

SUMMARY
Durable humoral immunity against epidemic infectious disease requires the survival of long-lived plasma cells (LLPCs). LLPC longevity is dependent on metabolic programs distinct from short-lived plasma cells (SLPCs); however, the mechanistic basis for this difference is unclear. We have previously shown that CD28, the prototypic T cell costimulatory receptor, is expressed on both LLPCs and SLPCs but is essential only for LLPC survival. Here we show that CD28 transduces pro-survival signaling specifically in LLPCs through differential SLP76 expression. CD28 signaling in LLPCs increased glucose uptake, mitochondrial mass/respiration, and reactive oxygen species (ROS) production. Unexpectedly, CD28-mediated regulation of mitochondrial respiration, NF-κB activation, and survival was ROS dependent. IRF4, a target of NF-κB, was upregulated by CD28 activation in LLPCs and decreased IRF4 levels correlated with decreased glucose uptake, mitochondrial mass, ROS, and CD28-mediated survival. Altogether, these data demonstrate that CD28 signaling induces a ROS-dependent metabolic program required for LLPC survival.
Graphical Abstract
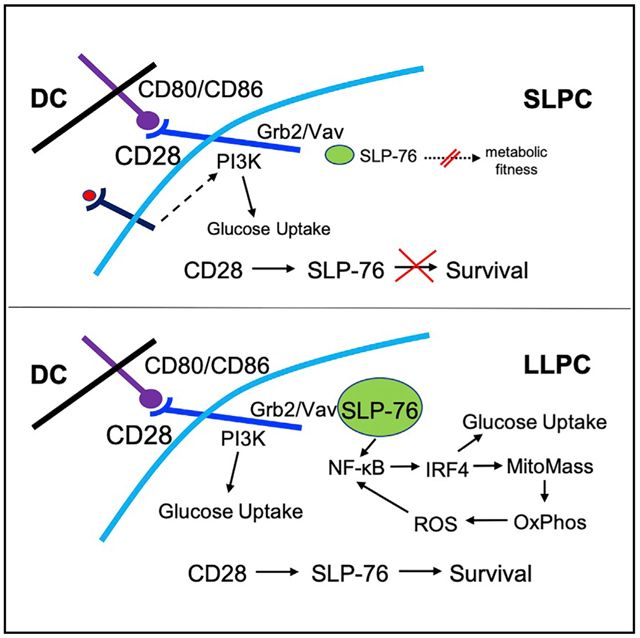
In Brief
Long-lived plasma cell survival requires a distinct metabolic program from their short-lived plasma cell counterparts. Utley et al. demonstrate that CD28 signaling through Grb2/Vav/SLP76 regulates LLPC survival and metabolic fitness through IRF4 upregulation and ROS-dependent signaling.
INTRODUCTION
Durable protective humoral immunity requires the continual production of antigen (Ag)-specific antibodies (Ab) by terminally differentiated plasma cells (PCs) (Bjorneboe et al., 1947). Given that the half-life of circulating Ab molecules is days to weeks (Fahey and Sell, 1965) while the half-life of Ab titers can be decades in humans (Amanna et al., 2007), sustained Ab levels directly reflect the maintenance of PC populations producing those Abs. These can be the short-lived PC (SLPC) subset (Slifka et al., 1998), which is replenished by memory B cells activated upon Ag re-exposure (Bernasconi et al., 2002). However, Ab titers can persist without continual Ag availability or B cells (Bhoj et al., 2016; Gray and Skarvall, 1988; Manz et al., 1998), and these are produced by the long-lived PC (LLPC) subset, which can survive for years to decades (Radbruch et al., 2006; Slifka et al., 1998).
LLPCs are not intrinsically long lived; rather, they are dependent upon access to and interaction with specific niches for their survival. LLPCs reside primarily in the bone marrow (BM) and SLPCs in secondary lymphoid organs such as the spleen (SP), although other sites exist (Radbruch et al., 2006). Stromal niche components that support LLPC survival include eosinophils, basophils, T regulatory cells, dendritic cells (DC), mesenchymal stromal cells, and megakaryocytes (Chu et al., 2011; Glatman Zaretsky et al., 2017; Minges Wols et al., 2002, 2007; Mohr et al., 2009; Rodriguez Gomez et al., 2010; Winter et al., 2010), as well as soluble factors such as APRIL, BAFF, and IL-6 (Benson et al., 2008; Minges Wols et al., 2002). There are also PC-intrinsic programs that specifically support LLPC survival, including a distinct and essential metabolic program of high glucose uptake and increased mitochondrial respiratory capacity (Lam et al., 2016, 2018; Milan et al., 2016). However, how this metabolic program is regulated, and why this is different from SLPCs, is unknown.
During B cell differentiation, genes necessary for PC survival and function are upregulated, including Irf4, Prdm1, Xpb1, and, interestingly, Cd28 (Delogu et al., 2006). CD28 is the prototypic T cell costimulatory receptor (Greenfield et al., 1998; June et al., 1987) that in conjunction with T cell receptor (TCR) augments activated T cell function and survival (Harding et al., 1992; Lindstein et al., 1989; Linsley et al., 1991; Shahinian et al., 1993; Vella et al., 1997). Importantly, CD28 co-stimulation enhances T cell metabolic fitness through induction of glycolysis and upregulation of mitochondrial respiration and fatty acid oxidation (FAO) (Buck et al., 2016; Frauwirth et al., 2002). CD28 co-stimulation is also essential for memory T cell generation through the reorganization of mitochondrial architecture and increased mitochondrial spare respiratory capacity (Klein Geltink et al., 2017).
Although CD28 is expressed on murine and human PCs (but not on B cells) (Halliley et al., 2015; Kozbor et al., 1987; Rozanski et al., 2011) and on the BMPC malignancy multiple myeloma (MM) (Pellat-Deceunynck et al., 1994; Robillard et al., 1998; Shapiro et al., 2001; Zhang et al., 1998), its function in PCs has been largely uncharacterized. Loss of CD28 in PCs was initially shown to inhibit early Ab responses (Delogu et al., 2006; Schebesta et al., 2007). We subsequently found that PC-intrinsic CD28 signaling (upon engaging its ligands CD80/CD86 on niche DCs, without a “signal 1” needed by T cells) was required for BM LLPC survival and sustained Ag-specific Ab titers (Rozanski et al., 2011, 2015). Although SLPCs express CD28, receptor activation did not induce pro-survival signaling seen in LLPCs. However, another study found that B lineage-specific loss of CD28 enhanced the generation of SLPCs, LLPCs, and resulting Ab responses (Njau et al., 2012), suggesting additional complexity of CD28’s role in PC biology.
The basis for the CD28 signaling differences between LLPCs and SLPCs is unknown but may represent a key determinant as to whether a PC can use the LLPC niche. We therefore sought to understand why CD28 transduces a pro-survival signal specifically in LLPCs and whether this regulates the LLPC metabolic program.
RESULTS
SLP76 Is Required for CD28-Mediated LLPC Survival
To understand how CD28 activation promotes LLPC but not SLPC survival, we first characterized the downstream signaling pathways. The CD28 cytoplasmic tail has two binding motifs that direct PI3K (tyrosine 170) or Grb2/Vav signaling (PYAP motif) (Boomer and Green, 2010). We have shown that inactivating mutations in the PYAP motif (CD28-AYAA-knockin [KI] mice; Boomer and Green, 2010) significantly diminished LLPC survival and sustained Ag-specific Ab production in vivo (Rozanski et al., 2015). In T cells, TCR and CD28 co-activation merge to recruit the adaptor protein SLP76 to the Grb2/Vav complex at the CD28 receptor tail (Michel and Acuto, 2002), and this links CD28 co-stimulation to phosphorylation/activation of PLCγ1 (Thaker et al., 2015). SLP76 is also expressed in B cells (Clements et al., 1998; Mizuno et al., 2005; Nagata et al., 1997; Raab et al., 2001; Su and Jumaa, 2003), but its role is not well characterized, as SLP65 assumes SLP76’s function for B cell receptor signaling (Koretzky et al., 2006). As CD28/Grb2/Vav signaling induced phosphorylation of PLCγ1 in LLPCs but not SLPCs (Rozanski et al., 2015), we examined whether LLPCs and SLPCs had differing SLP76 expression to explain the difference in CD28 signaling.
Initial transcriptomic analysis of human B cell differentiation subsets (Le Carrour et al., 2010) demonstrated that Lcp2 (encoding SLP76) was most highly expressed in BMPCs (Figure 1A). Note that the field has traditionally used BMPCs and LLPCs versus splenic PCs (SPPCs) and SLPCs interchangeably, and we do so throughout this report with the understanding that the subsets are clearly more complex. Consistent with these human data, comparison of Lcp2 expression in murine splenic B220+ B cells, B220+CD38+ plasmablasts (PBs), and B220−CD138+ PCs (Barwick et al., 2016) found highest expression in the PCs (Figure 1B). Analysis of a separate microarray dataset (Green et al., 2011) showed higher Lcp2 expression in murine BMPCs versus SPPCs (Figure 1C).
Figure 1. SLP76 Is Differentially Expressed in BMPCs versus SPPCs.

(A) Lcp2 mRNA expression in B cell and PC subsets (probe 205269_at) in the Amazonia database.
(B) Lcp2 expression in B220+ B cells from naive mice, SP B220+CD138+ plasmablasts, and B220−CD138+ PCs from LPS-treated mice (D3) by Illumina microarray. **p < 0.01, ***p < 0.001.
(C) Analysis from purified BMPC and SPPC samples using Affymetrix probe 1418641_at, normalized and annotated with the Affy and Limma Bioconductor packages in R. Statistical analysis was done between non-transformed expression values of BMPCs and SPPCs using Student’s t test. *p < .05.
(D) Total WT SP and BM mononuclear cells (MNCs) stained for CD138, B220, and CD3, then for intracellular SLP76, and analyzed using flow cytometry of the indicated populations versus FMO (full minus one control). Quantification of SLP76 expression in BMPCs and SPPCs as normalized mean fluorescence intensities (MFIs) (MFI of SLP76 minus FMO) pooled from two independent experiments is shown on the right. *p < .05.
Statistical analysis by two-way ANOVA; annotation key in STAR Methods. Error bars represent ± SD.
Transcriptomic analysis has the caveat that experimental conditions, technical approaches, and subset heterogeneity vary considerably across the available datasets. To directly assess SLP76 protein expression, we performed flow cytometric analysis and found intracellular SLP76 expression in both B220−CD138+ SPPCs and BMPCs (Figure 1D, upper left), but expression was significantly higher in BMPCs (Figure 1D, upper right). SLP76 expression in BMPCs is less but comparable with CD3+ T cells in these same samples (Figure 1D, lower). This is consistent with Lcp2 RNA expression and suggests a basis for CD28 signaling in LLPCs but not in SLPCs.
To directly assess this, we used SLP76-KI mice that have a disabling Y112/128F mutation in the SLP76 Vav binding domain (Jordan et al., 2008). Importantly, T cell help in these mice appears largely intact, as evidenced by their ability to clear acute lymphocytic choriomeningitis virus (LCMV) infection equivalently to wild-type (WT) mice (Smith-Garvin et al., 2010). BMPCs and SPPCs were purified from WT and SLP76-KI mice and treated in vitro with control versus activating anti-CD28 monoclonal antibody (mAb) in serum-free conditions. CD28 activation protected WT BMPCs from serum starvation-induced death, as previously reported (Murray et al., 2014; Rozanski et al., 2011, 2015) but could not rescue SLP76-KI BMPCs (Figure 2A), and as expected had no effect WT or SLP76-KI SPPC survival (Figure 2B). These findings indicate a LLPC-intrinsic role for SLP76 in CD28-mediated survival.
Figure 2. SLP76 Is Required for CD28-Mediated LLPC Survival.
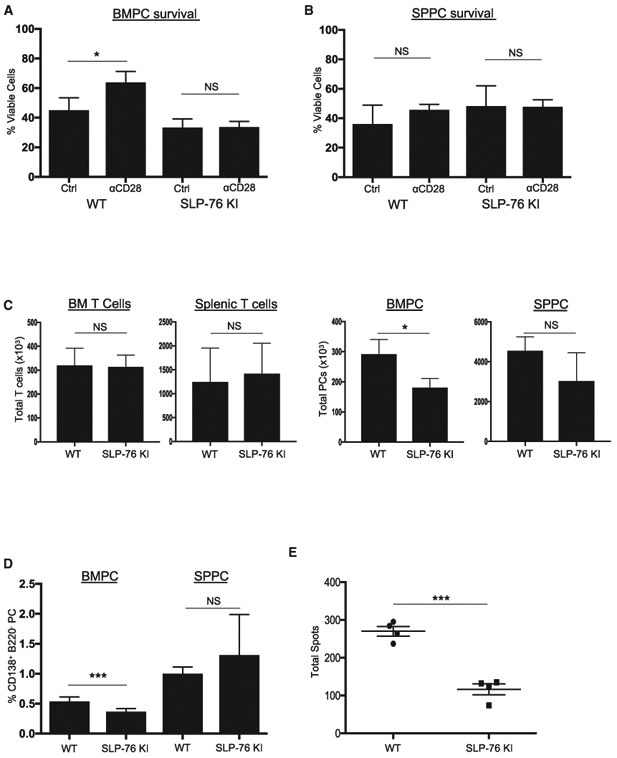
(A and B) Purified PCs from BM (A) or SP (B) of WT or SLP76-KI mice were treated with control Ig or anti-CD28 mAb-coated beads for 16 h in serum-free media, and viability was assessed using trypan blue exclusion. Representative of two independent experiments. *p < .05.
(C) Total numbers of CD3+ T cells and PCs from three individual WT or age-matched SLP76-KI mice were quantified using FACS. Representative of two independent experiments. *p < .05.
(D) BM and SP MNCs from three WT or age-matched SLP76-KI mice were stained for CD138, B220, and CD3. Percentage BMPCs or SPPCs on the basis of gating B220−CD138+ population. Data represent two independent experiments, with the percentage PCs for six individual mice quantified. ***p < 0.001.
(E) BM MNCs were harvested from four WT or SLP76-KI mice and total IgG-secreting cells quantified using ELISpot. Data are representative of two independent experiments. ***p < 0.001.
Statistical analysis by Student’s t test. Error bars represent ± SD.
If SLP76 links the CD28 receptor to downstream Grb2/Vav pro-survival signaling in LLPCs, loss of this linkage would mirror in vivo what is seen in the CD28-AYAA mice, in which CD28 is not capable of binding Grb2/Vav. Although WT and SLP76-KI mice had equivalent numbers of T cells from BM and SP (Figure 2C, left), there were significantly fewer BMPCs in the SLP76-KI mice (Figure 2C, right). There was no difference in the number of SPPCs, which also suggests intact T cell help in the SLP76-KI mice. The SLP76-KI mice also had lower percentages of BMPCs in the total mononuclear population, but no difference in the percentage of SPPCs (Figure 2D). The reduced SLP76-KI BMPC population was also reflected in a smaller number of BM IgG-secreting cells (Figure 2E). This selective loss of BM LLPCs in the SLP76-KI in vivo mice closely mirrors what is seen in the CD28-AYAA mice (Rozanski et al., 2015). Altogether, these data indicate that CD28’s pro-survival signal in LLPCs is dependent on SLP76 expression and suggest that lower SLP76 expression in SLPCs disables this signaling.
CD28 Regulates Glucose Uptake in LLPCs
In T cells, SLP76 levels regulate the balance between effector and memory CD4 T cells (Hussain et al., 2002), which have distinct metabolic programs (Buck et al., 2015). CD28 co-stimulation similarly regulates T cell metabolism by upregulating expression of the glucose transporter GLUT1 and glucose metabolism in effector T cells (Frauwirth et al., 2002) and enhancing mitochondrial respiration in memory T cells (Klein Geltink et al., 2017). The recent observation that LLPCs have higher glucose uptake than SLPCs (Lam et al., 2016, 2018) led us to examine if CD28 regulated glucose use in LLPC.
Using the fluorescent glucose analog 2-(N-[7-nitrobenz-2-oxa-1,3-diazol-4-yl]amino)-2-deoxyglucose (NBDG), we confirmed in unvaccinated mice that BMPCs take up more glucose than SPPCs (Figure 3A). To determine a role for CD28 signaling, we first examined if T cells from CD28-AYAA mice have a defect in glucose use, which is unknown but predicted given the reported defects in T cell function (Dodson et al., 2009). After 24 h of activation, CD28-AYAA T cells had significantly lower rates of glycolysis compared with WT T cells (Figure 3B). Given this defect, we assessed whether CD28-AYAA LLPCs also had diminished glucose uptake. NBDG uptake in freshly isolated CD28-AYAA BMPCs was significantly lower versus WT BMPCs (Figure 3C, left), which was also seen for the CD28-AYAA SPPCs (Figure 3C, right). In contrast, NBDG uptake in the BM B220+ B cells (Figure S1A) and SP B220+ B cells (Figure S1B) in these same samples was not different between WT and CD28-AYAA mice, which serves as an important internal technical control. CD28 activation in WT BMPCs also upregulated the cell surface expression GLUT1 (Figure 3D; also total GLUT1 by western blot, Figure S1C) and induced significantly more glucose uptake in BMPC compared with SPPC (Figure 3E). Similarly, CD28 activation increased GLUT1 expression in the human MM cell lines MM.1S, U266, and KMS11 (Figure S1D). Altogether these data demonstrate that CD28 activation upregulates glucose uptake in BMPCs.
Figure 3. CD28 Regulates Glucose Uptake in LLPCs.

(A) BM and SP MNCs from two WT mice were stained for CD138 and B220 and cultured in NBDG to assess glucose uptake using flow cytometry. Data are representative of three individual experiments. **p < 0.01.
(B) SP T cells were purified from WT or age-matched CD28-AYAA mice, activated with bead-bound anti-CD3 + anti-CD28 mAb + IL-2 ×24 h, and analyzed using Seahorse XF Glycostress test. Data are representative of three independent experiments. **p < 0.01.
(C) Level of NBDG uptake between WT and CD28KI-AYAA PCs evaluated as in (A). *p < .05.
(D) Purified BMPCs and SPPCs were treated with isotype control or anti-CD28 mAb × 16 h and evaluated for GLUT1 expression using flow cytometry. Results reflective of two independent experiments with values normalized to isotype control-treated MFI. Statistical analysis performed by analyzing raw MFI between control and anti-CD28 mAb-treated cells of SPPCs and BMPCs separately. **p < 0.01.
(E) Glucose uptake assessed by isolation and activation of SPPCs and BMPCs as in (C), evaluating NBDG uptake using flow cytometry. Analysis performed on pooled data from two independent experiments, normalizing to control-treated MFI between SPPCs and BMPCs. *p < .05.
Statistical analysis by Student’s t test. Error bars represent ± SD.
CD28 Regulates LLPC Mitochondrial Mass
CD28 co-stimulation in T cells also regulates FAO (Klein Geltink et al., 2017). Recent studies have shown that LLPCs primarily use FAO to generate ATP (Lam et al., 2016). To determine if CD28 regulated LLPC metabolism, we first analyzed gene expression in three human CD28+ MM cell lines following CD28 knockdown (KD) (Gavile et al., 2017), as CD28 activation in MM very closely mirrors what is seen in primary PCs (Boise et al., 2014). Several Gene Ontology pathways directly associated with mitochondrial metabolism were significantly downregulated by CD28 KD, including mitochondrial electron transport and the tricarboxylic acid cycle (Figure 4A). This led us to examine whether CD28 activation affected mitochondria in primary PCs. We found that freshly isolated WT BMPCs had significantly greater mitochondrial mass compared with SPPCs using the mitochondrial dye MitoTracker Green (Figure 4B). This difference was confirmed by using a different mitochondrial dye (MitoTracker Deep Red; Figure S2A) and separately by staining for the mitochondrial membrane protein voltage-dependent anion channel 1 (VDAC-1; Figure S2D), which is widely used as a measure mitochondrial mass (Scharping et al., 2016). Furthermore, these direct findings were supported by increased mRNA expression of genes for six mitochondrial membrane-resident proteins (vdac1 and 2, tomm20, tomm70a, ugcr10, and sucla2) in BMPCs versus SPPCs (Figure S2E, from the dataset used in Figure 1B). Direct involvement of CD28 in regulating LLPC mitochondrial mass was suggested by the significantly lower mitochondrial mass of CD28-AYAA BMPCs versus WT BMPCs (Figure 4C; Figure S2D by VDAC-1). Internal control staining of BM B220+ B cells in these same samples was not different between WT and CD28-AYAA mice (Figure S2B).
Figure 4. CD28 Regulates BMPC Mitochondrial Mass.

(A) RNA-seq data from MM.1S, RPMI8226, and KMS18MM cells transduced with empty vector versus shCD28. Metabolic Gene Ontologies significantly enriched in genes downregulated by shCD28 are shown on the left. The gene expression heatmap represents read count and Z score-normalized mRNA expression.
(B) BM and SP MNCs from WT mice were stained for CD138, B220, and MitoTracker Green and analyzed using flow cytometry. Quantification represents raw MFI values for BMPCs and SPPCs from individual mice pooled from three independent experiments. ****p < 0.0001.
(C) BM MNCs from two WT and CD28-AYAA mice were evaluated for mitochondrial mass as in (A). Data are representative of three independent experiments. *p<.05.
(D) PCs purified from BM of four WT mice were treated with isotype control or anti-CD28 mAb × 16 h, stained with MitoTracker Green, and analyzed using flow cytometry. Data are representative of three independent experiments. **p < 0.01.
Statistical analysis by Student’s t test. Error bars represent ± SD.
To directly assess CD28’s effect, WT BMPCs were activated by anti-CD28 mAb in vitro and mitochondrial mass quantified. Although cell viability was equivalent in all conditions, CD28 activation significantly increased mitochondrial mass in BMPCs (Figure 4D; Figure S2C for MitoTracker Deep Red staining), while this did not occur in SPPCs (data not shown). This finding that CD28 activation upregulated mitochondrial mass in LLPC predicts an increased capacity to use mitochondrial respiration.
CD28 Enhances LLPC Mitochondrial Respiration
Given the metabolic heterogeneity of the BMPC population (Lam et al., 2018), we used PC cell line models that we have found to recapitulate primary LLPC biology to more precisely interrogate a role for CD28 in LLPC metabolism (Rozanski et al., 2011). The murine PC line XXO responds to CD28 activation but has heterogeneous CD28 expression, so we began by flow-sorting into CD28high and CD28low populations (Figure 5A). Both expressed comparable levels of CD80 and CD86 (Figure S3A), which allows endogenous CD28 activation (Gavile et al., 2017; Murray et al., 2014) and comparison of CD28high versus CD28low XXO metabolic profiles without exogenous CD28 activation. We first examined glycolysis by real-time measurement of the extracellular acidification rate (ECAR), an approach that has been established for T cells and LLPCs (Lam et al., 2016; van der Windt et al., 2016). Both CD28high and CD28low XXO cells had equivalent baseline acidification prior to glucose addition (the baseline metabolic activity is typical for transformed cells), and there was no difference in the glycolysis rate upon glucose addition (Figure 5B). In contrast, the oxygen consumption rate (OCR) as a readout of mitochondrial respiration was significantly higher in CD28high (both basal and maximal OCR) versus CD28low XXO cells (Figure 5C). This was also seen under stress conditions caused by serum starvation, in which although the OCR values were an order of magnitude lower than in full-serum conditions, CD28high XXO cells maintained higher basal and maximal respiration (Figure 5D). To corroborate these findings using direct exogenous CD28 activation, we repeated these studies in the CD28-responsive murine PC cell line J558 (Rozanski et al., 2015) with high CD28 expression (Figure 5E) but very low or absent expression of CD80 and CD86 (Figure S3B). Direct CD28 activation with anti-CD28 mAb significantly increased the maximal respiratory capacity of J558 cells in both full-serum (Figure 5F, left) and serum-free (Figure 5F, right) conditions, while the effect on basal OCR was more variable. We similarly find that CD28 activation increases basal and maximal mitochondrial respiration in the human MM cell line MM.1S (Figure 5G). These finding demonstrate that CD28 activation significantly enhances the oxidative respiratory capacity of LLPCs.
Figure 5. CD28 Regulates PC Mitochondrial Respiration.

(A) CD28 expression of sorted XXO cells populations using flow cytometry and western blot (1 × 106 cells).
(B) CD28high and CD28low XXO cells plated × 16 h in full serum evaluated for glycolysis using the Seahorse XF Glycostress test.
(C) CD28high and CD28low XXO cells were plated in full serum as in (B), and mitochondrial respiration was assessed using the Seahorse XF Mitostress test. Data are representative of three independent experiments. **p < 0.01, ****p < 0.0001.
(D) CD28high and CD28low XXO cells plated in serum-free media × 16 h assessed for mitochondrial respiration as above. Data are representative of three independent experiments. **p < 0.01.
(E) CD28 expression of J558 using flow cytometry. *p < .05.
(F) J558 cells were treated with isotype control or anti-CD28 mAb × 16 h in full serum (left) or no serum (right), and mitochondrial respiration was determined as above. Data are representative of three independent experiments. **p < 0.01, ****p < 0.0001.
(G) MM.1S cells were treated with isotype control or anti-CD28 mAb × 16 h in full serum. Mitochondrial respiration was evaluated as above. Data are representative of three independent experiments. **p < 0.01.
Statistical analysis by Student’s t test. Error bars represent ± SD.
CD28 Induces ROS-Mediated Pro-survival Signaling
Increased mitochondrial mass/respiration should result in increased basal levels of reactive oxygen species (ROS), a major byproduct of oxidative phosphorylation. Consistent with the differences in mitochondrial mass, freshly isolated WT BMPCs had higher levels of total ROS versus SPPCs (Figure 6A), while B220+ B cells from the same samples had lower levels of ROS and no differences between BM and SP (Figure S4A). This was also seen using the mitochondrial ROS-specific dye MitoSOX (Figure S4B). Similarly, the percentage of ROShigh BMPCs from CD28-AYAA mice was significantly lower compared with WT BMPCs (Figure 6B), paralleling the differences in mitochondrial mass. Direct activation of CD28 increased ROS production in WT BMPCs (Figure 6C) while having no effect on SPPCs (Figure S4C). This was also consistent with observation that genes associated with cellular responses to hydrogen peroxide were most significantly downregulated by CD28 KD (Figure 4A).
Figure 6. CD28 Induces ROS for BMPC Survival.

(A) WT BM and SP MNCs were stained for CD138, B220, and cellular ROS with CM-DCFDA and analyzed using flow cytometry. Quantification represents raw MFI values for BM and SP PCs from individual mice pooled from three independent experiments. **p < 0.01.
(B) BM MNCs from two WT and CD28-AYAA mice were evaluated for ROS levels as in (A). *p < .05.
(C) BM and SP MNCs were treated with isotype control or anti-CD28 × 2 h in serum-free media and then stained for B220 CD138 and CM-DCFDA. Data are representative of three independent experiments. *p < .05.
(D) Purified BMPCs and SPPCs were treated with isotype control or anti-CD28 mAb × 16 h in serum-free media ± Euk 134. Survival was determined using trypan blue exclusion. Data are representative of three independent experiments. *p < .05.
(E) Purified BMPCs and SPPCs were treated with isotype control or anti-CD28 mAb ± Euk 134 × 30 m, stained for NF-κB RelA/p65 and DAPI, and analyzed using ImageStream for nuclear localization. Scale bar represents 10 μM.
(F) Change in ImageStream similarity score for NF-κB RelA/p65 nuclear localization with CD28 activation versus control (statistics above first bar) or CD28 activation ± Euk 134. Data pooled from three independent experiments. *p < .05.
(G) J558 cells were treated with isotype control or anti-CD28 mAb × 16 h in full serum ± Euk 134, and oxygen consumption rate (OCR) was determined using the Seahorse XF. Quantification is representative of maximal respiration. Data are representative of three independent experiments. *p < .05.
(H) CD28high and CD28lowXXO cells were plated × 16 h in full serum ± Euk 134, and OCR was measured as above. Data are representative of three independent experiments. Quantification is representative of maximal respiration. **p < 0.01.
(I) MM.1S cells were treated with isotype control or anti-CD28 mAb × 16h ± Euk 134, and OCR was evaluated as above. Quantification is representative of maximal respiration. Data are representative of two independent experiments. *p < .05.
Statistical analysis by Student’s t test, except for survival experiments, which were analyzed using ANOVA with Bonferroni analysis. Error bars represent ± SD.
Given the seeming paradox that CD28 enhances survival but ROS damage cells, we evaluated if ROS had any effect on CD28-mediated LLPC survival using the superoxide dismutase (a ROS scavenger) mimetic Euk 134. Unexpectedly, ROS scavenging completely abrogated the ability of CD28 activation to rescue WT BMPC survival from serum starvation (Figure 6D), which was also seen with the pan-ROS scavenger N-acetylcysteine (Figure S4D). ROS scavenging had no effect on SPPC survival (data not shown), indicating that the loss of ROS was not non-specifically inhibiting all PC pro-survival signals.
CD28 activation-induced ROS production activates NF-κB signaling in T cells (Los et al., 1995), and CD28 activation induces NF-κB signaling in both LLPCs and MM (Bahlis et al., 2007; Rozanski et al., 2011). We therefore evaluated whether CD28-induced ROS was involved in downstream NF-κB activation by assessing nuclear translocation of the NF-κB subunit RelA (which is activated by CD28 in LLPCs; Rozanski et al., 2011) in purified WT BMPCs ± anti-CD28 mAb ± Euk134. We found that ROS inhibition diminished the ability of CD28 activation to induce RelA nuclear translocation (Figures 6E and 6F), indicating that ROS are an important component of CD28-driven NF-κB signaling.
These data suggest that ROS from other sources (e.g., generated during B cell differentiation into PBs; Price et al., 2018) enables CD28 signaling through NF-κB in PC, and then the CD28-mediated increase in mitochondrial mass and ROS generation reinforces CD28 signaling (triggered by engagement with the LLPC niche) to maintain increased LLPC oxidative respiratory capacity. Consistent with signal reinforcement, ROS inhibition abrogated the CD28-mediated increase in maximal respiration in J558 (Figure 6G) and similarly decreased the maximal oxygen consumption in CD28high but not in CD28low XXO cells (Figure 6H), while not affecting glycolysis (data not shown). Furthermore, ROS scavenging inhibited the CD28 activation-induced maximal respiration in human MM.1S cells (Figure 6I) and blocked the ability of CD28 to rescue these cells from serum starvation-induced death (Figure S4E). Altogether, these findings suggest that the increased metabolic capacity induced by CD28 activation is reinforced by an increase in the metabolic byproduct (ROS).
CD28 Induces IRF4 Expression in BMPCs, and IRF4 Levels Are Associated with BMPC Metabolic Regulation and CD28-Mediated Survival
CD28 co-stimulation in T cells regulates a distinct set of transcriptional networks involved in glycolysis and mitochondrial respiration (Diehn et al., 2002; Riley et al., 2002). Interferon regulatory factor 4 (IRF4) is a transcription factor that regulates many of the same metabolic processes, including glucose uptake, mitochondrial mass, and oxidative phosphorylation in T cells (Mahnke et al., 2016; Man et al., 2013), although a direct connection in T cells between CD28 and IRF4 has not been reported. However, CD28 KD in MM cells results in decreased IRF4 mRNA and protein (Gavile et al., 2017). IRF4 is also a target of NF-κB in B cells (Grumont and Gerondakis, 2000) and is required for PC survival (Tellier et al., 2016). This raises the possibility that CD28 is programming LLPC metabolism through IRF4. Consistent with this, we found higher IRF4 expression in WT BMPCs versus SPPCs (Figure 7A) but no difference in IRF4 expression in the B220+ B cells in these same samples (Figure S5A).
Figure 7. CD28 Induces Irf4 Expression in BMPCs, and Irf4 Levels Are Associated with BMPC Metabolic Regulation.

(A) WT SP and BM MNC were stained for B220, CD138, and intracellular IRF4. Comparison of IRF4 expression between SP IRF4−/− B220+ cells and WT B220−CD138+ BMPCs and SPPCs. Representative of three independent experiments. ***p < 0.001.
(B) Purified BMPCs were treated with isotype control versus anti-CD28 mAb × 4 h in serum-free media. Irf4 mRNA expression was determined using qPCR with actin as a loading control; fold change in expression normalized to control-treated cells. Quantitation from pooled results from two independent experiments. *p < .05.
(C) Comparison of IRF4 protein expression between WT, IRF4+/− and IRF4−/− BMPCs. Data are representative of three independent experiments. **p < 0.01.
(D) BM (top panel) and SP (bottom panel) MNCs from age-matched WT, IRF4+/−, and IRF4−/− mice stained for B220 and CD138. Representative of three independent experiments.
(E) WT and IRF4+/− BM and SP MNCs were stained for B220, CD138, and NBDG. Representative of two independent experiments. *p < .05.
(F) Mitochondrial mass between WT and IRF4+/− BMPCs was evaluated by staining MitoTracker Green. Data are representative of three independent experiments. *p < .05.
(G) ROS levels between WT and IRF4+/− BMPCs determined by staining with CM-DCFDA. Data are representative of two independent experiments. **p < 0.01.
(H) BMPCs (2.5 × 103) of the indicated genotype were cocultured with WT BMDCs in a 1:2 ratio in serum-free media and assessed for viability using Live Dead Blue dye after 24 h. (Left) Total live PC quantification, (middle) percentage, and (right) total number of viable WT or IRF4 IRF4+/− BMPCs. ***p < 0.001, **p < 0.01, *p < .05.
(I) MM.1S cells transfected with either siRNA control 1 (siControl) or two different siRNA constructs (siIRF4-1, siIRF4-2). Left: IRF4 knockdown in three separate experiments; densitometry is relative to siControl for each experiment. Right: transfected cells were treated with control IgG or anti-CD28 mAb in full serum (FS) or no serum for 48 h, and survival was compared with FS siControl. *p < .05.
Statistical analysis by Student’s t test. Error bars represent ± SD.
To determine if CD28 can directly regulate IRF4 expression, we activated CD28 on purified WT BMPCs for 4 h and found significantly increased IRF4 mRNA expression by qPCR (Figure 7B). Graded IRF4 expression drives T cell and PC differentiation (Krishnamoorthy et al., 2017; Sciammas et al., 2006) and positively regulates the expression of the key PC transcription factor BLIMP1 (Kallies et al., 2004). We examined the effect of IRF4 expression levels by analyzing the gene dose effect in IRF4+/+ (WT), IRF4+/− (which have decreased IRF4 expression compared with WT; Figure 7C), and IRF4−/− mice. IRF4−/− mice had no PCs (in SP or BM), and IRF4+/− had fewer PCs compared with IRF4+/+ mice (Figure 7D), with no difference in CD28 expression between the IRF4+/− and IRF4+/+ BMPC (data not shown).
The dependence of PC on IRF4 for survival presents a major experimental obstacle of directly determining the role of CD28-induced IRF4 upregulation on LLPC metabolism. However, if IRF4 is involved in CD28 regulation of LLPC metabolism, decreased IRF4 expression in IRF4+/− BMPC should mirror what is seen in CD28-AYAA BMPCs. Consistent with this, IRF4+/− BMPC had lower glucose uptake (Figure 7E), mitochondrial mass (Figure 7F), and ROS levels (Figure 7G) versus IRF4+/+ BMPCs. If the changes in the IRF4+/− BMPCs reflect disruption of CD28 signaling, this predicts a diminution of the pro-survival effect of CD28 activation. To directly test this, WT or IRF4+/− BMPCs were co-cultured with WT DCs in vitro, which we have previously shown protects WT LLPCs from serum starvation-induced death through a mechanism mediated almost entirely by PC CD28 engagement with DC CD80/CD86 (Rozanski et al., 2011). In comparison with the significant rescue of WT BMPC survival with DC co-culture, rescue of IRF4+/− BMPC survival was significantly attenuated (Figure 7H). To better control the comparison between IRF4-replete and IRF4-deficient PCs, we knocked down IRF4 in human MM MM.1S cells using two different small interfering RNA (siRNA) constructs that consistently caused different degrees of IRF4 KD (Figure 7I, left), which did not affect CD28 expression (data not shown). IRF4 KD reduced MM.1S survival in both full-serum and serum-free conditions, which reflected the degree of KD and significantly abrogated the ability of direct CD28 activation to rescue MM.1S survival in serum-free conditions (Figure 7I, right). This is consistent with our previous findings in MM, in which increasing CD86 expression could not protect against IRF4 silencing-induced death (Gavile et al., 2017). Altogether these data indicate that CD28’s effects on LLPC metabolism and survival are mediated by further upregulation of IRF4 expression.
DISCUSSION
Durable protective humoral immunity is dependent on continual Ab production by LLPCs. LLPCs can survive for years, but the basis of their longevity is unclear. We have previously reported that activation of CD28 expressed on PCs is necessary for the survival of LLPCs but not SLPCs within their niches, even though both LLPCs and SLPCs have equivalent CD28 expression (Rozanski et al., 2011). However, another study showed that CD28 in the B lineage constrains the number of SPPCs and BMPCs generated after vaccination, in particular IgM+ PCs (and to a lesser extent IgG+ PCs) at notably early time points (days 7 and 18 after immunization), with loss of PC CD28 amplifying Ag-specific IgM and IgG responses (Njau et al., 2012). This suggests that intrinsic CD28 signaling regulates PC biology at two distinct stages, during initial PC generation and in the maintenance of LLPCs in peripheral niches. A role for CD28 in PC generation is supported by our finding that a threshold of SLP76 and ROS expression is needed to enable CD28 signaling (discussed below), and these thresholds are likely set early in B cell to PC differentiation.
We have now found a previously unrecognized role for SLP76 in linking the CD28 receptor to downstream pro-survival signaling in high-SLP76-expressing LLPCs but not low-expressing SLPCs. Our transcriptomic analysis revealed increased Lcp2 expression in BMPCs versus SPPCs and in a separate dataset that Lcp2 was highly upregulated in murine B220−CD138+ PBs in vivo. It should be noted that other available datasets do not show a clear upregulation of Lcp2 in LLPCs (Halliley et al., 2015; Lam et al., 2018), although this may be due to heterogeneity in the PC subsets analyzed (e.g., in Halliley et al., 2015, only 27% of the human BMPC subset containing LLPCs expressed CD28) or the difficulty in detecting low-level transcripts by single-cell RNA sequencing (RNA-seq) among the abundance of Ig transcripts (Lam et al., 2018). However, we found higher SLP76 protein expression in BMPCs versus SPPCs. Furthermore, functional analysis of SLP76-KI PCs directly demonstrated the requirement for SLP76 capable of binding Vav to transduce CD28’s pro-survival signal in BMPCs. This was reflected in vivo, where the selective loss of the BMPC population in the SLP76-KI mice closely mirrors the same loss in CD28-AYAA-KI mice that lack CD28/Grb2/Vav signaling. These in vivo studies, however, only narrowly interrogate the role of SLP76 in CD28 signaling in LLPCs and do not comprehensively examine SLP76’s broader function (either intrinsic or extrinsic) in overall PC biology.
Altogether these findings suggest that the conditions that “set” SLP76 expression during PC generation in the lymph node play a pivotal role in determining the fate of that PC, by determining whether it can use CD28 activation for survival once in the LLPC niche. Interestingly, the level of SLP76 expression alone likely does not explain why CD28 signals by itself (without a signal 1) in LLPCs but not in T cells, as T cells had higher SLP76 expression compared with BMPCs.
Recent studies have identified a distinct metabolic signature of increased glucose uptake and mitochondrial respiratory capacity in regulating LLPC survival and function (Lam et al., 2016, 2018). However, what initiates and sustains this metabolic program is unknown. Because of CD28’s regulation of glucose metabolism in T cells, we predicted that CD28 activation could regulate glucose in LLPCs. Consistent with this, we found that CD28 signaling upregulated glucose uptake in LLPCs. In the SPPC population, we found more variable glucose uptake that did not resolve distinct SPPC subpopulations as reported by others (Lam et al., 2016, 2018). However, this difference is likely technical in nature, as the NBDG uptake and staining in our in vitro conditions were significantly lower compared with the in vivo methods used by the previous studies. In vivo studies of CD28’s role in PC glucose uptake are experimentally challenging, given that loss of CD28 preferentially depletes the LLPC population that has been separately shown to be the PC subset with the highest glucose uptake. Thus, it cannot be differentiated whether any changes in in vivo PC NBDG uptake in the CD28-knockout (KO)/KI versus WT mice are due to changes in CD28-mediated PC-intrinsic glucose uptake versus loss of the NBDGhi LLPC subset through separate mechanisms. However, in vitro, we found that the defect in CD28-AYAA BMPC glucose uptake accounts for only about 20% of the total glucose uptake of the WT LLPCs and matches CD28-mediated increases in LLPC GLUT1 expression and glucose uptake in vitro. Thus, CD28 is likely one of multiple factors regulating LLPC glucose uptake, possibly playing a more important role under stress conditions, as we have seen in myeloma (Murray et al., 2014) and has been proposed for mitochondrial pyruvate metabolism (Lam et al., 2016). Interestingly, CD28-AYAA SPPCs also took up less glucose. This may be a consequence of the loss of ROS in the CD28-AYAA mutant, as ROS negatively regulate the phosphatase PTEN, which inhibits PI3K signaling (Leslie et al., 2003), and the resulting downregulation of PI3K signaling may suppress glucose uptake.
Unlike in T cells, we found that CD28 activation does not upregulate glycolysis in LLPCs, nor does CD28 KD result in significant changes in the glycolytic pathways in MM cells. Rather, CD28 activation significantly upregulated oxidative mitochondrial respiration, which dovetails with the observation that LLPCs primarily use fatty acids for basal respiration (Lam et al., 2016). CD28 co-stimulation in T cells has been elegantly shown to increase mitochondrial FAO and regulate mitochondrial mass/morphology necessary for memory T cell generation (Klein Geltink et al., 2017). Mirroring this biology, we found that mitochondrial mass in WT LLPCs was significantly greater than SLPCs and that CD28-AYAA LLPCs had significantly less mitochondrial mass compared with WT. Other reports have found no differences in mitochondrial mass between SLPCs and LLPCs (Lam et al., 2016), though these studies were in recently immunized mice, while ours were in steady-state unvaccinated mice. The molecular basis by which CD28 activation increases mitochondrial mass in LLPCs is unclear. Although expression of the master regulator of mitochondrial biogenesis PGC1α is higher in BMPCs versus other B lineage subsets (probe 219195_at; Le Carrour et al., 2010), we found that CD28 activation does not further increase PGC1α expression, similar to what has been reported in T cells (Menk et al., 2018). We have found that PGC1β is overexpressed in human MM (unpublished data), and we are pursuing this as a potential mediator of CD28’s mitochondrial effects.
The observation that CD28 induces ROS production in LLPCs led to the unexpected finding that ROS are necessary for CD28 signaling downstream to NF-κB, positioning ROS both upstream and downstream of CD28’s effect on the mitochondria. ROS have been shown to regulate the activation of the NF-κB for cell survival in other cell types (Schieber and Chandel, 2014), and CD28-mediated NF-κB activation is diminished by ROS inhibition in T cells (Los et al., 1995). Whether ROS are necessary to facilitate initial CD28 signaling in PCs, and where those ROS come from, is not known. It has been shown, however, that B cell differentiation into PBs requires a stepwise switch from glycolysis to oxidative phosphorylation with an accompanying increase in ROS (Price et al., 2018). One possibility is that multiple processes in B cell activation and differentiation to PBs/PCs generate ROS, with high levels of ROS enabling CD28 signaling in PCs. This in turn allows CD28 to regulate initial PC generation as well as transduce a pro-survival signal in PCs that access the LLPC niche. Once in the niche, ROS generated by the CD28-mediated increase in mitochondrial mass/metabolism reinforce niche-driven CD28 signaling to sustain LLPC metabolic fitness and survival. This model further suggests that other signals/feedbacks that regulate mitochondrial activity/ROS production can influence ROS levels and thus CD28 signaling. ROS levels may therefore be a convergent regulatory node for LLPC survival and function. It is also interesting to speculate that high ROS levels allow CD28 to signal in LLPCs without a signal 1, by lowering the threshold for downstream NF-κB activation.
The ability of CD28 to regulate glucose uptake and mitochondrial metabolism in LLPCs led us to hypothesize that IRF4, known to regulate these metabolic pathways in T cells (Mahnke et al., 2016), would be a downstream target of CD28 activation in LLPCs. We found that direct CD28 activation increased IRF4 expression on LLPCs. Similarly, CD28 KD in MM cell lines results in decreased IRF4 mRNA expression (Gavile et al., 2017). However, IRF4 is a challenging transcriptional factor to interrogate in PC biology, as conditional deletion leads to rapid cell death (Tellier et al., 2016). We therefore made use of the graded IRF4 protein expression in IRF4+/+ and IRF4+/− PCs and found that higher IRF4 expression correlated with CD28-responsive metabolic pathways in LLPCs, including glucose uptake, mitochondrial mass, and ROS. Consistent with this, the pro-survival effect of CD28 activation was significantly attenuated in IRF4+/− BM PCs as well as MM cell lines knocked down for IRF4. Direct measurement of metabolism are ongoing but are challenging given the small number of cells (IRF4+/− PC) and the rapid induction of death by IRF4 KD (in the MM cells). The finding that the IRF4+/− mice also have diminished SPPC populations clearly suggests that IRF4 is involved in other pathways that regulate PC metabolism and/or survival.
Altogether, our findings identify a previously unrecognized molecular network regulating LLPC survival, whereby CD28 activation on LLPCs signals through SLP76 to provide an essential pro-survival signal, and differences in SLP76 expression between LLPCs and SLPCs account for the inability of the latter to use this survival pathway. CD28 activation leads to increased LLPC glucose uptake, mitochondrial mass, and respiratory capacity to augment the metabolic fitness of LLPCs for survival. ROS production resulting from higher mitochondrial respiration then reinforces pro-survival/enhanced metabolism signaling downstream of CD28 activation in LLPCs. CD28-induced signaling then increases IRF4 expression to augment the metabolic program, which we propose crucially enhances the metabolic fitness of LLPCs in the hypoxic/nutrient-poor BM microenvironment.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kelvin Lee (kelvin.lee@roswellpark.org).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
This study did not generate any unique datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
The MM cell line MM.1S (human, female, RRID: CVCL_8792), XXO (murine, BALB/c, RRID: CVCL_4696), and J558 cells (murine, BALB/c, RRID: CVCL_3529) (all ATCC) were maintained in RPMI 1640 (CellGro, Corning NY), 10% fetal bovine serum (Thermo Fisher Scientific Hyclone, Waltham, MA), 100U/mL Penicillin, 100ug/mL streptomycin, and 2nM L-glutamine (all CellGro) at 37°C in 5% CO2. Cells were passaged every 2-3 days for a maximum of 30 days, with cells split to 2x105 cells/mL each passage.
Mice
Procedures were approved by the Institutional Animal Care and Use Committee at Roswell Park Comprehensive Cancer Center. Retired female C57Bl6/N (6-8 month old, Charles River) and age/sex matched CD28-AYAA knock-in (CD28-AYAA), IRF4+/−, and IRF4−/− mice were housed in pathogen-free facilities. Age matched 6-8 week old female littermates (WT C57Bl6/N and SLP76 Y112-128F knock-in mice) were housed at the University of Pennsylvania.
METHOD DETAILS
Tissue Processing and Plasma Cell Isolation
Bone marrow from femurs/tibias was flushed with cold PBS into 50mL conical tubes on ice. Spleens were similarly harvested into cold PBS. BM and spleens were homogenized by filtration through a 40 μM mesh screen and pelleted by spinning at 1000rpm for 5 minutes at 4°C. PBS was decanted, and 1mL ACK lysis buffer/1x108 cells (Thermo Fisher) was added and pipetted gently 3-4x. ACK buffer was diluted to 25mL with cold MACS buffer (PBS, 2% FBS, and 1mM EDTA). Cells were filtered with a 40 μM mesh screen and spun at 1000rpm for 5 minutes at 4°C. Plasma cell isolation was done as per manufacturer’s instructions (Mouse CD138+ Isolation Kit, Miltenyi).
ImageStream
Plasma cells were fixed in 4% formaldehyde for 30 minutes at 4°C, washed in PBS and permeabilized with 0.1% Triton X with 10% serum for blocking. A 1:100 dilution of RelA (p65) antibody from Cell Signaling (6956S) was added for 1 hr at 4°C for 1 hr, and washed in permeabilization buffer. 1:100 mouse anti-goat IgG-FITC (Santa Cruz sc-2356) was added for 15 minutes at room temp. Cells were washed in PBS, and resuspended in 30 μL PBS for ImageStream processing. DAPI (10 μM) was added to cells as a nuclear stain immediately prior to running through ImageStream machine. Analysis gated on single cells, in focus, double positive for nuclear stain and RelA/p65.
Analysis of RNA-seq data
Data from Gavile et al. was analyzed as previously described (Gavile et al., 2017). Briefly, genes differentially expressed between vector control and shCD28-infected cell lines were previously determined using edgeR (v3.12.1). Gene ontologies enriched in down- and upregulated genes were determined by GOstats (v2.36.0). Expression of mRNA used FPKM (fragments per kilobase per million reads) normalized read counts and heatmap representations were made using custom R (v3.6.0) code available upon request.
ELISPOT
Total IgG ASC was quantified by ELISpot assay as per manufacturer’s instructions (Mabtech). In brief, 96-well plates (Millipore) were precoated with 15 μg/ml anti-IgG capture antibody in PBS for 16hrs. 1x105 bone marrow mononuclear cells from either WT or SLP76 KI mice were then plated for 24hrs before assessing number of IgG secreting cells.
RNA Isolation and qPCR
The purification of total RNAs was prepared using the RNeasy mini kit (QIAGEN #74104) as per Purification of Total RNA, Including Small RNAs, from Animal Cells Protocol. Up to 1 × 107 of cultured cells are first centrifuged to obtain a cell pellet, then disrupted by resuspending the pellet in 700 ul of QIAzol Lysis Reagent and pipetting up and down. After addition of chloroform, the homogenate is then separated into aqueous and organic phases by centrifugation. RNA partitions to the upper, aqueous phase, while DNA partitions to the interphase and proteins to the lower, organic phase or the interphase. The upper, aqueous phase is extracted, and ethanol is added to provide appropriate binding conditions for all RNA molecules from 18 nucleotides upward. The sample is then applied to the RNeasy Mini spin column, where the total RNA binds to the membrane and phenol and other contaminants are efficiently washed away. On-column DNase digestion is also performed to remove any residual genomic DNA contamination followed by additional washes. High quality RNA is then eluted in 20 - 25 ul of RNase-free water. Quantitative assessment of the purified total RNA is then accomplished by using a Qubit high Sense RNA kit (Thermofisher #Q32854). The RNA is further evaluated qualitatively by a Tapestation Analysis (Agilent technologies).
Reverse Transcriptase (RT) reactions for each sample were processed with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems #4368814). 8.7 to 200ng of RNA (sample dependent) was mixed with 0.8uL 25x dNTP Mix, 2uL 10x Random Primers, 2uL 10x RT Buffer, 1uL Multiscribe Reverse Transcriptase, plus 4.2uL H2O for each sample in 0.2mL Thin-walled PCR tubes (Applied Biosystems #AB-1113) and the RT reactions were run on a Bio-Rad C1000 Touch Thermal Cycler with a four step program (Step 1 - 25°C 10 min, Step 2 - 37°C 120 min, Step 3 - 85°C 5 min, and Step 4 - 4°C hold).
Quantitative PCR (qPCR) was performed on a MicroAmp - Optical 384-well Reaction Plate (Applied Biosystems #4309849) with the Power SYBR Green Master Mix (Applied Biosystems #4368577). Reverse Transcription (RT) reactions were diluted then 2uL/well was loaded on the plate; samples were done in triplicate. A mix of Power SYBR Green Master Mix, forward and reverse primer mix, and water was combined for each target gene and 8uL aliquoted into wells for each set of samples, such that each well had 5uL of Power SYBR green master mix, 2.5uL of primer mix at 1uM, and 0.5uL H2O for a total volume with cDNA of 10uL/well. The qPCR was run on the Applied Biosystems QuantStudio 6 Flex Real-Time PCR System and analyzed with Applied Biosystems QuantStudio Real-Time PCR Software v1.2.
Extracellular Flux Assays
For all extracellular flux assays, cells were plated on Cell-Tak coated Seahorse XF96 cell culture microplates at a density of 8x104 cells per well. The assay plates were spin seeded for 5 minutes at 1,000 rpm and incubated at 37°C without CO2 prior to performing the assay on the Seahorse Bioscience XFe96. The Mitochondrial Stress Test was performed in XF Base Media containing 10 mM glucose, 1 mM sodium pyruvate, and 2 mM L-glutamine and the following inhibitors were added at the final concentrations: Oligomycin (2 μM), carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) (2 μM), Rotenone/Antimycin A (0.5 μM each). The Glycolytic Stress Test was performed in XF Base Media containing 2 mM L-glutamine and the following reagents were added at the final concentrations: Glucose (10 mM), Oligomycin (2 μM), and 2-deoxy-glycose (50 mM).
Flow Cytometry
MitoTracker Green FM (200 nM), MitoTracker Deep Red (100 nM), MitoSOX Red (5 μM), CM-DCFDA (10 μM Invitrogen, Molecular Probes), and 2NBDG (100 μg/mL), anti-B220-BV421 (clone RA3-6B2) and anti-CD138-APC (clone 281-2) from Biolegend, anti-VDAC1/Porin antibody (Abcam clone 20B12AF2, 0.2 μg/mL), Live Dead Blue Dye (Thermo Fisher), GLUT1 (Abcam ab15309). For MitoTracker Green/DR, MitoSox staining, BM and SP cells were plated in serum free media at 2x106/well and incubated at 37°C for 20 minutes with dyes. Cells were then stained washed and stained in PBS for CD138 and B220. Samples were analyzed using a Fortessa B for FACs analysis. Flow analysis was done using FlowJo.
Intracellular staining: BM and SP mononuclear cells were isolated and stained with for CD138-PE, B220-BV421, and CD3-APC. Following surface staining, the cells were resuspended in FoxP3 fixation/permeabilization solution (eBioscience), washed and stained with 1:100 dilution of primary sheep anti-mouse anti-SLP76 mAb (Santa Cruz, clone H300) followed by 1:100 dilution of goat anti-rabbit IgG-FITC (Santa Cruz sc-2012) in 1X buffer with 5% FBS. Samples were analyzed using an LSRII for FACS analysis, gating on CD3+ cells for T cells and B220low/CD138+ for plasma cells.
For ROS staining, BM and SP were plated in serum free media at 2x106/well and incubated at 37°C for 15 minutes prior to activation. Cells were treated with 25 μg/well of control hamster Ig or anti-CD28 mAb (PV1) for 2 hr, washed and stained for B220, CD138 and 10 μM CM-DCFDA. For analysis, CD138+/B220− cells were gated and the MFI was assessed on the FITC channel.
All cell sorting was done with the FACS Aria II by staining with CD28 (PE conjugate) and separating cells based on positive and negative expression levels. Cells were then cultured as described for 3 days prior to experimental use. Flow analysis was done using FlowJo.
BM-derived dendritic cell (BMDC) generation and co-culture viability assay
BM from C57BL/6 WT were used to generate BMDC. BM cells were taken from the tibia and femurs of age matched mice and put into culture in 6 well plates with 20ng/mL recombinant murine GM-CSF (Miltenyi mouse recombinant GM-CSF research grade 130-094-043) for 7 days at a concentration of 1e6 cells/mL, 2mL per well. On day 3 and 5, 1mL was taken from each well and replenished with fresh media with 20ng/mL of GM-CSF. Co-culture experiments: PC were purified from the bone marrow of C57BL/6 or IRF4+/− mice. Co-culture was plated in a 96 well plate at a 1:2 ratio, 20,000 PC with or without 40,000 BMDC in 0.2mL of culture medium/well (10% FBS RPMI) supplemented with 10ng/mL GM-CSF at 37°C with 5% CO2. DC were stained with Cell Trace Violet dye (ThermoFisher) to differentiate from PC. To assess viability, cells were taken directly from culture wells and stained with Live Dead Blue Fixable Viability Dye (Thermo Fisher) and CD138-APC (Biolegend). Viability was assessed both as percentage live and total live cells (based off cell number plated).
CD28 activation
Activation of human and murine CD28 was performed using 10 μg/mL anti-human CD28 mAb (Beckman Coulter, clone 28.2). For murine CD28, anti-murine CD28 mAb (BioXCell clone PV1), polyclonal control hamster-IgG {Genetex) were coated on polystyrene plates at 20 μg/mL for 12 hr at 4°C prior to cell addition.
Western blots
Cells were treated as indicated. Cell pellets were collected and frozen. Cells were lysed with Lysis Buffer (50 mM Tris HCl, 150 mM NaCl, 1% NP40, 1mM PMSF, 0.4 mg/ml NaF, 0.4 mg/ml NaVO4, 1 Pierce protease inhibitor tablet (Thermofisher, A32965)) and protein concentration was determined by BCA assay (Thermofisher, 23227). Equal amounts of protein were separated by SDS-PAGE and transferred to PVDF membranes (Millipore, IPVH00010) through wet transfer in Towbin buffer. Membranes were blocked in BSA and TBST for 30 minutes and incubated in primary antibodies for 16h. Membranes were then incubated in HRP-linked secondary antibodies for 1h and developed using Pierce ECL (Thermofisher, 32209) and the Li-Cor Odyssey Fc digital imaging system (Li-Cor). Protein concentration was estimated using the Image Studio software (Li-Cor).
IRF4 knockdown
MM1.S were transfected using the Amaxa Nucleofector II system from Lonza (Kit V, VVCA-1003). Cells were transfected with 1 μM ON-TARGETplus constructs: siControl 1 (Horizon Discovery D-001810-01-05, UGGUUUACAUGUCGACUAA), siIRF4 #1 (Horizon Discovery J-019668-07-0002, CAUCACAGCUCACGUAGAA) and siIRF4 #2 (Horizon Discovery J-019668-08-0002, CCACAGAU CUAUCCGCCAU). Transfected cells were incubated for 4 hours in complete media then used in viability assays as described. Cells were verified for knockdown efficiency after 48 hours by IRF4 western blot as described. Equal numbers of MM1.S cells for each transfection were incubated for 48 hours in complete media as described supplemented with 10% FBS or serum free media with 10 μg/ml of either anti-CD28 (Beckman Coulter B62001, clone 28.2) or Mouse IgG1 (BioXCell BE0083, clone MOPC-21). Cells were harvested and viability was determined by Trypan Blue exclusion. Survival percentage was determined by setting the full serum siControl to 100% and reflects an average of three trials.
T cell isolation/activation
Peripheral T cells were isolated from the spleens of 6-12 week old WT and CD28-AYAA female mice via negative selection using the EasySep Mouse T cell Isolation Kit (Stem Cell Technologies) and cultured in complete RPMI medium (10% FBS, 1% L-Glutamine, 1% Penicillin/Streptomycin, 0.5% HEPES). Cultured T cells were activated by αCD3, αCD28 antibodies (MiltenyiT Cell Activation/Expansion Kit) and 1ng/ml rIL-2.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analysis was done using GraphPad Prism 7. In Figure 1A, ordinary 2-way ANOVA was used for all cell types and genotypes. Student’s t test was performed for statistical analysis using two-tailed, non-equal variances, and 95% Confidence Interval. For statistical analysis of PC/T cell Seahorse data, final readings of basal oxygen consumption, maximal oxygen consumption, or glycolysis were compared between each of the technical replicates between treatments by Student’s t test. Error bars are ± SD. Notation in the figure legends: NS-not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| human anti-CD28 | Beckman Coulter | Cat#IM1376; Clone 28.2 |
| murine anti-CD28 | BioXCell | Cat#BE0015-5; Clone PV1 |
| polyclonal control hamster-IgG | Genetex | Cat#GTX00619 |
| B220 (BV421 conjugate) | Biolegend | Cat#103239; RRID: AB_10933424; Clone RA3-6B2 |
| CD138 (APC conjugate) | Biolegend | Cat#142505; RRID: AB_10960141; Clone 281-2 |
| VDAC1/Porin | Abcam | Cat#Ab14734; Clone 20B12AF2 |
| GLUT1 | Abcam | Cat#ab15309 |
| CD3 (APC conjugate) | Biolegend | Cat#100235; RRID: AB_2561455; Clone 17A2 |
| SLP76 | Santa Cruz | Cat#sc-6902; Clone H300 |
| anti-rabbit IgG-FITC | Santa Cruz | Cat#sc-2012 |
| IRF4 | Cell Signaling | Cat#4948S; Clone P173 |
| Actin | Sigma | Cat#A5441 |
| IRF4 (PE conjugate) | Biolegend | Cat#646403; Clone IRF4-3E4 |
| CD138 (PE conjugate) | Biolegend | Cat#142503; Clone 281-2; RRID: AB_10915989; |
| murine IgG1 | BioXCell | Cat#BE0083; Clone MOPC-21 |
| CD86 (PE conjugate) | Biolegend | Cat#105007; Clone GL-1; RRID# ; AB_313150 |
| CD80 (PE conjugate) | Biolegend | Cat#104707; Clone 16-10A1; RRID# AB_313128 |
| RelA (p65) | Cell Signaling | Cat#6956S; Clone L8F8; RRID# AB_10828935 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| MitoTracker Deep Red FM | ThermoFisher Scientific | Cat#M22426 |
| CM-H2DCFDA | ThermoFisher Scientific | Cat#C6827 |
| 2-NBDG | Caymen Chemicals | 11046 |
| Fixable Live/Dead Stain Blue | Invitrogen/ ThermoFisher Molecular Probes | Cat#L34962; Lot#2103470 |
| ACK lysing buffer | Thermo Scientific | A1049201 |
| MitoTracker Green FM | ThermoFisher Scientific | Cat#M7514 |
| MitoSOX Red | ThermoFisher Scientific | Cat#M36008 |
| Power SYBR Green Master Mix | Applied Biosystems, ThermoFisher Scientific | Cat#4367659 |
| Euk134 | Santa Cruz | Cat#sc-205321 |
| N-acetylcysteine amide | Sigma | Cat#A0737-5MG |
| Critical Commercial Assays | ||
| Mouse IgG ELISpot plus kit | MABTECH | Cat#3825-2HW-plus |
| Murine CD138 isolation kit | Miltenyi Biotech | Cat#130-092-530 |
| EasySep Mouse T cell isolation kit | STEMCELL Technologies | Cat3319851 |
| RNeasy mini kit | QIAGEN | Cat#74104 |
| Qubit dsDNA HS Assay Kit | Invitrogen, ThermoFisher Scientific | Cat#Q32854 |
| Deposited Data | ||
| RNaseq | Gavile et al., 2017 | NCBI GEO: GSE89511 |
| Microarray | Green et al., 2011 | NCBI GEO: GSE26408 |
| Microarray | Barwick et al., 2016 | NCBI GEO: GSE70294 |
| Experimental Models: Cell Lines | ||
| J558 | ATCC | Cat#TIB6; RRID: CVCL_3529 |
| MM.1S | ATCC | Cat#CRL-2975; RRID: CVCL_8792 |
| XXO | ATCC | Cat#TIB-20; RRID: CVCL_4696 |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6.129X1-Cd28tm2.1Jmg/Mmjax | Jackson | Stock 012305, 32040-JAX |
| Mouse: C57BL/6NCrl | Charles River | Strain Code 027 |
| Mouse: B6.129X1-Cd28tm1Jmg/Mmjax “CD28AYAA” | Jackson | Stock 012302, 32039-JAX |
| Oligonucleotides | ||
| Irf4 Forward: AGATTCCAGGTGACTCTGTG | IDT | N/A |
| Irf4 Reverse: CTGCCCTGTCAGAGTATTTC | IDT | N/A |
| actin Forward: TACCCAGGCATTGCTGACAGG | IDT | N/A |
| actin Reverse: ACTTGCGGTGCACGATGGA | IDT | N/A |
| Software and Algorithms | ||
| FlowJo 10.4.1 | FlowJo | https://www.flowjo.com/solutions/flowjo/downloads |
| Prism 7 | Graphpad | https://www.graph-pad.com/scientificsoftware/prism/ |
Highlights.
CD28 specifically induces LLPC survival through the adaptor protein SLP76
CD28 activation induces ROS production, required for CD28-mediated LLPC survival
CD28 signaling increases LLPC glucose uptake, mitochondrial mass for metabolic fitness
CD28 activation increases LLPC metabolic fitness through increased IRF4 expression
ACKNOWLEDGMENTS
This research was supported by NIH grants CA121044 (K.P.L.), T32CA085183 (A.U.), CA127910, CA192844 (L.H.B.), and AI100157 (S.H.O.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107815.
REFERENCES
- Amanna IJ, Carlson NE, and Slifka MK (2007). Duration of humoral immunity to common viral and vaccine antigens. N. Engl. J. Med 357, 1903–1915. [DOI] [PubMed] [Google Scholar]
- Bahlis NJ, King AM, Kolonias D, Carlson LM, Liu HY, Hussein MA, Terebelo HR, Byrne GE Jr., Levine BL, Boise LH, and Lee KP (2007). CD28-mediated regulation of multiple myeloma cell proliferation and survival. Blood 109, 5002–5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barwick BG, Scharer CD, Bally APR, and Boss JM (2016). Plasma cell differentiation is coupled to division-dependent DNA hypomethylation and gene regulation. Nat. Immunol 17, 1216–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson MJ, Dillon SR, Castigli E, Geha RS, Xu S, Lam KP, and Noelle RJ (2008). Cutting edge: the dependence of plasma cells and independence of memory B cells on BAFF and APRIL. J. Immunol 180, 3655–3659. [DOI] [PubMed] [Google Scholar]
- Bernasconi NL, Traggiai E, and Lanzavecchia A (2002). Maintenance of serological memory by polyclonal activation of human memory B cells. Science 298,2199–2202. [DOI] [PubMed] [Google Scholar]
- Bhoj VG, Arhontoulis D, Wertheim G, Capobianchi J, Callahan CA, Ellebrecht CT, Obstfeld AE, Lacey SF, Melenhorst JJ, Nazimuddin F, et al. (2016). Persistence of long-lived plasma cells and humoral immunity in individuals responding to CD19-directed CAR T-cell therapy. Blood 128, 360–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorneboe M, Gormsen H, and Lundquist F (1947). Further experimental studies on the role of the plasma cells as antibody producers. J. Immunol 55, 121–129. [PubMed] [Google Scholar]
- Boise LH, Kaufman JL, Bahlis NJ, Lonial S, and Lee KP (2014). The Tao of myeloma. Blood 124, 1873–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boomer JS, and Green JM (2010). An enigmatic tail of CD28 signaling. Cold Spring Harb. Perspect. Biol 2, a002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck MD, O’Sullivan D, and Pearce EL (2015). T cell metabolism drives immunity. J. Exp. Med 212, 1345–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck MD, O’Sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE, Qiu J, Kretz O, Braas D, van der Windt GJ, et al. (2016). Mitochondrial dynamics controls T cell fate through metabolic programming. Cell 166, 63–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu VT, Fröhlich A, Steinhauser G, Scheel T, Roch T, Fillatreau S, Lee JJ, Löhning M, and Berek C (2011). Eosinophils are required for the maintenance of plasma cells in the bone marrow. Nat. Immunol 12, 151–159. [DOI] [PubMed] [Google Scholar]
- Clements JL, Ross-Barta SE, Tygrett LT, Waldschmidt TJ, and Koretzky GA (1998). SLP-76 expression is restricted to hemopoietic cells of monocyte, granulocyte, and T lymphocyte lineage and is regulated during T cell maturation and activation. J. Immunol 161, 3880–3889. [PubMed] [Google Scholar]
- Delogu A, Schebesta A, Sun Q, Aschenbrenner K, Perlot T, and Busslinger M (2006). Gene repression by Pax5 in B cells is essential for blood cell homeostasis and is reversed in plasma cells. Immunity 24, 269–281. [DOI] [PubMed] [Google Scholar]
- Diehn M, Alizadeh AA, Rando OJ, Liu CL, Stankunas K, Botstein D, Crabtree GR, and Brown PO (2002). Genomic expression programs and the integration of the CD28 costimulatory signal in T cell activation. Proc. Natl. Acad. Sci. U S A 99, 11796–11801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson LF, Boomer JS, Deppong CM, Shah DD, Sim J, Bricker TL, Russell JH, and Green JM (2009). Targeted knock-in mice expressing mutations of CD28 reveal an essential pathway for costimulation. Mol. Cell. Biol 29, 3710–3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahey JL, and Sell S (1965). The Immunoglobulins of Mice. V. The Metabolic (Catabolic) Properties of Five Immunoglobulin Classes. J. Exp. Med 122, 41–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, Elstrom RL, June CH, and Thompson CB (2002). The CD28 signaling pathway regulates glucose metabolism. Immunity 16, 769–777. [DOI] [PubMed] [Google Scholar]
- Gavile CM, Barwick BG, Newman S, Neri P, Nooka AK, Lonial S, Lee KP, and Boise LH (2017). CD86 regulates myeloma cell survival. Blood Adv. 1, 2307–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatman Zaretsky A, Konradt C, Dépis F, Wing JB, Goenka R, Atria DG, Silver JS, Cho S, Wolf AI, Quinn WJ, et al. (2017). T regulatory cells support plasma cell populations in the bone marrow. Cell Rep. 18, 1906–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray D, and Skarvall H (1988). B-cell memory is short-lived in the absence of antigen. Nature 336, 70–73. [DOI] [PubMed] [Google Scholar]
- Green MR, Monti S, Dalla-Favera R, Pasqualucci L, Walsh NC, Schmidt-Supprian M, Kutok JL, Rodig SJ, Neuberg DS, Rajewsky K, et al. (2011). Signatures of murine B-cell development implicate Yy1 as a regulator of the germinal center-specific program. Proc. Natl. Acad. Sci. U S A 108, 2873–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfield EA, Nguyen KA, and Kuchroo VK (1998). CD28/B7 costimulation: a review. Crit. Rev. Immunol 18, 389–418. [DOI] [PubMed] [Google Scholar]
- Grumont RJ, and Gerondakis S (2000). Rel induces interferon regulatory factor 4 (IRF-4) expression in lymphocytes: modulation of interferon-regulated gene expression by rel/nuclear factor kappaB. J. Exp. Med 191, 1281–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliley JL, Tipton CM, Liesveld J, Rosenberg AF, Darce J, Gregoretti IV, Popova L, Kaminiski D, Fucile CF, Albizua I, et al. (2015). Long-lived plasma cells are contained within the CD19(−)CD38(hi)CD138(+) subset in human bone marrow. Immunity 43, 132–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding FA, McArthur JG, Gross JA, Raulet DH, and Allison JP (1992). CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature 356, 607–609. [DOI] [PubMed] [Google Scholar]
- Hussain SF, Anderson CF, and Farber DL (2002). Differential SLP-76 expression and TCR-mediated signaling in effector and memory CD4 T cells. J. Immunol 168, 1557–1565. [DOI] [PubMed] [Google Scholar]
- Jordan MS, Smith JE, Burns JC, Austin JE, Nichols KE, Aschenbrenner AC, and Koretzky GA (2008). Complementation in trans of altered thymocyte development in mice expressing mutant forms of the adaptor molecule SLP76. Immunity 28, 359–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- June CH, Ledbetter JA, Gillespie MM, Lindsten T, and Thompson CB (1987). T-cell proliferation involving the CD28 pathway is associated with cyclosporine-resistant interleukin 2 gene expression. Mol. Cell. Biol 7, 4472–4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallies A, Hasbold J, Tarlinton DM, Dietrich W, Corcoran LM, Hodgkin PD, and Nutt SL (2004). Plasma cell ontogeny defined by quantitative changes in blimp-1 expression. J. Exp. Med 200, 967–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein Geltink RI, O’Sullivan D, Corrado M, Bremser A, Buck MD, Buescher JM, Firat E, Zhu X, Niedermann G, Caputa G, et al. (2017). Mitochondrial priming by CD28. Cell 171, 385–397.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koretzky GA, Abtahian F, and Silverman MA (2006). SLP76 and SLP65: complex regulation of signalling in lymphocytes and beyond. Nat. Rev. Immunol 6, 67–78. [DOI] [PubMed] [Google Scholar]
- Kozbor D, Moretta A, Messner HA, Moretta L, and Croce CM (1987). Tp44 molecules involved in antigen-independent T cell activation are expressed on human plasma cells. J. Immunol 138, 4128–4132. [PubMed] [Google Scholar]
- Krishnamoorthy V, Kannanganat S, Maienschein-Cline M, Cook SL, Chen J, Bahroos N, Sievert E, Corse E, Chong A, and Sciammas R (2017). The IRF4 gene regulatory module functions as a read-write integrator to dynamically coordinate T helper cell fate. Immunity 47, 481–497.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam WY, Becker AM, Kennerly KM, Wong R, Curtis JD, Llufrio EM, McCommis KS, Fahrmann J, Pizzato HA, Nunley RM, et al. (2016). Mitochondrial pyruvate import promotes long-term survival of antibody-secreting plasma cells. Immunity 45, 60–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam WY, Jash A, Yao CH, D’Souza L, Wong R, Nunley RM, Meares GP, Patti GJ, and Bhattacharya D (2018). Metabolic and transcriptional modules independently diversify plasma cell lifespan and function. Cell Rep. 24, 2479–2492.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie NR, Bennett D, Lindsay YE, Stewart H, Gray A, and Downes CP (2003). Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J. 22, 5501–5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindstein T, June CH, Ledbetter JA, Stella G, and Thompson CB (1989). Regulation of lymphokine messenger RNA stability by a surface-mediated T cell activation pathway. Science 244, 339–343. [DOI] [PubMed] [Google Scholar]
- Linsley PS, Brady W, Grosmaire L, Aruffo A, Damle NK, and Ledbetter JA (1991). Binding of the B cell activation antigen B7 to CD28 costimulates T cell proliferation and interleukin 2 mRNA accumulation. J. Exp. Med 173, 721–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Los M, Schenk H, Hexel K, Baeuerle PA, Dröge W, and Schulze-Osthoff K (1995). IL-2 gene expression and NF-kappa B activation through CD28 requires reactive oxygen production by 5-lipoxygenase. EMBO J. 14, 3731–3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahnke J, Schumacher V, Ahrens S, Käding N, Feldhoff LM, Huber M, Rupp J, Raczkowski F, and Mittrücker HW (2016). Interferon regulatory factor 4 controls TH1 cell effector function and metabolism. Sci. Rep 6, 35521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man K, Miasari M, Shi W, Xin A, Henstridge DC, Preston S, Pellegrini M, Belz GT, Smyth GK, Febbraio MA, et al. (2013). The transcription factor IRF4 is essential for TCR affinity-mediated metabolic programming and clonal expansion of T cells. Nat. Immunol 14, 1155–1165. [DOI] [PubMed] [Google Scholar]
- Manz RA, Löhning M, Cassese G, Thiel A, and Radbruch A (1998). Survival of long-lived plasma cells is independent of antigen. Int. Immunol 10, 1703–1711. [DOI] [PubMed] [Google Scholar]
- Menk AV, Scharping NE, Rivadeneira DB, Calderon MJ, Watson MJ, Dunstane D, Watkins SC, and Delgoffe GM (2018). 4-1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J. Exp. Med 215, 1091–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel F, and Acuto O (2002). CD28 costimulation: a source of Vav-1 for TCR signaling with the help of SLP-76? Sci. STKE 2002, pe35. [DOI] [PubMed] [Google Scholar]
- Milan E, Fabbri M, and Cenci S (2016). Autophagy in plasma cell ontogeny and malignancy. J. Clin. Immunol 36 (Suppl 1), 18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minges Wols HA, Underhill GH, Kansas GS, and Witte PL (2002). The role of bone marrow-derived stromal cells in the maintenance of plasma cell longevity. J. Immunol 169, 4213–4221. [DOI] [PubMed] [Google Scholar]
- Minges Wols HA, Ippolito JA, Yu Z, Palmer JL, White FA, Le PT, and Witte PL (2007). The effects of microenvironment and internal programming on plasma cell survival. Int. Immunol 19, 837–846. [DOI] [PubMed] [Google Scholar]
- Mizuno K, Tagawa Y, Watanabe N, Ogimoto M, and Yakura H (2005). SLP-76 is recruited to CD22 and dephosphorylated by SHP-1, thereby regulating B cell receptor-induced c-Jun N-terminal kinase activation. Eur. J. Immunol 35, 644–654. [DOI] [PubMed] [Google Scholar]
- Mohr E, Serre K, Manz RA, Cunningham AF, Khan M, Hardie DL, Bird R, and MacLennan IC (2009). Dendritic cells and monocyte/macrophages that create the IL-6/APRIL-rich lymph node microenvironments where plasmablasts mature. J. Immunol 182, 2113–2123. [DOI] [PubMed] [Google Scholar]
- Murray ME, Gavile CM, Nair JR, Koorella C, Carlson LM, Buac D, Utley A, Chesi M, Bergsagel PL, Boise LH, and Lee KP (2014). CD28-mediated pro-survival signaling induces chemotherapeutic resistance in multiple myeloma. Blood 123, 3770–3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata K, Nakamura T, Kitamura F, Kuramochi S, Taki S, Campbell KS, and Karasuyama H (1997). The Ig alpha/Igbeta heterodimer on mu-negative proB cells is competent for transducing signals to induce early B cell differentiation. Immunity 7, 559–570. [DOI] [PubMed] [Google Scholar]
- Njau MN, Kim JH, Chappell CP, Ravindran R, Thomas L, Pulendran B, and Jacob J (2012). CD28-B7 interaction modulates short- and long-lived plasma cell function. J. Immunol 189, 2758–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellat-Deceunynck C, Bataille R, Robillard N, Harousseau JL, Rapp MJ, Juge-Morineau N, Wijdenes J, and Amiot M (1994). Expression of CD28 and CD40 in human myeloma cells: a comparative study with normal plasma cells. Blood 84, 2597–2603. [PubMed] [Google Scholar]
- Price MJ, Patterson DG, Scharer CD, and Boss JM (2018). Progressive upregulation of oxidative metabolism facilitates plasmablast differentiation to a T-independent antigen. Cell Rep. 23, 3152–3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raab M, Pfister S, and Rudd CE (2001). CD28 signaling via VAV/SLP-76 adaptors: regulation of cytokine transcription independent of TCR ligation. Immunity 15, 921–933. [DOI] [PubMed] [Google Scholar]
- Radbruch A, Muehlinghaus G, Luger EO, Inamine A, Smith KG, Dörner T, and Hiepe F (2006). Competence and competition: the challenge of becoming a long-lived plasma cell. Nat. Rev. Immunol 6, 741–750. [DOI] [PubMed] [Google Scholar]
- Riley JL, Mao M, Kobayashi S, Biery M, Burchard J, Cavet G, Gregson BP, June CH, and Linsley PS (2002). Modulation of TCR-induced transcriptional profiles by ligation of CD28, ICOS, and CTLA-4 receptors. Proc. Natl. Acad. Sci. U S A 99, 11790–11795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robillard N, Jego G, Pellat-Deceunynck C, Pineau D, Puthier D, Mellerin MP, Barillé S, Rapp MJ, Harousseau JL, Amiot M, and Bataille R (1998). CD28, a marker associated with tumoral expansion in multiple myeloma. Clin. Cancer Res 4, 1521–1526. [PubMed] [Google Scholar]
- Rodriguez Gomez M, Talke Y, Goebel N, Hermann F, Reich B, and Mack M (2010). Basophils support the survival of plasma cells in mice. J. Immunol 185, 7180–7185. [DOI] [PubMed] [Google Scholar]
- Rozanski CH, Arens R, Carlson LM, Nair J, Boise LH, Chanan-Khan AA, Schoenberger SP, and Lee KP (2011). Sustained antibody responses depend on CD28 function in bone marrow-resident plasma cells. J. Exp. Med 208, 1435–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozanski CH, Utley A, Carlson LM, Farren MR, Murray M, Russell LM, Nair JR, Yang Z, Brady W, Garrett-Sinha LA, et al. (2015). CD28 promotes plasma cell survival, sustained antibody responses, and BLIMP-1 upregulation through its distal PYAP proline motif. J. Immunol 194, 4717–4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, Ferris RL, and Delgoffe GM (2016). The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity 45, 701–703. [DOI] [PubMed] [Google Scholar]
- Schebesta A, McManus S, Salvagiotto G, Delogu A, Busslinger GA, and Busslinger M (2007). Transcription factor Pax5 activates the chromatin of key genes involved in B cell signaling, adhesion, migration, and immune function. Immunity 27, 49–63. [DOI] [PubMed] [Google Scholar]
- Schieber M, and Chandel NS (2014). ROS function in redox signaling and oxidative stress. Curr. Biol 24, R453–R462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciammas R, Shaffer AL, Schatz JH, Zhao H, Staudt LM, and Singh H (2006). Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell differentiation. Immunity 25, 225–236. [DOI] [PubMed] [Google Scholar]
- Shahinian A, Pfeffer K, Lee KP, Kündig TM, Kishihara K, Wakeham A, Kawai K, Ohashi PS, Thompson CB, and Mak TW (1993). Differential T cell costimulatory requirements in CD28-deffcient mice. Science 261, 609–612. [DOI] [PubMed] [Google Scholar]
- Shapiro VS, Mollenauer MN, and Weiss A (2001). Endogenous CD28 expressed on myeloma cells up-regulates interleukin-8 production: implications for multiple myeloma progression. Blood 98, 187–193. [DOI] [PubMed] [Google Scholar]
- Slifka MK, Antia R, Whitmire JK, and Ahmed R (1998). Humoral immunity due to long-lived plasma cells. Immunity 8, 363–372. [DOI] [PubMed] [Google Scholar]
- Smith-Garvin JE, Burns JC, Gohil M, Zou T, Kim JS, Maltzman JS, Wherry EJ, Koretzky GA, and Jordan MS (2010). T-cell receptor signals direct the composition and function of the memory CD8+ T-cell pool. Blood 116, 5548–5559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su YW, and Jumaa H (2003). LAT links the pre-BCR to calcium signaling. Immunity 19, 295–305. [DOI] [PubMed] [Google Scholar]
- Le Carrour T, Assou S, Tondeur S, Lhermitte L, Lamb N, Reme T, Pantesco V, Hamamah S, Klein B, and De Vos J (2010). Amazonia!: an online resource to Google and visualize public human whole genome expression data. Open Bioinform. J. 4, 5–10. [Google Scholar]
- Tellier J, Shi W, Minnich M, Liao Y, Crawford S, Smyth GK, Kallies A, Busslinger M, and Nutt SL (2016). Blimp-1 controls plasma cell function through the regulation of immunoglobulin secretion and the unfolded protein response. Nat. Immunol 17, 323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaker YR, Schneider H, and Rudd CE (2015). TCR and CD28 activate the transcription factor NF-κB in T-cells via distinct adaptor signaling complexes. Immunol. Lett 163, 113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Windt GJW, Chang CH, and Pearce EL (2016). Measuring bioenergetics in T cells using a seahorse extracellular flux analyzer. Curr. Protoc. Immunol 113, 3.16b.11–13.16b.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vella AT, Mitchell T, Groth B, Linsley PS, Green JM, Thompson CB, Kappler JW, and Marrack P (1997). CD28 engagement and proinflammatory cytokines contribute to T cell expansion and long-term survival in vivo. J. Immunol 158, 4714–4720. [PubMed] [Google Scholar]
- Winter O, Moser K, Mohr E, Zotos D, Kaminski H, Szyska M, Roth K, Wong DM, Dame C, Tarlinton DM, et al. (2010). Megakaryocytes constitute a functional component of a plasma cell niche in the bone marrow. Blood 116, 1867–1875. [DOI] [PubMed] [Google Scholar]
- Zhang XG, Olive D, Devos J, Rebouissou C, Ghiotto-Ragueneau M, Ferlin M, and Klein B (1998). Malignant plasma cell lines express a functional CD28 molecule. Leukemia 12, 610–618. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.