in patients with CML treated with imatinib is virtually always accompanied by clinical resistance, and mutations in the ATP phosphate-binding loop (P-loop) are associated with a poor prognosis. Blood 102, 276–283 (2003) [DOI] [PubMed] [Google Scholar]
- 16.Nardi V, Azam M, Daley GQ: Mechanisms and implications of imatinib resistance mutations in BCR-ABL. Curr Opin Hematol 11, 35–43 (2004) [DOI] [PubMed] [Google Scholar]
- 17.Soverini S, Martinelli G, Rosti G, Giannini B, Trabacchi E, Castagnetti F, Testoni N, Luatti S, de vivo A, Cilloni D, Izzo B, Fava M, abruzzese E, Alberti D, Pane F, Saglio G, Baccarani M: ABL mutations in late chronic phase chronic myeloid leukemia patients with up-front cytogenetic resistance to imatinib are associated with a greater likelihood of progression to blast crisis and shorter survival: a study by the GIMEMA Working Party on Chronic Myeloid Leukemia. J Clin Oncol 23,4100–4109 (2005) [DOI] [PubMed] [Google Scholar]
- 18.Graham SM, Jorgensen HG, Allan E, Pearson C, Alcorn MJ, Richmond L, Holyoake TL: Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood 99, 319–325 (2002) [DOI] [PubMed] [Google Scholar]
- 19.Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL: Overriding imatinib resistance with a novel ABL kinase inhibitor. Science 305, 399–401 (2004) [DOI] [PubMed] [Google Scholar]
- 20.Weisberg E, Manley PW, Breitenstein W, Bruggen J, Cowan-Jacob SW, Ray A, Huntly B, Fabbro D, Feudrich G, Hall-Meyers E, Kung AL, Mestan J, Daley GQ, Callahan L, Catley L, Cavazza C, Azam M, Neuberg D, Wright RD, Gilliland DC, Griffin JD: Characterization of AMN107, a selective inhibitor of native and mutant Bcr- Abl. Cancer Cell 7, 129–141 (2005) [DOI] [PubMed] [Google Scholar]
- 21.O’Hare T, Eide CA, Deininger MW: Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood 110, 2242–2249 (2007) [DOI] [PubMed] [Google Scholar]
- 22.O’ Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F, Adrian LT, Zhou T, Huang WS, Wu Q, Metcalf CA, Tyner JW, Loriaux MM, Corbin AS, Wardwell S, Ning Y, Keats JA, Wang Y, Sundaramoothi R, Thomas M, Zhou D, Snodgrass J, Commodore L, Sayer TK, Dalgano DC, Deininger MW, Druker BJ, Clackson T: AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell 16, 401– 412 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang X, Zhao Y, Smith C, Gasparetto M, Turhan A, Eaves A, Eaves C: Chronic myeloid leukemia stem cells possess multiple unique features of resistance to BCR-ABL targeted therapies. Leukemia 21,926–935 (2007) [DOI] [PubMed] [Google Scholar]
- 24.Copland M, Hamilton A, Elrick LJ, Baird JW, Allan EK, Jordanides N, Barow M, Mountford JC, Holyoake TL: Dasatinib (BMS-354825) targets an earlier progenitor population than imatinib in primary CML but does not eliminate the quiescent fraction. Blood 107,4532– 453 (2006) [DOI] [PubMed] [Google Scholar]
- 25.Jorgensen HG, Allan EK, Jordanides NE, Mountford JC, Holyoake TL: Nilotinib exerts equipotent antiproliferative effects to imatinib and does not induce apoptosis in CD34+ CML cells. Blood 109, 4016–4019 (2007) [DOI] [PubMed] [Google Scholar]
- 26.Konig H, Holtz M, Modi H, Manley P, Holyoake TL, Forman SJ, Bhatia R: Enhanced BCR-ABL kinase inhibition does not result in increased inhibition of downstream signaling pathways or increased growth suppression in CML progenitors. Leukemia 22,748–755 (2008) [DOI] [PubMed] [Google Scholar]
- 27.Konig H, Holyoake TL, Bhatia R: Effective and selective inhibition of chronic myeloid leukemia primitive hematopoietic progenitors by the dual Src/Abl kinase inhibitor SKI-606. Blood 111, 2329–2338 (2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quintas-Cardama A, Cortes J: Molecular biology of bcr/abl1-positive chronic myeloid leukemia. Blood 113, 1619–1630 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW, Druker BJ: Chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR- ABL activity. J. Clin. Invest December 13,2010. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kroemer G, Jaattela M: Lysosomes and autophagy in cell death control. Nat Rev Cancer 2005; 5:886–897. [DOI] [PubMed] [Google Scholar]
- 31.Klionsky DJ: Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 8, 931–937 (2007) [DOI] [PubMed] [Google Scholar]
- 32.Baehrecke EH: Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol 6, 505–510 (2005) [DOI] [PubMed] [Google Scholar]
- 33.Kroemer G, Levine B: Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol 9,1004–1010 (2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kondo Y, Kanzawa T, Sawaya R, Kondo S: The role of autophagy in cancer development and response to therapy. Nat Rev Cancer 5,726–734 (2005) [DOI] [PubMed] [Google Scholar]
- 35.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G: Self- eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 8,741–752 (2007) [DOI] [PubMed] [Google Scholar]
- 36.Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB: Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 120, 237–248 (2005) [DOI] [PubMed] [Google Scholar]
- 37.Maclean KH, Dorsey FC, Cleveland JL, Kastan MB: Targeting lysosomal degradation induces p53-dependent cell death and prevents cancer in mouse models of lymphomagenesis. J. Clin Invest 118, 79–88 (2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lum JJ, DeBerardinis RJ, Thompson CB: Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol 6, 439–448 (2005) [DOI] [PubMed] [Google Scholar]
- 39.Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB: Autophagy inhibition enhances therapy- induced apoptosis in a Myc-induced model of lymphoma. J. Clin. Invest 117,326–336 (2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Degtyarev M, De Maziere A, Orr C, Lin J, Lee RB, Tien JY, Prior WW, van Dijk S, Wu H, Gray DC, Davis DP, Stern HM, Murray LJ, Hoeflich KP, Klumperman J, Friedman LS, Lin K: Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J. Cell. Biol 183,101–116 (2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fan QW, Cheng C, Hackett C, Feldmn M, Houseman BT, Nicolaides T, Haas-Kogan D, Jones CD, Oakes SA, Debnath J, Schokat KM, Weiss WA: Akt and autophagy cooperate to promote survival of drug-resistant glioma. Sci. Signal 3 (147):ra81(2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gelinas C, Fan Y, Nelson DA, Jin S, White E: Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 10, 51–64 (2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paglin S, Hollister T, Delohery T, Hackett N, McMahill M, Sphicas E, Domingo D, Yahalom J: A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res 61, 439–444 (2001) [PubMed] [Google Scholar]
- 44.Ito H, Daido S, Kanzawa T, Kondo S, Kondo Y: Radiation-induced autophagy is associated with LC3 and its inhibition sensitizes malignant glioma cells. Int J Oncol 26, 1401–1410 (2005) [PubMed] [Google Scholar]
- 45.Abedin MJ, Wang D, McDonnell MA, Lehmann U, Kelekar A: Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ 14, 500– 510 (2007) [DOI] [PubMed] [Google Scholar]
- 46.Kanzawa T, Germano IM, Komata T, Ito H, Kondo Y, Kondo S: Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ 11, 448–457 (2004) [DOI] [PubMed] [Google Scholar]
- 47.Kanzawa T, Kondo Y, Ito H, Kondo S, Germano I: Induction of autophagic cell death in malignant glioma cells by arsenic trioxide. Cancer Res 63, 2103–2108 (2003) [PubMed] [Google Scholar]
- 48.Lu Z, Luo RZ, Lu Y, Zhandg X, Yu Q, Khare S, Kondo S, Yu Y, Mills GB, Liao WS-L, Bast RC; The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J. Clin. Invest 118, 3917–3929 (2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Amaravadi RK.: Autophagy-induced tumor dormancy in ovarian cancer. J. Clin. Invest 118, 3837–3840 (2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Colell A, Ricci JE, Tait S, Milasta S, Maurer U, Boucher-Hayes L, Fitzgerald P, Guio-Carrion A, Waterhouse NJ, Li CW, Mari B, Barbry P, Newmeyer DD, Beere HM, Green DR: GAPDH and autophagy preserve survival after apoptotic cytochrome c release in the absence of caspase activation. Cell 129, 983–997 (2007) [DOI] [PubMed] [Google Scholar]
- 51.Dikic I, Johansen T, Kirkin T,V: Selective autophagy in cancer development and therapy. Cancer Res. 70, 3431– 3434 (2010) [DOI] [PubMed] [Google Scholar]
- 52.Calabretta B, Perrotti D: The biology of CML blast crisis. Blood 103, 4010–4022 (2004) [DOI] [PubMed] [Google Scholar]
- 53.Sharma SV, Gajowniczek P, Way IP: A common signaling cascade may underlie “addiction” to the Src, BCR-ABL, and EGF receptor oncogenes. Cancer Cell 10, 425–435 (2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bellodi C, Lidonnici MR, Hamilton A, Helgason A,GV, Soliera AR, Ronchetti M, Galavotti S, Young KW, Selmi T, Yacobi R, Van Etten RA, Donato N, Hunter A, Dinsdale D, Tirro E, Vigneri P, Nicotera P, Dyer MJ, Holyoake T, Salomoni P, Calabretta B: Targeting autophagy potentiates tyrosine kinase inhibitor -induced cell death in Philadelphia chromosome -positive cells, including primary CML stem cells. J. Clin. Invest 119, 1109–1123 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eisenberg-Lerner A, Bialik S, Kimchi A: Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ 16, 966–975 (2009) [DOI] [PubMed] [Google Scholar]
- 56.Wullschleger S, Loewith R, Hall MN: TOR signaling in growth and metabolism. Cell 124, 471–484 (2006) [DOI] [PubMed] [Google Scholar]