

The role of the lectin pathway (LP) of complement has not been explored in the thrombotic microangiopathies (TMA). We examined levels of MASP2, the effector enzyme of the LP, in three major forms of TMA and assessed the effect of the anti‐MASP2 human monoclonal antibody narsoplimab on TMA plasma‐induced activation of caspase 8 in microvascular endothelial cells. MASP2 levels were highly elevated in all TMA patients versus controls. Caspase 8 activation was suppressed by clinically relevant levels of narsoplimab. In conclusion, we found that the LP of complement is activated in TMAs of diverse etiology and LP inhibition may be of therapeutic benefit in these disorders.
Keywords: complement, hemolytic uremic syndrome, lectin pathway, MASP, thrombotic microangiopathy
Summary
Involvement of the alternative complement pathway (AP) in microvascular endothelial cell (MVEC) injury characteristic of a thrombotic microangiopathy (TMA) is well documented. However, the role of the lectin pathway (LP) of complement has not been explored. We examined mannose‐binding lectin associated serine protease (MASP2), the effector enzyme of the LP, in thrombotic thrombocytopenic purpura, atypical hemolytic uremic syndrome and post‐allogeneic hematopoietic stem cell transplantation (alloHSCT) TMAs. Plasma MASP2 and terminal complement component sC5b‐9 levels were assessed by enzyme‐linked immunosorbent assay (ELISA). Human MVEC were exposed to patient plasmas, and the effect of the anti‐MASP2 human monoclonal antibody narsoplimab on plasma‐induced MVEC activation was assessed by caspase 8 activity. MASP2 levels were highly elevated in all TMA patients versus controls. The relatively lower MASP2 levels in alloHSCT patients with TMAs compared to levels in alloHSCT patients who did not develop a TMA, and a significant decrease in variance of MASP2 levels in the former, may reflect MASP2 consumption at sites of disease activity. Plasmas from 14 of the 22 TMA patients tested (64%) induced significant MVEC caspase 8 activation. This was suppressed by clinically relevant levels of narsoplimab (1·2 μg/ml) for all 14 patients, with a mean 65·7% inhibition (36.8–99.4%; P < 0·0001). In conclusion, the LP of complement is activated in TMAs of diverse etiology. Inhibition of MASP2 reduces TMA plasma‐mediated MVEC injury in vitro. LP inhibition therefore may be of therapeutic benefit in these disorders.
Introduction
The three major complement pathways, classical (CP), alternative (AP) and lectin (LP), are involved in many homeostatic processes distinct from their role in innate immunity [1]. Although all three systems can be initiated independently by diverse stimuli, the proteolytic cascades they induce converge at C3, leading to formation of inflammatory fragments C3a and C5a and assembly of the lytic membrane attack complex C5b‐9 [2]. A fourth ‘extrinsic’ complement pathway has also been proposed, as activated by tissue factor and thrombin [3]. It includes plasmin and thrombin, which can cleave C3 and C5, directly activating complement, at least in vitro [4]. The importance of dysregulation of the AP in the activation and injury of microvascular endothelium and platelets, characteristic of the two major thrombotic microangiopathies (TMA) thrombotic thrombocytopenic purpura (TTP) and atypical hemolytic uremic syndrome (aHUS), has been well documented, and is supported by clinical responses to inhibitors of complement C5 (reviewed in [5]). New data suggest that interactions among all four pathways – labeled ‘immunothrombosis’ [6] – may be critical in TMA initiation and/or progression; however, this scenario is under‐explored in relationship to the LP. Dissection of such cross‐talk may offer new avenues for intervention in the TMAs.
The LP could be involved in a variety of pr‐thrombotic disorders through the binding of pattern recognition molecules such as mannose‐binding lectin (MBL), ficolins and collectins to carbohydrate patterns present on microbial pathogens and injured cells, enabling complex formation and activation of the MBL‐associated serine proteases 1 and 2 (MASP1, MASP2) [7]. Activated MASP2 then cleaves C4 and C2 to form C3 convertase (C4bC2a) [7]. Positive feedback loops arise in the setting of either excessive complement activation or acquired or congenital defects in complement regulatory proteins, the latter characteristic of an aHUS‐type of TMA [5]. AP amplification is quantitatively responsible for the magnitude of complement activation initiated by the LP or CP [8], whereas MASP1 and MASP2 are critical for efficient LP activation and its amplification [7].
Clinical associations between end‐products of the AP and TMAs are well documented. Circulating C3 breakdown products C3c and C3d are increased in acute aHUS [9], and C3a, C5a and soluble (s)C5b‐9 are elevated in the plasma and urine of patients with acute TTP and aHUS [10, 11, 12]. In tissues, deposition of sC5b‐9 on glomerular, dermal and intestinal microvasculature has been demonstrated in acute aHUS [13, 14]. However, involvement of the LP has not been assessed in the TMAs. Cumulative evidence suggests that MASP2 is redundant in human defense, as individuals with primary MASP2 deficiency are not prone to infectious or autoimmune diseases [15]. In contrast, we hypothesized that over‐activity of MASP2 in the LP is important in three major TMAs, TTP, aHUS and ‘secondary’ aHUS‐type TMAs, occurring in the setting of infections, autoimmune disease or allogeneic hematopoietic stem cell transplantation [alloHSCT, also known as transplant‐associated TMA (TA‐TMA)]. We also postulated that this will be reflected by changes in plasma MASP2 levels. We then hypothesized that interference with MASP2 activity in vitro, using the human monoclonal antibody (mAb) narsoplimab (OMS721), currently in a Phase 2 program in alloHSCT‐associated TMA, would suppress activation of microvascular endothelial cells (MVEC) induced by acute TMA patient plasmas, an initial step in models of TMA plasma and serum‐induced MVEC injury [16].
Methods
TMA patients
TMAs occurring in the absence of disseminated intravascular coagulation were diagnosed by standard clinical criteria [17] including: microangiopathic changes based upon an increase over baseline of peripheral schistocytes or histological evidence of microangiopathy on kidney or skin biopsy; serum lactate dehydrogenase (LDH) levels exceeding the upper limit of normal; and de‐novo thrombocytopenia, based on a platelet count < 150 × 109/l or a ≥ 25% decrease from baseline. They were then differentiated by distintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13) activity and inhibitor assays as either acquired TTP (< 5% ADAMTS13 activity and presence of an ADAMTS13 inhibitor) or a non‐TTP form of TMA (> 10% ADAMTS13 activity). All patients with diarrhea also had a negative culture and polymerase chain reaction (PCR)‐based test for shigatoxin. Thirteen individuals with acute, acquired TTP and 18 individuals with a non‐TTP form of TMA resembling acute aHUS, of whom five had intercurrent cancer and cancer chemotherapy, three autoimmune disease, one idiopathic and nine following an alloHSCT for a hematologic malignancy, were included into this study (Table 1).
Table 1.
Clinical and complement pathway data for patients
| Patient code | Patient Dx | TMA factor | Platelet | LDH | Creatinine | sC5b‐9 | MASP‐2 |
|---|---|---|---|---|---|---|---|
| (×109/l) | (U/l) | (mg/dl) | (ng/ml) | (ng/ml) | |||
| 1 | aHUS | Cancer/chemoTx | 12 | 365 | 0·6 | 1079 | 118·08 |
| 13 | aHUS | Cancer/chemoTx | 70 | n.d. | 1·1 | 3845 | 142·81 |
| 15 | aHUS | Cancer/chemoTx | n.d. | n.d. | n.d. | 2676 | 80·66 |
| 18 | aHUS | Cancer/chemoTx | 12 | 861 | 4 | 4310 | 97·89 |
| 24 | aHUS | Cancer/chemoTx | 9 | 1974 | 1·9 | 5916 | 273·73 |
| 2 | TTP | None | 9 | 1959 | 1·3 | 770 | 174·66 |
| 3 | TTP | None | 9 | 861 | 1·45 | 1399 | 150·08 |
| 4 | TTP | None | 13 | 532 | 1·2 | 1187 | 224·8 |
| 5 | TTP | None | 12 | 817 | 1·7 | 963 | 253·58 |
| 6 | TTP | None | 18 | 1400 | 6 | 4636·92 | 210·38 |
| 10 | TTP | None | 7 | 1485 | 1·2 | 4406 | 175·43 |
| 11 | TTP | None | 35 | 1599 | 0·9 | 1352 | 144·9 |
| 12 | TTP | None | 7 | 1143 | 1·4 | 3638 | 274·67 |
| 16 | TTP | None | 78 | 6534 | 1·3 | 2439 | 122·24 |
| 17 | TTP | None | 14 | 2343 | 0·7 | 3406·06 | 363·79 |
| 25 | TTP | None | n.d. | n.d. | n.d. | 8109·8 | 331·91 |
| 34 | aHUS | None | 46 | 1983 | 4 | 926·82 | 450·66 |
| 41 | TTP | None | 7 | 1388 | 0·8 | 2081 | 142·49 |
| 45 | TTP | None | 37 | 776 | 0·68 | 5332 | 341·5 |
| 46 | aHUS | SLE | 138 | 216 | 0·7 | 5915 | 139·57 |
| 52 | aHUS | SLE | 7 | 1143 | 1·4 | 1952 | 176·43 |
| 42 | aHUS | Autoimmune | 14 | 694 | 8·6 | 3426 | 225·11 |
| alloHSCT patient code | |||||||
| 9 | TMA | HSCT | 56 | 413 | 1·51 | 795·6 | 189·51 |
| 23 | TMA | HSCT | 13 | 932 | 1·57 | 778·19 | 157·05 |
| 37 | TMA | HSCT | 39 | 186 | 2·1 | 17·9 | 101·88 |
| 42 | TMA | HSCT | 35 | 605 | 1·18 | 1194·76 | 131·6 |
| 44 | TMA | HSCT | 48 | 440 | 1·89 | 197·99 | 131·6 |
| 50 | TMA | HSCT | 115 | 269 | 2·03 | 297·67 | 284·23 |
| 52 | TMA | HSCT | 84 | 377 | 1·64 | 208·72 | 173 |
| 57 | TMA | HSCT | 90 | 304 | 1·04 | 341·5 | 130 |
| 78 | TMA | HSCT | 107 | 200 | 1·08 | 277·46 | 208·94 |
MASP2 = mannose‐binding lectin associated serine protease 2; HSCT = hematopoietic stem cell transplantation; SLE = systemic lupus erythematosus; aHUS = atypical hemolytic uremic syndrome; TMA = thrombotic microangiopathy; n.d. = not done; LDH = lactate dehydrogenase.
The nine patients with an acute TMA following alloHSCT were diagnosed based on the consensus criteria of Cho et al. [18]. Their TMAs persisted after graft‐versus‐host disease prophylaxis with calcineurin inhibitors or mTOR inhibitors were stopped and glucocorticoids and mycophenolate mofetil substituted. None had an active infection at the time of TMA diagnosis. Those subjects were part of a prospective study of TMA incidence and course in the alloHSCT setting among adults and registered with the US National Clinical Trials network (NCT02604420). One hundred transplant patients were enrolled, 97 of whom had hematological malignancies. Twenty subjects met study criteria for a HSCT‐TMA. Three resolved following discontinuation of mTOR or calcineurin inhibitors. Seven had a precipitating infection, six of whom expired with ongoing severe TMA. TMAs persisted in the remaining 10 subjects, nine of whom had samples available for our study.
Control subjects included 45 healthy adults, age‐matched to our alloHSCT TMA cohort, as well as the 80 individuals post‐alloHSCT who did not develop a TMA. Studies were approved by the Weill Cornell Institutional Review Board.
Clinical and laboratory monitoring
All subjects were HIV seronegative at baseline. Among the alloHSCT patients, who were followed serially for at least 1·5 years, any intervention for TMA, typically involving plasma exchange or use of the anti‐C5 mAb eculizumab, was initiated at the discretion of the patient’s primary physician. For determination of complement components, whole blood was collected by peripheral venipuncture into heparin sodium tubes on ice to minimize ex‐vivo complement activation, centrifuged within 30 min, and plasmas stored at −80°C in 200 μl aliquots. Commercial enzyme‐linked immunosorbent assays for MASP2 (MyBioSource, San Diego, CA, USA) and sC5b‐9 (Quidel, San Diego, CA, USA) were performed as per the manufacturer’s directions. ADAMTS13 activity was measured by fluorescence resonance energy transfer (FRET), utilizing a synthetic substrate.
Endothelial cell cultures
Primary human neonatal MVEC of dermal origin were purchased from ScienCell Research Labs (San Diego, CA, USA) and Clonetics (San Diego, CA, USA). The identity of all EC had been confirmed by phenotypical and genotypical analyses, as described over the past decade by our laboratory [16]. EC were maintained in T‐25 flasks (Falcon, Becton Dickinson Labware, Lincoln Park, NJ, USA), coated with 50 µg/ml human plasma fibronectin (Chemicon International, Temecula, CA, USA) in phosphate‐buffered saline (PBS), in ECM 1001 medium (ScienCell Research Labs) containing a proprietary endothelial cell growth supplement, penicillin, streptomycin and 15% fetal bovine serum. All EC were used in passages 2–6. Subcultures involved a 5–10 min exposure to 0·25% trypsin–ethylenediamine tetraacetic acid (EDTA), followed by washing with PBS, pH 7.2.
Caspase 8 assays
MVECs, 1·5 × 105/well, were subcultured in 24‐well fibronectin‐coated plates in the presence of control or active TMA plasmas (2%, v/v) overnight at 37°C in the presence or absence of narsoplimab or an isotype‐matched control. Activation of caspase 8 was evaluated by a functional assay based on hydrolysis of Acetyl‐Ile‐Glu‐Thr‐Asp‐p‐nitraniline, performed according to the manufacturer’s instructions (caspase 8 assay kit, colorimetric; Sigma, St Louis, MO, USA), with measurement of optical density (OD) absorbance at 405 nm.
Results
MASP2 plasma levels
As shown in Table 2, median MASP2 levels were elevated in all acute TTP patients [n = 13; 210·4 ng/ml (P < 0·0001)] and all non‐TTP TMA patients [n = 18; 150·0 ng/ml (P < 0·0001)] compared with healthy controls [n = 39; 70·8 ng/ml (range = 26·9–210·9)]. For the nine individuals with non‐TTP TMAs occurring apart from an alloHSCT, MASP2 levels were markedly elevated [142·8 ng/ml; P = 0·0005)] (Table 2). These levels were also significantly elevated in the subset of nine non‐TTP TMA cases occurring post‐alloHSCT, assessed at the time of development of a persistent TMA (154·0 ng/ml; P = 0·0005). These plasmas were obtained during the acute TMA episode, and no individual was receiving active treatment for a TMA at the time of their collection.
Table 2.
Plasma levels of MASP2 and C5b‐9, biomarkers for complement activation, in patients with acquired TTP, non‐TTP TMAs and controls
| TMA diagnosis | n | MASP2, ng/ml (median; range) | sC5b‐9, ng/ml (median; range) | P |
|---|---|---|---|---|
| TTP | 13 | 210·4 (122·2–363·8) | < 0·0001 | |
| 2439·0 (963–8109·8) | 0·0007 | |||
| All non‐TTP | 18 | 150·0 (80·7–450·7) | < 0·0001 | |
| 1002·9 (17·9–5916) | 0·0015 | |||
| Non‐TTP, no HSCT | 9 | 142·8 (80·7–450·7) | 0·0005 | |
| 3426·0 (926·8–5916) | 0·002 | |||
| HSCT‐TMA | 9 | 154·0 (101·9–284·2) | 0·0005 | |
| 297·7 (17·9–1194·8) | 0·26 | |||
| Healthy controls | 39 | 70·8 (26·9–210·9) | – | |
| Healthy controls | 6 | 400·0 (100–1136) | – | |
| HSCT, no TMA | 80 | 348·6 (25·1–2507·6) | – |
MASP2 = mannose‐binding lectin associated serine protease 2; HSCT = hematopoietic stem cell transplantation; SLE = systemic lupus erythematosus; aHUS = atypical hemolytic uremic syndrome; TMA = thrombotic microangiopathy; TMA = thrombotic microangiopathy; TTP = thrombotic thrombocytopenic purpura.
MASP2 levels in alloHSCT patients who did not develop a TMA, assessed at day 100 ± 28 days post‐transplant, which represented the mean time to development of a TMA in our study, were also significantly elevated versus controls [n = 80, 113·5 ng/ml (range = 56–430·3) (P < 0·0001)] (Fig. 1). Lack of a significant rise in MASP2 levels in patients with persistent TMAs versus those who did not develop a TMA (P = 0·25) (Fig. 1), combined with a significant decrease in variance of MASP2 levels in the former group (P = 0·005), may reflect consumption of MASP2 at sites of disease activity, i.e. the microvasculature. This possibility is supported by MASP2 levels available at the first time‐point of TMA recognition from five of the 10 subjects with persistent TMAs post‐alloHSCT. As shown in Fig. 2, three of the five individuals (patients 057, 009 and 037) had declines in plasma MASP2 levels at the time of TMA diagnosis and two had stable elevated levels of the enzyme (patients 052 and 023), but none showed further elevation in MASP2 levels at that time‐point. Subsequent MASP2 levels are not interpretable, as they may reflect changes related to treatment, including plasma exchange and, in four of the five patients, use of the anti‐C5 mAb eculizumab.
Fig. 1.
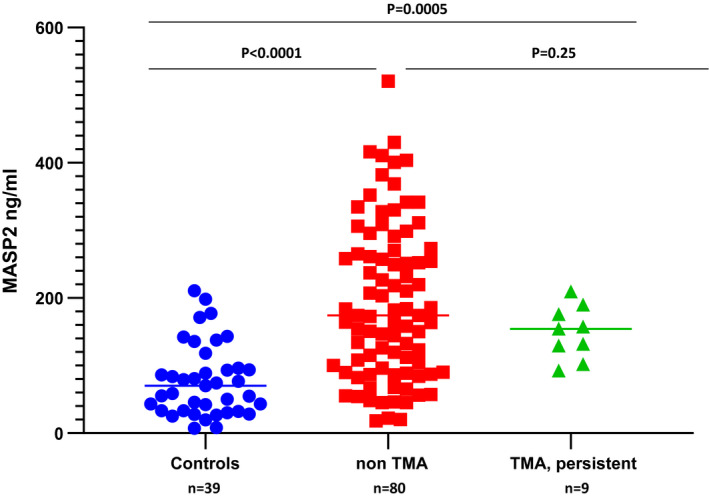
Plasma mannose‐binding lectin‐associated serine protease 2 (MASP2) levels in the setting of allogeneic hematopoietic stem cell transplantation. Plasma MASP2 levels were measured in 39 healthy controls, nine individuals developing a non‐thrombotic thrombocytopenic purpura (TTP) type of thrombotic microangiopathy (TMA) post‐alloHSCT, and 80 individuals undergoing an alloHSCT who did not develop a TMA but were assessed at the mean time‐point for TMA development in the former group (day 100 ± 28 days).
Fig. 2.
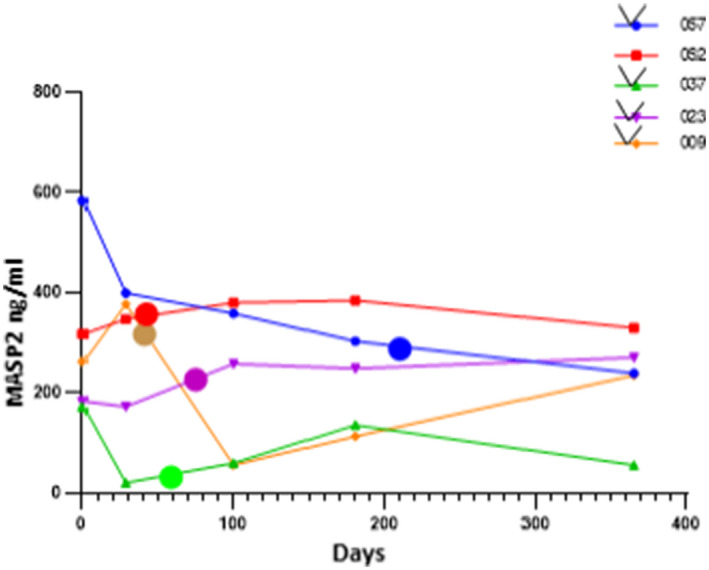
Longitudinal assessment of mannose‐binding lectin‐associated serine protease 2 (MASP2) plasma levels in five individuals developing a persistent thrombotic microangiopathy (TMA) post‐alloHSCT. The large dots represent plasma levels at the time of TMA recognition, in addition to levels obtained at regularly scheduled visits (five times/year) for all individuals post‐transplant. Four of the five patients received anti‐C5 monoclonal antibody (mAb) eculizumab at the time of TMA diagnosis (the exception is patient 052).
Plasma markers of terminal complement component activation and correlations with MASP2 levels
As summarized in Table 2, median levels of sC5b‐9 were elevated in all acute TTP patients [n = 13; 2439·0 ng/ml (P = 0·0007)] and all non‐TTP TMA patients [n = 18; 1002·9 ng/ml) (P = 0·0015)], compared with healthy controls. Median sC5b‐9 levels were also significantly elevated in the subset of non‐TTP TMA patients apart from the context of an alloHSCT [n = 9; 3426·0 ng/ml (P = 0·002)]. However, sC5b‐9 levels in the subset of patients with TMAs post‐alloHSCT did not reach statistical significance, either when compared to levels recorded in individuals post‐alloHSCT who did not develop a TMA (n = 80; P = 0·40) or versus a healthy control group for which fewer individuals were available (n = 6; P = 0·26; Table 2).
Associations were then sought between MASP2 and sC5b‐9 plasma levels. There was a positive correlation between sC5b‐9 and MASP2 (R 2 = 0·419) for TTP patients (n = 12; complete data missing for one patient). However, no correlation between these two types of complement products was observed for the non‐TTP types of TMA, either in the non‐transplant setting [R 2 = 0·150; n = 8 (complete data absent for one patient)] or in the context of an alloHSCT‐linked TMA (R 2 = 0·035; n = 9).
Caspase 8 activation of human MVEC by TMA patient plasmas and its inhibition by the anti‐MASP2 mAb narsoplimab
Caspase 8 exists in the cell as an inactive proenzyme. It is converted into an active form, consisting of 40 and 23 kD subunits, upon recruitment to the cytoplasmic domain of activated death receptors just prior to apoptotic cell injury [19]. We have previously reported that acute TTP plasmas induced caspase 8 activation in MVEC as a prelude to apoptosis [16]. We now hypothesized that plasmas from patients with an acute TMA arising in a variety of clinical settings would also activate this enzyme. An initial dose response for narsoplimab‐mediated inhibition of caspase 8 activation involved concentrations of 0·24, 1·2, 6, 30 and 150 μg/ml. Plateau inhibition was noted at the 1·2‐μg/ml dose, which was then utilized in subsequent experiments, together with an isotype control antibody at 10 μg/ml. This concentration of narsoplimab represents the lower end of that achievable in ongoing clinical trials of the drug in the adult HSCT setting [20].
Of the 22 TTP and non‐alloHSCT TMA patients listed in Table 1 and available for this study, 14 (64%) induced significant caspase 8 activation (> 20% over baseline control plasmas). Caspase 8 activation in MVEC was re‐examined with these 14 plasmas in the presence of narsoplimab (1·2 µg/ml) or control antibody (10 µg/ml). As shown in Fig. 3, blockade of MASP2 activity led to a decrease in caspase 8 activity for all 14 patient plasmas, with a mean 65·7% inhibition (range = 36·8–99·4%) (P < 0·0001).
Fig. 3.
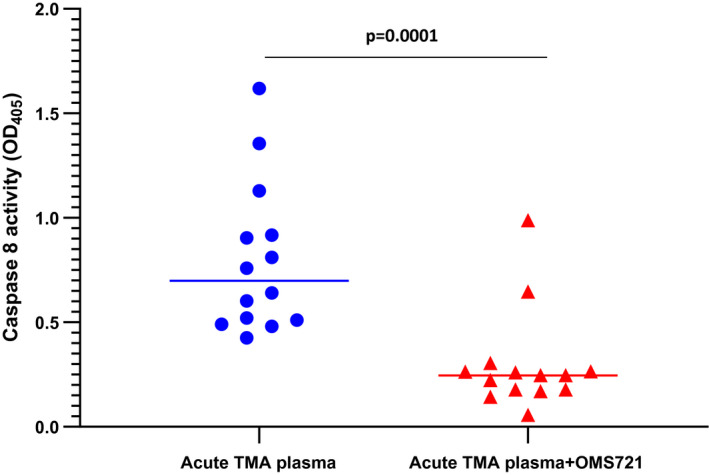
Acute thrombotic microangiopathy (TMA) plasmas induce caspase 8 activation in human microvascular endothelial cell (MVEC), which is blocked by anti‐mannose‐binding lectin‐associated serine protease 2 (MASP2) monoclonal antibody (mAb) narsoplimab. Cultures of primary human neonatal dermal microvascular endothelial cells were exposed to plasmas from patients with acute thrombotic thrombocytopenic purpura (TTP) or non‐TTP types of TMA not associated with transplantation for 24 h in the presence or absence of anti‐MASP2 mAb narsoplimab (1·2 μg/ml). Caspase 8 activity was analyzed in cell lysates.
Discussion
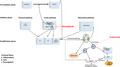
Excessive or unregulated complement activation contributes to a myriad of inflammatory, infectious, autoimmune, neoplastic and degenerative diseases, as activated complement components are produced in response to tissue damage, amplifying and exacerbating that injury. Multiple studies have examined the role of the AP in a variety of TMAs, its distinction from CP involvement and the role of congenital defects in ADAMTS13 versus complement and complement regulatory factors in susceptibility to TTP versus aHUS, respectively [16]. For example, in TTP C3a and sC5b‐9 levels are elevated in the absence of changes in CP markers [12]. Apart from a suggestion of LP involvement in TTP, as C4d levels decreased following remission induction [12], this pathway has not been similarly investigated in the TMAs. Similarly, known congenital defects in LP and particularly MASP2 have not been studied in this context, but should be examined. However, host‐cell surfaces can exhibit altered glycan structures during inflammation and oxidative stress, and therefore would be expected to serve as targets for MBL, ficolin and collectin binding. This is the first step in LP activation, and part of a positive feedback loop leading to pathological AP activation and inflammation [2], as illustrated in Fig. 4. Our observation of increased MASP2 in two major types of TMA, acquired TTP and non‐TTP types of TMA linked to a variety of associated conditions, is consistent with the fact that these disorders are characterized by extensive MVEC activation and injury. Our finding that clinically relevant levels of the MASP2 inhibitor narsoplimab significantly suppressed TMA plasma‐mediated caspase 8 activation in MVEC supports the concept that LP activation is integral to the pathophysiology of these disorders, rather than just a consequence of AP activation.
Fig. 4.
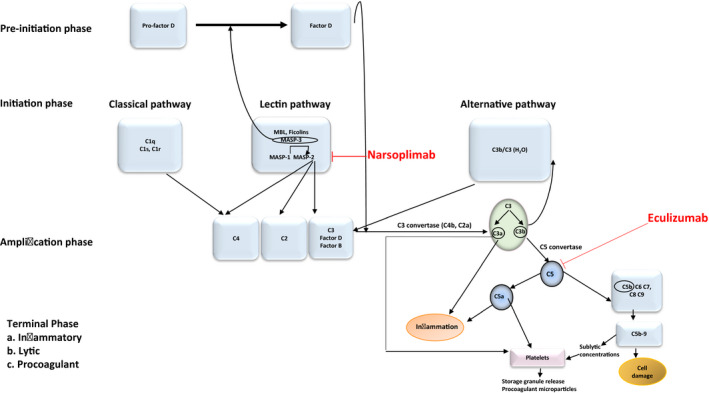
Interaction among components of the complement cascade and the coagulation system, and points of intervention with anti‐complement therapeutics. Multiple points of interaction among the three principal pathways of the complement cascade, the classical (CP), alternative (AP) and lectin (LP) pathways, are illustrated. They include pre‐initiation, initiation and amplification phases ultimately leading to a terminal phase involving inflammatory, lytic and procoagulant outcomes. Points susceptible to blockade by narsoplimab, the anti‐mannose‐binding lectin‐associated serine protease 2 (MASP2) monoclonal antibody (mAb) and eculizumab, the anti‐C5 mAb, are illustrated. In terms of specific interactions of the LP with the coagulation cascade, MASP2 can directly cleave C4 as well as C3, activating prothrombin [7]. MASP1 and MASP2 contribute to in‐vitro fibrin clot formation, and MASP1, the exclusive activator of MASP2 [7], is essential for obstructive thrombosis in a murine model of arterial injury [1]. Plasmin and thrombin can cleave C3 and C5, directly activating complement, at least in vitro [4] Reciprocal interactions with thrombin‐induced complement activation in vivo may require platelet involvement, as the amount of thrombin generated in vivo is insufficient to directly induce C activation, at least in the fluid phase [34, 35]. Complement components also have a direct effect on platelets, which express complement receptors [C1qR, C5aR, C3aR, P‐selectin (which acts as a C3b receptor)] and complement regulatory molecules [C1‐I, CD55, CFH, CD46 (MCP), CD59] [3, 5, 36]. C3a and C5a directly activate resting platelets and potentiate platelet activation induced by multiple agonists, and sublytic concentrations of sC5b‐9 are potent platelet agonists, leading to platelet storage granule secretion and release of procoagulant.
The importance of complement activity in TMAs linked to alloHSCT is well documented, based on alterations in circulating sC5b‐9 and C5a and deposition of sC5b‐9 on tissue microvasculature [13, 14], as well as dramatic responses to the anti‐C5 mAb eculizumab, at least in pediatric populations [21]. These are critical findings, as alloHSCT‐associated TMAs complicate some 20% of these procedures, and 3‐year survival rates for those with persistent TMAs are a dismal 11% [13]. The LP had not been explored in this setting. Chemotherapy in association with autologous HSCT for malignancy does correlate with a marked increase in serum MASP2 levels, persisting for approximately 4 weeks post‐transplant [22], but TMAs are extremely rare in adults following an autologous, as opposed to an allogeneic, transplant. Higher serum MASP2 levels are associated with longer event‐free survival in children with lymphoma [23], and this is thought to be secondary to consumption of MASP2 on tissue microvasculature. We found a significant increase in MASP2 levels, with a wide variance, in alloHSCT patients evaluated at a time post‐transplantation typical of HSCT‐TMA development, regardless of whether or not a TMA occurred. At the time of development of a persistent HSCT‐TMA, MASP2 levels remained elevated over healthy controls, but with a trend towards lower levels, and a highly significant lower variance, than in those individuals not developing a TMA post‐alloHSCT
The clinical relevance of these findings is strengthened by studies of the role of the LP in other thrombotic states. Increased MASP2 levels correlate with chronic cardiovascular disease risk factors, including dyslipidemia, hypertension and obesity [1], and these levels may be significantly reduced over those baselines following cardiac surgery with cardiopulmonary bypass [1] and in the setting of an acute myocardial infarction [24]. These latter changes are thought to be secondary to LP activation, with MASP2 consumption in tissue during myocardial ischemia. Indeed, strong MBL staining limited to the coronary vascular endothelium is characteristic of the early phase of myocardial ischemia/reperfusion injury in rats [25]. MBL deposition at lesional sites, in the absence of concurrent deposition of immunoglobulin (Ig)G IgM, or C1q, supports the concept of LP activation without CP involvement in these disorders [8]. Targeting the LP by knock‐out of MASP2 or MBL, or administration of anti‐MASP2 or anti‐MBL mAbs, protects against ischemic reperfusion injuries of the heart, brain and gastrointestinal tract in mice (reviewed in [1]). Mice treated with an anti‐rat MASP2 mAb had reduced myocardial infarct size following transient ligation of the left anterior descending coronary artery [26]. A recent review concluded that further research is required to more clearly define the role of the LP in other thrombotic conditions [27]. Inhibition of MASP2, the key effector enzyme of the LP, blocks this pathway at its initial steps, an advantage given the amplifying nature of the complement cascade. It may represent a novel therapeutic strategy for a variety of thrombotic disorders, distinct from eculizumab which, in blocking C5, acts at a later point (Fig. 4).
It is also known that platelets exposed to sera from acute TTP and aHUS patients show C3 and sC5b‐9 deposition, accompanied by their release of tissue factor‐enriched microparticles [5, 28]. This supports the involvement of platelets in cross‐talk between the coagulation and complement systems (Fig. 4). Deposition of sC5b‐9 on transformed human MVEC exposed to acute aHUS sera has also been demonstrated [29], but our studies are the first, to our knowledge, to document involvement of the LP in TMA serum or plasma‐induced EC pathology.
Narsoplimab was granted US Food and Drug Administration (FDA) breakthrough therapy designation for patients who have high‐risk HSCT‐TMAs, and in the European Union it has been designated an orphan medicinal product in HSCT, based on improved survival compared to historical controls in a Phase 2 study [20] and salutary case reports [30]. Blockade of MASP2 should not be accompanied by a high risk of infectious or autoimmune disorders, as it appears redundant in human defenses. Even very low levels do not appear to predispose to infectious or autoimmune diseases [31]. In terms of the broad benefits of this research, it was recently shown that lung injury related to infection with SARS‐CoV‐2, the etiological agent of the novel coronavirus‐2019 (COVID‐19) pandemic, is a systemic microvascular thrombopathy, characterized by deposition of MASP2 and C5b‐9 in pulmonary as well as cutaneous microvasculature [32]. Given positive feedback loops between activation of the complement and coagulation pathways (Fig. 4) and the hypercoagulable state characteristic of COVID‐19, consideration of intervention with anti‐AP and anti‐LP complement strategies has been raised [32].
Limitations of our study include lack of demonstration of MASP2 in tissues of TMA patients. As complement activation occurs at the surface of cells, circulating levels of MASP2 might be an inadequate surrogate for disease activity or therapeutic efficacy, similar to limited correlations of TMA activity with plasma C5a and sC5b‐9 levels [33]. Measurement of MASP2 levels in our TTP and non‐alloHSCT aHUS patients following remission induction would also be of interest.
Disclosures
J. L. received a research grant and a non‐restricted educational grant from Omeros Corp., the manufacturer of narsoplimab.
Author contributions
J. L., S. E., J. C. and J. A. designed the study and analyzed the data. S. E., J. C. and J. A. carried out the experiments. K. V. B. provided transplant patients from an observational study that he and J. L. designed and conducted. J. L. drafted the paper. All authors approved the final version of the manuscript.
Acknowledgements
Discussions with Thomas Dudler PhD, are gratefully acknowledged. This work was supported by a grant from Omeros, Inc. and the Angelo Donghia Foundation (J. L.) and by grants from the National Institutes of Health (HL148123, HL123605, GM114731), and partly supported by OCASCR, PHF, and OMRF grants (to J.A.).
Data availability statement
All data generated or analysed for the purposed of this study are included in this published article.
References
- 1. Frauenknecht V, Thiel S, Storm L et al Plasma levels of mannan‐binding lectin (MBL)‐associated serine proteases (MASPs) and MBL‐associated protein in cardio‐ and cerebrovascular diseases. Clin Exp Immunol 2013; 173:112–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Beltrame MH, Catarino SJ, Goeldner I, Boldt ABW, de Messias Reason IJ. The lectin pathway of complement and rheumatic heart disease. Front Ped 2015; 2:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fumagalli S, De Simoni M‐G. Lectin complement pathway and its bloody interactions in brain ischemia. Stroke 2016; 47:3067–73. [DOI] [PubMed] [Google Scholar]
- 4. Amara U, Flierl MA, Rittirsch D et al Molecular intercommunication between the complement and coagulation systems. J Immunol 2010; 185:5628–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Noris M, Mescia F, Remuzzi G. STEC‐HUS, atypical HUS and TTP are all diseases of complement activation. Nat Rev Nephrol 2012; 8:622–33. [DOI] [PubMed] [Google Scholar]
- 6. Kawecki C, Lenting PJ, Denis CV. Von Willebrand factor and inflammation. J Thromb Hemost 2017; 15:1285–94. [DOI] [PubMed] [Google Scholar]
- 7. Dodo J, Kocsis A, Gal P. Be on target: strategies of targeting alternative and lectin pathway components in complement‐mediated diseases. Front Immunol 2018; 9:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Harboe M, Garred P, Karlstrom E, Lindstad JK, Stahl GL, Mollnes TE. The down‐stream effects of mannan‐induced lectin complement pathway activation depend quantitatively on alternative pathway amplification. Mol Immunol 2009; 47:373–80. [DOI] [PubMed] [Google Scholar]
- 9. Kim Y, Miller K, Michael AF. Breakdown products of C3 and Factor B in hemolytic‐uremic syndrome. J Lab Clin Med 1977; 89:845–50. [PubMed] [Google Scholar]
- 10. Cateland SR, Holers VM, Geyer S, Yang S, Wu HM. Biomarkers of terminal complement activation confirm the diagnosis of aHUS and differentiate aHUS from TTP. Blood 2014; 123:3733–8. [DOI] [PubMed] [Google Scholar]
- 11. Reti M, Farkas P, Scuka D et al Complement activation in thrombotic thrombocytopenic purpura. J Thromb Hemost 2012; 10:791–8. [DOI] [PubMed] [Google Scholar]
- 12. Prufer F, Scheiring J, Sautter S et al Terminal complement complex (C5b–9) in children with recurrent hemolytic uremic syndrome. Sem Thromb Hemost 2006; 32:121–7. [DOI] [PubMed] [Google Scholar]
- 13. Chapin J, Shore T, Forsberg P, Desman G, Van Besien K, Laurence J. Hematopoietic transplant‐associated microangiopathy: case report and review of diagnosis and treatment. Clin Adv Hematol Oncol 2014; 12:565–73. [PubMed] [Google Scholar]
- 14. Laskin BL, Maisel J, Goebel J et al Renal arteriolar C4d deposition: a novel characteristic of hematopoietic stem cell transplantation‐associated thrombotic microangiopathy. Transplantation 2013; 96:217–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Garcia‐Laorden MI, Hernandez‐Brito E, Munoz‐Almagro C et al Should MASP‐2 deficiency be considered a primary immunodeficiency? Relevance of the lectin pathway. J Clin Immunol 2020; 40:203–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stefanescu R, Bassett D, Modarresi R, Santiago F, Fakruddin M, Laurence J. Synergistic interactions between interferon‐γ and TRAIL modulate c‐FLIP in endothelial cells, mediating their lineage‐specific sensitivity to thrombotic thrombocytopenic purpura plasma‐associated apoptosis. Blood 2008; 112:340–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Laurence J, Haller H, Mannucci PM, Nangaku M, Praga M, de Cordoba SR. Atypical hemolytic uremic syndrome (aHUS): essential aspects of an accurate diagnosis. Clin Adv Hematol Oncol 2016; 14(11 S1):2–15. [PubMed] [Google Scholar]
- 18. Cho BS, Yahng SA, Lee SE et al Validation of recently proposed consensus criteria for thrombotic microangiopathy after allogeneic hematopoietic stem‐cell transplantation. Transplantation 2010; 90:918–26. [DOI] [PubMed] [Google Scholar]
- 19. Li JH, Kirkiles‐Smith NC, McNiff JM, Pober JS. TRAIL induces apoptosis and inflammatory gene expression in human endothelial cells. J Immunol 2003; 171:1526–33. [DOI] [PubMed] [Google Scholar]
- 20. Rambaldi A, Khaled S, Smith M, Zecca M, Kwong YL, Claes K, et al Improved survival following OMS721 treatment of hematopoietic stem cell transplant‐associated thrombotic microangiopathy (HCT‐TMA). Eur Hematol Assoc, Stockholm, Sweden, June 14–17, 2018, Abst PF724. [Google Scholar]
- 21. Jodele S, Dandoy CE, Myers KC et al New approaches in the diagnosis, pathophysiology, and treatment of pediatric hematopoietic stem cell transplantation‐associated thrombotic microangiopathy. Transf Apheresis Sci 2016; 54:181–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Swierzko AS, Michalski M, Sokolowska A et al The role of complement activating collectins and associated serine proteases in patients with hematologic malignancies, receiving high‐dose chemotherapy, and autologous hematopoietic stem cell transplantations (auto‐HSCT). Front Immunol 2018; 9:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zehnder A, Fisch U, Hirt A et al Prognosis in pediatric hematologic malignancies is associated with serum concentration of mannose‐binding lectin‐associated serine protease‐2 (MASP‐2). Pediatr Blood Cancer 2009; 53:53–7. [DOI] [PubMed] [Google Scholar]
- 24. Zhang M, Hou Y, Cavusoglu E et al MASP‐2 activation is involved in ischemia‐related necrotic myocardial injury in humans. Int J Cardiol 2013; 166:499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Collard CD, Vakeva A, Morissey MA et al Complement activation after oxidative stress. Role of the lectin complement pathway. Am J Pathol 2000; 156:1549–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Clark JE, Dudler T, Marber MS, Schwaeble W. Cardioprotection by an anti‐MASP‐2 antibody in a murine model of myocardial infarction. Open Heart 2018; 5:e000652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Larsen JB, Hvas CL, Hvas AM. The lectin pathway in thrombotic conditions – a systematic review. Thromb Hemost 2018; 118:1141–66. [DOI] [PubMed] [Google Scholar]
- 28. Stahl A‐I, Vaziri‐Sani F, Heinen S et al Factor H dysfunction in patients with atypical hemolytic uremic syndrome contributes to complement deposition on platelets and their activation. Blood 2008; 111:5307–15. [DOI] [PubMed] [Google Scholar]
- 29. Galbusera M, Noris M, Gastoldi S et al An ex vivo test of complement activation on endothelium for individualized eculizumab therapy in hemolytic uremic syndrome. Am J Kid Dis 2019; 74:56–72. [DOI] [PubMed] [Google Scholar]
- 30. Zecca M, Comoli P, Mina T et al Resolution of acute kidney injury secondary to TA‐TMA by the anti‐MASP‐2 monoclonal antibody OMS721 in a pediatric HSCT recipient [Abstract EBMT 17]. Eur Soc Blood Marrow Transplant; Marseille, France, 26–29 March, 2017. [Google Scholar]
- 31. Garcia‐Laorden MI, Hernandez‐Brito E, Munoz‐Almagro C et al Should MASP‐2 deficiency be considered a primary immunodeficiency? Relevance of the lectin pathway. J Clin Immunol 2020; 40:203–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Magro C, Mulvey JJ, Berlin D et al Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID‐19 infection: a report of five cases. Transl Res 2020; 220:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bohl S, Kuchenbauer F, von Harsdorf S et al Thrombotic microangiopathy after allogeneic stem cell transplantation: a comparison of eculizumab therapy and conventional therapy. Biol Blood Marrow Transplant 2017; 23:2172–7. [DOI] [PubMed] [Google Scholar]
- 34. Schmidt CQ, Verschoor A. Complement and coagulation: so close, yet so far. Blood 2017; 130:2581–2. [DOI] [PubMed] [Google Scholar]
- 35. Subramaniam S, Jurk K, Hobohm L et al Distinct contributions of complement factors to platelet activation and fibrin formation in venous thrombus development. Blood 2017; 129:2291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Speth C, Rambach G, Wurzner R et al Complement and platelets: mutual interference in the immune network. Mol Immunol 2015; 67:108–18. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analysed for the purposed of this study are included in this published article.