

Abstract
The COVID‐2019 pandemic is the most severe acute public health threat of the twenty‐first century. To properly address this crisis with both robust testing and novel treatments, we require a deep understanding of the life cycle of the causative agent, the SARS‐CoV‐2 coronavirus. Here, we examine the architecture and self‐assembly properties of the SARS‐CoV‐2 nucleocapsid protein, which packages viral RNA into new virions. We determined a 1.4 Å resolution crystal structure of this protein's N2b domain, revealing a compact, intertwined dimer similar to that of related coronaviruses including SARS‐CoV. While the N2b domain forms a dimer in solution, addition of the C‐terminal spacer B/N3 domain mediates formation of a homotetramer. Using hydrogen‐deuterium exchange mass spectrometry, we find evidence that at least part of this putatively disordered domain is structured, potentially forming an α‐helix that self‐associates and cooperates with the N2b domain to mediate tetramer formation. Finally, we map the locations of amino acid substitutions in the N protein from over 38,000 SARS‐CoV‐2 genome sequences. We find that these substitutions are strongly clustered in the protein's N2a linker domain, and that substitutions within the N1b and N2b domains cluster away from their functional RNA binding and dimerization interfaces. Overall, this work reveals the architecture and self‐assembly properties of a key protein in the SARS‐CoV‐2 life cycle, with implications for both drug design and antibody‐based testing.
Keywords: coronavirus, COVID‐19, crystal structure, nucleocapsid, SARS‐CoV‐2
Short abstract
1. INTRODUCTION
Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) 1 , 2 is the third coronavirus to cross from an animal reservoir to infect humans in the twenty‐first century, after SARS‐CoV 3 , 4 and Middle‐East respiratory syndrome (MERS) coronavirus. 5 Isolation and sequencing of SARS‐CoV‐2 were reported in January 2020, and the virus was found to be highly related to SARS and share a probable origin in bats. 2 , 6 Since its emergence in December 2019 in Wuhan, China, the virus has infected over 10.5 million people and caused more than 500,000 deaths as of July 1, 2020 (https://coronavirus.jhu.edu). The high infectivity of SARS‐CoV‐2 and the worldwide spread of this ongoing outbreak highlight the urgent need for public health measures and therapeutics to limit new infections. Moreover, the severity of the atypical pneumonia caused by SARS‐CoV‐2 (COVID‐2019), often requiring multiweek hospital stays and the use of invasive ventilators, 7 , 8 , 9 highlights the need for therapeutics to lessen the severity of individual infections.
Current therapeutic strategies against SARS‐CoV‐2 target major points in the life cycle of the virus. The antiviral remdesivir, first developed against Ebola virus, 10 , 11 inhibits the viral RNA‐dependent RNA polymerases of a range of coronaviruses including SARS‐CoV‐2 12 , 13 , 14 and has shown promise against SARS‐CoV‐2 in small‐scale trials in both primates and humans. 15 , 16 Another target is the viral protease (Mpro/3CLpro), which is required to process viral polyproteins into their active forms. 17 Finally, the transmembrane spike (S) glycoprotein mediates binding to host cells through the angiotensin converting enzyme 2 and transmembrane protease, serine 2 proteins, and mediates fusion of the viral and host cell membranes. 18 , 19 , 20 , 21 As the most prominent surface component of the virus, the spike protein is the major target of antibodies in patients, and is the focus of several current efforts at SARS‐CoV‐2 vaccine development. Initial trials using antibody‐containing plasma of convalescent COVID‐19 patients has also shown promise in lessening the severity of the disease. 22
While the above efforts target viral entry, RNA synthesis, and protein processing, there has so far been less emphasis on other steps in the viral life cycle. One critical step in coronavirus replication is the assembly of the viral genomic RNA and nucleocapsid (N) protein into a ribonucleoprotein (RNP) complex, which interacts with the membrane (M) protein and is packaged into virions. Electron microscopy analysis of related betacoronaviruses has suggested that the RNP complex adopts a helical filament structure, 23 , 24 , 25 , 26 , 27 , 28 but recent cryoelectron tomography analysis of intact SARS‐CoV‐2 virions has revealed a beads‐on‐a‐string like arrangement of globular RNP complexes that sometimes assemble into stacks resembling helical filaments. 29 Despite its location within the viral particle rather than on its surface, patients infected with SARS‐CoV‐2 show higher and earlier antibody responses to the nucleocapsid protein than to the surface spike protein. 30 , 31 As such, a better understanding of the SARS‐CoV‐2 N protein's structure, and structural differences between it and N proteins of related coronaviruses including SARS‐CoV, may aid the development of sensitive and specific immunological tests.
Coronavirus N proteins possess a shared domain structure with an N‐terminal RNA‐binding domain and a C‐terminal domain responsible for dimerization. The assembly of the N protein into higher‐order RNP complexes is not well understood, but likely involves cooperative interactions between the dimerization domain and other regions of the protein, plus the bound RNA. 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 , 40 Here, we present a high‐resolution structure of the SARS‐CoV‐2 N dimerization domain, revealing an intertwined dimer similar to that of related betacoronaviruses. We also analyze the self‐assembly properties of the SARS‐CoV‐2 N protein, and show that higher‐order assembly requires both the dimerization domain and the extended, disordered C‐terminus of the protein. Together with other work revealing the structure and RNA‐binding properties of the nucleocapsid N‐terminal domain, these results lay the groundwork for a comprehensive understanding of SARS‐CoV‐2 nucleocapsid assembly and architecture.
2. RESULTS
2.1. Structure of the SARS‐CoV‐2 nucleocapsid dimerization domain
Betacoronavirus nucleocapsid (N) proteins share a common overall domain structure, with ordered RNA‐binding (N1b or N‐terminal domain/NTD) and dimerization (N2b or C‐terminal domain/CTD) domains separated by short regions with high predicted disorder (N1a, N2a, and spacer B/N3; Figure 1a). Self‐association of the full‐length SARS‐CoV N protein and the isolated C‐terminal region (domains N2b plus spacer B/N3; residues 210–422) was first demonstrated by yeast two‐hybrid analysis, 32 and the purified full‐length protein was shown to self‐associate into predominantly dimers in solution. 33 The structures of the N2b domain of SARS‐CoV and several related coronaviruses confirmed the obligate homodimeric structure of this domain, 34 , 35 , 36 , 37 , 38 , 39 , 40 and other work showed that the region C‐terminal to this domain mediates further self‐association into tetramer, hexamer, and potentially higher oligomeric forms. 41 , 42 , 43 Other studies have suggested that the protein's N‐terminal region, including the RNA‐binding N1b domain, can also self‐associate, 44 , 45 highlighting the possibility that assembly of full‐length N into helical filaments is mediated by cooperative interactions among several interfaces.
FIGURE 1.
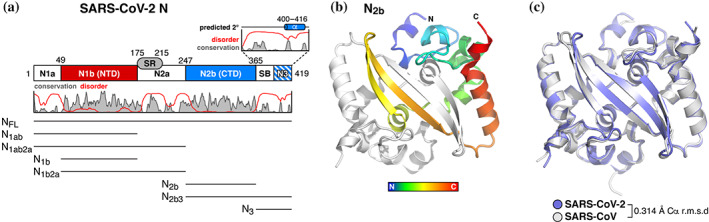
Structure of the severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) nucleocapsid dimerization domain. (a) Domain structure of the SARS‐CoV‐2 nucleocapsid protein, as defined previously, 46 , 47 with plot showing the Jalview alignment conservation score (three‐point smoothed; gray) 61 and DISOPRED3 disorder propensity (red) 62 for nine related coronavirus N proteins (SARS‐CoV, SARS‐CoV‐2, Middle‐East respiratory syndrome [MERS]‐CoV, HCoV‐OC43, HCoV‐HKU1, HCoV‐NL63, and HCoV‐229E, infectious bronchitis virus [IBV], and Murine Hepatitis virus [MHV]). SR, serine/arginine rich domain; SB, spacer B. The boundary between SB and N3 is not well defined due to low identity between SARS‐CoV/SARS‐CoV‐2 and MHV N proteins. 47 All purified truncations are noted at bottom. (b) Top‐down view of the SARS‐CoV‐2 N2b dimer, with one monomer colored as a rainbow (N‐terminus blue, C‐terminus red) and the other colored white. See Figure S1a for comparison with other structures of this domain. (c) Structural overlay of the SARS‐CoV‐2 N2b dimer (blue) and the equivalent domain of SARS‐CoV‐N (PDB ID 2CJR) 34
To characterize the structure and self‐assembly properties of the SARS‐CoV‐2 nucleocapsid, we first cloned and purified the protein's N2b dimerization domain (N2b; residues 247–364). 46 , 47 We crystallized and determined two high‐resolution crystal structures of N2b; a 1.45 Å resolution structure of His6‐tagged N2b at pH 8.5, and a 1.42 Å resolution structure of N2b after His6‐tag cleavage, at pH 4.5 (see Section 4 and Table S1). These structures reveal a compact, tightly intertwined dimer with a central four‐stranded β‐sheet comprising the bulk of the dimer interface (Figure 1b). This interface is composed of two β‐strands and a short α‐helix from each protomer that extend toward the opposite protomer and pack against its hydrophobic core. The asymmetric units of both structures contain two N2b dimers, giving four crystallographically independent views of the N2b dimer. These four dimers differ only slightly, showing overall Cα r.m.s.d values of 0.15–0.19 Å and with most variation arising from loop regions (Figure S1a). Our structures also overlay closely with four other recently deposited structures of the SARS‐CoV‐2 N2b domain (PDB IDs 6WJI, 6YUN, 6ZCO, and 7C22; all unpublished). One of these structures (PDB ID 7C22) adopts the same space group and unit cell parameters as our structure of untagged N2b. Including all of these structures, there are now nine independent crystallographic views of the SARS‐CoV‐2 N2b domain dimer (17 total protomers; the 6ZCO dimer is assembled from crystal symmetry) in five crystal forms at pH 4.5, 7.5, 7.8, and 8.5 (crystallization pH for 6ZCO is not reported). All of these structures overlay closely, with an overall Cα r.m.s.d of 0.15–0.31 Å (Figure S1a).
The structure of N2b closely resembles that of related coronaviruses, including SARS‐CoV, infectious bronchitis virus, MERS‐CoV, and HCoV‐NL63. 34 , 35 , 36 , 37 , 38 , 39 The structure is particularly similar to that of SARS‐CoV, with which the N2b domain shares 96% sequence identity; only five residues differ between these proteins' N2b domains (SARS‐CoV Gln268 ➔ SARS‐CoV‐2 A267, D291 ➔ E290, H335 ➔ Thr334, Gln346 ➔ Asn345, and Asn350 ➔ Gln349), and the structures are correspondingly similar with an overall Cα r.m.s.d of 0.314 Å across the N2b dimer (Figure 1c).
A crystal structure of the SARS‐CoV N protein revealed a helical assembly of N2b domain dimers that was proposed as a possible structure for the observed helical nucleocapsid filaments in virions. 34 We therefore examined the packing of N2b domain dimers in the six crystal structures of this domain, five of which show distinct space groups and unit cell parameters. We identified two dimer–dimer packing modes that appear in multiple crystal forms, with packing mode 1 appearing in five structures, and packing mode 2 appearing in four (Figure S1b). Neither of these packing modes would result in the assembly of a helical filament if repeated, nor do the dimer–dimer interfaces strongly correlate with conserved surfaces on the N2b domain. This evidence, combined with our finding that N2b forms solely dimers in solution (see below), suggests that packing of N2b domain dimers does not underlie higher‐order assembly of SARS‐CoV‐2 N protein filaments.
2.2. N protein variation in SARS‐CoV‐2 patient samples
Since the first genome sequence of SARS‐CoV‐2 was reported in January 2020, 2 , 6 over 38,000 full genomic sequences have been deposited in public databases (as of June 3, 2020). To examine the variability of the N protein in these sequences, we downloaded a comprehensive list of reported mutations within the SARS‐CoV‐2 N gene in a set of 38,318 genome sequences from the China National Center for Bioinformation, 2019 Novel Coronavirus Resource. Among these sequences, there are 10,979 instances of amino acid substitutions spread across 250 of the 419 amino acids of the N protein (Figure 2a, Table S2). While many of these substitutions arise only once in our dataset and may therefore reflect errors in sequencing or sequence assembly, most likely reflect true variation among circulating strains of SARS‐CoV‐2. As a whole, the reported substitutions are enriched in the three intrinsically disordered domains (N1a, N2a, and spacer B/N3), with a particularly high density of substitutions in the serine/arginine‐rich subdomain of N2a (SR in Figure 2a). The most common substitutions are R203K and G204R, which occur together as the result of a common trinucleotide substitution in genomic positions 28,881–28,883, from GGG to AAC (~4,100 of the 38,318 sequences in our dataset; Figure S2a,b). While positions 203 and 204 accounted for over two‐thirds of the total individual amino acid substitutions in this dataset, the N2a domain shows a strong enrichment of mutations even when these positions are not considered (Figure 2a). In contrast to the enrichment of missense mutations in the N2a domain, synonymous mutations were distributed relatively equally throughout the protein (Figure S2c, Table S2). Thus, these data suggest that the N2a domain is uniquely tolerant of mutations, in keeping with its likely structural role as a disordered linker between the RNA‐binding N1b domain and the N2b dimerization domain.
FIGURE 2.
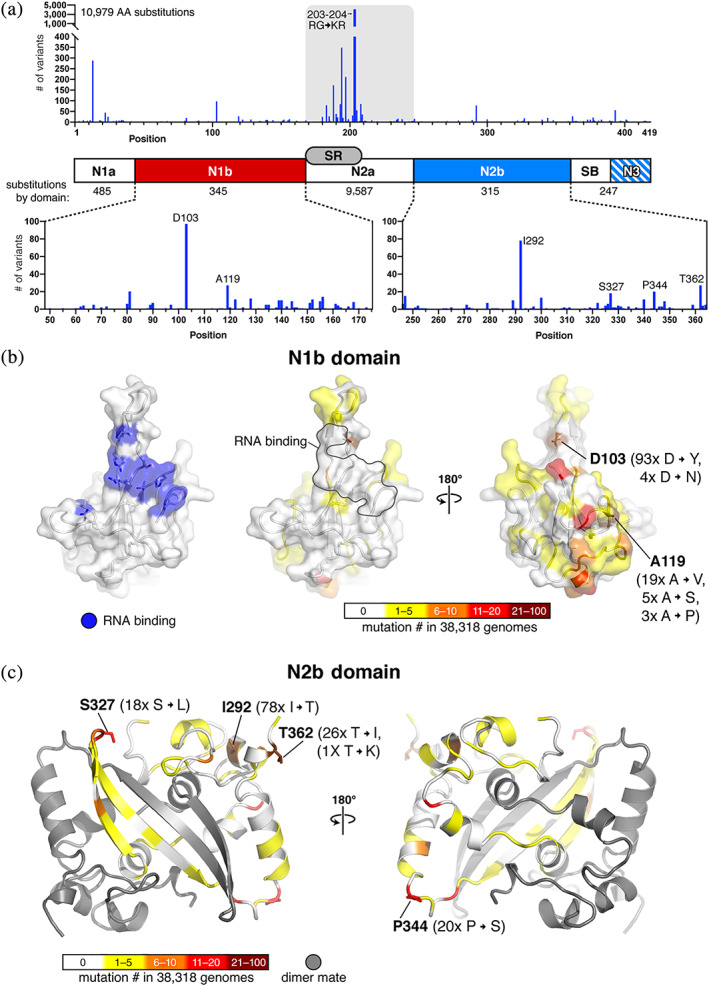
N protein variability in severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) patient sequences. (a) Top: Plot showing the number of observed amino acid variants at each position in the N gene in 38,318 SARS‐CoV‐2 genomes (details in Table S2). The most highly‐mutated positions are R203 and G204, which are each mutated more than 4,000 times due to a prevalent trinucleotide substitution (Figure S2a,b). Bottom: Plots showing amino acid variants in the N1b and N2b domains. (b) Surface views of the N protein N1b domain (PDB ID 6VYO; Center for Structural Genomics of Infectious Diseases [CSGID], unpublished). At left, blue indicates RNA‐binding residues identified by NMR peak shifts (A50, T57, H59, R92, I94, S105, R107, R149, and Y172). 49 At right, two views colored by the number of variants at each position observed in a set of 38,318 SARS‐CoV‐2 genomes. The two most frequently mutated residues are shown in stick view and labeled. Only one mutation (A50E, observed in one sequence) overlaps the putative RNA binding surface. (c) Cartoon view of the N protein N2b domain, with one monomer colored gray and the other colored by the number of variants at each position observed in a set of 38,318 SARS‐CoV‐2 genomes. The four most frequently mutated residues are shown in stick view and labeled
While the majority of N protein mutations are in the N2a domain, we nonetheless identified 345 instances of amino acid variants in the RNA‐binding N1b domain, and 315 instances in the N2b domain. We mapped these onto high‐resolution structures of both domains (Figure 2b,c). Two high‐resolution crystal structures of the SARS‐CoV‐2 N1b domain have been determined (PDB ID 6M3M and 6VYO), 48 and a recent NMR study determined a solution structure of the domain and defined its likely RNA binding surface (Figure 2b). 49 In keeping with its functional importance, the identified RNA binding surface shows only a single mutation in this dataset (Figure 2b; middle panel). In the N2b domain, most mutations occur on surface residues, particularly in loop regions, while the functionally important dimer interface is largely invariant (Figure 2c).
Finally, the 38,318 SARS‐CoV‐2 genome sequences contain nine sequences with reported nonsense/premature stop codons in the N protein. Two of these are located at position 256 within the N2b domain, while the remaining seven are located in the spacer B/N3 region between positions 372–418 (Table S2).
2.3. Self‐association of the SARS‐CoV‐2 N protein
Our structures of the SARS‐CoV‐2 N protein N2b domain reveal that, as in related coronaviruses, this domain mediates homodimer formation. We next systematically investigated the molecular basis for higher‐order self‐assembly of the SARS‐CoV‐2 nucleocapsid. We first purified the full‐length N protein (NFL) for analysis of its oligomeric state. While our initial attempts at purification yielded large aggregates significantly contaminated with nucleic acid (Figure S3a), purification of the protein in high‐salt buffer (1 M NaCl) and in the presence of both DNase and RNase yielded pure NFL (Figure S3b). Size exclusion chromatography coupled to multiangle light scattering (SEC‐MALS) of purified NFL revealed a heterogeneous population that is predominantly homotetrameric (Figure 3a).
FIGURE 3.
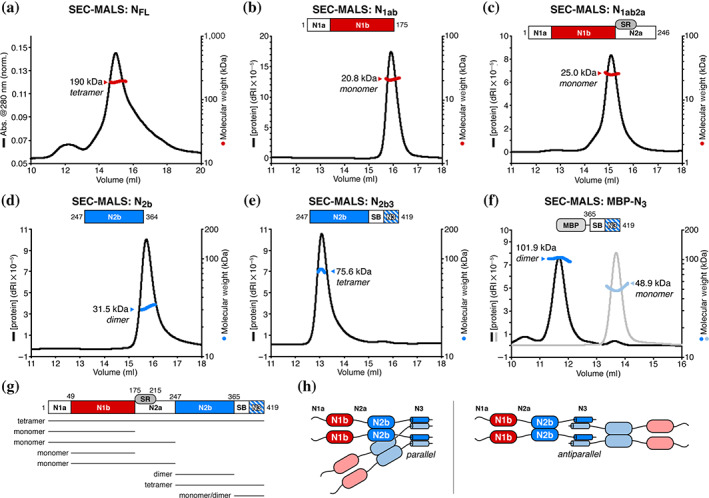
The C‐terminus of N mediates tetramer formation. (a) Size exclusion chromatography (Superose 6 Increase 10/300 GL; void volume = 8.4 ml, total volume = 20.5 ml) coupled to multiangle light scattering (SEC‐MALS) analysis of full‐length severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) N. The measured MW of 190.0 kDa closely matches that of a tetramer (182.5 kDa). See Figure S3b for SDS‐PAGE analysis of all purified proteins. (b) SEC‐MALS (Superdex 200 Increase 10/300 GL; void volume = 7.3 ml, total volume = 20.6 ml; used for Panels (b–f)) analysis of SARS‐CoV‐2 N1ab (residues 2–175). The measured MW of 20.8 kDa closely matches that of a monomer (18.9 kDa). dRI, differential refractive index. (c) SEC‐MALS analysis of SARS‐CoV‐2 N1ab2a (residues 2–246). The measured MW of 25.0 kDa is slightly less than that of a monomer (26.2 kDa), reflecting partial proteolysis within the N2a domain (Figure S3b). (d) SEC‐MALS analysis of SARS‐CoV‐2 N2b. The measured MW (31.5 kDa) closely matches that of a homodimer (26.5 kDa). (e) SEC‐MALS analysis of SARS‐CoV‐2 N2b3. The measured MW (75.6 kDa) closely matches that of a homotetramer (77.4 kDa). (f) SEC‐MALS analysis of maltose binding protein (MBP)‐SARS‐CoV‐2 N3 (“Peak 1” black/dark blue; “Peak 2” gray/light blue) The measured MW of Peak 1 (101.9 kDa) and Peak 2 (48.9 kDa) closely match those of a homodimer (101.7 kDa) and a monomer (50.9 kDa). The small peak at 10.5 ml suggests higher‐order self‐assembly. (g) Schematic summary of size exclusion and SEC‐MALS results on N protein constructs. See Figure S3c,d for SEC‐MALS analysis of N1b (residues 49–174) and N1b2a (residues 49–246). (h) Possible configurations of a SARS‐CoV‐2 N protein tetramer. Dimerization is mediated by the N2b domain, and these dimers self‐associate through the N3 region to form homotetramers. Left: Parallel arrangement of the putative N3 domain α‐helices; Right: antiparallel arrangement
To determine the molecular basis for homotetramer assembly, we purified a series of truncation constructs encompassing the ordered N1b and N2b domains and their associated linker domains (N1a, N2a, and spacer B/N3; Figure 1a). We characterized four truncations encompassing the protein's N‐terminal regions, including N1ab (residues 2–175), N1b (residues 49–175), N1ab2a (residues 2–246), and N1b2a (residues 49–246). All four of these truncations are monomeric in solution as determined by SEC‐MALS (Figures 3b,c and S3c,d). We next analyzed N2b, which forms a homodimer in our crystal structures. As expected, N2b is dimeric in solution (Figure 3d).
Finally, we analyzed the contribution of the C‐terminal spacer B/N3 region to N protein self‐assembly. Prior work with the Murine Hepatitis Virus N protein showed that this region can, on its own, incorporate into nucleocapsid structures that lack the associated Membrane (M) protein, suggesting that the region mediates a homotypic interaction between N proteins. 47 Other work with SARS‐CoV and HCoV‐229E N proteins also found that the C‐terminal spacer B/N3 region is required for higher‐order assembly of tetramers and larger oligomers. 41 , 42 , 43 We purified a construct encoding N2b and the spacer B/N3 region (N2b3, residues 247–419) and found that it forms a homotetramer (Figure 3e). We also analyzed self‐assembly of the spacer B/N3 region on its own by performing SEC‐MALS analysis on this isolated region (N3, residues 365–419) fused to a His6‐maltose binding protein (MBP) tag. Initial purification of His6‐MBP‐N3 yielded two peaks on the final size exclusion column, which we separately pooled and analyzed by SEC‐MALS. We found that these two peaks correspond to a monomer and a dimer, respectively (Figure 3f). The pooled dimer population also showed a small population of potentially higher‐order oligomers (Figure 3f). Together, these data suggest that assembly of betacoronavirus N protein filaments likely proceeds through at least three steps, each mediated by different oligomerization interfaces: (a) dimerization mediated by the N2b domain; (b) tetramerization mediated by the spacer B/N3 region (Figure 3g,h); and (c) oligomer/filament assembly mediated by cooperative RNA binding and potential higher‐order self‐association of N homotetramers.
To gain structural insight into how the spacer B/N3 region mediates N protein self‐association, we performed hydrogen‐deuterium exchange mass spectrometry (HDX‐MS) on N2b and N2b3 (Figure 4). By probing the rate of exchange of amide hydrogen atoms with deuterium atoms in a D2O solvent, HDX‐MS provides information on the level of secondary structure and solvent accessibility across an entire protein. We found that H‐D exchange rates within N2b largely agreed with our crystal structure: regions in β‐strands or α‐helices showed low exchange rates consistent with high order, while loop regions showed increased exchange rates consistent with their likely flexibility (Figure 4a,c,d).
FIGURE 4.
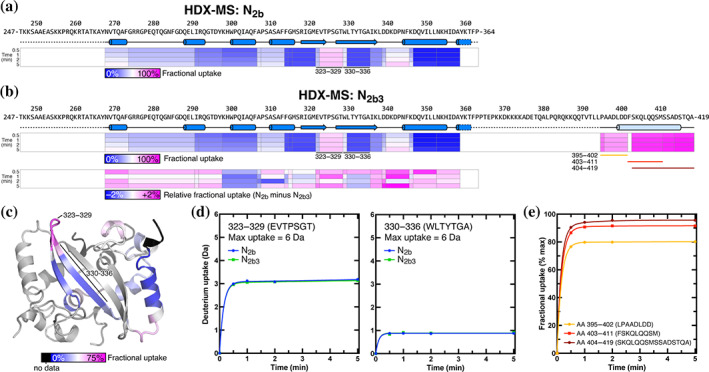
Hydrogen‐deuterium exchange mass spectrometry (HDX‐MS) analysis of N2b and N2b3. (a) Schematic showing the N2b sequence and structure, plus protein regions detected by HDX‐MS. Each peptide is colored by its fractional deuterium uptake during the course of the experiment (blue–white–magenta = 0–100% fractional uptake). (b) Schematic showing the N2b3 sequence and inferred structure (the α‐helix spanning residues 400–416 is predicted by PSI‐PRED), plus protein regions detected by HDX‐MS. Two sets of exchange rates are shown: fractional deuterium uptake in N2b3 (upper box) colored as in Panel (a), and relative uptake comparing N2b and N2b3 (lower box). (c) Structure of the N2b dimer, with one monomer colored by fractional deuterium uptake (blue–white–magenta = 0–75% fractional uptake). (d) Uptake plots for two peptides within the ordered N2b domain, with uptake in N2b indicated in blue and uptake in N2b3 indicated in green. The peptide covering residues 323–329 (located within a loop) is relatively exposed, while the peptide covering residues 330–336 (within a β‐strand) is strongly protected from H‐D exchange. (e) Uptake plots for three peptides in the C‐terminal region of N2b3, plotted by fractional deuterium uptake. Peptides covering residues 395–402 (yellow) and 403–411 (red) show more protection than residues 404–419, suggesting that this region is partially structured. See Figure S4a for each peptide plotted by absolute deuterium uptake
Compared to N2b, N2b3 contains an additional 56 amino acids (residues 365–419). While residues 360–394 were not detected in our HDX‐MS analysis, we detected spectra for seven overlapping peptides spanning residues 395–419 at the protein's extreme C‐terminus (Figure 4b). While all of these peptides exhibited higher levels of exchange than the ordered N2b domain, peptides spanning the N‐terminal part of this region (particularly residues 395–402) showed a degree of protection compared to those at the extreme C‐terminus (residues 404–419; Figure 4e). This finding suggests that at least part of the spacer B/N3 domain possesses secondary structure and may mediate N2b3 tetramer formation. Indeed, a recent molecular dynamics simulation of the N3 domain suggests the existence of an α‐helix spanning residues 400–411 in this domain, 50 and our own analysis using the PSI‐PRED server 51 suggests an α‐helix spanning residues 400–416 (Figure 1a).
We next compared HDX‐MS exchange rates of N2b versus N2b3 for peptides within the N2b domain. We reasoned that if the C‐terminus of N2b3 mediates tetramer formation, it may do so by docking against a surface in the N2b domain, which may be detectable by reduced deuterium uptake in the involved region. Contrary to this expectation, we found that the H‐D exchange rates within the N2b domain were nearly identical between the two constructs, varying at most 2% in fractional deuterium uptake in individual peptides (Figure 4b,d). While these data do not rule out the possibility that the spacer B/N3 region docks against N2b, they nonetheless support our SEC‐MALS data showing that spacer B/N3 independently self‐associates to mediate N protein tetramer formation. We attempted to test this idea by measuring the association of His6‐MBP‐N3 with N2b, N2b3, and NFL in a pull‐down assay (Figure S4b). We were unable to detect binding of His6‐MBP‐N3 to any of these three constructs. As both N2b3 and NFL likely exist as pre‐formed tetramers, their failure to interact with additional His6‐MBP‐N3 is not surprising. The inability of N2b to interact with His6‐MBP‐N3, however, is consistent with the idea that the spacer B/N3 domain self‐interacts rather than binding the N2b domain.
3. DISCUSSION
Given the severity of the ongoing COVID‐19 pandemic, a deep understanding of the SARS‐CoV‐2 life cycle is urgently needed. Here, we examine the architecture and self‐assembly properties of the SARS‐CoV‐2 nucleocapsid protein, a key player in viral replication responsible for packaging viral RNA into new virions. Through two high‐resolution crystal structures, we show that this protein's N2b domain forms a compact, strand‐swapped dimer similar to that of related betacoronaviruses. While the N2b domain mediates dimer formation, we find that addition of the C‐terminal spacer B/N3 domain mediates formation of a robust homotetramer. We envision two possible modes of N protein tetramer assembly based on either parallel or antiparallel arrangement of the putative α‐helices in the N3 domain (Figure 3h). How these tetramers interact with viral RNA and self‐assemble into either helical filaments or the more recently observed globular viral RNP complexes 29 will require higher‐level reconstitution and/or high‐resolution analysis of the internal structure of SARS‐CoV‐2 virions.
Given the importance of nucleocapsid‐mediated RNA packaging to the viral life cycle, small molecules that inhibit nucleocapsid self‐assembly or mediate aberrant assembly may be effective at reducing the severity of infections and the infectivity of patients. The high resolution of our crystal structures will enable their use in virtual screening efforts to identify such nucleocapsid assembly modulators. Given the high conservation of the N2b domain in betacoronaviruses, these assembly modulators may also be effective at countering related viruses including SARS‐CoV. As SARS‐CoV‐2 is unlikely to be the last betacoronavirus to jump from an animal reservoir to humans, the availability of such treatments could drastically alter the course of future epidemics.
The SARS‐CoV‐2 genome has been subject to unprecedented scrutiny, with over 38,000 individual genome sequences deposited in public databases as of early June 2020. We used this set of genome sequences to identify over 10,000 instances of amino acid substitutions in the N protein, and showed that these variants are strongly clustered in the protein's N2a linker domain. The ~650 substitutions we identified in the N1b and N2b domains were clustered away from these domains' RNA binding and dimerization interfaces, reflecting the functional importance of these surfaces.
Given the early and strong antibody responses to the nucleocapsid displayed by SARS‐CoV‐2 infected patients, the distribution of mutations within this protein should be carefully considered as antibody‐based tests are developed. The high variability of the N2a domain means that individual patient antibodies targeting this domain may not be reliably detected with tests using the reference N protein; especially if these antibodies recognize residues 203 and 204, which are mutated in a large fraction of infections. At the same time, patient antibodies targeting the conserved N1b and N2b domains may in fact cross‐react with nucleocapsids of related coronaviruses like SARS‐CoV. The availability of a panel of purified N protein constructs now makes it possible to systematically examine the epitopes of both patient‐derived and commercial anti‐nucleocapsid antibodies.
4. MATERIALS AND METHODS
4.1. Cloning and protein purification
SARS‐CoV‐2 N protein constructs (NFL [residues 2–419], N1ab [2–175], N1ab2a [2–246], N1b [49–175], N1b2a [49–246], N2b [247–364], N2b3 [247–419]) were amplified by PCR from the IDT 2019‐nCoV N positive control plasmid (IDT cat. # 10006625; NCBI RefSeq YP_009724397) and inserted by ligation‐independent cloning into UC Berkeley Macrolab vector 2B‐T (AmpR, N‐terminal His6‐fusion; Addgene #29666) for expression in Escherichia coli. N3 (residues 365–419) was similarly inserted into UC Berkeley Macrolab vector 2C‐T (AmpR, N‐terminal His6‐MBP fusion; Addgene #29706). Plasmids were transformed into E. coli strain Rosetta 2(DE3) pLysS (Novagen), and grown in the presence of ampicillin and chloramphenical to an OD600 of 0.8 at 37°C, induced with 0.25 mM IPTG, then grown for a further 16 hr at 18°C prior to harvesting by centrifugation. Harvested cells were resuspended in buffer A (25 mM Tris–HCl pH 7.5, 5 mM MgCl2 10% glycerol, 5 mM β‐mercaptoethanol, 1 mM NaN3) plus 500 mM NaCl (1M NaCl for NFL) and 5 mM imidazole pH 8.0. For purification, cells were lysed by sonication, then clarified lysates were loaded onto a Ni2+ affinity column (Ni‐NTA Superflow; Qiagen), washed in buffer A plus 300 mM NaCl and 20 mM imidazole pH 8.0, and eluted in buffer A plus 300 mM NaCl and 400 mM imidazole. For cleavage of His6‐tags, proteins were buffer exchanged in centrifugal concentrators (Amicon Ultra, EMD Millipore) to buffer A plus 300 mM NaCl and 20 mM imidazole, then incubated 16 hr at 4°C with TEV protease. 52 Cleavage reactions were passed through a Ni2+ affinity column again to remove uncleaved protein, cleaved His6‐tags, and His6‐tagged TEV protease. Proteins were concentrated in centrifugal concentrators and purified by size‐exclusion chromatography (Superdex 200; GE Life Sciences) in gel filtration buffer (25 mM Tris–HCl pH 7.5, 300 mM NaCl, 5 mM MgCl2, 10% glycerol, 1 mM DTT). Purified proteins were concentrated and stored at 4°C for analysis.
4.2. Size exclusion chromatography coupled to multiangle light scattering
For SEC‐MALS, 100 μl purified proteins at 2–5 mg/ml were injected onto a Superdex 200 Increase 10/300 GL column or Superose 6 Increase 10/300 GL column (GE Life Sciences) in gel filtration buffer. Light scattering and refractive index profiles were collected by miniDAWN TREOS and Optilab T‐rEX detectors (Wyatt Technology), respectively, and molecular weight was calculated using ASTRA v. 6 software (Wyatt Technology).
4.3. Hydrogen‐deuterium exchange mass spectrometry
H‐D exchange experiments were conducted with a Waters Synapt G2S system. Total volume of 5 μl samples containing 10 μM protein in gel filtration buffer were mixed with 55 μl of the same buffer made with D2O for several deuteration times (0 s, 1 min, 2 min, 5 min, 10 min) at 15°C. The exchange was quenched for 2 min at 1°C with an equal volume of quench buffer (3 M guanidine HCl, 0.1% formic acid). Proteins were cleaved with pepsin and separated by reverse‐phase chromatography, then directed into a Waters SYNAPT G2s quadrupole time‐of‐flight mass spectrometer. Peptides were identified using PLGS version 2.5 (Waters, Inc.), deuterium uptake was calculated using DynamX version 2.0 (Waters Corp.), and uptake was corrected for back‐exchange using DECA. 53 Uptake plots were generated in Prism version 8.
4.4. Crystallization and structure determination
For crystallization of untagged N2b, protein (40 mg/ml) in crystallization buffer (25 mM Tris–HCl pH 7.5, 200 mM NaCl, 5 mM MgCl2, and 1 mM Tris(2‐carboxyethyl)phosphine) was mixed 1:1 with well solution containing 100 mM sodium acetate pH 4.5, 50 mM sodium/potassium tartrate, and 34% polyethylene glycol (PEG) 3350 at 20°C in hanging drop format. For crystallization of His6‐tagged N2b, protein (40 mg/ml) in crystallization buffer was mixed 1:1 with well solution containing 100 mM Tris–HCl pH 8.5, 50 mM ammonium sulfate, and 38% PEG 3350 at 20°C in hanging drop format. Both untagged and His6‐tagged N2b formed large shard‐like crystals, and were frozen in liquid nitrogen directly from the crystallization drop without further cryoprotection.
Diffraction data were collected at beamline 24ID‐E at the Advanced Photon Source. Diffraction datasets were processed with the RAPD pipeline (https://github.com/RAPD/RAPD/), which uses XDS 54 for indexing and data reduction, and the CCP4 programs AIMLESS 55 and TRUNCATE 56 for scaling and conversion to structure factors. The structure of untagged N2b was determined by molecular replacement in PHASER 57 using a dimer of the SARS‐CoV N2b domain (PDB ID 2GIB) 35 as a template. The resulting model was manually rebuilt in COOT 58 and refined in phenix.refine 59 using positional, isotropic B‐factor, and TLS (one group per chain) refinement. The structure of His6‐tagged N2b was determined by molecular replacement from the structure of untagged N2b, then manually rebuilt and refined as above. Data collection statistics, refinement statistics, and database accession numbers for diffraction data and final structures can be found in Table S1. All structural figures were generated with PyMOL version 2.3.
4.5. Nickel pull‐down
Nickel pull‐down assays were performed in buffer A plus 300 mM NaCl and 10 mM imidazole pH 8.0. Then, 10 μg bait (His6‐MBP‐N3) was mixed with equal weights of each prey protein in 50 μl total reaction volume and incubated on ice for 30 min (5 μl “load” sample removed). Total volume of 30 μl of a 50% slurry of Ni‐NTA Superflow beads (Qiagen) was added and the mixture was incubated with occasional mixing on ice for 30 min. Beads were washed three times with 1 ml buffer, then bound proteins were eluted with the addition of 30 μl buffer A plus 300 mM NaCl and 250 mM imidazole pH 8.0. Then, 10 μl of each eluate was analyzed by SDS‐PAGE alongside the load samples.
4.6. Bioinformatics
To examine variation in SARS‐CoV‐2 sequences, we downloaded a list of variants in the N gene in 38,318 genome sequences from China National Center for Bioinformation, 2019 Novel Coronavirus Resource (https://bigd.big.ac.cn/ncov?lang=en; downloaded June 3, 2020). We tabulated all mis‐sense and nonsense mutations, and graphed the number of amino acid variants at each codon in Prism version 8 (all variant frequencies are listed in Table S2). To examine the prevalence of the trinucleotide substitution at genome positions 28,881–28,883, we downloaded a set of 200 SARS‐CoV‐2 genomes from the NCBI Virus Resource: https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/virus?SeqType_s=Nucleotide&VirusLineage_ss=SARS‐CoV‐2%20taxid:2697049). We selected genomes with and without the substitution to show in Figure S2a. We used the NextStrain database 60 to visualize the prevalence of the N protein G204R mutation, which is diagnostic of the GGG ➔ AAC trinucleotide substitution in positions 28,881–28,883. To visualize SARS‐CoV‐2 clade identity, we used the URL “https://nextstrain.org/ncov/global?c=clade_membership&l=unrooted.” To color by N protein residue 204 identity, we used the URL “https://nextstrain.org/ncov/global?c=gt-N_204&l=unrooted” (screenshots taken July 2, 2020).
AUTHOR CONTRIBUTIONS
Qiaozhen Ye: Investigation; methodology. Alan West: Investigation. Steve Silletti: Investigation. Kevin Corbett: Conceptualization; investigation; methodology; project administration; visualization; writing‐original draft; writing‐review and editing.
Supporting information
Appendix S1: Supporting information
ACKNOWLEDGMENTS
The authors thank the staff of Advanced Photon Source NE‐CAT beamline 24ID‐E for assistance with diffraction data collection, E. Komives for advice on HDX‐MS interpretation, R. Lumpkin for assistance with the DECA software, and J. Pogliano, M. Daugherty, A. Schmidt, and members of the Corbett lab for helpful discussions. K. D. C. acknowledges generous institutional support from UC San Diego. The Waters Synapt HDX‐MS at the UCSD BPMS Facility is supported by NIH S10 OD016234. The Northeastern Collaborative Access Team beamlines at the Advanced Photon Source are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). The Eiger 16M detector on the 24‐ID‐E beam line is funded by a NIH‐ORIP HEI grant (S10OD021527). The Advanced Photon Source is a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE‐AC02‐06CH11357.
Ye Q, West AMV, Silletti S, Corbett KD. Architecture and self‐assembly of the SARS‐CoV‐2 nucleocapsid protein. Protein Science. 2020;29:1890–1901. 10.1002/pro.3909
Funding information Argonne National Laboratory, Grant/Award Number: DE‐AC02‐06CH11357; NIH‐ORIP HEI, Grant/Award Number: S10OD021527; National Institute of General Medical Sciences; National Institutes of Health, Grant/Award Numbers: P30 GM124165, S10 OD016234
REFERENCES
- 1. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382:727–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Drosten C, Günther S, Preiser W, et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348:1967–1976. [DOI] [PubMed] [Google Scholar]
- 4. Ksiazek TG, Erdman D, Goldsmith CS, et al. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003;348:1953–1966. [DOI] [PubMed] [Google Scholar]
- 5. Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus ADME, Fouchier RAM. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367:1814–1820. [DOI] [PubMed] [Google Scholar]
- 6. Zhou P, Lou YX, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Goyal P, Choi JJ, Pinheiro LC, et al. Clinical characteristics of Covid‐19 in New York City. N Engl J Med. 2020;382:2372–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guan W, Ni Z, Hu Y, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 2020;382:1708–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bialek S, Boundy E, Bowen V, et al. Severe outcomes among patients with coronavirus disease 2019 (COVID‐19)—United States, February 12–March 16, 2020. Morb Mortal Wkly Rep. 2020;69:343–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Warren TK, Jordan R, Lo MK, et al. Therapeutic efficacy of the small molecule GS‐5734 against Ebola virus in rhesus monkeys. Nature. 2016;531:381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Siegel D, Hui HC, Doerffler E, et al. Discovery and synthesis of a phosphoramidate prodrug of a pyrrolo[2,1‐f][triazin‐4‐amino] adenine C‐nucleoside (GS‐5734) for the treatment of Ebola and emerging viruses. J Med Chem. 2017;60:1648–1661. [DOI] [PubMed] [Google Scholar]
- 12. Yin W, Mao C, Luan X, et al. Structural basis for inhibition of the RNA‐dependent RNA polymerase from SARS‐CoV‐2 by remdesivir. Science. 2020;368:1499–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Agostini ML, Andres EL, Sims AC, et al. Coronavirus susceptibility to the antiviral remdesivir (GS‐5734) is mediated by the viral polymerase and the proofreading exoribonuclease. MBio. 2018;9:e00221–e00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gordon CJ, Tchesnokov EP, Feng JY, Porter DP, Götte M. The antiviral compound remdesivir potently inhibits RNA‐dependent RNA polymerase from Middle East respiratory syndrome coronavirus. J Biol Chem. 2020;295:4773–4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Grein J, Ohmagari N, Shin D, et al. Compassionate use of remdesivir for patients with severe Covid‐19. N Engl J Med. 2020;382:2327–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Williamson BN, Feldmann F, Schwarz B, et al. Clinical benefit of remdesivir in rhesus macaques infected with SARS‐CoV‐2. bioRxiv. 2020. 10.1038/s41586-020-2423-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang L, Lin D, Sun X, et al. Crystal structure of SARS‐CoV‐2 main protease provides a basis for design of improved α‐ketoamide inhibitors. Science. 2020;368:409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS‐CoV‐2 spike glycoprotein. Cell. 2020;181:281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wrapp D, Wang N, Corbett KS, et al. Cryo‐EM structure of the 2019‐nCoV spike in the prefusion conformation. Science. 2020;367:1260–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of SARS‐CoV‐2 by full‐length human ACE2. Science. 2020;367:1444–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hoffmann M, Kleine‐Weber H, Schroeder S, et al. SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Duan K, Liu B, Li C, et al. Effectiveness of convalescent plasma therapy in severe COVID‐19 patients. Proc Natl Acad Sci U S A. 2020;117:9490–9496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Davies HA, Dourmashkin RR, Macnaughton MR. Ribonucleoprotein of avian infectious bronchitis virus. J Gen Virol. 1981;53:67–74. [DOI] [PubMed] [Google Scholar]
- 24. Macnaughton MR, Davies HA, Nermut MV. Ribonucleoprotein‐like structures from coronavirus particles. J Gen Virol. 1978;39:545–549. [DOI] [PubMed] [Google Scholar]
- 25. Masters PS. The molecular biology of coronaviruses. Adv Virus Res. 2006;65:193–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. de Haan CAM, Rottier PJM. Molecular interactions in the assembly of coronaviruses. Adv Virus Res. 2005;64:165–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fehr AR, Perlman S. Coronaviruses: An overview of their replication and pathogenesis Coronaviruses: Methods and protocols. Volume 1282 New York, NY: Springer, 2015; p. 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bárcena M, Oostergetel GT, Bartelink W, et al. Cryo‐electron tomography of mouse hepatitis virus: Insights into the structure of the coronavirion. Proc Natl Acad Sci U S A. 2009;106:582–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Klein S, Cortese M, Winter SL, et al. SARS‐CoV‐2 structure and replication characterized by in situ cryo‐electron tomography. bioRxiv. 2020. 10.1101/2020.06.23.167064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hachim A, Kavian N, Cohen CA, et al. Beyond the spike: Identification of viral targets of the antibody response to SARS‐CoV‐2 in COVID‐19 patients. medRxiv. 2020. 10.1101/2020.04.30.20085670. [DOI] [Google Scholar]
- 31. Burbelo PD, Riedo FX, Morishima C, et al. Sensitivity in Detection of Antibodies to Nucleocapsid and Spike Proteins of Severe Acute Respiratory Syndrome Coronavirus 2 in Patients With Coronavirus Disease 2019. The Journal of Infectious Diseases. 2020;222(2):206–213. 10.1093/infdis/jiaa273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Surjit M, Liu B, Kumar P, Chow VTK, Lal SK. The nucleocapsid protein of the SARS coronavirus is capable of self‐association through a C‐terminal 209 amino acid interaction domain. Biochem Biophys Res Commun. 2004;317:1030–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Luo H, Ye F, Sun T, et al. In vitro biochemical and thermodynamic characterization of nucleocapsid protein of SARS. Biophys Chem. 2004;112:15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen CY, Chang C‐K, Chang YW, et al. Structure of the SARS coronavirus nucleocapsid protein RNA‐binding dimerization domain suggests a mechanism for helical packaging of viral RNA. J Mol Biol. 2007;368:1075–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yu IM, Oldham ML, Zhang J, Chen J. Crystal structure of the severe acute respiratory syndrome (SARS) coronavirus nucleocapsid protein dimerization domain reveals evolutionary linkage between Corona‐ and Arteriviridae. J Biol Chem. 2006;281:17134–17139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Takeda M, Chang C‐K, Ikeya T, et al. Solution structure of the C‐terminal dimerization domain of SARS coronavirus nucleocapsid protein solved by the SAIL‐NMR method. J Mol Biol. 2008;380:608–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jayaram H, Fan H, Bowman BR, et al. X‐Ray structures of the N‐ and C‐terminal domains of a coronavirus nucleocapsid protein: Implications for nucleocapsid formation. J Virol. 2006;80:6612–6620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. van Nguyen TH, Lichière J, Canard B, et al. Structure and oligomerization state of the C‐terminal region of the Middle East respiratory syndrome coronavirus nucleoprotein. Acta Crystallogr. 2019;D75:8–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Szelazek B, Kabala W, Kus K, et al. Structural characterization of human coronavirus NL63 N protein. J Virol. 2017;91:e02503–e02516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ma Y, Tong X, Xu X, Li X, Lou Z, Rao Z. Structures of the N‐ and C‐terminal domains of MHV‐A59 nucleocapsid protein corroborate a conserved RNA‐protein binding mechanism in coronavirus. Protein Cell. 2010;1:688–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Luo H, Chen J, Chen K, Shen X, Jiang H. Carboxyl terminus of severe acute respiratory syndrome coronavirus nucleocapsid protein: Self‐association analysis and nucleic acid binding characterization. Biochemistry. 2006;45:11827–11835. [DOI] [PubMed] [Google Scholar]
- 42. Chang C, Chen C‐MM, Chiang M, Hsu Y, Huang T. Transient oligomerization of the SARS‐CoV N protein—Implication for virus ribonucleoprotein packaging. PLoS One. 2013;8:e65045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lo Y‐S, Lin S‐Y, Wang S‐M, et al. Oligomerization of the carboxyl terminal domain of the human coronavirus 229E nucleocapsid protein. FEBS Lett. 2013;587:120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fan H, Ooi A, Tan YW, et al. The nucleocapsid protein of coronavirus infectious bronchitis virus: Crystal structure of its N‐terminal domain and multimerization properties. Structure. 2005;13:1859–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cong Y, Kriegenburg F, de Haan CAM, Reggiori F. Coronavirus nucleocapsid proteins assemble constitutively in high molecular oligomers. Sci Rep. 2017;7:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hurst KR, Ye R, Goebel SJ, Jayaraman P, Masters PS. An interaction between the nucleocapsid protein and a component of the replicase‐transcriptase complex is crucial for the infectivity of coronavirus genomic RNA. J Virol. 2010;84:10276–10288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hurst KR, Koetzner CA, Masters PS. Identification of in vivo‐interacting domains of the murine coronavirus nucleocapsid protein. J Virol. 2009;83:7221–7234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kang S, Yang M, Hong Z, et al. Crystal structure of SARS‐CoV‐2 nucleocapsid protein RNA binding domain reveals potential unique drug targeting sites. Acta Pharm Sin B. 2020. 10.1016/j.apsb.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dinesh DC, Chalupska D, Silhan J, Veverka V, Boura E. Structural basis of RNA recognition by the SARS‐CoV‐2 nucleocapsid phosphoprotein. bioRxiv. 2020. 10.1101/2020.04.02.022194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cubuk J, Alston JJ, Jeremías Incicco J, et al. The SARS‐CoV‐2 nucleocapsid protein is dynamic, disordered, and phase separates with RNA. bioRxiv. 2020. 10.1101/2020.06.17.158121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Buchan DWA, Minneci F, Nugent TCO, Bryson K, Jones DT. Scalable web services for the PSIPRED protein analysis workbench. Nucleic Acids Res. 2013;41:W349–W357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tropea JE, Cherry S, Waugh DS. Expression and purification of soluble His(6)‐tagged TEV protease. Methods Mol Biol. 2009;498:297–307. [DOI] [PubMed] [Google Scholar]
- 53. Lumpkin RJ, Komives EA. DECA, a comprehensive, automatic post‐processing program for HDX‐MS data. Mol Cell Proteomics. 2019;18:2516–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kabsch W. XDS. Acta Crystallogr. 2010;D66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Evans PR, Murshudov GN. How good are my data and what is the resolution? Acta Cryst. 2013;D69:1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Winn MD, Ballard CC, Cowtan KD, et al. Overview of the CCP4 suite and current developments. Acta Cryst. 2011;D67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of coot. Acta Cryst. 2010;D66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Afonine PV, Grosse‐Kunstleve RW, Echols N, et al. Towards automated crystallographic structure refinement with phenix.refine . Acta Crystallogr. 2012;D68:352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hadfield J, Megill C, Bell SM, et al. Nextstrain: Real‐time tracking of pathogen evolution. Bioinformatics. 2018;34:4121–4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Livingstone CD, Barton GJ. Protein sequence alignments: A strategy for the hierarchical analysis of residue conservation. Comput Appl Biosci. 1993;9:745–756. [DOI] [PubMed] [Google Scholar]
- 62. Jones DT, Cozzetto D. DISOPRED3: Precise disordered region predictions with annotated protein‐binding activity. Bioinformatics. 2015;31:857–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting information