

In the title Schiff base compound, the hydroxy group forms a intramolecular hydrogen bond to the imine N atom generating an S(6) ring motif. The 3-chlorobenzene ring is inclined to the phenol ring by 9.38 (11)°. The configuration about the C=N bond is E.
Keywords: crystal structure, 3-chloro-4-methylaniline, 2-hydroxy-5-methylbenzaldehyde, Schiff base
Abstract
The title compound, C15H14ClNO, was synthesized by condensation reaction of 2-hydroxy-5-methylbenzaldehyde and 3-chloro-4-methylaniline, and crystallizes in the monoclinic space group P21/c. The 3-chlorobenzene ring is inclined to the phenol ring by 9.38 (11)°. The configuration about the C=N bond is E and an intramolecular O—H⋯N hydrogen bond forms an S(6) ring motif. The Hirshfeld surface analysis of the crystal structure indicates that the most important contributions for the packing arrangement are from H⋯H (43.8%) and C⋯H/H⋯C (26.7%) interactions. The density functional theory (DFT) optimized structure at the B3LYP/ 6–311 G(d,p) level is compared with the experimentally determined molecular structure and the HOMO–LUMO energy gap is provided.
Chemical context
Schiff bases contain the azomethine moiety (–RCH=N–R′) and are prepared by condensation reactions between amines and active carbonyl compounds. Schiff bases are employed as catalyst carriers (Grigoras et al., 2001 ▸), thermo-stable materials (Vančo et al., 2004 ▸), metal–cation complexing agents and in biological systems (Taggi et al., 2002 ▸). Schiff bases show biological activities including antibacterial, antifungal, anticancer, antiviral and herbicidal activities (Desai et al., 2001 ▸; Singh & Dash, 1988 ▸; Karia & Parsania, 1999 ▸; Siddiqui et al., 2006 ▸). Moreover, Schiff base ligands are potentially capable of forming stable complexes by coordination of metal ions with their nitrogen atoms as donors (Ebrahimipour et al., 2012 ▸). They are important for their photochromic properties and have applications in various fields such as the measurement and control of radiation intensities in imaging systems, optical computers, electronics, optoelectronics and photonics (Iwan et al., 2007 ▸). The present work is a part of an ongoing structural study of Schiff bases and their utilization in the synthesis of quinoxaline derivatives (Faizi et al., 2018 ▸), fluorescence sensors (Faizi et al., 2016 ▸; Mukherjee et al., 2018 ▸; Kumar et al., 2017 ▸, 2018 ▸) and non-linear optical properties (Faizi et al., 2020 ▸). We report herein on the synthesis (from 2-hydroxy-5-methylbenzaldehyde and 3-chloro-4-methylaniline), crystal structure, Hirshfeld surface analysis and DFT computational calculations of the title compound, (I). The results of calculations by density functional theory (DFT) carried out at the B3LYP/6–311 G(d,p) level are compared with the experimentally determined molecular structure in the solid state.
Structural commentary
The molecular structure of the title compound (I) is shown in Fig. 1 ▸. An intramolecular O—H⋯N hydrogen bond is observed (Table 1 ▸ and Fig. 1 ▸). This is a relatively common feature in analogous imine–phenol compounds (see Database survey section). The imine group, which displays a C9—C8— N1—C5 torsion angle of −177.49 (18)°, contributes to the general non-planarity of the molecule. The chlorobenzene ring (C2–C7) is inclined by 9.38 (11)° to the phenol ring (C9–C14). The configuration of the C7=N1 bond of this Schiff base is E, and the intramolecular O1—H1⋯N1 hydrogen bond forms an S(6) ring motif (Fig. 1 ▸ a and Table 1 ▸). The C14—O1 distance [1.354 (2) Å] is close to normal values reported for single C—O bonds in phenols and salicylideneamines (Ozeryanskii et al., 2006 ▸). The N1—C8 bond is short at 1.281 (3) Å, indicating the existence of an imine bond, while the long C8—C9 bond [1.446 (3) Å] implies a single bond. All these data support the existence of the phenol–imine tautomer for (I) in its crystalline state. These features are similar to those observed in related 4-dimethylamino-N-salicylideneanilines (Wozniak et al., 1995 ▸; Pizzala et al., 2000 ▸). The C—N, C=N and C—C bond lengths are normal and close to the values observed in related structures (Faizi et al., 2017 ▸).
Figure 1.

The molecular structure of the title compound (I), showing the atom labelling and the intermolecular O—H⋯N hydrogen bond as a dashed line. Displacement ellipsoids are drawn at the 40% probability level.
Table 1. Hydrogen-bond geometry (Å, °).
Cg1 is the centroid of the C2–C7 ring.
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| O1—H1⋯N1 | 0.79 (4) | 1.89 (3) | 2.625 (3) | 153 (3) |
| C1—H1C⋯Cl1 | 0.96 | 2.91 | 3.072 (3) | 91 |
| C1—H1A⋯N1i | 0.96 | 2.86 | 3.734 (3) | 152 |
| C1—H1C⋯Cg1ii | 0.96 | 2.92 | 3.617 (2) | 131 |
Symmetry codes: (i) 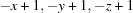 ; (ii)
; (ii) 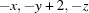 .
.
Supramolecular features
In the crystal packing of (I), the molecules are linked by C1—H1A⋯N1 [H1A⋯N1(−x + 1, −y + 1, −z + 1) = 2.86 Å] interactions, forming sheets propagating along the a-axis direction (Fig. 2 ▸ a). Weak C—H⋯π interactions [C1—H1C⋯Cg1(−x, −y + 2, −z) = 2.92 Å] are observed (Table 1 ▸ and Fig. 2 ▸ b). Notably, weak π–π stacking interactions between chlorobenzene rings [Cg1⋯Cg1(−x + 1, −y + 1, −z + 1) = 3.7890 (2) Å, where Cg1 is the centroid of the C2–C7 ring] along the a axis lead to the formation of a three-dimensional network.
Figure 2.

A view along the a axis of the crystal packing of title compound (I) showing (a) the C1—H1C⋯Cg1 interactions and (b) the most important interactions as dashed lines.
Hirshfeld surface analysis
The intermolecular interactions were investigated quantitatively and visualized with Crystal Explorer 17.5 (Turner et al., 2017 ▸; Spackman et al., 2009 ▸). The shorter and longer contacts are indicated as red and blue spots, respectively, on the Hirshfeld surfaces, and contacts with distances approximately equal to the sum of the van der Waals radii are represented as white spots. The d norm (a–d) and shape index (e) surface mappings are shown in Fig. 3 ▸. The most important red spots on the d norm surface represent O1⋯Cl1 interactions (Fig. 3 ▸ b) and C1—H1C⋯Cg1 interactions (Fig. 3 ▸ c). Some additional interactions indicated by light-red spots are corresponding to contacts around phenolic and chlorobenzene rings (Fig. 3 ▸ d). The red and blue triangles are absent on the shape-index surface, which indicates there are no strong π–π stacking interactions in the crystal structure.
Figure 3.
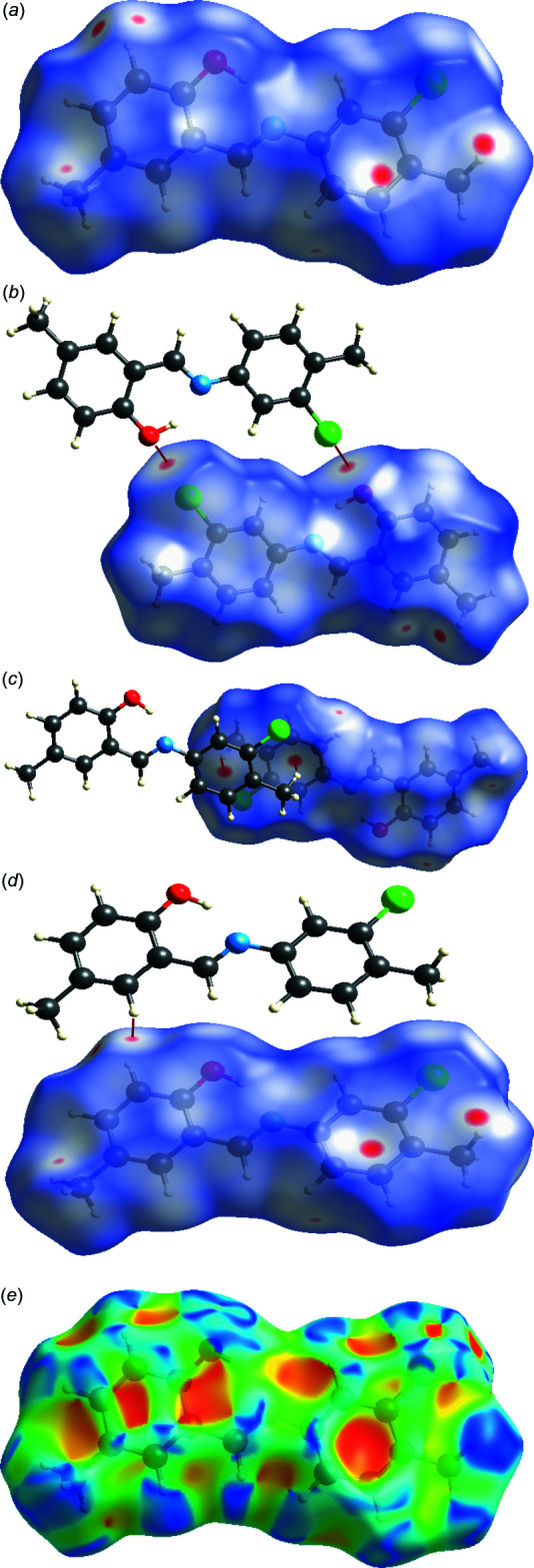
A view of the three-dimensional Hirshfeld surface for (I), plotted over (a)–(d) d norm and (e) shape-index.
Analysis of the two-dimensional fingerprint plots (Fig. 4 ▸ a–f) indicates that the H⋯H (43.8%) interactions are the major factor in the crystal packing with C⋯H/H⋯C (26.7%) interactions making the next highest contribution. The percentage contributions of other weak interactions are: Cl⋯H/H⋯Cl (12.4%), O⋯H/H⋯O (6.6%) and N⋯H/H⋯N (3.8%).
Figure 4.

(a) The overall two-dimensional fingerprint plot for the title compound and (b)–(f) those delineated into H⋯H, C⋯H/H⋯C, Cl⋯H/H⋯Cl, O⋯H/H⋯O and N⋯H/H⋯N contacts, respectively.
DFT calculations
The optimized structure in the gas phase of compound (I) was generated theoretically via density functional theory (DFT) using standard B3LYP functional and 6–311 G(d,p) basis-set calculations (Becke, 1993 ▸) as implemented in GAUSSIAN 09 (Frisch et al., 2009 ▸). The theoretical and experimental results are in good agreement (Table 2 ▸). The highest-occupied molecular orbital (HOMO), acting as an electron donor, and the lowest-unoccupied molecular orbital (LUMO), acting as an electron acceptor, are very important parameters for quantum chemistry. When the energy gap is small, the molecule is highly polarizable and has high chemical reactivity (Fukui, 1982 ▸; Khan et al., 2015 ▸). The DFT calculations provide some important information on the reactivity and site selectivity of the molecular framework, E HOMO and E LUMO, which clarify the inevitable charge-exchange collaboration inside the studied material, electronegativity (χ), hardness (η), electrophilicity (ω), softness (σ) and fraction of electron transferred (ΔN). These data are recorded in Table 3 ▸. The significance of η and σ is for the evaluation of both the reactivity and stability. The electron transition from the HOMO to the LUMO energy level is shown in Fig. 5 ▸. The HOMO and LUMO are localized in the plane extending from the whole 2-{[(3-chloro-4-methylphenyl)imino]methyl}-4-methylphenol ring. The energy band gap [ΔE = E LUMO − E HOMO] of the molecule is 4.0023 eV, the frontier molecular orbital energies E HOMO and E LUMO being −5.9865 eV and −1.9842 eV, respectively. The dipole moment of (I) is estimated to be 4.30 Debye.
Table 2. Comparison of observed (X-ray data) and calculated (DFT) geometric parameters (Å, °).
| Parameter | X-ray | B3LYP/6–311G(d,p) |
|---|---|---|
| O1—C14 | 1.354 (2) | 1.354 |
| C7—Cl1 | 1.735 (2) | 1.735 |
| N1—C8 | 1.281 (3) | 1.281 |
| C8—C9 | 1.446 (3) | 1.446 |
| N1—C5 | 1.418 (3) | 1.418 |
| C2—C7 | 1.385 (3) | 1.385 |
| C13—C14—C9 | 119.36 (19) | 119.4 |
| C9—C8—N1 | 121.82 (19) | 121.8 |
| C8—N1—C5 | 122.08 (19) | 122.1 |
Table 3. Calculated molecular energies for (I).
| Molecular Energy (a.u.) (eV) | Compound (I) |
|---|---|
| Total Energy TE (eV) | −31841.0844 |
| E HOMO (eV) | −5.9865 |
| E LUMO (eV) | −1.9842 |
| Gap, ΔE (eV) | 4.0023 |
| Dipole moment, μ (Debye) | 4.30 |
| Ionization potential, I (eV) | 5.9865 |
| Electron affinity, A | 1.9842 |
| Electronegativity, χ | 3.985 |
| Hardness, η | 2.001 |
| Electrophilicity index, ω | 3.968 |
| Softness, σ | 0.250 |
| Fraction of electron transferred, ΔN | 0.754 |
Figure 5.

Molecular orbitals showing the HOMO–LUMO electronic transition in the title compound.
Database survey
A search of the Cambridge Structural Database (CSD, version 5.39; Groom et al., 2016 ▸) gave 13 hits for the 2-{[(3-chloro-4-methylphenyl)imino]methyl}-4-methylphenol moiety. Out of 13, only a few are very closely related to the title compound. In (E)-4-methoxy-2-{[(4-methylphenyl)imino]methyl}phenol (DUPGOL; Koşar et al., 2010 ▸), the methyl group is replaced by a methoxy group and the dihedral angle between the benzene rings is 5.46 (2)°. In 2-[(E)-(5-chloro-2-methylphenyl)iminomethyl]-4-methylphenol (AFILAE; Zheng, 2013 ▸), the dihedral angle between the planes of the chlorophenyl and methylphenol rings is 35.0 (3)°. In 2-{(E)-[(3-chloro-4-methylphenyl)imino]methyl}-4-(trifluoromethoxy)phenol (TERTUI; Atalay et al., 2017 ▸), the dihedral angle between the benzene rings is 8.3 (2)° and an intramolecular O—H⋯N hydrogen bond closes an S(6) ring. In 2-{(E)-[(3-iodo-4-methylphenyl)imino]methyl}-4-(trifluoromethoxy)phenol (XEBCOY; Pekdemir et al., 2012 ▸), the dihedral angle between the two benzene rings is 12.4 (2)°. For 4-[(2-hydroxy-5-methoxybenzylidene)amino]benzonitrile (XIGNEI; Chiang et al., 2013 ▸), a complex with zinc is reported. In N-(5-hydroxysalicylidene)-2,4,6-trimethylaniline (ZIKNOW; Tenon et al., 1995 ▸), the angle between the planes of the benzene rings is 74.5 (1)° and chlorine is absent.
Synthesis and crystallization
The title compound was prepared by refluxing mixed solutions of 2-hydroxy-5-methylbenzaldehyde (34.0 mg, 0.25 mmol) in ethanol (15 ml) and 3-chloro-4-methylaniline (35.4 mg, 0.25 mmol) in ethanol (15 ml). The reaction mixture was stirred for 5 h under reflux. Single crystals of the title compound suitable for X-ray analysis were obtained by slow evaporation of an ethanol solution (yield 65%, m.p. 383–386 K).
Refinement
Crystal data, data collection and structure refinement details are summarized in Table 4 ▸. The hydroxy H atom was located in a difference-Fourier map and its positional parameters were refined freely with U iso(H) = 1.5U eq(O). Other H atoms were fixed geometrically and treated as riding with C—H = 0.96 Å (methyl) or 0.93 Å (aromatic), and U iso(H) = 1.2U eq(C) for aromatic H atoms or U iso(H) = 1.5U eq(C) for methyl H atoms.
Table 4. Experimental details.
| Crystal data | |
| Chemical formula | C15H14ClNO |
| M r | 259.72 |
| Crystal system, space group | Monoclinic, P21/c |
| Temperature (K) | 296 |
| a, b, c (Å) | 8.0534 (5), 6.3764 (3), 25.3657 (16) |
| β (°) | 96.392 (5) |
| V (Å3) | 1294.47 (13) |
| Z | 4 |
| Radiation type | Mo Kα |
| μ (mm−1) | 0.28 |
| Crystal size (mm) | 0.65 × 0.37 × 0.21 |
| Data collection | |
| Diffractometer | Stoe IPDS 2 |
| Absorption correction | Integration (X-RED32; Stoe & Cie, 2002 ▸) |
| T min, T max | 0.885, 0.958 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 7752, 2414, 1801 |
| R int | 0.040 |
| (sin θ/λ)max (Å−1) | 0.606 |
| Refinement | |
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.045, 0.137, 1.03 |
| No. of reflections | 2414 |
| No. of parameters | 169 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement |
| Δρmax, Δρmin (e Å−3) | 0.22, −0.26 |
Supplementary Material
Crystal structure: contains datablock(s) I. DOI: 10.1107/S2056989020009421/vm2236sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989020009421/vm2236Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989020009421/vm2236Isup3.cml
CCDC reference: 2015356
Additional supporting information: crystallographic information; 3D view; checkCIF report
Acknowledgments
Langat Singh College, B. R. Bihar University India, is thanked for the use of laboratory facilities.
supplementary crystallographic information
Crystal data
| C15H14ClNO | F(000) = 544 |
| Mr = 259.72 | Dx = 1.333 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| a = 8.0534 (5) Å | Cell parameters from 9569 reflections |
| b = 6.3764 (3) Å | θ = 1.6–30.3° |
| c = 25.3657 (16) Å | µ = 0.28 mm−1 |
| β = 96.392 (5)° | T = 296 K |
| V = 1294.47 (13) Å3 | Stick, orange |
| Z = 4 | 0.65 × 0.37 × 0.21 mm |
Data collection
| Stoe IPDS 2 diffractometer | 2414 independent reflections |
| Radiation source: sealed X-ray tube, 12 x 0.4 mm long-fine focus | 1801 reflections with I > 2σ(I) |
| Plane graphite monochromator | Rint = 0.040 |
| Detector resolution: 6.67 pixels mm-1 | θmax = 25.5°, θmin = 1.6° |
| rotation method scans | h = −9→9 |
| Absorption correction: integration (X-RED32; Stoe & Cie, 2002) | k = −7→7 |
| Tmin = 0.885, Tmax = 0.958 | l = −30→30 |
| 7752 measured reflections |
Refinement
| Refinement on F2 | 0 restraints |
| Least-squares matrix: full | Hydrogen site location: mixed |
| R[F2 > 2σ(F2)] = 0.045 | H atoms treated by a mixture of independent and constrained refinement |
| wR(F2) = 0.137 | w = 1/[σ2(Fo2) + (0.0845P)2 + 0.059P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.03 | (Δ/σ)max < 0.001 |
| 2414 reflections | Δρmax = 0.22 e Å−3 |
| 169 parameters | Δρmin = −0.26 e Å−3 |
Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Cl1 | 0.74526 (10) | 0.21146 (12) | 0.56404 (2) | 0.0820 (3) | |
| O1 | 0.3423 (3) | 0.0128 (3) | 0.32974 (7) | 0.0729 (5) | |
| N1 | 0.5050 (2) | 0.3352 (3) | 0.37421 (7) | 0.0546 (4) | |
| C9 | 0.3492 (2) | 0.3538 (3) | 0.28875 (8) | 0.0495 (5) | |
| C5 | 0.6021 (3) | 0.4244 (3) | 0.41869 (8) | 0.0517 (5) | |
| C10 | 0.2953 (3) | 0.4790 (3) | 0.24498 (8) | 0.0532 (5) | |
| H10 | 0.333321 | 0.616621 | 0.244352 | 0.064* | |
| C6 | 0.6276 (3) | 0.2994 (3) | 0.46336 (8) | 0.0558 (5) | |
| H6 | 0.582700 | 0.164970 | 0.462883 | 0.067* | |
| C8 | 0.4537 (3) | 0.4436 (3) | 0.33310 (8) | 0.0540 (5) | |
| H8 | 0.484407 | 0.583899 | 0.331729 | 0.065* | |
| C7 | 0.7199 (3) | 0.3742 (4) | 0.50893 (8) | 0.0560 (5) | |
| C14 | 0.2942 (3) | 0.1445 (3) | 0.28904 (8) | 0.0537 (5) | |
| C11 | 0.1881 (3) | 0.4072 (3) | 0.20271 (8) | 0.0544 (5) | |
| C2 | 0.7902 (3) | 0.5727 (4) | 0.51180 (8) | 0.0569 (5) | |
| C3 | 0.7630 (3) | 0.6946 (4) | 0.46627 (9) | 0.0624 (6) | |
| H3 | 0.808124 | 0.828887 | 0.466715 | 0.075* | |
| C4 | 0.6720 (3) | 0.6247 (4) | 0.42059 (9) | 0.0606 (6) | |
| H4 | 0.657040 | 0.711090 | 0.390901 | 0.073* | |
| C12 | 0.1343 (3) | 0.1995 (4) | 0.20491 (9) | 0.0603 (5) | |
| H12 | 0.061070 | 0.146941 | 0.177099 | 0.072* | |
| C13 | 0.1860 (3) | 0.0705 (4) | 0.24686 (9) | 0.0618 (6) | |
| H13 | 0.148288 | −0.067273 | 0.246965 | 0.074* | |
| C15 | 0.1266 (3) | 0.5478 (4) | 0.15702 (9) | 0.0699 (7) | |
| H15A | 0.110835 | 0.687133 | 0.169929 | 0.105* | |
| H15B | 0.022293 | 0.495293 | 0.140088 | 0.105* | |
| H15C | 0.207326 | 0.550746 | 0.131900 | 0.105* | |
| C1 | 0.8890 (3) | 0.6569 (4) | 0.56097 (9) | 0.0722 (7) | |
| H1A | 0.817191 | 0.672248 | 0.588488 | 0.108* | |
| H1B | 0.935229 | 0.790941 | 0.553389 | 0.108* | |
| H1C | 0.977895 | 0.561373 | 0.572449 | 0.108* | |
| H1 | 0.399 (4) | 0.082 (5) | 0.3505 (14) | 0.099 (12)* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Cl1 | 0.1001 (5) | 0.0819 (5) | 0.0596 (4) | 0.0149 (4) | −0.0115 (3) | 0.0193 (3) |
| O1 | 0.0944 (13) | 0.0595 (10) | 0.0617 (10) | −0.0062 (9) | −0.0051 (9) | 0.0144 (8) |
| N1 | 0.0546 (10) | 0.0596 (11) | 0.0481 (9) | 0.0028 (8) | −0.0013 (7) | 0.0010 (8) |
| C9 | 0.0467 (11) | 0.0521 (11) | 0.0489 (11) | 0.0015 (9) | 0.0018 (8) | 0.0018 (8) |
| C5 | 0.0497 (11) | 0.0586 (12) | 0.0458 (10) | 0.0081 (9) | 0.0014 (8) | 0.0017 (8) |
| C10 | 0.0564 (12) | 0.0506 (11) | 0.0514 (11) | −0.0007 (9) | 0.0007 (9) | 0.0043 (9) |
| C6 | 0.0575 (12) | 0.0535 (12) | 0.0553 (11) | 0.0105 (10) | 0.0008 (9) | 0.0040 (9) |
| C8 | 0.0542 (12) | 0.0529 (12) | 0.0528 (11) | 0.0025 (9) | −0.0034 (9) | 0.0023 (9) |
| C7 | 0.0569 (12) | 0.0617 (13) | 0.0481 (11) | 0.0185 (10) | −0.0001 (9) | 0.0050 (9) |
| C14 | 0.0601 (13) | 0.0513 (11) | 0.0496 (11) | 0.0027 (10) | 0.0062 (9) | 0.0060 (9) |
| C11 | 0.0532 (12) | 0.0616 (12) | 0.0477 (11) | 0.0017 (10) | 0.0021 (9) | 0.0008 (9) |
| C2 | 0.0531 (12) | 0.0654 (14) | 0.0510 (11) | 0.0124 (10) | 0.0010 (9) | −0.0064 (10) |
| C3 | 0.0648 (14) | 0.0625 (14) | 0.0583 (13) | −0.0030 (11) | 0.0000 (10) | −0.0016 (10) |
| C4 | 0.0646 (13) | 0.0642 (13) | 0.0515 (11) | −0.0047 (11) | −0.0003 (10) | 0.0060 (10) |
| C12 | 0.0586 (12) | 0.0700 (14) | 0.0507 (11) | −0.0053 (11) | −0.0003 (9) | −0.0081 (10) |
| C13 | 0.0692 (14) | 0.0540 (12) | 0.0621 (13) | −0.0097 (11) | 0.0067 (11) | −0.0030 (10) |
| C15 | 0.0756 (16) | 0.0780 (17) | 0.0524 (12) | 0.0013 (13) | −0.0093 (11) | 0.0077 (11) |
| C1 | 0.0725 (15) | 0.0864 (18) | 0.0545 (13) | 0.0117 (13) | −0.0067 (11) | −0.0140 (12) |
Geometric parameters (Å, º)
| Cl1—C7 | 1.735 (2) | C11—C12 | 1.397 (3) |
| O1—C14 | 1.354 (2) | C11—C15 | 1.505 (3) |
| O1—H1 | 0.79 (4) | C2—C3 | 1.389 (3) |
| N1—C8 | 1.281 (3) | C2—C1 | 1.502 (3) |
| N1—C5 | 1.418 (3) | C3—C4 | 1.374 (3) |
| C9—C10 | 1.397 (3) | C3—H3 | 0.9300 |
| C9—C14 | 1.406 (3) | C4—H4 | 0.9300 |
| C9—C8 | 1.446 (3) | C12—C13 | 1.372 (3) |
| C5—C6 | 1.382 (3) | C12—H12 | 0.9300 |
| C5—C4 | 1.394 (3) | C13—H13 | 0.9300 |
| C10—C11 | 1.378 (3) | C15—H15A | 0.9600 |
| C10—H10 | 0.9300 | C15—H15B | 0.9600 |
| C6—C7 | 1.388 (3) | C15—H15C | 0.9600 |
| C6—H6 | 0.9300 | C1—H1A | 0.9600 |
| C8—H8 | 0.9300 | C1—H1B | 0.9600 |
| C7—C2 | 1.385 (3) | C1—H1C | 0.9600 |
| C14—C13 | 1.385 (3) | ||
| C14—O1—H1 | 105 (2) | C7—C2—C1 | 123.1 (2) |
| C8—N1—C5 | 122.08 (19) | C3—C2—C1 | 120.7 (2) |
| C10—C9—C14 | 118.46 (18) | C4—C3—C2 | 122.6 (2) |
| C10—C9—C8 | 119.64 (19) | C4—C3—H3 | 118.7 |
| C14—C9—C8 | 121.86 (18) | C2—C3—H3 | 118.7 |
| C6—C5—C4 | 118.5 (2) | C3—C4—C5 | 120.1 (2) |
| C6—C5—N1 | 116.1 (2) | C3—C4—H4 | 120.0 |
| C4—C5—N1 | 125.41 (19) | C5—C4—H4 | 120.0 |
| C11—C10—C9 | 122.7 (2) | C13—C12—C11 | 122.0 (2) |
| C11—C10—H10 | 118.6 | C13—C12—H12 | 119.0 |
| C9—C10—H10 | 118.6 | C11—C12—H12 | 119.0 |
| C5—C6—C7 | 120.1 (2) | C12—C13—C14 | 120.4 (2) |
| C5—C6—H6 | 119.9 | C12—C13—H13 | 119.8 |
| C7—C6—H6 | 119.9 | C14—C13—H13 | 119.8 |
| N1—C8—C9 | 121.82 (19) | C11—C15—H15A | 109.5 |
| N1—C8—H8 | 119.1 | C11—C15—H15B | 109.5 |
| C9—C8—H8 | 119.1 | H15A—C15—H15B | 109.5 |
| C2—C7—C6 | 122.4 (2) | C11—C15—H15C | 109.5 |
| C2—C7—Cl1 | 119.58 (17) | H15A—C15—H15C | 109.5 |
| C6—C7—Cl1 | 118.04 (18) | H15B—C15—H15C | 109.5 |
| O1—C14—C13 | 118.7 (2) | C2—C1—H1A | 109.5 |
| O1—C14—C9 | 121.97 (19) | C2—C1—H1B | 109.5 |
| C13—C14—C9 | 119.36 (19) | H1A—C1—H1B | 109.5 |
| C10—C11—C12 | 117.03 (19) | C2—C1—H1C | 109.5 |
| C10—C11—C15 | 121.7 (2) | H1A—C1—H1C | 109.5 |
| C12—C11—C15 | 121.2 (2) | H1B—C1—H1C | 109.5 |
| C7—C2—C3 | 116.2 (2) | ||
| C8—N1—C5—C6 | 170.44 (19) | C9—C10—C11—C15 | 177.6 (2) |
| C8—N1—C5—C4 | −9.5 (3) | C6—C7—C2—C3 | 0.1 (3) |
| C14—C9—C10—C11 | 1.3 (3) | Cl1—C7—C2—C3 | −179.31 (17) |
| C8—C9—C10—C11 | −176.3 (2) | C6—C7—C2—C1 | 179.4 (2) |
| C4—C5—C6—C7 | 0.4 (3) | Cl1—C7—C2—C1 | 0.0 (3) |
| N1—C5—C6—C7 | −179.51 (18) | C7—C2—C3—C4 | −0.1 (3) |
| C5—N1—C8—C9 | −177.49 (18) | C1—C2—C3—C4 | −179.4 (2) |
| C10—C9—C8—N1 | 179.5 (2) | C2—C3—C4—C5 | 0.2 (4) |
| C14—C9—C8—N1 | 2.0 (3) | C6—C5—C4—C3 | −0.4 (3) |
| C5—C6—C7—C2 | −0.3 (3) | N1—C5—C4—C3 | 179.5 (2) |
| C5—C6—C7—Cl1 | 179.12 (16) | C10—C11—C12—C13 | −0.6 (3) |
| C10—C9—C14—O1 | 179.3 (2) | C15—C11—C12—C13 | −178.4 (2) |
| C8—C9—C14—O1 | −3.1 (3) | C11—C12—C13—C14 | 0.4 (4) |
| C10—C9—C14—C13 | −1.5 (3) | O1—C14—C13—C12 | 179.9 (2) |
| C8—C9—C14—C13 | 176.1 (2) | C9—C14—C13—C12 | 0.6 (3) |
| C9—C10—C11—C12 | −0.3 (3) |
Hydrogen-bond geometry (Å, º)
Cg1 is the centroid of the C2–C7 ring.
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1—H1···N1 | 0.79 (4) | 1.89 (3) | 2.625 (3) | 153 (3) |
| C1—H1C···Cl1 | 0.96 | 2.91 | 3.072 (3) | 91 |
| C1—H1A···N1i | 0.96 | 2.86 | 3.734 (3) | 152 |
| C1—H1C···Cg1ii | 0.96 | 2.92 | 3.617 (2) | 131 |
Symmetry codes: (i) −x+1, −y+1, −z+1; (ii) −x, −y+2, −z.
Funding Statement
This work was funded by University Grants Commission grant . Université Sidi Mohamed Ben Abdallah (Morocco) grant . University of Science and Technology, Ibb Branch (Yemen) grant .
References
- Atalay, Ş., Gerçeker, S., Meral, S. & Bülbül, H. (2017). IUCrData, 2, x171725.
- Becke, A. D. (1993). J. Chem. Phys. 98, 5648–5652.
- Chiang, H.-W., Su, Y.-T. & Wu, J.-Y. (2013). Dalton Trans. 42, 15169–15182. [DOI] [PubMed]
- Desai, S. B., Desai, P. B. & Desai, K. R. (2001). Heterocycl. Commun. 7, 83–90.
- Ebrahimipour, S. Y., Mague, J. T., Akbari, A. & Takjoo, R. (2012). J. Mol. Struct. 1028, 148–155.
- Faizi, M. S. H., Ahmad, M., Kapshuk, A. A. & Golenya, I. A. (2017). Acta Cryst. E73, 38–40. [DOI] [PMC free article] [PubMed]
- Faizi, M. S. H., Alam, M. J., Haque, A., Ahmad, S., Shahid, M. & Ahmad, M. (2018). J. Mol. Struct. 1156, 457–464.
- Faizi, M. S. H., Gupta, S., Mohan, V. K., Jain, K. V. & Sen, P. (2016). Sens. Actuators B Chem. 222, 15–20.
- Faizi, M. S. H., Osório, F. A. P. & Valverde, C. (2020). J. Mol. Struct. 1210, 128039–464.
- Farrugia, L. J. (2012). J. Appl. Cryst. 45, 849–854.
- Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G. A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H. P., Izmaylov, A. F., Bloino, J., Zheng, G., Sonnenberg, J. L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery, J. A. Jr, Peralta, J. E., Ogliaro, F., Bearpark, M., Heyd, J. J., Brothers, E., Kudin, K. N., Staroverov, V. N., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A., Burant, J. C., Iyengar, S. S., Tomasi, J., Cossi, M., Rega, N., Millam, J. M., Klene, M., Knox, J. E., Cross, J. B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R. E., Yazyev, O., Austin, A. J., Cammi, R., Pomelli, C., Ochterski, J. W., Martin, R. L., Morokuma, K., Zakrzewski, V. G., Voth, G. A., Salvador, P., Dannenberg, J. J., Dapprich, S., Daniels, A. D., Farkas, Ö., Foresman, J. B., Ortiz, J. V., Cioslowski, J. & Fox, D. J. (2009). GAUSSIAN 09. Gaussian Inc., Wallingford, CT, USA.
- Fukui, K. (1982). Science, 218, 747–754. [DOI] [PubMed]
- Grigoras, M., Catanescu, O. & Simonescu, C. I. (2001). Rev. Roum. Chim. 46, 927–939.
- Groom, C. R., Bruno, I. J., Lightfoot, M. P. & Ward, S. C. (2016). Acta Cryst. B72, 171–179. [DOI] [PMC free article] [PubMed]
- Iwan, A., Kaczmarczyk, B., Janeczek, H., Sek, D. & Ostrowski, S. (2007). Spectrochim. Acta A Mol. Biomol. Spectrosc. 66, 1030–1041. [DOI] [PubMed]
- Karia, F. D. & Parsania, P. H. (1999). Asian J. Chem. 11, 991–995.
- Khan, E., Shukla, A., Srivastava, A., Shweta, P. & Tandon, P. (2015). New J. Chem. 39, 9800–9812.
- Koşar, B., Özek, A., Albayrak, Ç. & Büyükgüngör, O. (2010). Acta Cryst. E66, o469. [DOI] [PMC free article] [PubMed]
- Kumar, M., Kumar, A., Faizi, M. S. H., Kumar, S., Singh, M. K., Sahu, S. K., Kishor, S. & John, R. P. (2018). Sens. Actuators B Chem. 260, 888–899.
- Kumar, S., Hansda, A., Chandra, A., Kumar, A., Kumar, M., Sithambaresan, M., Faizi, M. S. H., Kumar, V. & John, R. P. (2017). Polyhedron, 134, 11–21.
- Mukherjee, P., Das, A., Faizi, M. S. H. & Sen, P. (2018). Chemistry Select, 3, 3787–3796.
- Ozeryanskii, V. A., Pozharskii, A. F., Schilf, W., Kamieński, B., Sawka-Dobrowolska, W., Sobczyk, L. & Grech, E. (2006). Eur. J. Org. Chem. pp. 782–790.
- Pekdemir, M., Işık, Ş. & Alaman Ağar, A. (2012). Acta Cryst. E68, o2148. [DOI] [PMC free article] [PubMed]
- Pizzala, H., Carles, M., Stone, W. E. E. & Thevand, A. (2000). J. Chem. Soc. Perkin Trans. 2, pp. 935–939.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Sheldrick, G. M. (2015a). Acta Cryst. A71, 3–8.
- Sheldrick, G. M. (2015b). Acta Cryst. C71, 3–8.
- Siddiqui, J. I., Iqbal, A., Ahmad, S. & Weaver, W. (2006). Molecules, 11, 206–211. [DOI] [PMC free article] [PubMed]
- Singh, W. M. & Dash, B. C. (1988). Pesticides, 22, 33–37.
- Spackman, M. A. & Jayatilaka, D. (2009). CrystEngComm, 11, 19–32.
- Stoe & Cie (2002). X-AREA, X-RED32 and X-SHAPE. Stoe & Cie GmbH, Darmstadt, Germany.
- Taggi, A. E., Hafez, A. M., Wack, H., Young, B., Ferraris, D. & Lectka, T. (2002). J. Am. Chem. Soc. 124, 6626–6635. [DOI] [PubMed]
- Tenon, J. A., Carles, M. & Aycard, J.-P. (1995). Acta Cryst. C51, 2603–2606. [DOI] [PubMed]
- Turner, M. J., McKinnon, J. J., Wolff, S. K., Grimwood, D. J., Spackman, P. R., Jayatilaka, D. & Spackman, M. A. (2017). Crystal Explorer 17. The University of Western Australia.
- Vančo, J., Švajlenová, O., Račanská, E. J., Muselík, J. & Valentová, J. (2004). J. Trace Elem. Med. Biol. 18, 155–161. [DOI] [PubMed]
- Wozniak, K., He, H., Klinowski, J., Jones, W., Dziembowska, T. & Grech, E. (1995). J. Chem. Soc. Faraday Trans. 91, 7–85.
- Zheng, Y.-F. (2013). Acta Cryst. E69, o1349. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I. DOI: 10.1107/S2056989020009421/vm2236sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989020009421/vm2236Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989020009421/vm2236Isup3.cml
CCDC reference: 2015356
Additional supporting information: crystallographic information; 3D view; checkCIF report