

Abstract
Coronavirus disease 2019 (COVID-19) is a mild to moderate respiratory tract infection, however, a subset of patients progress to severe disease and respiratory failure. The mechanism of protective immunity in mild forms and the pathogenesis of severe COVID-19 associated with increased neutrophil counts and dysregulated immune responses remain unclear. In a dual-center, two-cohort study, we combined single-cell RNA-sequencing and single-cell proteomics of whole-blood and peripheral-blood mononuclear cells to determine changes in immune cell composition and activation in mild versus severe COVID-19 (242 samples from 109 individuals) over time. HLA-DRhiCD11chi inflammatory monocytes with an interferon-stimulated gene signature were elevated in mild COVID-19. Severe COVID-19 was marked by occurrence of neutrophil precursors, as evidence of emergency myelopoiesis, dysfunctional mature neutrophils, and HLA-DRlo monocytes. Our study provides detailed insights into the systemic immune response to SARS-CoV-2 infection and reveals profound alterations in the myeloid cell compartment associated with severe COVID-19.
Keywords: COVID-19, SARS-CoV-2, monocytes, neutrophils, dysfunctional neutrophils, emergency myelopoiesis, immune profiling, scRNA-seq, mass cytometry
Graphical Abstract
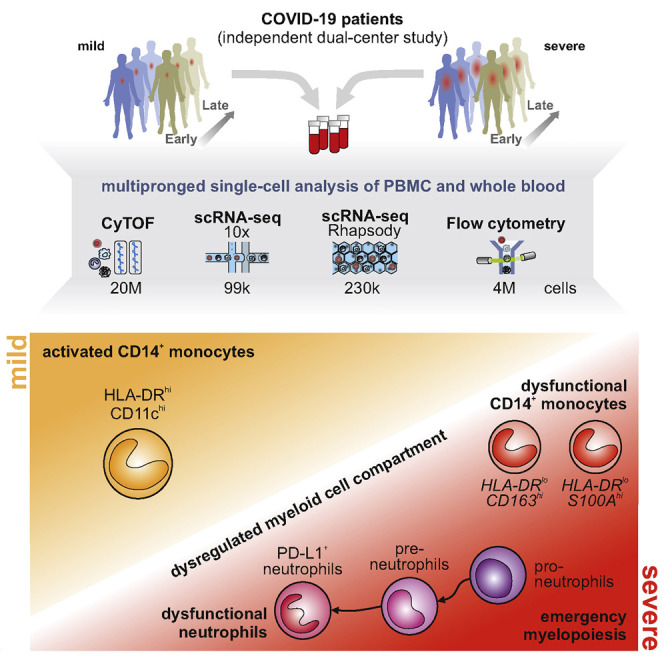
Analysis of patients with mild and severe COVID-19 reveals the presence of dysfunctional neutrophils in the latter that is linked to emergency myelopoiesis.
Introduction
Clinical presentations of COVID-19 are highly variable, and while the majority of patients experiences mild to moderate symptoms, 10%–20% of patients develop pneumonia and severe disease (Huang et al., 2020a; Wang et al., 2020; Zhou et al., 2020a). Clinical deterioration with respiratory failure and acute respiratory distress syndrome (ARDS) typically develops in the second week of disease. This kinetic may suggest a role for secondary immune responses in the development of severe COVID-19 (Ong et al., 2020). However, the exact mechanisms that govern the pathophysiology of the different disease courses of COVID-19 remain ill-defined.
Single-cell studies of bronchoalveolar lavage samples have suggested a complex dysregulation of the pulmonary immune response in severe COVID-19 (Chua et al., 2020; Liao et al., 2020). Overall, systemic inflammation is linked to an unfavorable clinical course of disease and the development of severe COVID-19 (Giamarellos-Bourboulis et al., 2020; Lucas et al., 2020; Ong et al., 2020). SARS-CoV-2 infection induces specific T cell and B cell responses, which is reflected by elevation of SARS-CoV-2 peptide-specific T cells (Braun et al., 2020; Grifoni et al., 2020) and the production of SARS-CoV-2-specific antibodies (Long et al., 2020; Ni et al., 2020; Robbiani et al., 2020). Patients with severe COVID-19 have high systemic levels of inflammatory cytokines, particularly interleukin (IL)-6 and IL-1β (Chen et al., 2020; Giamarellos-Bourboulis et al., 2020; Lucas et al., 2020; Ong et al., 2020), whereas interferon (IFN) responses appear blunted, as shown by whole blood transcriptomics (Hadjadj et al., 2020) and plasma profiling (Trouillet-Assant et al., 2020). A number of studies and regular clinical observations indicate an increase of neutrophils and a decrease of non-classical (CD14loCD16hi) monocytes in severe COVID-19 (Hadjadj et al., 2020; Merad and Martin, 2020; Sanchez-Cerrillo et al., 2020). Profound immune dysregulation is commonly observed in severe infections and sepsis, characterized by a progression from hyperinflammatory states to immunosuppression (Remy et al., 2020; Ritchie and Singanayagam, 2020), and similar mechanisms have been proposed for severe COVID-19 (Giamarellos-Bourboulis et al., 2020). Yet, comprehensive insights into the immunopathology of severe COVID-19 are still missing. Exacerbated immune responses played a major role in the pathophysiology of SARS, leading to severe lung injury and respiratory failure (Perlman and Dandekar, 2005). Mitigation of immunodysregulation is therefore viewed as a major therapeutic avenue for the treatment and prevention of severe COVID-19 (Dimopoulos et al., 2020; Jamilloux et al., 2020). In support of this view, a recent multicenter study reported that dexamethasone treatment significantly reduced mortality in hospitalized patients with COVID-19, particularly in patients on mechanical ventilation (Horby et al., 2020). Previous studies of peripheral blood mononuclear cell (PBMC) transcriptomes in a small number of patients with COVID-19 revealed changes in several cellular compartments, including monocytes, natural killer (NK) cells, dendritic cells (DCs), and T cells (Lee et al., 2020; Wilk et al., 2020).
The heterogeneity of clinical manifestations and the complexity of immune responses to COVID-19 highlight the need for detailed analyses using high-resolution techniques and well-characterized clinical cohorts. We hypothesized that distinct responses, particularly within the innate immune system, underlie the different clinical trajectories of COVID-19 patients (Chua et al., 2020; Kuri-Cervantes et al., 2020; Mathew et al., 2020; McKechnie and Blish, 2020). Here, we used single-cell transcriptomics and single-cell proteomics to analyze immune responses in blood samples in two independent cohorts of COVID-19 patients.
Activated HLA-DRhiCD11chiCD14+ monocytes were increased in patients with mild COVID-19, similar to patients with SARS-CoV-2 negative flu-like illness (“FLI”). In contrast, monocytes characterized by low expression of HLA-DR, and marker genes indicative of anti-inflammatory functions (e.g., CD163 and PLAC8) appeared in patients with severe COVID-19. The granulocyte compartment was profoundly altered in severe COVID-19, marked by the appearance of neutrophil precursors due to emergency myelopoiesis, dysfunctional neutrophils expressing PD-L1, and exhibiting an impaired oxidative burst response. Collectively, our study links highly dysregulated myeloid cell responses to severe COVID-19.
Results
Dual Center Cohort Study to Assess Immunological Alterations in COVID-19 Patients
In order to probe the divergent immune responses in mild versus severe COVID-19, we analyzed blood samples collected from independent patient cohorts at two university medical centers in Germany. Samples from the Berlin cohort (cohort 1) (Kurth et al., 2020), were analyzed by mass cytometry (CyTOF) and single-cell RNA-sequencing (scRNA-seq) using a droplet-based single-cell platform (10x Chromium), while samples from the Bonn cohort (cohort 2) were analyzed by multi-color flow cytometry (MCFC) and on a microwell-based scRNA-seq system (BD Rhapsody). We analyzed a total of 24 million cells by their protein markers and >328,000 cells by scRNA-seq in 242 samples from 53 COVID-19 patients and 56 controls, including 8 patients with FLI (Figures 1 A, 1B, and S1 A; Table S1).
Figure 1.

Cohort Definition and Single-Cell Multi-omics Analysis Strategy
(A) Pipeline for control and COVID-19 blood samples of the two cohorts (see also Table S1). Whole blood samples were subjected to red blood cell (RBC) lysis and processed for CyTOF mass cytometry (two antibody panels), multi-color flow cytometry (MCFC), or scRNA-seq (BD Rhapsody). PBMCs were isolated by density centrifugation and processed directly or after frozen storage, labeled with cell hashing antibodies and loaded on droplet-based (10x) or microwell-based (BD Rhapsody) scRNA-seq platforms. Box (bottom left): number of subjects in each cohort. Boxes (on the right): number of samples analyzed with each technique.
(B) Number of samples per technique summarized across cohorts, divided by disease severity according to WHO ordinal scale and by the time after onset of first symptoms (early: days 0–10, late: >day 11).
(C) UMAP of CD45+ leukocytes, down-sampled to 70,000 cells, from mass cytometry using antibody panel 2 (30 markers, Table S2). Cells are colored according to donor origin (blue, age-matched controls; gray, FLI; yellow, mild COVID-19; red, severe COVID-19) and major lineage subtypes.
(D) Box and whisker (10–90 percentile) plots of major cell lineage composition in whole blood from FLI (n = 8), COVID-19 patients with mild (n = 8) or severe disease (n = 9), age-matched controls measured by mass cytometry (ctrl CyTOF, n = 9) or by flow cytometry (ctrl flow, n = 19) (Kverneland et al., 2016). Kruskal-Wallis and Dunn’s multiple comparison test ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. n.a., not available.
Figure S1.

Overview of Sample Analysis Pipeline, Major Leukocyte Lineages Definition, and Quantification by CyTOF and MCFC, Related to Figure 1
A, Overview of the analysis pipeline for scRNA-seq and proteomics of COVID-19 samples.
B, High resolution SPADE analysis with 400 target nodes and individual nodes aggregated to the indicated major immune cell lineages according to the expression of lineage specific cell marker such as CD14 for monocytes and CD15 for neutrophils of whole blood samples collected from FLI patients, COVID-19 patients and controls and stained with CyTOF panel 1 and 2, respectively.
C, Boxplots of the composition of total granulocytes and non-classical monocytes within whole blood samples from the second cohort of COVID-19 patients showing either mild (n = 3) or severe disease (n = 7) as well as controls (n = 11) measured by flow cytometry. Statistical analysis was performed using unpaired t test. ∗∗p < 0.01, ∗∗∗p < 0.001.
We first characterized alterations of the major leukocyte lineages by mass cytometry on whole blood samples from 20 COVID-19 patients collected between day 4 and day 29 after symptom onset and compared them to 10 age- and gender-matched controls and 8 FLI patients. We designed two antibody panels to specifically capture alterations in mononuclear leukocytes (lymphocytes, monocytes, and DCs, panel 1), and in granulocytes (Table S2, panel 2). High-resolution SPADE analysis was performed with 400 target nodes and individual nodes were aggregated into cell subsets based on lineage-specific markers, such as CD14 for monocytes and CD15 for neutrophils (Figure S1B). Uniform manifold approximation and projection (UMAP) analysis revealed distinct clustering of samples from COVID-19 patients, FLI, and healthy controls, with marked changes of the monocyte and granulocyte compartment (Figure 1C). Leukocyte lineages were compared in the earliest available samples in COVID-19 patients (days 4–13), FLI, and controls (Figure 1D; Table S1). Because leukocyte counts were not available for all control samples, we compared the control samples for CyTOF (“ctrl CyTOF”) to data from our recently published healthy control cohorts (“ctrl flow”) (Kverneland et al., 2016; Sawitzki et al., 2020). The proportions of all major lineages were highly similar, irrespective of the methodology (Figure 1D). Cell counts of the published cohort could therefore be used as a reference to report absolute cell counts for leukocyte lineages in COVID-19 samples. In line with recent reports (Barnes et al., 2020; Xia et al., 2020), we observed elevated leukocytes and increased proportions of neutrophils in patients with severe COVID-19 (Figure 1D), whereas only proportional increases in neutrophils were evident in FLI and mild COVID-19 patients (Figure 1D). Total lymphocytes and T cells were strongly reduced in all COVID-19 and FLI patients, whereas non-classical monocytes were specifically depleted in COVID-19 (Figure 1D). Increased neutrophils in severe COVID-19 and loss of non-classical monocytes in both mild and severe disease were validated in cohort 2 by MCFC (Figure S1C; Tables S1 and S3).
Thus, SARS-CoV-2 infection is associated with lymphopenia and profound alterations of the myeloid compartment.
Severity-Dependent Alterations of the Myeloid Cell Compartment in COVID-19
Given the dramatic changes in various immune cell populations (Figures 1C and 1D), we next assessed their composition and activation state by droplet-based scRNA-seq in 27 samples from 18 COVID-19 patients (8 mild and 10 severe, cohort 1, Table S1) collected between day 3 and day 20 after symptom onset. A total of 48,266 single-cell transcriptomes of PBMCs were analyzed together with 50,783 PBMCs from publicly available control datasets (21 control donors, Table S1). UMAP and high-resolution cell type classification identified all cell types expected in the mononuclear compartment of blood with a high granularity in the monocytes, identifying five distinct clusters (clusters 0–4) (Figures 2 A and S2 A; Table S4). Monocytes in clusters 0–3 expressed CD14, and cluster 4 comprised the non-classical monocytes marked by FCGR3A (encoding CD16a) and low expression of CD14. Separate visualization of cells in mild and severe cases revealed highly disease severity-specific clusters (Figure 2B). A distinct subset of CD14 + monocytes (cluster 1) (Figure 2A) marked by high expression of HLA-DRA, HLA-DRB1, and co-stimulatory molecule CD83 (Figure S2D), the engagement of which has been linked to prolonged expansion of antigen-specific T cells (Hirano et al., 2006), was selectively detected in mild COVID-19 (Figure 2C). In addition, we identified another closely related CD14 + HLA-DR hi monocyte population (cluster 2), which was characterized by high expression of IFN-stimulated genes (ISGs). However, upon closer analysis, this cluster was found to originate from a single donor with mild COVID-19 (Figures 2A–2C and S2D). Both cluster 1 and cluster 2 expressed high levels of ISGs IFI6 and ISG15 (Figure S2D). In patients with severe COVID-19, monocytes showed low expression of HLA-DR and high expression of alarmins S100A8/9/12 (cluster 3, Figures 2A–2C and S2D). The most prominent change in severe COVID-19 was the appearance of two distinct cell populations (cluster 5+6), absent in PBMCs of patients with mild COVID-19 and control donors (Figure 2A). Published markers (Kwok et al., 2020; Ng et al., 2019) identified clusters 5 and 6 as neutrophils and immature neutrophils, respectively (Figures 2A and 2B). Immature neutrophils (cluster 6) expressed CD24, PGLYRP1, DEFA3, and DEFA4, whereas neutrophil cluster 5 expressed FCGR3B (CD16b), CXCL8, and LCN2 (lipocalin 2) (Figures 2C and S2A). Their migration within the PBMC fraction on a density gradient marked these cells as low-density neutrophils (LDNs).
Figure 2.

scRNA-Seq of PBMC from Patients of the Two Independent Cohorts
(A) UMAP visualization of scRNA-seq profiles (10x, cohort 1) of 99,049 PBMC from 49 samples (8 mild, 10 severe patients, different time points) and 22 control samples colored according to cell type classification (Louvain clustering), reference-based cell-type annotation, and marker gene expression (Table S4).
(B) UMAP shown in (A) colored according to disease severity (yellow, mild COVID-19; red, severe COVID-19).
(C) Dot plots of the intersection of the top 20 marker genes sorted by average log fold change determined for the indicated myeloid cell subsets in the PBMC datasets of both cohorts.
(D) UMAP visualization of scRNA-seq profiles (BD Rhapsody, cohort 2) of 139,848 PBMCs (50 samples of 8 mild, 9 severe COVID-19; 14 samples of 13 controls; different time points), coloring as in (A) (see also Figure S2A and Table S4).
(E) Box and whisker plots (25–75 percentile) of percentages of cell subsets of total PBMC (per patient). Boxes are colored according to disease group and dots according to the respective cohort of the sample. Dirichlet-multinomial regression adjusted with the Benjamini-Hochberg method, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
See also Table S1.
Figure S2.

Cluster-Specific Marker Gene Expression Shows Inflammatory Activation Signatures of Monocyte Subsets and the Appearance of Neutrophil Subsets in the PBMC Fraction, Related to Figure 2
(A), Dot plots of the top 5 marker genes sorted by average log fold change determined for the clusters depicted in Figure 2A.
(B), Dot plot representation of the top 5 marker genes sorted by average log fold change determined for the clusters depicted in Figure 2D.
C: Heatmap of the Spearman correlation coefficients between myeloid cell subsets in two cohorts, based on the union of top 50 marker genes per cluster.
In the second cohort, PBMCs from 17 COVID-19 patients (8 mild, 9 severe, Table S1), sampled between 2 and 25 days after symptom onset, and 13 controls, were collected for scRNA-seq on a microwell-based platform (BD Rhapsody). High-quality single-cell transcriptomes for 139,848 PBMCs were assessed and their population structure was visualized using UMAP (Figure 2D; Table S4). Data-driven cell-type classification (Aran et al., 2019) and cluster-specific marker gene expression identified all cell types expected in the PBMC compartment and revealed additional clusters and substructures (Figures 2D and S2B). Similar to cohort 1, monocytes exhibited significant plasticity and were subclassified into 5 clusters (Figure 2D, clusters 0–4). Disease-severity-associated changes seen in cohort 1 were validated in cohort 2 (Figure 2E). Immature and mature neutrophil clusters were detected in both cohorts (clusters 5–6) and showed near identical marker gene expression (Figure 2C). Similar to cohort 1, a prominent shift in subpopulation occupancy was observed in the monocyte clusters (Figures 2D and 2E).
Based on the union of the top 50 genes for monocyte and neutrophil clusters, we found a high correlation between the independently defined functional states within the monocyte compartment, and mature and immature neutrophils in cohort 1 and cohort 2 (Figure S2C). Violin plot representation of important marker genes illustrated distinct phenotypic states and underscored the high similarity of the two cohorts (Figure S2D).
Disease-severity-dependent alterations of the monocyte compartment and the appearance of two LDN populations were detected in two cohorts of COVID-19 patients.
Predominance of HLA-DRhiCD11chi Inflammatory Monocytes in Mild and HLA-DRloCD11cloCD226+CD69+ Monocytes in Severe COVID-19
The monocyte compartment is particularly affected by COVID-19, indicated by a loss of CD14loCD16hi non-classical monocytes (Figures 1C and 1D). Disease-severity-dependent shifts in monocyte activation were identified by scRNA-seq (Figure 2). We further explored the phenotypic alterations of the monocyte compartment using mass cytometry (Table S2, panel 1) on whole blood samples from COVID-19 patients with a mild or severe disease (n = 8+9), patients with FLI (n = 8), and age- and gender-matched controls (n = 9, all collected within cohort 1, Table S1). Unsupervised cluster analysis using 15 surface antigens and the proliferation marker Ki67 separated the monocyte and DC compartment into 12 main cell clusters (Figures 3A and 3B). Classical CD14hiCD16− monocytes displayed high heterogeneity and separated into seven main subclusters. Most classical monocytes showed high expression of activation markers CD38, CD95, and CXCR3. The four most prevalent clusters (1, 2, 5, and 6) varied according to CD62L, HLA-DR, CD11c, and Ki67 expression, with CD62L and HLA-DR showing a reverse expression pattern (Figure 3A). Cluster 1 displayed an activated inflammatory phenotype with high co-expression of CD11c and HLA-DR (Bernardo et al., 2018; Janols et al., 2014). In addition, we observed classical monocyte cell clusters (7, 9, and 10) with high CD226 and CD69 but low HLA-DR expression and thus signs of altered or alternative activation (Davison et al., 2017; Reymond et al., 2004; Vo et al., 2016). Among the HLA-DRlo clusters, particularly cluster 7 showed high expression of CD34 indicative of a more immature phenotype. In contrast, the majority of CD14hiCD16+ intermediate monocyte cell clusters showed high CD11c and HLA-DR expression.
Figure 3.

CD11clo and HLA-DRlo but CD226+CD69+ Monocytes in Severe COVID-19
(A) Heatmap of CyTOF data (antibody panel 1, cohort 1) covering monocytes and DCs. Main cell, as defined by the numbers 1 to 12, and individual cell clusters are displayed in columns and marker identity is indicated in rows. MSI, marker staining intensity respective expression level, significance level for the following comparisons: (1) controls (ctrl, n = 9) versus COVID-19 (mild and severe, n = 17, first row), (2) mild (n = 8) versus severe (n = 9, second row), (3) FLI (n = 8) versus mild COVID-19 (n = 8, third row), as well as (4) controls (ctrl, n = 9) versus FLI (n = 8) are indicated using a gray scale on top of the heatmap (p value scale next to heatmap). COVID-19 samples collected between days 4 and 13 post-symptom onset ( = first day of sample collection per patient). Abundance testing via generalized mixed effects models and multiple comparison adjustment using the Benjamini-Hochberg procedure and a false discovery rate (FDR) cutoff of 5% across all clusters/subsets and between-group comparisons.
(B) UMAP of monocytes and DCs, down-sampled to 70,000 cells, (39 markers, Table S2). Cells are colored according to main cell clusters (1 to 12, colors as in A) as defined in the table, donor origin (blue, controls; gray, FLI; yellow, mild COVID-19; red, severe COVID-19) and expression intensity of HLA-DR, CD11c, CD226, and CD69.
(C) Box and whisker (10–90 percentile) plots of main monocyte clusters 1, 10 (CD14hiCD16− classical monocytes), 11, and 3 (CD14hiCD16+ intermediate monocytes) determined by mass cytometry (whole blood, cohort 1): controls (n = 9), FLI patients (n = 8), COVID-19 patients (mild, n = 8; severe, n = 9). Abundance testing via R multcomp and lsmeans packages adjusted using the Benjamini-Hochberg procedure and an FDR-cutoff of 5% across all clusters/subsets and between-group comparisons.
(D) Box and whisker (10–90 percentile) plots of CXCR3+, HLA-DRhiCD11chi, and CD226+CD69+ monocytes measured by mass cytometry (whole blood, cohort 1): controls (n = 9), FLI patients (n = 8), and COVID-19 patients (mild, n = 8; severe, n = 9). Kruskal-Wallis and Dunn’s multiple comparison tests.
(E) Boxplot of HLA-DRhiCD11chi monocytes (cohort 2) measured by flow cytometry: COVID-19 (mild, n = 3; severe, n = 7) and age-matched controls (n = 11). Unpaired t test.
∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Monocytes from COVID-19 patients separated from those of FLI patients and controls (Figure 3B), mainly based on elevated CD226 and CD69 expression in COVID-19. Monocytes in mild and severe COVID-19 clustered separately, and monocytes from mild COVID-19 clustered closer to monocytes in FLI. FLI patients and mild COVID-19 contained higher proportions of HLA-DRhiCD11chi cells (clusters 3 and 11), and total HLA-DRhiCD11chi monocytes were higher compared to controls and severe COVID-19, reflecting blunted monocyte activation in severe COVID-19, reminiscent of observations in sepsis (Janols et al., 2014) (Figures 3A, 3C, and 3D). Increased levels of activated HLA-DRhiCD11chi monocytes in mild COVID-19 patients were confirmed by MCFC in cohort 2 (Figure 3E). In severe COVID-19, we detected increased expression of CD226 and CD69 (cluster 10) and/or decreased expression of HLA-DR, and total CD226+CD69+ monocytes were elevated compared to controls. Cluster 10 expressed high levels of CD10, which is induced during macrophage differentiation (Huang et al., 2020b). Thus, an alternative activation pattern of classical monocytes appeared to be COVID-19-specific and was associated with severe disease. Besides activated lymphocytes, monocytes also upregulate CD69 expression (Davison et al., 2017), which promotes tissue infiltration and retention (Cibrián and Sánchez-Madrid, 2017). Similarly, CD226 expression on alternatively activated monocytes might also promote diapedesis and tissue infiltration (Reymond et al., 2004). Together, this activation pattern may contribute to the reduction of circulating monocytes in COVID-19.
HLA-DRlo Monocytes Persist in Severe COVID-19
Next, we dissected COVID-19-associated phenotypic alterations of monocytes by scRNA-seq. Marker genes of the monocyte clusters derived from Figure 2A showed that mild COVID-19 associated clusters 1 and 2 were characterized by an ISG-driven transcriptional program (Figure S3 A), and gene ontology enrichment analysis (GOEA) assigned these clusters to “type I interferon signaling pathway” (Figure S3B). A monocyte cluster marked by low expression of HLA-DR and high expression of S100A12 and CXCL8 (cluster 3, HLA-DR lo S100A hi) was strongly associated with severe COVID-19 (Figures 2B, S2D, and S3A). For further in-depth analysis, we subclustered the monocyte compartment of the PBMC dataset of cohort 2 (Figures 2D and S3C; Table S1) resulting in 7 subclusters (Figure 4 A). Cluster 1 was marked by high expression of HLA-DRA and HLA-DRB1 and co-stimulatory molecule CD83 and was therefore designated HLA-DR hi CD83 hi-activated inflammatory monocytes (Figures 4A, 4B, S3D, and S3E). We identified two major clusters (0 and 2) and a smaller cluster (6) with low HLA-DR expression, which were associated with severe COVID-19 (Figures 4B, S3D, and S3E). Low HLA-DR expression is an established surrogate marker of monocyte dysfunction (Venet et al., 2020) which results in reduced responsiveness to microbial stimuli (Veglia et al., 2018), suggesting that clusters 0 and 6 are composed of dysfunctional monocytes. Genes of the S100A family were expressed in both HLA-DR lo clusters (Figure 4B), albeit to a higher degree in cluster 0 (HLA-DR lo S100A hi, e.g., S100A12) (Figure S2, Figure S3D and S3E; Table S4). Cluster 2 monocytes expressed high levels of SELL (CD62L) and CD163 (HLA-DR lo CD163 hi) (Figure 4B), associated with anti-inflammatory macrophage functions (Fischer-Riepe et al., 2020; MacParland et al., 2018), as well as pre-maturation markers like MPO and PLAC8 (Figure 4B), recently linked to immature monocyte states in sepsis patients (Reyes et al., 2020). In line with these findings, clusters 0, 2, and 6 were significantly enriched in a gene signature derived from sepsis-associated monocytes (Figure 4C) (Reyes et al., 2020). Moreover, blood monocytes isolated from COVID-19 patients showed a blunted cytokine response to LPS stimulation, particularly monocytes from patients with severe COVID-19 (Figure 4D). Accordingly, HLA-DR lo monocyte clusters (0, 2, and 6) were detected almost exclusively in severe COVID-19 (Figure 4E). We next analyzed time-dependent cluster occupancies per patient in cohort 2 (Figures 4E and 4F). Activated HLA-DR hi CD83 hi monocytes (cluster 1) were found in all cases of mild COVID-19, even at late time points (Figures 4E and 4F). In contrast, HLA-DR lo CD163 hi monocytes (cluster 2) were present mainly early in severe disease, while HLA-DR lo S100A hi monocytes (cluster 0) dominated the late phase of disease (Figures 4E and 4F). Violin plots of ISG (Figure S3D) and visualization of marker genes (Figure S3E) indicated differential expression patterns of IFN signature genes in individual monocyte clusters. To reveal the kinetics of ISG expression, we plotted the expression of ISG15 and IFI6 in the complete monocyte population for all patients that had been sampled at least twice (Figure 4G). Expression levels were highest at early time points and consistently decreased over time, clearly indicating that the IFN response in COVID-19 is inversely linked to disease severity and time (Figures S3F and S3G). In contrast, decreased expression of HLA-DRA and HLA-DRB1 in severe COVID-19 is evident early on and sustained over time.
Figure S3.

Transcriptional Differences of Monocytes from Mild and Severe COVID-19, Related to Figure 4
A, Dot plot of the top 10 marker genes sorted by average log fold change of the clusters within the monocyte space of cohort 1 (related to Figure 2, Table S4).
B, Gene ontology enrichment analysis based on the complete marker genes obtained for each monocyte cluster of cohort 1, showing the top 10 significant terms enriched in each cluster ranked by adjusted p values.
C, Back-mapping of monocyte clusters of cohort 2 (Figure 4C) onto the PBMC UMAP of cohort 2 (Figure 2D). The legend shows the association of the colors to the clusters together with the labeling of the clusters based on expressed marker genes (according to Figures 2 and S3D–S3F).
D, Violin plots of marker gene expression in the monocyte clusters identified in the complete PBMC space of cohort 2 (Figures 2C and 2D)
E, Dot plot of the top 10 marker genes sorted by average log fold change calculated for the monocyte clusters (Figure 4C).
F, Violin plots of the IFI6 and ISG15 expression in cells of mild and severe patients, additionally divided into early (1-10 days after disease onset) and late (> 10 days after disease onset). Statistical analysis was performed using Wilcoxon Rank Sum test adjusted with the Bonferroni method, ∗∗∗∗p < 0.0001.
G, Violin plots showing the time-dependent change of HLA-DRA and HLA-DRB1 expression in the monocyte population of cohort 1 (mild: n = 4; severe: n = 4) and cohort 2 (mild: n = 5; severe: n = 7). Mild samples are colored in yellow, severe samples in red and controls in blue, with the latter shown as reference violin plots representing the expression of all control monocytes in the respective cohort (cohort 1: n = 22, cohort 2: n = 6).
Figure 4.

Disease-Related Longitudinal Changes in Monocyte Transcriptomes
(A) UMAP visualization of monocytes (43,772 cells; from Figure 2C, cohort 2); 46 samples from controls (n = 6) and COVID-19 (mild, n = 7; severe, n = 8). Cells are colored according to the identified monocyte clusters (Louvain clustering, Table S4).
(B) Visualization of scaled expression of selected genes (monocyte markers, Figures 2 and S3E) using the UMAP defined in (A). Three main clusters defining monocytes in COVID-19 (HLA-DRloCD163hi, HLA-DRloS100Ahi, and HLA-DRhiCD83hi monocytes) indicated by dashed areas.
(C) AUCell-based enrichment of a gene signature from sepsis-associated monocytes (MS1 cells) (Reyes et al., 2020), violin plots of the area under the curve (AUC) scores. Horizontal lines: median of the respective AUC scores per cluster.
(D) Cytokine detection of IL-1β, tumor necrosis factor alpha (TNF)-α, and IL-12 in supernatants of purified monocytes (controls, ctrl, n = 3; COVID-19, mild, n = 3, and severe, n = 3) after 8 h in vitro incubation with or without 1 ng/mL LPS. Mean ± standard deviation. Kruskal-Wallis test adjusted with Benjamini-Hochberg method, ∗p < 0.05.
(E) Mapping of monocytes derived from COVID-19 patients (mild early, mild late, severe early, and severe late) onto UMAP from (A), coloring according to monocyte cluster identity.
(F) Cluster occupancy over time for patients with longitudinal scRNA-seq data (mild, n = 5; severe, n = 7), coloring according to (A). Vertical dashed lines: time points of sampling. Red bar, WHO ordinal scale; X, patient deceased. Patient IDs on the right side, grouping according to disease severity. Bold dotted line (right): patients classified as mild at initial sampling developing severe disease over time.
(G) Time-dependent change of IFI6 and ISG15 expression (violin-plots) in monocytes of cohort 1 (mild [yellow], n = 4; severe [red], n = 4), cohort 2 (mild [yellow], n = 5; severe [red], n = 7), and controls (cohort 1, n = 22, cohort 2, n = 6).
(H) Network representation of marker genes and their predicted upstream transcriptional regulators for monocyte clusters 0, 1, 2, and 3. Edges: predicted transcriptional regulation. Transcription factors (TFs, inner circle) and predicted target genes (outer circle) represented as nodes sized and colored according to the scaled expression level across all clusters. Selected TFs and genes labeled based on connectivity and literature mining. Numbers in the center refer to clusters defined in (A).
Transcription factor prediction indicated a STAT signaling-driven gene expression program in monocytes in COVID-19 (Figure 4H), with additional regulation by CEBPD and CEBPE, which have been implicated in gene expression programs of sepsis-associated monocytes (Reyes et al., 2020). STAT3 was predicted as a specific regulator of genes enriched in HLA-DR lo CD163 hi and HLA-DR lo S100A hi monocytes (clusters 2 and 0), in line with their immunosuppressive phenotype.
Taken together, dynamic changes of monocyte phenotypes were associated with COVID-19 disease severity and time after onset of disease.
Low-Density Neutrophils Emerge in Severe COVID-19 Patients Indicative of Emergency Myelopoiesis
PBMCs derived from blood samples of patients with severe COVID-19 contained two distinct clusters of LDNs (Figures 2A, clusters 5 and 6, and 2D, clusters 5 and 6). LDNs were slightly more frequent in cohort 1, and we analyzed these cells in more detail. Subsampling of all LDNs (Figure 5 A; Table S1) and re-clustering the cells revealed 8 transcriptionally distinct cell clusters (Figures 5A and 5B; Table S4). Based on published markers for pro- and pre-neutrophils, and mature neutrophils (Kwok et al., 2020; Ng et al., 2019; Scapini et al., 2016) we identified clusters 4 and 6 as CD81 + SPN(CD43)+ FUT4(CD15)+ CD63 + CEACAM8(CD66b)+ pro-neutrophils, clusters 3 and 5 as ITGAM(CD11b)+ CEACAM8(CD66b)+ CD101 +/− pre-neutrophils, and the remaining clusters as mature neutrophils (Figure S4 A). Accordingly, pro- and pre-neutrophils were enriched for transcriptional signatures of neutrophil progenitors derived from published single-cell data (Figure 4C) (Pellin et al., 2019; Popescu et al., 2019), and pro-neutrophils in clusters 4 and 6 showed the highest proportion of cells with a proliferative signature (Figure S4B). Clusters 0, 1, and 2 (originally in cluster 4 in Figure 2A) expressed mature neutrophil markers FCGR3B (CD16) and MME (CD10) (Figure S4A).
Figure 5.

Immature and Dysfunctional Low-Density Neutrophils Emerge in PBMC
(A) UMAP representation and clustering of low-density neutrophils (LDNs, 3,154 cells) in PBMCs (cohort 1, clusters 5/6, Figure 2A) from 21 samples (6 mild, 10 severe COVID-19). Left panel: cluster affiliation in Figure 2A. Right panel: data-driven clustering and cell type nomenclature based on marker genes (Table S4).
(B) Dot plot of the top 10 marker genes sorted by average log fold change associated with the neutrophil clusters identified in (A).
(C) Signature enrichment scores of single-cell data from neutrophil progenitors (Pellin et al., 2019; Popescu et al., 2019) in LDN clusters, plotted as violin plots. The lines in the violin plots represent the median of the respective AUC scores per cluster and the 0.25 and 0.75 quantiles. The ribosomalhi-specific cluster 7 was excluded from this analysis.
(D) Violin plots of expression of selected activation genes across the neutrophil clusters identified in (A). The panel of genes was chosen based on their described role in neutrophil extracellular trap formation (PRTN3, ELANE, MPO, and PADI4) and neutrophil activation and dysregulation (CD24, OLFM4, LCN2, BPI, CD274 [PD-L1], Arginase 1 [ARG1], and ANXA1).
(E) Expression of ARG1 and CD274(PD-L1) projected on the UMAP from (A).
Figure S4.

Additional Analysis of Dysfunctional Neutrophils in PBMC Fraction, Related to Figure 5
A, Dot plot of marker genes associated with immature neutrophils (pro- and pre-neutrophils), and mature neutrophils.
B, Pie charts showing the proportion of cells predicted to be in a given cell cycle stage. The numbers refer to the cell clusters presented in panel A.
Differential gene expression analysis for each cluster revealed extensive phenotypic heterogeneity within the LDN compartment (Figure 5B). LDNs mainly arise under pathological conditions, such as severe infection and sepsis in the context of emergency myelopoiesis (Schultze et al., 2019), and they have been associated with dysfunctional immune responses, marked by combined immunosuppression and inflammation (Silvestre-Roig et al., 2019). While LDN in cluster 1 expressed numerous ISGs (ISG15, IFITM1/3, and RSAD2), cluster 4 (pro-neutrophils) expressed genes (e.g., MPO, ELANE, and PRTN3) that are involved in neutrophil extracellular trap formation (Stiel et al., 2018; Thomas et al., 2014; You et al., 2019) among other functions and that have been associated with sepsis (Ahmad et al., 2019; Carbon et al., 2019; Silvestre-Roig et al., 2019). Both pre-neutrophil clusters expressed PADI4, another co-factor in NETosis (Leshner et al., 2012) (Figure 5D). NETs have recently been implicated in the pathogenesis of COVID-19 (Barnes et al., 2020; Zuo et al., 2020). Both pre-neutrophils (clusters 3 and 5) and pro-neutrophils expressed genes including CD24, OLFM4, LCN2, and BPI, previously associated with poor outcome in sepsis (Figures 5B and S4A) (Kangelaris et al., 2015).
All LDNs also expressed high levels of alarmins S100A8 and S100A9 (Figure 5D), whereas other S100 genes (e.g., S100A4 and S100A12) were strongly induced in selected neutrophil clusters. Finally, known inhibitors of T cell activation, namely CD274 (PD-L1) and Arginase 1 (ARG1) (Bronte et al., 2003; Li et al., 2018) were highly expressed in neutrophils in COVID-19 patients (Figure 5E). ARG1 + neutrophils in sepsis patients were shown to deplete arginine and constrain T cell function in septic shock (Darcy et al., 2014) and were predictive of the development of nosocomial infections (Uhel et al., 2017). Mature CD274(PD-L1)+ neutrophils (cluster 0) have been attributed suppressive functions in various conditions including HIV-1 infection (Bowers et al., 2014), cancer (Chun et al., 2015) and in lymph nodes (Castell et al., 2019), spleen (Langereis et al., 2017), and blood after LPS exposure (de Kleijn et al., 2013). ARG1 + cells were mainly immature neutrophils (clusters 3–6) and did not overlap with CD274 (PD-L1) expressing cells, indicating different populations of dysfunctional and potentially suppressive neutrophils in severe COVID-19.
LDNs recovered from PBMC fractions of COVID-19 patients revealed the presence of dysfunctional neutrophils and pointed toward multiple potentially deleterious pathways activated in severe COVID-19.
Persistent Increase of Activated Neutrophil Precursors and PD-L1+ Neutrophils in Severe COVID-19
Alterations of the neutrophil compartment were further interrogated by mass cytometry of whole blood samples of COVID-19 patients (n = 8 mild + 9 severe, cohort 1), FLI patients (n = 8), and age- and gender-matched controls (n = 9) (Table S1), using a panel designed to detect myeloid cell maturation and activation states as well as markers of immunosuppression or dysfunction (Table S2). Unsupervised clustering analysis of all neutrophils in all samples revealed 10 major clusters (Figure 6 A) of immature (clusters 2, 5, 6, and 7), mature (clusters 1, 3, and 4), and remaining clusters of low abundancy (clusters 8, 9, and 10). Based on their differential expression of CD11b, CD16, CD24, CD34, and CD38, clusters 5 and 6 were identified as pro-neutrophils and cluster 2 as pre-neutrophils (Kwok et al., 2020; Ng et al., 2019). The fourth immature cell cluster (7) showed very low expression of CD11b and CD16, reminiscent of pro-neutrophils, but lacking CD34, CD38, and CD24 (Figure 6A), suggesting a hitherto unappreciated pro-neutrophil-like population. The mature neutrophils segregated into non-activated (cluster 1), partially activated (cluster 3), and highly activated cells (cluster 4), based on the loss of CD62L and upregulation of CD64, as well as signs of proliferative activity (Ki67+) (Figure 6A).
Figure 6.

Appearance of Immature and PD-L1+ Neutrophils in Severe COVID-19
(A) Heatmap revealing differences in marker expression determined by mass cytometry (antibody panel 2, cohort 1) of main neutrophil cell cluster (1 to 10). Main individual neutrophil cell clusters are displayed in columns and marker identity is indicated in rows. MSI, marker staining intensity respective expression level. Significance level for the following comparisons: (1) controls (ctrl, n = 9) versus COVID-19 (mild and severe, n = 17, first row), (2) mild (n = 8) versus severe (n = 9, second row), (3) FLI (n = 8) versus mild COVID-19 (n = 8, third row), as well as (4) controls (ctrl, n = 9) versus FLI (n = 8) are indicated using a gray scale on top of the heatmap (see also p value scale next to the heatmap). Samples of COVID-19 patients collected between day 4 and 13 post-symptom onset (= first day of sample collection per patient). Abundance testing via generalized mixed effects models and multiple comparison adjustment using the Benjamini-Hochberg procedure and an FDR-cutoff of 5% across all clusters/subsets and between-group comparisons
(B) UMAP of neutrophils, down-sampled to 70,000 cells (30 markers, Table S2). Cells are colored according to main cell clusters (1 to 10, see table). Donor origin (blue, controls; gray, FLI; yellow, mild COVID-19; red, severe COVID-19).
(C) UMAP (from (B) with cells colored according to expression intensity of CD38, CD34, CD16, CD11b, CD33, CD64, CD62L, and CD45.
(D) Box and whisker (10–90 percentile) plots of main neutrophil cell clusters 1 to 7, reaching proportions of over 1%, measured by mass cytometry (whole blood, cohort 1): controls (n = 9), FLI (n = 8), and COVID-19 (mild, n = 8; severe, n = 9). Abundance testing via generalized mixed effects models and multiple comparison adjustment using the Benjamini-Hochberg procedure and an FDR-cutoff of 5% across all clusters/subsets and between-group comparisons.
(E) Box and whisker (10–90 percentile) plots of proportions of CD34+, CD11blo/−CD16−, CD64+, CD62L+, CD10−CD11blo/−CD16− (reported from panel 1) and PD-L1+ neutrophils (whole blood, cohort 1): controls (n = 9), FLI (n = 8), and COVID-19 (mild, n = 8; severe, n = 9). Kruskal-Wallis and Dunn’s multiple comparison tests.
∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Neutrophils from COVID-19 patients clearly separated from those of controls and also FLI patients in UMAP analysis (Figure 6B), and neutrophils in patients with severe COVID-19 were distinct from those of patients with mild disease (Figure 6B). Cells from control donors accumulated in areas enriched for mature non-activated cells (cluster 1) and immature pre-neutrophil-like cells (cluster 2). In contrast, neutrophils from FLI patients were mainly mature non-activated (cluster 1) and mature highly activated (cluster 4) cells. Neutrophils from COVID-19, particularly from patients with severe disease, primarily occupied immature pre- and pro-neutrophil-like clusters. Plotting cell cluster-specific surface marker expression onto the UMAPs (Figure 6C) as well as statistical analyses of cell cluster distribution and surface marker expression among different patient groups supported these observations (Figures 6D and 6E). Samples from FLI patients contain a high proportion of highly activated mature neutrophils, but barely any immature neutrophils. In contrast, severe COVID-19 is associated with the appearance of immature pre- and pro-neutrophils (Figures 6D and 6E). Interestingly, immature cell clusters in severe COVID-19 showed signs of recent activation like upregulation of CD64 (Mortaz et al., 2018), RANK, and RANKL (Riegel et al., 2012), as well as reduced CD62L expression (Mortaz et al., 2018). In addition to loss of CD62L, immature and mature neutrophils from severe COVID-19 showed elevated PD-L1 expression compared to control samples (Figure 6E). Indeed, CD62L downregulation and high PD-L1 expression has been frequently associated with suppressive function of neutrophils and granulocytic myeloid-derived suppressor cells (gMDSCs) (Bronte et al., 2016; Cassetta et al., 2019; Kamp et al., 2012; Pillay et al., 2012; Tak et al., 2017; Testa et al., 2004; Younos et al., 2015). Interestingly, a recent study described a high abundance of similar immature and dysfunctional CD64+ and PD-L1+ neutrophils in sepsis patients (Meghraoui-Kheddar et al., 2020).
Thus, SARS-CoV-2 infection induces major alterations in the neutrophil compartment. While neutrophils in FLI patients display a mature activated phenotype, a release of immature neutrophils with phenotypic signs of immunosuppression and dysfunction is a hallmark of severe COVID-19.
We next assessed the dynamics of the changes within the myeloid cell compartment over time. We grouped samples according to collection time as “early” (within the first 10 days) or late (during the following 20 days) after onset of symptoms. In both cohorts, we observed a tendency toward (cohort 1) or significantly higher (cohort 2) proportions of granulocytes in severe versus mild COVID-19 patients, both at early and late time points (Figure S5 A). We observed a persistent release of immature neutrophils (e.g., cluster 6) in severe COVID-19 (Figure S5B) showing high expression of CD64 and PD-L1, but downregulation of CD62L as a sign of activation, dysfunction, and immunosuppression (Figure S5C). In addition, severe COVID-19 patients show further increased frequencies of mature, partially activated neutrophils (cluster 3) at later time periods (Figure S5B). Thus, the neutrophil compartment of severe COVID-19 patients is characterized by a combination of persistent signs of inflammation and immunosuppression, which is reminiscent of long-term post-traumatic complications (Hesselink et al., 2019).
Figure S5.

Longitudinal Analysis of Neutrophil and Monocyte Cell Populations, Related to Figure 6
A, Box and whisker (10-90 percentile) plots of time-dependent differences in total granulocytes and monocytes, non-classical monocytes and correlation analysis between days post-symptom onset and proportion of non-classical monocytes.
B, Box and whisker (10-90 percentile) plots of time-dependent differences in main neutrophil cell cluster 3, 5, 6 and 7 in cohort 1.
C, Box and whisker (10-90 percentile) plots of time-dependent differences in proportions of CD34+, CD11blo/-CD16-, CD64+, CD62L+, CD10-CD11blo/-CD16- (reported from panel 1) and PD-L1+ neutrophils in cohort 1.
D, Box and whisker (10-90 percentile) plots of time-dependent differences in main monocyte cluster 1, 10 (belonging to CD14hiCD16- classical monocytes), cluster 11 and 3 (belonging to CD14hiCD16+ intermediate monocytes) in cohort 1.
E, Box and whisker (10-90 percentile) plots of time-dependent differences in CXCR3+, HLA-DRhiCD11chi and CD226+CD69+ monocytes.
(F), Box and whisker (10-90 percentile) plot showing time-dependent differences in HLA-DRhiCD11chi monocytes in cohort 2.
Measurements in cohort 1 were done applying mass cytometry on whole blood samples distinguishing between COVID-19 patients with mild (days 0-10: n = 6, days 11-30: n = 12) or severe disease (days 0-10: n = 9, days 11-30: n = 13) course. Mixed-effect-analysis and Sidak’s multiple comparison test was used to calculate significant differences
Measurements in cohort 2 were done with flow cytometry on 26 whole blood samples from COVID-19 patients showing either mild (n = 8) or severe disease (n = 18) course as well as 11 samples from age-matched controls (n = 10).
∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001
We also analyzed time-dependent phenotypic changes in the monocyte compartment by mass cytometry. Non-classical monocytes started to recover in COVID-19 patients during the later stages of the disease (Figure S5A). HLA-DRhiCD11chi monocyte cell clusters also declined at later time points in mild COVID-19 (Figures S5D–S5F), which correlates well with the longitudinal changes of IFI6 and ISG15 as well as HLA-DRA and HLA-DRB1 expression profiles (Figures 4G and S3F). In contrast, overall proportions of HLA-DRhiCD11chi monocytes in severe COVID-19 remained low throughout the course of the disease. Proportions of CD10hi macrophage-like cluster 10 and CD226+CD69+ monocytes were generally higher at later stages in severe COVID-19, which resembled the kinetics of HLA-DR lo S100A hi monocytes identified by scRNA-seq (Figure 4F). This indicates a prolonged alternative activation of monocytes in severe COVID-19 (Figure S5E).
Single-Cell Transcriptomes of Whole Blood Reveal Suppressive-like Neutrophils in Severe COVID-19
Whole blood CyTOF analysis (cohort 1) clearly indicated very distinct phenotypic alterations of the neutrophil compartment in mild and severe forms of COVID-19. To further delineate the underlying transcriptional programs, we performed scRNA-seq analysis on fresh whole blood samples of 23 individuals (34 samples, cohort 2, Table S1). Integrated visualization of all samples of cohort 2 (fresh/frozen PBMCs, fresh whole blood, 229,731 cells, Figure S6 A) revealed the expected blood leukocyte distribution, including granulocytes (Figures 7 A and S6A; Table S4). Cell-type distribution identified by scRNA-seq profiles (Figure S6B) strongly correlated with MCFC characterization of the same samples (Figure S6C). For further analysis of the granulocyte compartment, we first combined the whole blood samples with the fresh PBMCs to guide the clustering of all major immune cells resulting in a total of 122,954 cells (Figure 7A). From these samples, we identified all neutrophil clusters and extracted the cells derived from whole blood for subsampling, which revealed a structure of 9 clusters (n = 58,383 cells) (Figures 7B and 7C).
Figure S6.

Overview of scRNA-Seq Dataset from Cohort 2 and Additional Characterization of Neutrophils, Related to Figure 7
(A), UMAP (on the left) of the complete scRNA-seq dataset from cohort 2 (frozen PBMC, fresh PBMC, fresh whole blood), encompassing 98 samples from 16 controls, 8 mild, and 10 severe COVID-19 patients. Right panel: violin plots of the number of genic reads, transcripts and genes expressed in the PBMC (right) versus the granulocyte fraction (left) across the different datasets of cohort 2. The UMAP is split by experimental condition and the classified granulocyte and PBMC fractions are marked separately. The table below indicates the number of cells per experimental condition separated by control, COVID-19 mild and COVID-19 severe. The numbers of samples are indicated in brackets.
(B), Box and whisker plots (25–75 percentile) of cell type frequencies identified by scRNA-seq in fresh whole blood samples after erythrocyte lysis comparing 16 samples from 15 controls, 6 from 5 mild COVID-19 and 12 from 4 severe COVID-19 patients.
(C), Comparison between cell frequencies identified by scRNA-seq and MCFC. Pearson’s correlation between the mean of each cell population measured in MCFC (y axis) and by scRNA-seq of R2 = 0.96 with p = 0.0098 (left). The stacked bar chart sorted by disease severity shows the cell type frequency for controls (n = 16), mild (n = 5) and severe COVID-19 samples (n = 18) split by scRNA-seq and MCFC.
(D), Dot plot of literature-based marker genes classifying different neutrophil subsets.
(E), UMAP of neutrophils showing the scaled expression of MME(CD10) and CXCR4 with enrichment in the control-specific clusters 0.
(F), UMAP of AUCell-based enrichment of gene signatures derived from the neutrophil clusters from cohort 2 on the UMAP visualization of cohort 1. The UMAP is colored by the ‘Area Under the Curve’ (AUC) scores of each cell.
(G), Dot plot visualization of selected significantly enriched Gene Ontology terms and KEGG pathways for each cluster from the neutrophil space. The dots are colored by their adjusted p value and the size of the dots is defined by the number of genes found in the Gene Ontology term.
(H), Network representation of marker genes and their predicted upstream transcriptional regulators for neutrophil clusters 6 (pre-Neutrophils) and 8 (pro-Neutrophils). Edges represent predicted transcriptional regulation. Transcription factors in the inner circle and their predicted target genes in the outer circle are represented as nodes sized and colored according to the scaled expression level across all clusters. Selected genes and transcription factors were labeled based on connectivity and literature mining.
(I), Diffusion map dimensionality reduction of the main neutrophil clusters 8, 6, 2, and 1 from the severe COVID-19 patients (top) and diffusion pseudotime visualized on the diffusion map indicating the transition probability of the different clusters in the following order: 8 - 6 - 2 - 1 (bottom).
(J), Genes specific for each cluster (HSP90AA1, CD274(PD-L1), CD177, MME(CD10), ARG1) visualized along the diffusion pseudotime (top) with the density of each cluster along the pseudotime (bottom) highlighting the proposed order of differentiation of the different neutrophil subsets.
Figure 7.

Immature and Dysfunctional Whole-Blood Neutrophils in Severe COVID-19
(A) UMAP of 35 fresh blood samples from cohort 2 (122,954 cells, PBMCs, and whole blood): controls (n = 17), mild COVID-19 (early, n = 3; late, n = 3) and severe COVID-19 (early, n = 3, late = 9). Clusters defined by Louvain clustering. Cell types assigned based on reference-based cell type classification (Aran et al., 2019) and marker gene expression (Table S4).
(B) UMAP visualization of neutrophils (58,383 cells; 34 whole blood samples, cohort 2): controls (n = 16), mild COVID-19 (early, n = 3; late, n = 3), and severe COVID-19 (early, n = 3; late, n = 9). Clusters defined by Louvain clustering (Table S4).
(C) Nomenclature and marker genes for each neutrophil cluster from (B).
(D) Dot plot of selected marker genes for each neutrophil cluster from (B).
(E) Dot plot of genes from different functional classes (based on literature research). Clusters 8, 6, 1, and 2 are specific for severe COVID-19, cluster 0 represents homeostatic mature neutrophils from controls.
(F) Heatmap divided by disease severity and stage (early versus late) showing the proportion of each patient group for each cluster.
(G) Density plot of cell frequency by disease severity and stage (early versus late) overlaid on the UMAP of the neutrophil space.
(H) UMAP visualization showing scaled expression of CD274 (PD-L1) and FCGR1A (CD64).
(I) Violin plots showing AUCell-based enrichment as AUC scores of gene signature from granulocytic myeloid-derived suppressor cells (Bayik et al., 2020) and PD-L1hi neutrophils after LPS exposure (de Kleijn et al., 2013) in neutrophil clusters from (B). Horizontal lines: median of the respective AUC scores per cluster and 0.25 and 0.75 quantiles.
(J) Network representation of marker genes and their predicted upstream transcriptional regulators for neutrophil clusters 1 (mature/COVID-19 severe-specific) and 0 (mature/control-specific). Edges in cluster color: predicted transcriptional regulation. TFs (inner circle) and their predicted target genes (outer circle): nodes, sized, and colored according to scaled expression level across all clusters. Selected genes and TFs labeled based on connectivity and literature mining.
(K) Box and whisker (10-90 percentile) plots representing the hematological analyses (whole blood, cohort 1): mild (n = 11), severe (n = 21) COVID-19. Analytes, measured by flow cytometry in white blood cell differential channel, included absolute counts of immature granulocytes (IG, dotted line: upper limit of reference range) and width of neutrophil cytometric dispersions (NE-WX, dispersion of side scatter; NE-WY, dispersion of side fluorescence light; NE-WZ, dispersion of forward scatter). Mann Whitney test applied to IG count analysis and mixed-effect-analysis and Sidak’s multiple comparison test to NE-WX, NE-WY, and NE-WZ analyses.
(L) Box and whisker (10–90 percentile) plots of E. coli- and PMA-induced neutrophil oxidative burst (reactive oxygen species [ROS] production) and phagocytosis of whole blood samples (cohort 1; mild, n = 10; severe [n = 8] COVID-19) in comparison to controls measured by flow cytometry. Dotted line: relative level of controls run in the assay. Mixed-effect-analysis and Sidak’s multiple comparison test. ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Using marker- and data-driven approaches as applied to LDN (Figure 5D and S4A), we identified FUT4(CD15) + CD63 + CD66b + pro-neutrophils, ITGAM(CD11b)+ CD101 + pre-neutrophils, along with 7 mature neutrophil clusters (Figures 7B–7D and S6D; Table S4). Heterogeneous expression of various markers involved in mature neutrophil function including CXCR2, FCGR2A (CD32), FCGR1A (CD64), or MME (CD10), pointed toward distinct functionalities within the neutrophil compartment (Figures 7E, S6D, and S6E). Seven of the nine neutrophil clusters identified in whole blood in cohort 2 could also be mapped to the fresh PBMC transcriptomes in cohort 1 (Figure S6F), indicating that scRNA-seq of fresh PBMC in COVID-19 patients reveals relevant parts of the neutrophil space. The transcriptional phenotype of pro- and pre-neutrophils (cluster 8+9) was corroborated in cohort 2 (Figures 7B–7D and S6D).
Heatmap and UMAP visualization of the cell type distribution identified pro- and pre-neutrophils mainly at late time points in severe COVID-19 (Figures 7F and 7G). Furthermore, mature neutrophils with a high IFN-signature (cluster 1) were associated with severe COVID-19 (Figures 7E and S6G). This cluster was also enriched for markers identified by CyTOF as differentially expressed in patients with severe COVID-19 (Figure 6), such as elevated expression of CD274 (PD-L1) and FCGR1A (CD64) (Figure 7H). In addition to CD274, cells in cluster 1 expressed genes indicative of a potentially suppressive or anti-inflammatory state, including ZC3H12A (Figure 7E), which is known to suppress hepatitis C virus replication and virus-induced pro-inflammatory cytokine production (Lin et al., 2014). Cluster 2 was also enriched for cells from COVID-19 patients, mainly from severe but also mild cases (Figures 7F and 7G).
Gene signatures from granulocytic MDSC (Bayik et al., 2020) and CD274(PD-L1)+ neutrophils after LPS exposure (de Kleijn et al., 2013), both shown to be immunosuppressive, were enriched in clusters 1, 2, and 6, which mainly harbor cells from severe COVID-19. This indicates a suppressive functionality of these cells in severe COVID-19 (Figure 7I). Predictions of transcription factor (TF)-based regulation of the cluster-specific gene signatures separated mature neutrophils from patients with severe COVID-19 (cluster 1) and control patients (cluster 0) (Figure 7J). IFN-response genes are mainly controlled by STAT and IRF TFs, whereas the transcriptional signature of cluster 0 is mainly driven by the CEBP TF family. The TF network underlying the transcriptional difference in pro-neutrophils is mainly driven by E2F family members and pre-neutrophils mainly depend on ETS TFs (Figure S6H).
Pseudotime analysis strongly supported the differentiation trajectory from pro-neutrophils (cluster 8) via pre-neutrophils (cluster 6) to mature neutrophils in cluster 2 and 1 (Figures S6I and S6J). Particularly CD274 (PD-1L) was enriched in cluster 1 compared to cluster 2, supporting the potential of neutrophils to progress toward a suppressive phenotype in severe COVID-19 (Figure S6J). Interestingly, CD177 is expressed in pre-neutrophils and persisting in cluster 1 further highlighting the newly emerging character of this cluster (Volkmann et al., 2020).
Finally, we studied whether the persistent emergence of immature, potentially dysfunctional neutrophils in severe COVID-19 patients can be captured under routine diagnostic conditions. Therefore, samples of 32 COVID-19 patients (Table S1, cohort 1) were characterized by routine hematology analyses using a clinical flow cytometry system (Sysmex analyzer). Indeed, the assumption of rescue myelopoiesis in severe COVID-19 was supported by significantly higher counts in the population of immature granulocytes (IG, representing promyelocytes, myelocytes, and metamyelocytes) in this patient group (Figure 7K). We also found significant differences in the neutrophil compartment, when analyzing the width of dispersion with respect to granularity, activity, and cell volume defined as NE-WX, NE-WY, and NE-WZ, respectively. As compared to patients with mild course, severely ill patients displayed increases in width of dispersion of activity and cell volume as surrogates for increased cellular heterogeneity, immaturity, and dysregulation in severe COVID-19 (Figure 7K), resembling previously described alterations in sepsis patients (Stiel et al., 2016). Furthermore, neutrophils of severe COVID-19 patients were partially dysfunctional, because their oxidative burst upon stimulation with standardized stimuli (E. coli or PMA) was strongly impaired in comparison to control and mild COVID-19 neutrophils, whereas phagocytic activity was preserved (Figure 7L; Table S1).
Collectively, the neutrophil compartment in peripheral blood of severe COVID-19 patients is characterized by the appearance of LDN, FUT4(CD15)+ CD63 + CD66b + pro-neutrophils, and ITGAM(CD11b)+ CD101 + pre-neutrophils, reminiscent of emergency myelopoiesis, as well as CD274(PD-L1)+ ZC3H12A + mature neutrophils reminiscent of gMDSC-like cells, which might exert suppressive or anti-inflammatory functions.
Discussion
SARS-CoV-2 infection generally causes mild disease in the majority of individuals, however, ∼10%–20% of COVID-19 patients progress to severe disease with pneumonia and respiratory failure. The reported case-fatality rates among patients with critical illness and respiratory failure vary, with a mean of ∼25% (Quah et al., 2020). Dysregulated immune responses have been described in patients with severe COVID-19 (Chua et al., 2020; Giamarellos-Bourboulis et al., 2020; Lucas et al., 2020; Merad and Martin, 2020; Messner et al., 2020; Wei et al., 2020; Zhou et al., 2020b). Hence, detailed knowledge of the cellular and molecular processes that drive progression from mild disease to potentially fatal courses of COVID-19 is urgently needed to identify predictive biomarkers and therapeutic targets.
Here, we employed four complementary technologies at single-cell resolution to assess alterations in the systemic immune response in mild or severe courses of COVID-19. We analyzed a total of 53 patients (161 samples) from two independent cohorts collected at two university medical centers in Germany (Kurth et al., 2020). Combining single-cell transcriptomics with single-cell proteomics, using different technological platforms in two independent patient cohorts, provided a detailed view of the systemic immune responses in COVID-19 and allowed for cross-validation and in-depth interrogation of key findings. The results were further supported by additional routine diagnostics lab measurements and functional assays, linking the results of the exploratory investigations to functional phenotypes and clinically relevant diagnostics.
This multipronged approach revealed drastic changes within the myeloid cell compartment during COVID-19, particularly in patients with a severe course of disease. Early activation of HLA-DRhiCD11chi/HLA-DR hi CD83 hi monocytes with a strong antiviral IFN-signature was a hallmark of mild COVID-19, which receded during the natural course of disease. In contrast, HLA-DRlo dysfunctional monocytes along with clear evidence of emergency myelopoiesis with release of immature neutrophils including pro- and pre-neutrophils into the circulation marked severe COVID-19. Furthermore, we identified neutrophils in severe COVID-19 with transcriptional programs reminiscent of dysfunction and immunosuppression not observed in controls or patients with mild COVID-19. Thus, defective monocyte activation and dysregulated myelopoiesis may contribute to severe disease course and ARDS development (Middleton et al., 2020).
Previous immunophenotyping studies have reported an increase of inflammatory monocytes with a strong IFN-response in COVID-19 (Liao et al., 2020; Merad and Martin, 2020; Zhou et al., 2020b). Mononuclear phagocytes and neutrophils appear to dominate inflammatory infiltrates in the lungs, and resident alveolar macrophages are replaced by inflammatory monocyte-derived macrophages in patients with severe COVID-19 (Chua et al., 2020; Liao et al., 2020). Here, we report substantial time- and disease severity-dependent alterations of the monocyte compartment in COVID-19. Marked depletion of CD14loCD16hi non-classical monocytes observed in all COVID-19 patients, but not in patients with SARS-CoV-2 negative FLI (Figure 1D), is in line with previous reports on COVID-19, and other severe viral infections (Lüdtke et al., 2016; Naranjo-Gómez et al., 2019). Single-cell proteomics and transcriptomics revealed a transient increase in highly activated CD14+HLA-DRhiCD11chi (HLA-DRA hi CD83 hi) monocytes in mild COVID-19. This was similar in patients presenting with common cold or FLI, but absent in severe COVID-19 (Figures 3 and 4). In contrast, in severe COVID-19, monocytes showed low expression of HLA-DR, and high levels of MAFB, PLBD1, and CD163, all of which are associated with anti-inflammatory macrophage functions (Bronte et al., 2016; Cuevas et al., 2017; Fischer-Riepe et al., 2020; MacParland et al., 2018). Low HLA-DR expression on monocytes is an established surrogate marker of immunosuppression in sepsis (Venet et al., 2020). Elevated HLA-DRlo monocytes have been associated with an increased risk of infectious complications after trauma (Hoffmann et al., 2017) and fatal outcome in septic shock (Monneret et al., 2006). Indeed, the HLA-DR lo CD163 + monocytes showed enrichment of genes associated with poor prognosis in sepsis patients, including PLAC8 (Maslove et al., 2019) and MPO (Schrijver et al., 2017) (Figure 4B). In line with this dysfunctional phenotype, PLAC8 was recently shown to suppress production of IL-1β and IL-18 (Segawa et al., 2018). In fact, we observed that inflammatory cytokine production, including IL-1β release, was impaired in monocytes from patients with severe COVID-19 (Figure 4). CD14+HLA-DRlo monocytes have also been implicated with immunosuppression in cancer patients (Bronte et al., 2016; Mengos et al., 2019; Meyer et al., 2014). While exhibiting anti-inflammatory features, especially in the early stages of severe disease (Figures S3C–S3F), persistently high expression of CD226 and CD69 may promote tissue infiltration and organ dysfunction (Davison et al., 2017; Reymond et al., 2004; Vo et al., 2016).
Acute pathological insults, such as trauma or severe infections, trigger a process referred to as emergency myelopoiesis to replenish functional granulocytes and other hematopoietic cells. Emergency myelopoiesis is characterized by the mobilization of immature myeloid cells, which are often linked to immunosuppressive functions (Loftus et al., 2018; Schultze et al., 2019). In fact, emergence of suppressive myeloid cells including neutrophils, often referred to as granulocytic MDSCs, has been observed during sepsis and severe influenza (Darcy et al., 2014; Loftus et al., 2018; Sander et al., 2010; De Santo et al., 2008). LDN in PBMC fractions in severe COVID-19 contained immature neutrophils, including pro- and pre-neutrophils, which was not observed in mild cases (Figure 5). These immature LDNs showed a surface marker and gene expression profile reminiscent of granulocytic MDSCs including genes such as S100A12, S100A9, MMP8, ARG1 (Uhel et al., 2017), and OLFM4, which has been recently associated with immunopathogenesis in sepsis (Alder et al., 2017). Emergence of pro-neutrophils in severe COVID-19 was also detected by single-cell proteomics on whole blood samples. Strikingly, both immature and the mature neutrophils showed increased expression of CD64 and PD-L1 (Figures 6 and S5), similar to recently described alterations in sepsis (Meghraoui-Kheddar et al., 2020). In addition to the altered phenotype, we also observed an altered functionality. Neutrophils from patients with severe COVID-19 showed an impaired oxidative burst response, while their phagocytic capacity was preserved (Figure 7).
Single-cell transcriptomics of whole blood samples revealed mature activated neutrophils in both mild and severe COVID-19 (Figure 7B, cluster 2), however, expression of CD274 (PD-L1) was only found in severe COVID-19 (cluster 1), and it increased in later stages of the disease. Expression of PD-L1 on neutrophils has been associated with T cell suppression (Bowers et al., 2014; Castell et al., 2019; de Kleijn et al., 2013; Langereis et al., 2017), suggesting that neutrophils in severe COVID-19 might exert suppressive functions. Furthermore, the expression of CD177 on mature activated neutrophils and the identification of genes associated with anti-inflammatory functions (CD274 and ZC3H12A) suggest a model in which neutrophils emerging prematurely from the bone marrow are programmed toward an anti-inflammatory or even suppressive phenotype in severe COVID-19. The transcriptional programs induced in immature neutrophils, including pro- and pre-neutrophils, as well as in COVID-19-associated mature neutrophil clusters, align with other observations in severe COVID-19 patients, including increased NET formation (Barnes et al., 2020; Zuo et al., 2020), coagulation (Klok et al., 2020; Pfeiler et al., 2014), and immunothrombosis (Stiel et al., 2018; Xu et al., 2020). In contrast, these transcriptional programs were not observed in patients with mild COVID-19 or in SARS-CoV-2 negative controls, even though the latter exhibited a range of comorbidities (e.g., COPD, type II diabetes).
Thus, defective or repressed monocyte activation combined with dysregulated myelopoiesis may cause a deleterious loop of continuous tissue inflammation and ineffective host defense.
Limitations of Study
The pathophysiological consequences of the dysfunctional phenotype of myeloid cells in severe COVID-19 remain unclear at this stage. It is, however, highly likely that they contribute to immunosuppression in critically ill patients, potentially leading to insufficient host defense, disbalanced inflammation, and increased susceptibility to superinfections. While our dual cohort study design provided robust and reproducible results concerning the alterations within the myeloid compartment in COVID-19, it is too early to speculate on the underlying mechanisms driving this response, such as genetics, lifestyle, comorbidities, environmental factors, or initial viral load (Ellinghaus et al., 2020). Utilizing the herein established transcriptional and functional phenotypes of the myeloid cell compartment, it will likely be possible to estimate the potential contribution of the causes mentioned above in larger clinical studies in the future and to address potential upstream events of immune dysregulation in preclinical model systems as they become available (Bao et al., 2020; Cohen, 2020). Indeed, in future studies it will be interesting to dissect whether the myeloid subsets in COVID-19 are anti-inflammatory or even capable of suppressing other immune cells, and which pathways might be mainly involved. Clearly, PD-L1 is a prime candidate (Bowers et al., 2014; Castell et al., 2019; de Kleijn et al., 2013; Langereis et al., 2017).
Collectively, our data link a striking appearance of immature and dysfunctional cells, in both the monocyte and neutrophil compartment, to disease severity in COVID-19. Consequently, the development of treatments and prevention strategies for severe COVID-19 may benefit from insights gained in other fields such as oncology, which have successfully applied therapies targeting suppressive myeloid cells.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| HLA-DR BV421 (L243) | Biolegend | Cat# 307635; RRID:AB_10897449 |
| CD4 BV510 (OKT4) | Biolegend | Cat# 317444; RRID:AB_2561866 |
| CD16 BV605 (3G8) | Biolegend | Cat# 302039; RRID:AB_2561354 |
| CD45 BV711 (HI30) | Biolegend | Cat# 304050; RRID:AB_2563466 |
| CD8 BV785 (SK1) | Biolegend | Cat# 344740; RRID:AB_2566202 |
| CD66b FITC (G10F5) | Biolegend | Cat# 305104; RRID:AB_314496 |
| CD14 PerCp-Cy5.5 (MφP9) | Becton Dickinson | Cat# 562692; RRID:AB_2737726 |
| CD56 PE (MY31) | Becton Dickinson | Cat# 345810; RRID:AB_396511 |
| CD3 PE/Dazzle (UCHT1) | Biolegend | Cat# 300450; RRID:AB_2563618 |
| CD11c PE/Cy5 (B-ly6) | Becton Dickinson | Cat# 551077; RRID:AB_394034 |
| Siglec8 PE/Cy7 (7C9) | Biolegend | Cat# 347112; RRID:AB_2629720 |
| CD203c APC (NP4D6) | Biolegend | Cat# 324609; RRID:AB_2099774 |
| CD1c AlexaFluor700 (L161) | Biolegend | Cat# 331530; RRID:AB_2563657 |
| CD19 APC/Fire 750 (HIB19) | Biolegend | Cat# 302258; RRID:AB_2629691 |
| CD45 89Y (HI30) | Fluidigm | Cat# 3089003; RRID:AB_2661851 |
| HLA-DR purified (L243) | Biolegend | Cat# 307602; RRID:AB_314680 |
| CD3 purified (UCHT1) | Biolegend | Cat# 300443; RRID:AB_2562808 |
| CD196 141Pr (G034E3) | Fluidigm | Cat# 3141003A; RRID:AB_2687639 |
| CD19 142Nd (HIB-19) | Fluidigm | Cat# 3142001; RRID:AB_2651155 |
| CD123 143Nd (6H6) | Fluidigm | Cat# 3143014B; RRID:AB_2811081 |
| CD15 144Nd (W6D3) | Fluidigm | Cat# 3144019B |
| CD138 145Nd (DL101) | Fluidigm | Cat# 3145003B |
| CD64 146Nd (10.1) | Fluidigm | Cat# 3146006; RRID:AB_2661790 |
| CD21 purified (Bu32) | Biolegend | Cat# 354902; RRID:AB_11219188 |
| CD226 purified (REA1040) | Miltenyi Biotec | Produced at request |
| IgD purified (IgD26) | Biolegend | Cat# 348235; RRID:AB_2563775 |
| ICOS 148Nd (C398.4A) | Fluidigm | Cat# 3148019B; RRID:AB_2756435 |
| CD206 purified (152) | Biolegend | Cat# 321127; RRID:AB_2563729 |
| CD96 purified (REA195) | Miltenyi Biotec | Produced at request |
| KLRG1 purified (REA261) | Miltenyi Biotec | Produced at request |
| TCRgd purified (11F2) | Miltenyi Biotec | Produced at request |
| FceRI 150Nd (AER-37) | Fluidigm | Cat# 3150027B |
| CD155 purified (REA1081) | Miltenyi Biotec | Produced at request |
| CD95 purified (DX2) | Biolegend | Cat# 305631; RRID:AB_2563766 |
| TIGIT 153Eu (MBSA43) | Fluidigm | Cat# 3153019B; RRID:AB_2756419 |
| CD62L 153Eu (DREG56) | Fluidigm | Cat# 3153004B; RRID:AB_2810245 |
| CD62L purified (DREG56) | Biolegend | Cat# 304835; RRID:AB_2563758 |
| CD1c purified (L161) | Biolegend | Cat# 331502; RRID:AB_1088995 |
| CD27 155Gd (L128) | Fluidigm | Cat# 3155001B; RRID:AB_2687645 |
| CXCR3 156Gd (G025H7) | Fluidigm | Cat# 3156004B; RRID:AB_2687646 |
| KLRF1 purified (REA845) | Miltenyi Biotec | Produced at request |
| CD10 158Gd (HI10a) | Fluidigm | Cat# 3158011B |
| CD33 158Gd (WM53) | Fluidigm | Cat# 3158001; RRID:AB_2661799 |
| CD14 160Gd (RMO52) | Fluidigm | Cat# 3160006; RRID:AB_2661801 |
| CD28 purified (L293) | BD Bioscience | Cat# 348040; RRID:AB_400367 |
| CD69 162Dy (FN50) | Fluidigm | Cat# 3162001B |
| CD294 163Dy (BM16) | Fluidigm | Cat# 3163003B; RRID:AB_2810253 |
| RANKL APC | Miltenyi Biotec | Cat# 130-098-511; RRID:AB_2656691 |
| Anti-APC 163Dy | Fluidigm | Cat# 3163001B; RRID:AB_2687636 |
| CXCR5 164Dy (51505) | Fluidigm | Cat# 3164016B; RRID:AB_2687858 |
| Siglec 8 164Dy (7C9) | Fluidigm | Cat# 3164017B |
| CD34 166Er (581) | Fluidigm | Cat# 3166012B; RRID:AB_2756424 |
| CD38 167Er (HIT2) | Fluidigm | Cat# 3167001B; RRID:AB_2802110 |
| Ki67 168Er (Ki-67) | Fluidigm | Cat# 3168007B; RRID:AB_2800467 |
| CD25 169Tm (2A3) | Fluidigm | Cat# 3169003; RRID:AB_2661806 |
| CD24 169Tm (ML5) | Fluidigm | Cat# 3169004B; RRID:AB_2688021 |
| Lag3 purified (11C3C65) | Biolegend | Cat# 369302; RRID:AB_2616876 |
| RANK purified (80704) | R&D Systems | Cat# MAB683; RRID:AB_2205330 |
| CD161 purified (HP-3G10) | Biolegend | Cat# 339919; RRID:AB_2562836 |
| CD11b purified (ICRF44) | Biolegend | Cat# 301337; RRID:AB_2562811 |
| CD45RO purified (4G11) | DRFZ Berlin | N/A |
| CD44 purified (BJ18) | Biolegend | Cat# 338811; RRID:AB_2562835 |
| CD137 173Yb (4B4-1) | Fluidigm | Cat# 3173015B |
| PD-1 175Lu (EH12.2H7) | Fluidigm | Cat# 3175008; RRID:AB_2687629 |
| PD-L1 175Lu (29.E2A3) | Fluidigm | Cat# 3175017B; RRID:AB_2687638 |
| CD56 176Yb (NCAM16.2) | Fluidigm | Cat# 3176008; RRID:AB_2661813 |
| CD8A purified (GN11) | DRFZ Berlin | N/A |
| IgM purified (MHM-88) | Biolegend | Cat# 314502; RRID:AB_493003 |
| CD11c purified (Bu15) | Biolegend | Cat# 337221; RRID:AB_2562834 |
| B2M purified (2M2) | Biolegend | Cat# 316302; RRID:AB_492835 |
| CD16 209Bi (3G8) | Fluidigm | Cat# 3209002B; RRID:AB_2756431 |
| A0251 anti-human Hashtag 1 | Biolegend | Cat# 394601; RRID:AB_2750015 |
| A0252 anti-human Hashtag 2 | Biolegend | Cat# 394603; RRID:AB_2750016 |
| A0253 anti-human Hashtag 3 | Biolegend | Cat# 394605; RRID:AB_2750017 |
| A0254 anti-human Hashtag 4 | Biolegend | Cat# 394607; RRID:AB_2750018 |
| A0255 anti-human Hashtag 5 | Biolegend | Cat# 394609; RRID:AB_2750019 |
| A0256 anti-human Hashtag 6 | Biolegend | Cat# 394611; RRID:AB_2750020 |
| A0257 anti-human Hashtag 7 | Biolegend | Cat# 394613; RRID:AB_2750021 |
| CD235ab Biotin (HIR2) | Biolegend | Cat# 306618; RRID:AB_2565773 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| BD Horizon Brilliant Stain Buffer | Becton Dickinson | Cat# 563794 |
| RBC lysis buffer 10X | Biolegend | Cat# 420301 |
| Pierce 16% Formaldehyde (w/v), Methanol-free | Thermo Fisher | Cat# 28908 |
| RPMI 1640 Medium | GIBCO | Cat# 11875093 |
| Fetal Bovine Serum | PAN Biotec | Cat# 3302 |
| Stain Buffer (FBS) | Becton Dickinson | Cat# 554656 |
| Pancoll human, Density: 1.077 g/ml | Pan Biotech | Cat# P04-601000 |
| Dulbecco’S Phosphate Buffered Saline, MO | Sigma-Aldrich | Cat# D8537 |
| FcR Blocking Reagent, human | Miltenyi | Cat# 130-059-901 |
| Cell-ID Intercalator-Ir | Fluidigm | Cat# 201192A |
| Permeabilization buffer 10X | eBioscience | Cat# 00-8333-56 |
| Maxpar PBS | Fluidigm | Cat# 201058 |
| Maxpar Cell Staining buffer | Fluidigm | Cat# 201068 |
| Maxpar X8 Multimetal Labeling Kit | Fluidigm | Cat# 201300 |
| Proteomic stabilizer | Smart Tube Inc. | Cat# PROT1 |
| Nuclease-Free Water | Invitrogen | Cat# AM9937 |
| KAPA HiFi HotStart Ready Mix | Roche | Cat# KK2601 |
| Human Tru Stain FcX | Biolegend | Cat# 422301 |
| TE Buffer, pH8.0, 1mM disodium EDTA | Thermo Fisher | Cat# 12090015 |
| SPRIselect | Beckmann Coulter | Cat# B23318 |
| 10% Tween 20 | BIO-RAD | Cat# 1662404 |
| Buffer EB | QIAGEN | Cat# 19086 |
| Ethanol, Absolute | Fisher Bioreagents | Cat# BP2818-500 |
| Glycerol, 85% | Merck | Cat# 1040941000 |
| Bovine Serum Albumin (IgG-Free, Protease-Free) | Jackson Immuno Research | Cat# 001-000-161 |
| LPS, TLRpure | Innaxon | Cat# IAX-100-010 |
| Tween20 | Sigma-Aldrich | Cat# P1379-500M |
| MagniSort™ Negative Selection Beads | Thermo Fisher | Cat# MSNB-6002-74 |
| Lysercell WDF | Sysmex | Cat# AL-337-564 |
| Fluorocell™ WDF | Sysmex | Cat# CV-377-552 |
| Critical Commercial Assays | ||
| LIVE/DEAD Fixable Yellow Dead Cell Stain Kit | Thermo Fisher | Cat# L34967 |
| LEGENDplex™ Human Inflammation Panel 1 (Mix&Match) | Biolegend | Cat# 740809 |
| Human Single-Cell Multiplexing Kit | Becton Dickinson | Cat# 633781 |
| BD Rhapsody WTA Amplification Kit | Becton Dickinson | Cat# 633801 |
| BD Rhapsody Cartridge Kit | Becton Dickinson | Cat# 633733 |
| BD Rhapsody cDNA Kit | Becton Dickinson | Cat# 633773 |
| High Sensitivity D5000 ScreenTape | Agilent | Cat# 5067-5592 |
| Qubit dsDNA HS Assay Kit | ThermoFisher | Cat# Q32854 |
| Chromium Next GEM Single Cell 3′ GEM, Library & Gel Bead Kit v3.1 | 10x genomics | Cat# 1000121 |
| Chromium Next GEM Chip G Single Cell Kit | 10x genomics | Cat# 1000120 |
| Single Index Kit T Set A | 10x genomics | Cat# 1000213 |
| High Sensitivity DNA Kit | Agilent | Cat# 5067-4626 |
| NovaSeq 6000 S1 Reagent Kit (100 cycle) | Illumina | Cat# 200012865 |
| NovaSeq 6000 S2 Reagent Kit (100 cycle) | Illumina | Cat# 20012862 |
| NovaSeq 6000 S2 Reagent Kit (200 cycles) | Illumina | Cat# 20040326 |
| NovaSeq 6000 S2 Reagent Kit (200 cycles) | Illumina | Cat# 20040326 |
| NextSeq 500/550 High Output Kit v2.5 (150 Cycles) | Illumina | Cat# 20024907 |
| Pan Monocyte Isolation Kit, human | Miltenyi | Cat# 130-096-537 |
| CE/IVD Phagoburst | BD Biosciences | Cat# 341058 |
| CD/IVD PHAGOTEST | BD Biosciences | Cat# 341060 |
| Deposited Data | ||
| scRNA-seq raw data | This paper | EGAS00001004571 |
| Processed scRNA-seq count data and code | This paper | http://fastgenomics.org |
| Supplemental Table S1, Table S2, Table S3, Table S4 | This paper | https://data.mendeley.com/datasets/hwxhw2sxys/1 |
| Oligonucleotides | ||
| SI-PCR primer | IDT | AATGATACGGCGACCACCGAGATCTA CACTCTTTCCCTACACGACGC∗T∗C |
| HTO additive primer | IDT | GTGACTGGAGTTCAGACGTGTGC∗T∗C |
| D701_S | IDT | CAAGCAGAAGACGGCATACGAGA TCGAGTAATGTGACTGGAGTTCAGACGTGT∗G∗C |
| D702_S | IDT | CAAGCAGAAGACGGCATACGAGA TTCTCCGGAGTGACTGGAGTTCAGACGTGT∗G∗C |
| D703_S | IDT | CAAGCAGAAGACGGCATACGAGATA ATGAGCGGTGACTGGAGTTCAGACGTGT∗G∗C |
| D705_S | IDT | CAAGCAGAAGACGGCATACGAGATTTC TGAATGTGACTGGAGTTCAGACGTGT∗G∗C |
| Software and Algorithms | ||
| CellRanger | 10x genomics | v3.1.0 |
| Bcl2fastq2 | Illumina | v2.20 |
| STAR | Dobin et al., 2013 | v2.6.1b |
| Cutadapt | Martin, 2011 | v1.16 |
| Dropseq-tools | https://github.com/broadinstitute/Drop-seq/ | v2.0.0 |
| R | https://www.cran.r-project.org | v3.6.2 |
| Seurat (R package) | Butler et al., 2018; Hafemeister and Satija, 2019; Stuart et al., 2019 | v3.1.4, v3.1.2 (CRAN) |
| Harmony (R package) | Korsunsky et al., 2019 (https://github.com/immunogenomics/harmony) | v1.0 |
| Destiny (R package) | Angerer et al., 2016 | v 3.0.1 |
| ClusterProfiler (R package) | Yu et al., 2012 | v3.10.1 (CRAN) |
| SingleR (R package) | Aran et al., 2019 | v1.0.5 (Bioconductor) |
| DirichletReg (R package) | Maier, 2014 | v0.6.3.1 (CRAN) |
| AUCell (R package) | Aibar et al., 2017 | v1.6.1 (CRAN) |
| Cytobank | Kotecha et al., 2010https://www.cytobank.org | https://doi.org/10.1002/0471142956.cy1017s53 |
| SPADE (Cytobank) | Qiu et al., 2011 | Cytobank is running a version of SPADE derived from v1.10.2 |
| flowCore (R package) | https://www.bioconductor.org/packages/release/bioc/html/flowCore.html | v1.48.1 (Bioconductor), 10.18129/B9.bioc.flowCore |
| CytoML (R package) | https://github.com/RGLab/CytoML | v1.8.1 (Bioconductor), 10.18129/B9.bioc.CytoML |
| CytofBatchAdjust (R package) | https://github.com/CUHIMSR/CytofBatchAdjust | https://doi.org/10.3389/fimmu.2019.02367 |
| uwot (R package) | https://cran.r-project.org/web/packages/uwot/index.html | v0.1.8 (CRAN) |
| ComplexHeatmap (R package) | Gu et al., 2016 | v1.20.0 (Bioconductor) |
| lme4 (R package) | Nowicka et al., 2017 | v1.1-21 (CRAN) |
| multcomp (R package) | Hothorn et al., 2008 | v1.4-13 (CRAN) |
| lsmeans (R package) | Lenth, 2016 | v2.30-0 (CRAN) |
| Prism (software) | https://www.graphpad.com | v8 |
| FlowJo | https://www.flowjo.com | v10.6.1 |
| Cytoscape | https://www.cytoscape.org | v3.7.1 (https://doi.org/10.1101/gr.1239303) |
| iRegulon | Janky et al., 2014 | v1.3 |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Joachim L. Schultze (j.schultze@uni-bonn.de).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
ScRNA-seq data generated during this study are deposited at the European Genome-phenome Archive (EGA) under access number EGAS00001004571, which is hosted by the EBI and the CRG.
Experimental Model and Subject Details
Samples from patients with COVID-19 were collected within two cohort studies (Kurth et al., 2020) designed to allow deep molecular and immunological transcriptomic and proteomic profiling of COVID-19 in blood. Patients for which sufficient material was available for scRNA-seq, CyTOF or flow cytometry analysis, were included in this study. This study was designed to describe immunological deviations in COVID-19 patients without intention of the development of new treatments or new diagnostics, and therefore sample size estimation was not included in the original study design.
Cohort 1 / Berlin cohort
This study includes a subset of patients enrolled between March 2 and July 02 2020 in the Pa-COVID-19 study, a prospective observational cohort study assessing pathophysiology and clinical characteristics of patients with COVID-19 at Charité Universitätsmedizin Berlin (Kurth et al., 2020). The study is approved by the Institutional Review board of Charité (EA2/066/20). Written informed consent was provided by all patients or legal representatives for participation in the study. The patient population included in all analyses of cohort 1 consists of 10 control donors (samples collected in 2019 before SARS-CoV2 outbreak), 8 patients presenting with flu-like illness but tested SARS-CoV-2-negative, 25 mild and 29 severe COVID-19 patients (Figures 1A and 1B; Table S1). Information on age, sex, medication, and co-morbidities is listed in Table S1. All COVID-19 patients were tested positive for SARS-CoV-2 RNA in nasopharyngeal swabs and allocated to mild (WHO 2-4) or severe (5-7) disease according to the WHO clinical ordinal scale. We also included publicly available single-cell transcriptome data derived from 22 control samples into the analysis; 3 samples were derived from 10x Genomics, San Francisco, CA 94111, USA (5k_pbmc_v3: https://support.10xgenomics.com/single-cell-gene-expression/datasets/3.0.2/5k_pbmc_v3, pbmc_10k_v3: https://support.10xgenomics.com/single-cell-gene-expression/datasets/3.0.0/pbmc_10k_v3, pbmc_1k_v3: https://support.10xgenomics.com/single-cell-gene-expression/datasets/3.0.0/pbmc_1k_v3), 19 samples derived from Reyes et al. (2020).
Cohort 2 / Bonn cohort
This study was approved by the Institutional Review board of the University Hospital Bonn (073/19 and 134/20). After providing written informed consent, 19 control donors and 22 COVID-19 patients (Figures 1A and 1B; Table S1) were included in the study. In patients who were not able to consent at the time of study enrollment, consent was obtained after recovery. Information on age, sex, medication, and co-morbidities are listed in Table S1. COVID-19 patients who tested positive for SARS-CoV-2 RNA in nasopharyngeal swabs were recruited at the Medical Clinic I of the University Hospital Bonn between March 30 and June 17, 2020 and allocated to mild (WHO 2-4) or severe (5-7) disease according to the WHO clinical ordinal scale. Controls in cohort 2 were collected from healthy people or from otherwise hospitalized patients with a wide range of diseases and comorbidities including chronic inflammatory immune responses. These individuals were either tested negative for SARS-CoV-2, serologically negative or had no indication for acute COVID-19 disease based on clinical or laboratory parameters.
Method Details
Isolation of blood cells for scRNA-seq
Cohort 1 / Berlin cohort
PBMC were isolated from heparinized whole blood by density centrifugation over Pancoll (density: 1.077 g/ml; PAN-Biotech). If the pellet was still slightly red, remaining CD235ab+ cells (Erythrocytes) were depleted by Negative Selection (MagniSort Thermo Fisher). Subsequently the PBMC were prepared for 3′scRNA-seq (10xGenomics) or cryopreserved and analyzed later.
Cohort 2 / Bonn cohort
In the Bonn cohort, scRNA-seq was performed on fresh whole blood, fresh PBMC and frozen PBMC. Briefly, PBMC were isolated from EDTA-treated or heparinized peripheral blood by density centrifugation over Pancoll or Ficoll-Paque density centrifugation (density: 1.077 g/ml). Cells were then washed with DPBS, directly prepared for scRNA-seq (BD Rhapsody) or cryopreserved in RPMI1640 with 40% FBS and 10% DMSO. Whole blood was prepared by treatment of 1ml peripheral blood with 10ml of RBC lysis buffer (Biolegend). After one wash in DPBS cells were directly processed for scRNA-seq (BD Rhapsody) or multi-color flow cytometry (MCFC). Frozen PBMC were recovered by rapidly thawing frozen cell suspensions in a 37°C water bath followed by immediate dilution in pre-warmed RPMI-1640+10% FBS (GIBCO) and centrifugation at 300 g for 5min. After centrifugation, the cells were resuspended in RPMI-1640+10% FBS and processed for scRNA-seq. Antibody cocktails were cryopreserved as described before (Schulz and Mei, 2019).
Antibodies used for mass cytometry
All anti-human antibodies pre-conjugated to metal isotopes were obtained from Fluidigm Corporation (San Francisco, US). All remaining antibodies were obtained from the indicated companies as purified antibodies and in-house conjugation was done using the MaxPar X8 labeling kit (Fluidigm). Table S2 shows a detailed list of all antibodies used for panel 1 and panel 2.
Sample processing, antigen staining and data analysis of mass cytometry-based immune cell profiling
500μl of whole blood (heparin) was fixed in 700μl of proteomic stabilizer (Smart Tube Inc., San Carlos, US) as described in the user manual and stored at −80°C until further processing. Whole blood samples were thawed in Thaw/Lyse buffer (Smart Tube Inc.). For barcoding antibodies recognizing human beta-2 microglobulin (B2M) were conjugated in house to 104Pd, 106Pd, 108Pd, 110Pd, 198Pt (Mei et al., 2015, 2016; Schulz and Mei, 2019). Up to 10 individual samples were stained using a staining buffer from Fluidigm with a combination of two different B2M antibodies for 30min at 4°C. Cells were washed and pooled for surface and intracellular staining.
For surface staining the barcoded and pooled samples were equally divided into two samples. Cells were resuspended in antibody staining cocktails for panel 1 or panel 2 respectively (Table S2) and stained for 30min at 4°C. For secondary antibody staining of panel 2, cells were washed and stained with anti-APC 163Dy for 30min at 4°C. After surface staining cells were washed with PBS and fixed overnight in PFA solution diluted in PBS to 2%.
For intracellular staining cells were washed twice with a permeabilization buffer (eBioscience, San Diego, US) and stained with the respective antibodies diluted in a permeabilization buffer for 30min at room temperature. After washing, cells were stained with iridium intercalator (Fluidigm) diluted in 2% PFA for 20min at room temperature. Cells were washed once with PBS and then twice with ddH2O and kept at 4°C until mass cytometry measurement.
A minimum of 100,000 cells per sample and panel were acquired on a CyTOF2/Helios mass cytometer (Fluidigm). For normalization of the fcs files 1:10 EQ Four Element Calibration Beads (Fluidigm) were added. Cells were analyzed using a CyTOF2 upgraded to Helios specifications, with software version 6.7.1014, using a narrow bore injector. The instrument was tuned according to the manufacturer’s instructions with tuning solution (Fluidigm) and measurement of EQ four element calibration beads (Fluidigm) containing 140/142Ce, 151/153Eu, 165Ho and 175/176Lu served as a quality control for sensitivity and recovery. Directly prior to analysis, cells were resuspended in ddH2O, filtered through a 20-μm cell strainer (Celltrics, Sysmex), counted and adjusted to 5-8 x105 cells/ml. EQ four element calibration beads were added at a final concentration of 1:10 v/v of the sample volume to be able to normalize the data to compensate for signal drift and day-to-day changes in instrument sensitivity. Samples were acquired with a flow rate of 300-400 events/s. The lower convolution threshold was set to 400, with noise reduction mode turned on and cell definition parameters set at event duration of 10-150 pushes (push = 13μs). The resulting flow cytometry standard (FCS) files were normalized and randomized using the CyTOF software’s internal FCS-Processing module on the non-randomized (‘original’) data. The default settings in the software were used with time interval normalization (100 s/minimum of 50 beads) and passport version 2. Intervals with less than 50 beads per 100 s were excluded from the resulting FCS file.
Blood processing for multi-color flow cytometry
1ml of fresh blood from control or COVID-19 donors was treated with 10ml of RBC lysis buffer (Biolegend). After RBC lysis, cells were washed with DPBS and 1-2 million cells were used for flow cytometry analysis. Cells were then stained for surface markers (Table S3) in DPBS with BD Horizon Brilliant Stain Buffer (Becton Dickinson) for 30min at 4°C. To distinguish live from dead cells, the cells were incubated with LIVE/DEAD Fixable Yellow Dead Cell Stain Kit (1:1000 – Thermo Scientific). Following staining and washing, the cell suspension was fixed with 4% PFA for 5min at room temperature to prevent any possible risk of contamination during acquisition of the samples. Flow cytometry analysis was performed on a BD Symphony instrument (Becton Dickinson) configured with 5 lasers (UV, violet, blue, yellow-green, red).
Ex vivo functional analysis of neutrophils
Determination of neutrophil oxidative burst and phagocytosis was performed by flow cytometry using the CE/IVD PHAGOBURST and PHAGOTEST assay (BD Biosciences, Heidelberg, Germany) according to the manufacturer’s instructions.
Briefly, heparinized whole blood was incubated with PMA, unlabeled opsonized E. coli bacteria or washing solution (negative control) at 37°C for 10min. Dihydrorhodamine (DHR 123) was then added for 10min, erythrocytes were lysed and DNA staining solution was added. The freely cell permeable nonfluorescent Dihydrorhodamine 123 becomes fluorescent when oxidized by reactive oxygen species. Stained samples were analyzed on a Navios flow cytometer (Beckman Coulter, Krefeld, Germany) within less than 30min. The respiratory burst intensity in neutrophils was determined by analysis of increase in the mean fluorescence intensity (MFI) in the FL1 Channel in the stimulated samples compared to the unstimulated control.
For analysis of neutrophil phagocytic activity, heparinized whole blood was incubated with FITC-labeled opsonized E. coli bacteria for 10min at 37°C or 0°C (negative control). After incubation, the reaction was stopped, erythrocytes were lysed and the DNA staining solution was added. Stained samples were analyzed on a Navios flow cytometer (Beckman Coulter, Krefeld, Germany) within less than 30min. The phagocytic activity of neutrophils was determined by the increase in MFI in the FL1 Channel in the stimulated sample compared to the unstimulated control. Data were analyzed using prism version 8. Mixed-effect-analysis and Sidak’s multiple comparison test was applied to report statistical differences of E.coli- and PMA-induced ROS production as well as phagocytosis between mild and severe COVID-19 patients.
Ex vivo functional analysis of monocytes
Monocytes were isolated from frozen PBMCs by negative selection using the Pan Monocyte Isolation Kit (Miltenyi, Bergisch Gladbach, Germany). The purity of isolated cells was assessed by BD Canto 2 flow cytometer, and preparations with > 85% purity were used for experiments. Monocytes were resuspended in complete RPMI1640 medium (GIBCO) supplemented with 10% heat-inactivated fetal bovine serum (Pan Biotech), 10 U/ml penicillin and 10 μg/ml streptomycin (Sigma-Aldrich, USA), and stimulated for 8h with LPS (1 ng/ml; TLRpure; Innaxon, UK). After stimulation, cell-free supernatants were collected and tested for IL-1β, IFNγ, and TNFα, respectively, using the cytokine bead assay Legend-Plex Mix&Match inflammation panel 1 kit (Biolegend, USA). Cytokine-bound beads were measured with a BD Canto 2 flow cytometer and analyzed using Legend-Plex Software 8.0 (Biolegend, USA).
Hematological analyses of the granulocyte compartment
Blood samples were collected into K3EDTA evacuated plastic tubes (Greiner Bio-One GmbH, Frickenhausen, Germany) and were subsequently analyzed using Sysmex XN-10 hematology analyzers within a XN-2000 or XN-9100 configuration (Sysmex Corporation, Kobe, Japan) as previously described (Stiel et al., 2016). Immature granulocytes (IG) were quantified by automated flow cytometry using the Sysmex WBC differential channel in XN-10 hematology analyzers within a XN-2000 or XN-9100 configuration (Sysmex Corporation, Kobe, Japan). Whole blood was treated with LysercellWDF for cell permeabilization and stained with the nucleic acid polymethine fluorescent dye FluorocellWDF according to Sysmex-proprietary protocols without modifications. Subsequent differentiation of white blood cells into lymphocytes, monocytes, neutrophils, eosinophils, and immature granulocytes was achieved by flow cytometry using a 663 nm laser. The cell populations’ distinct forward-scattered light (FSC = NE-WZ, i.e., cell volume), side-scattered light (SSC = NE-WX, i.e., granularity), and side-fluorescent light (SFL = NE-WY, i.e., nucleic acid content) properties allowed determining the width of neutrophil cytometric dispersions applying Sysmex-proprietary algorithms. Data were analyzed using prism version 8. Mann Whitney test was used to report differences in IG count, whereas mixed-effect-analysis and Sidak’s multiple comparison test was applied to report statistical differences of NE-WX, NE-WY and NE-WZ between mild and severe COVID-19 patients.
10x Genomics Chromium single-cell RNA-seq
PBMC were isolated and prepared as described above. Afterward, patient samples were hashtagged with TotalSeq-A antibodies (Biolegend) according to the manufacturer’s protocol for TotalSeq™-A antibodies and cell hashing with 10x Single Cell 3′ Reagent Kit v3.1. 50μL cell suspension with 1x106 cells were resuspended in staining buffer (2% BSA, Jackson Immuno Research; 0.01% Tween-20, Sigma-Aldrich; 1x DPBS, GIBCO) and 5 μL Human TruStain FcX™ FcBlocking (Biolegend) reagent were added. The blocking was performed for 10min at 4°C. In the next step 1μg unique TotalSeq-A antibody was added to each sample and incubated for 30min at 4°C. After the incubation time 1.5mL staining buffer were added and centrifuged for 5min at 350g and 4°C. Washing was repeated for a total of 3 washes. Subsequently, the cells were resuspended in an appropriate volume of 1x DPBS (GIBCO), passed through a 40μm mesh (Flowmi™ Cell Strainer, Merck) and counted, using a Neubauer Hemocytometer (Marienfeld). Cell counts were adjusted and hashtagged cells were pooled equally. The cell suspension was super-loaded, with 50,000 cells, in the Chromium™ Controller for partitioning single cells into nanoliter-scale Gel Bead-In-Emulsions (GEMs). Single Cell 3′ reagent kit v3.1 was used for reverse transcription, cDNA amplification and library construction of the gene expression libraries (10x Genomics) following the detailed protocol provided by 10x Genomics. Hashtag libraries were prepared according to the cell hashing protocol for 10x Single Cell 3′ Reagent Kit v3.1 provided by Biolegend, including primer sequences and reagent specifications. Biometra Trio Thermal Cycler was used for amplification and incubation steps (Analytik Jena). Libraries were quantified by Qubit™ 2.0 Fluorometer (ThermoFisher) and quality was checked using 2100 Bioanalyzer with High Sensitivity DNA kit (Agilent). Sequencing was performed in paired-end mode with a S1 and S2 flow cell (2 × 50 cycles) using NovaSeq 6000 sequencer (Illumina).
BD Rhapsody single-cell RNA-seq
Whole transcriptome analyses, using the BD Rhapsody Single-Cell Analysis System (BD, Biosciences) were performed on PBMC and whole blood samples prepared as described above. Cells from each sample were labeled with sample tags (BD Human Single-Cell Multiplexing Kit) following the manufacturer’s protocol. Briefly, a total number of 1x106 cells were resuspended in 180μl of Stain Buffer (FBS) (BD PharMingen). The sample tags were added to the respective samples and incubated for 20min at room temperature. After incubation, 200μl stain buffer was added to each sample and centrifuged for 5min at 300 g and 4°C. Samples were washed one more time. Subsequently cells were resuspended in 300μl of cold BD Sample Buffer and counted using Improved Neubauer Hemocytometer (INCYTO). Labeled samples were pooled equally in 650μl cold BD Sample Buffer. For each pooled sample two BD Rhapsody cartridges were super-loaded with approximately 60,000 cells each. Single cells were isolated using Single-Cell Capture and cDNA Synthesis with the BD Rhapsody Express Single-Cell Analysis System according to the manufacturer’s recommendations (BD Biosciences). cDNA libraries were prepared using the BD Rhapsody Whole Transcriptome Analysis Amplification Kit following the BD Rhapsody System mRNA Whole Transcriptome Analysis (WTA) and Sample Tag Library Preparation Protocol (BD Biosciences). The final libraries were quantified using a Qubit Fluorometer with the Qubit dsDNA HS Kit (ThermoFisher) and the size-distribution was measured using the Agilent high sensitivity D5000 assay on a TapeStation 4200 system (Agilent technologies). Sequencing was performed in paired-end mode (2∗75 cycles) on a NovaSeq 6000 and NextSeq 500 System (Illumina) with NovaSeq 6000 S2 Reagent Kit (200 cycles) and NextSeq 500/550 High Output Kit v2.5 (150 Cycles) chemistry, respectively.
Quantification and Statistical Analysis
Data pre-processing of 10x Genomics Chromium scRNA-seq data
CellRanger v3.1.0 (10x Genomics) was used to process scRNA-seq. To generate a digital gene expression (DGE) matrix for each sample, we mapped their reads to a combined reference of GRCh38 genome and SARS-CoV-2 genome and recorded the number of UMIs for each gene in each cell.
Data pre-processing of BD Rhapsody scRNA-seq data
After demultiplexing of bcl files using Bcl2fastq2 V2.20 from Illumina and quality control, paired-end scRNA-seq reads were filtered for valid cell barcodes using the barcode whitelist provided by BD. Cutadapt 1.16 was then used to trim NexteraPE-PE adaptor sequences where needed and to filter reads for a PHRED score of 20 or above (Martin, 2011). Then, STAR 2.6.1b was used for alignment against the Gencode v27 reference genome (Dobin et al., 2013). Dropseq-tools 2.0.0 were used to quantify gene expression and collapse to UMI count data (https://github.com/broadinstitute/Drop-seq/). For hashtag-oligo based demultiplexing of single-cell transcriptomes and subsequent assignment of cell barcodes to their sample of origin the respective multiplexing tag sequences were added to the reference genome and quantified as well.
ScRNA-seq data analysis of 10x Chromium data of cohort 1
ScRNA-seq UMI count matrices were imported to R 3.6.2 and gene expression data analysis was performed using the R/Seurat package 3.1.4 (Butler et al., 2018; Hafemeister and Satija, 2019). Demultiplexing of cells was performed using the HTODemux function implemented in Seurat.
Data quality control
We excluded cells based on the following quality criteria: more than 25% mitochondrial reads, more than 25% HBA/HBB gene reads, less than 250 expressed genes or more than 5,000 expressed genes and less than 500 detected transcripts. We further excluded genes that were expressed in less than five cells. In addition, mitochondrial genes have been excluded from further analysis.
Normalization
LogNormalization (Seurat function) was applied before downstream analysis. The original gene counts for each cell were normalized by total UMI counts, multiplied by 10,000 (TP10K) and then log transformed by log10(TP10k+1).
Data integration
After normalization, the count data was scaled regressing for total UMI counts and principal component analysis (PCA) was performed based on the 2,000 most variable features identified using the vst method implemented in Seurat. Subsequently, the scRNA-seq data from cohort 1 was integrated with publicly available 10x scRNaseq data of healthy controls using the ‘harmony’ algorithm (Korsunsky et al., 2019) based on the first 20 principal components to correct for technical differences in the gene expression counts of the different data sources (Control samples from Reyes et al. (2020), 10x v3.1 PBMC benchmarking data from healthy controls and 10x v3.1 scRNA-seq data from cohort 1). We downloaded the count matrices for the publicly available scRNA-seq data and filtered the cells using the above-mentioned quality criteria prior to data integration. For two-dimensional data visualization we performed UMAP based on the first 20 dimensions of the ‘harmony’ data reduction. The cells were clustered using the Louvain algorithm based on the first 20 ‘harmony” dimensions with a resolution of 0.4.
Differential expression tests and cluster marker genes
Differential expression (DE) tests were performed using FindMarkers/FindAllMarkers functions in Seurat with Wilcoxon Rank Sum test. Genes with > 0.25 log-fold changes, at least 25% expressed in tested groups, and Bonferroni-corrected p values < 0.05 were regarded as significantly differentially expressed genes (DEGs). Cluster marker genes were identified by applying the DE tests for upregulated genes between cells in one cluster to all other clusters in the dataset. Top ranked genes (by log-fold changes) from each cluster of interest were extracted for further illustration. The exact number and definition of samples used in the analysis are specified in the legend of Figure 2A and summarized in Table S1.
Cluster annotation
Clusters were annotated based on a double-checking strategy: 1) by comparing cluster marker genes with public sources, and 2) by directly visualizing the expression pattern of CyTOF marker genes.
GO enrichment analysis
Significant DEGs between each monocyte cluster and the rest of monocyte subpopulations were identified by FindMarkers function from the Seurat package using Wilcoxon Rank Sum test statistics for genes expressed in at least 25% of all monocyte clusters. P values were corrected for multiple testing using Bonferroni correction and genes with corrected p values lower or equal 0.05 have been taken as significant DEGs for GO enrichment test by R package/ClusterProfiler v.3.10.1 (Yu et al., 2012).
Correlation analysis of marker genes for monocyte and neutrophils between cohort 1 and 2
To systematically compare the similarity of marker gene expression in the identified monocyte/neutrophils subpopulations between the two cohorts, the Spearman correlation coefficients were calculated based on the union of the top 50 marker genes of each cluster sorted by fold change in the two cohorts, based on their average expression of all cells in the specific subpopulation. The pairwise comparisons were performed, and the correlation coefficients were displayed using a heatmap.
Subset analysis of the neutrophils within the PBMC dataset of cohort 1
The neutrophil space was investigated by subsetting the PBMC dataset to those clusters identified as neutrophils and immature neutrophils (cluster 5 and 6). Within those subsets, we selected top 2,000 variable genes and repeated the clustering using the SNN-graph based Louvain algorithm mentioned above with a resolution of 0.6. The dimensionality of the data was then reduced to 10 PCs, which served as input for the UMAP calculation. To categorize the observed neutrophil clusters into the respective cell cycle states, we applied the CellCycleScoring function of Seurat and visualized the results as pie charts.
A gene signature enrichment analysis using the ‘AUCell’ method (Aibar et al., 2017) was applied to link observed neutrophil clusters to existing studies and neutrophils of cohort 2. We set the threshold for the calculation of the area under the curve (AUC) to marker genes from collected publications and top 30 of the ranked maker genes from each of neutrophil clusters from cohort 2. The resulting AUC values were normalized the maximum possible AUC to 1 and subsequently visualized in violin plots or UMAP plots.
ScRNA-seq data analysis of Rhapsody data of cohort 2
General steps for Rhapsody data downstream analysis
ScRNA-seq UMI count matrices were imported to R 3.6.2 and gene expression data analysis was performed using the R/Seurat package 3.1.2 (Butler et al., 2018). Demultiplexing of cells was performed using the HTODemux function implemented in Seurat. After identification of singlets, cells with more than 25% mitochondrial reads, less than 250 expressed genes or more than 5,000 expressed genes and less than 500 detected transcripts were excluded from the analysis and only those genes present in more than 5 cells were considered for downstream analysis. The following normalization, scaling and dimensionality reduction steps were performed independently for each of the data subsets used for the different analyses as indicated respectively. In general, gene expression values were normalized by total UMI counts per cell, multiplied by 10,000 (TP10K) and then log transformed by log10(TP10k+1). Subsequently, the data was scaled, centered and regressed against the number of detected transcripts per cell to correct for heterogeneity associated with differences in sequencing depth. For dimensionality reduction, PCA was performed on the top 2,000 variable genes identified using the vst method implemented in Seurat. Subsequently, UMAP was used for two-dimensional representation of the data structure. Cell type annotation was based on the respective clustering results combined with data-driven cell type classification algorithms based on reference transcriptome data (Aran et al., 2019) and expression of known marker genes.
scRNA-seq analysis of the complete BD Rhapsody dataset of cohort 2 including data from frozen and fresh PBMC and whole blood
ScRNA-seq count data of 229,731 cells derived from fresh and frozen PBMC samples purified by density gradient centrifugation and whole blood after erythrocyte lysis of cohort 2 (Bonn, BD Rhapsody) were combined, normalized and scaled as described above (see Figure S6A). After variable gene selection and PCA, UMAP was performed based on the first 20 principal components (PCs). No batch correction or data integration strategies were applied to the data. Visualization of the cells (Figure S6A) showed overlay of cells of the same type (e.g., T cells clustered within the same cluster, irrespective of cell isolation procedure). In other words, cell type distribution was unaffected by the technical differences in sample handling. Data quality and information content was visualized as violin plots showing the number of detected genes, transcripts (UMIs) and genic reads per sample handling strategy split by PBMC and granulocyte fraction.
scRNA-seq analysis of fresh and frozen PBMC samples
ScRNA-seq count data of 139,848 cells derived from fresh and frozen PBMC samples of cohort 2 (Bonn, BD Rhapsody) purified by density gradient centrifugation were normalized and scaled as described above. After variable gene selection and PCA, UMAP was performed and the cells were clustered using the Louvain algorithm based on the first 20 PCs and a resolution of 0.4. Cluster identities were determined by reference-based cell classification and inference of cluster-specific marker genes using the Wilcoxon rank sum test using the following cutoffs: genes have to be expressed in more than 20% of the cells of the respective cluster, exceed a logarithmic fold change cutoff to at least 0.2, and exhibited a difference of > 10% in the detection between two clusters. The exact number and definition of samples used in the analysis are specified in the legend of Figure 2D and summarized in Table S1.
Quantification of the percentages of cell clusters in the PBMC scRNA-seq data of both cohorts separated by disease group
To compare shifts in the monocyte and neutrophil populations in the PBMC compartment of COVID-19 patients, the percentages of the cellular subsets - as identified by clustering and cluster annotation explained above for the two independent scRNA-seq datasets (cohort 1 and cohort 2) - of the total number of PBMC in each dataset were quantified per sample and visualized together in boxplots. To determine the statistical significance of differences in cell proportions between the different conditions, a Dirichlet regression model was used, due to the fact that the proportions are not independent of one another. The R/RDirichletReg (Maier, 2014) package was used. The p values were corrected for multiple testing using the Benjamini-Hochberg procedure.
Subset analysis of the monocytes within the PBMC dataset of cohort 2
The monocyte space was investigated by subsetting the PBMC dataset to those clusters identified as monocytes (cluster 0-4), removing cells with strong multi-lineage marker expressions, and repeating the variable gene selection (top 2,000 variable genes), regression for the number of UMIs and scaling as described above. The dimensionality of the data was then reduced to 8 PCs, which served as input for the UMAP calculation. The SNN-graph based Louvain clustering of the monocytes was performed using a resolution of 0.2. Marker genes per cluster were calculated using the Wilcoxon rank sum test using the following cutoffs: genes have to be expressed in > 20% of the cells, exceed a logarithmic fold change cutoff to at least 0.25, and exhibited a difference of > 10% in the detection between two clusters. The exact number and definition of samples used in the analysis are specified in the legend of Figure 4A and summarized in Table S1.
Time kinetics analysis of identified monocyte clusters
For each patient and time point of sample collection, the proportional occupancy of the monocyte clusters was calculated, and the relative proportions were subsequently visualized as a function of time.
Analysis of scRNA-seq data from fresh PBMC and whole blood samples of cohort 2
ScRNA-seq count data derived from fresh PBMC samples purified by density gradient centrifugation and whole blood after erythrocyte lysis of cohort 2 (BD Rhapsody) were normalized, scaled, and regressed for the number of UMI per cell as described above. After PCA based on the top 2,000 variable genes, UMAP was performed using the first 30 PCs. Cell clusters were determined using Louvain clustering implemented in Seurat based on the first 30 principle components and a resolution of 0.8. Cluster identities were assigned as detailed above using reference-based classification and marker gene expression. Subsequently, the dataset was subsetted for whole blood samples after erythrocyte lysis and clusters identified as neutrophils and immature neutrophils, and re-scaled and regressed. After PCA on the top 2,000 variable genes, the neutrophil subset data was further processed using the data integration approach implemented in Seurat (Stuart et al., 2019) based on the first 30 PCs removing potential technical biases of separate experimental runs. UMAP and clustering were performed as described above on the top 12 PCs using a resolution of 0.3. Differentially expressed genes between clusters were defined using a Wilcoxon rank sum test for differential gene expression implemented in Seurat. Genes had to be expressed in > 10% of the cells of a cluster, exceed a logarithmic threshold > 0.1. The exact number and definition of samples used in the analysis are specified in the legend of Figure 7A and summarized in Table S1.
Quantification of percentages of cell subsets in whole blood scRNA-seq data of cohort 2
After cell type classification of the combined scRNA-seq dataset of fresh PBMC and whole blood samples of cohort 2 described above, 89,883 cells derived from whole blood samples after erythrocyte lysis were subsetted. Percentages of cell subsets in those whole blood samples of the total number of cells were quantified per sample and visualized in boxplots separated by disease stage and group.
Confusion matrix
For each cluster of neutrophils, the relative proportion across disease severity and time point was visualized as a fraction of samples from the respective condition contributing to the cluster.
GO enrichment
Gene set enrichment was performed on gene sets from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Kanehisa, 2019), Hallmark gene sets (Liberzon et al., 2015) and Gene Ontology (GO) (Ashburner et al., 2000; Carbon et al., 2019) using the R package/ClusterProfiler v.3.10.1 (Yu et al., 2012).
Cell cycle state analysis of scRNA-seq data
To categorize the cells within the neutrophil clusters into the respective cell cycle states, we applied the CellCycleScoring function of Seurat and visualized the results as pie charts.
Trajectory analysis
Trajectory analysis was performed using the destiny algorithm v3.0.1 (Angerer et al., 2016). In brief, the neutrophil space was subsetted to only severe patients (early and late) and only the most prominent clusters of the latter (clusters 1,2,6,8). The normalized data were scaled and regressed for UMIs and a diffusion map was calculated based on the top 2,000 variable genes with a sum of at least 10 counts over all cells. Based on the diffusion map, a diffusion pseudo time was calculated to infer a transition probability between the different cell states of the neutrophils. Subsequently, the density of the clusters along the pseudotime and marker gene expression for each cluster were visualized.
Enrichment of gene sets was performed using the ‘AUCell’ method (Aibar et al., 2017) implemented in the package (version 1.4.1) in R. We set the threshold for the calculation of the AUC to the top 3% of the ranked genes and normalized the maximum possible AUC to 1. The resulting AUC values were subsequently visualized in violin plots or UMAP plots.
Transcription factor prediction analysis
The Cytoscape (version v3.7.1, https://doi.org/10.1101/gr.1239303) plug-in iRegulon (Janky et al., 2014) (version 1.3) was used to predict the transcription factors potentially regulating cluster-specifically expressed gene sets in the neutrophil and monocyte subset analysis in cohort 2. The genomic regions for TF-motif search were limited to 10kb around the respective transcriptional start sites and filtered for predicted TFs with a normalized enrichment score > 4.0. Next, we filtered for TFs, which exceeded a cumulative normalized expression cutoff of 50 in the respective cluster. Subsequently, we selected transcription factors of known relevance in the context of neutrophil and monocyte biology and constructed a network linking target genes among the cluster-specifically expressed marker genes and their predicted and expressed regulators for visualization in Cytoscape.
Mass cytometry data analysis
Cytobank.org was used for de-barcoding of individual samples and manually gating of cell events to remove doublets, normalization beads and dead cells (Kotecha et al., 2010). Per channel intensity ranges were aligned between batches of measurements using a reference sample acquired across all batches and the BatchAdjust function to compute scaling factors at the 95th event percentiles (Schuyler et al., 2019). For semi-automated gating of populations of interest, high-resolution SPADE clustering was conducted on all indicated asinh-transformed markers (Table S2) with 400 target nodes (Qiu et al., 2011). Individual SPADE nodes were then aggregated and annotated to cell subsets (bubbles) according to the expression of lineage-specific differentiation markers. Clustering results and FCS files were subsequently loaded into the R CytoML/flowCore environment (10.18129/B9.bioc.CytoML, 10.18129/B9.bioc.flowCore) for further downstream analyses. To generate UMAP representations all events of a given population of interest were down-sampled to 70,000 cells and then embedded using the tumap function (R uwot package, https://cran.r-project.org/web/packages/uwot/index.html) parameterized by local neighborhood 50, learning rate 0.5, and using the indicated markers (Table S2) as input dimensions. For statistical analysis of cell population abundances, we fitted a generalized linear mixed-effects model (GLMM) for each population using the lme4 package (Nowicka et al., 2017). P values resulting from differential abundance testing (via R multcomp and lsmeans packages) were adjusted using the Benjamini-Hochberg procedure and an FDR-cutoff of 5% across all clusters/subsets and between-group comparisons (Hothorn et al., 2008; Lenth, 2016). Additionally, indicated surface marker positive populations were exported from Cytobank and analyzed using prism 8. Kruskal-Wallis and Dunn’s multiple comparison test was used to compare differences in proportions between patient groups, whereas mixed-effect-analysis and Sidak’s multiple comparison test was applied to report time-dependent differences. The exact numbers and definitions of samples used in the analyses are specified in the respective figure legends and summarized in Table S1.
Data Analysis of Flow Cytometry Data
Flow cytometry data analysis was performed with FlowJo V10.6.1. Cell type was defined as granulocytes (CD45+, CD66b+), non-classical monocytes (CD45+, CD66b-, CD19-, CD3-, CD56-, CD14lo, CD16+). Relative cell percentage or mean fluorescence intensity (MFI) was used for visualization and statistical analysis was done using unpaired t test.
Data visualization
In general, the R packages Seurat and the ggplot2 package (version 3.1.0) (Wickham, 2016) were used to generate figures. For visualization of mass cytometry data, cluster minimum-spanning trees were rendered using Cytobank, the ComplexHeatmap package (Gu et al., 2016) was used to display subset phenotypes and GraphPad Prism to generate boxplots of quantitative data.
Additional Resources
Part of the patients included in this study have been recruited within the clinical trial DRKS00021688, registered at the German registry for clinical studies (Kurth et al., 2020).
In addition to the deposition of the raw sequencing data on EGA, we provide an interactive platform for data inspection and analysis via FASTGenomics. The FASTGenomics platform (fastgenomics.org) provides processed count tables of the datasets generated in this study as well as key analytical results, such as UMAP coordinates and cluster identities, and the code written to analyze the respective data.
Consortia
The members of the Deutsche COVID-19 Omics Initiative (DeCOI) are Angel Angelov, Robert Bals, Alexander Bartholomäus, Anke Becker, Daniela Bezdan, Ezio Bonifacio, Peer Bork, Thomas Clavel, Maria Colome-Tatche, Andreas Diefenbach, Alexander Dilthey, Nicole Fischer, Konrad Förstner, Julia-Stefanie Frick, Julien Gagneur, Alexander Goesmann, Torsten Hain, Michael Hummel, Stefan Janssen, Jörn Kalinowski, René Kallies, Birte Kehr, Andreas Keller, Sarah Kim-Hellmuth, Christoph Klein, Oliver Kohlbacher, Jan O. Korbel, Ingo Kurth, Markus Landthaler, Yang Li, Kerstin Ludwig, Oliwia Makarewicz, Manja Marz, Alice McHardy, Christian Mertes, Markus Nöthen, Peter Nürnberg, Uwe Ohler, Stephan Ossowski, Jörg Overmann, Silke Peter, Klaus Pfeffer, Anna R. Poetsch, Alfred Pühler, Nikolaus Rajewsky, Markus Ralser, Olaf Rieß, Stephan Ripke, Ulisses Nunes da Rocha, Philip Rosenstiel, Antoine-Emmanuel Saliba, Leif Erik Sander, Birgit Sawitzki, Philipp Schiffer, Eva-Christina Schulte, Joachim L. Schultze, Alexander Sczyrba, Oliver Stegle, Jens Stoye, Fabian Theis, Janne Vehreschild, Jörg Vogel, Max von Kleist, Andreas Walker, Jörn Walter, Dagmar Wieczorek, and John Ziebuhr.
Acknowledgments
We thank Michael Kraut, Heidi Theis, Gudrun Hack, Claudia Finnemann, Magdalena Bürkle, Moritz Vollgraf, and Wibke Groenewald for perfect technical assistance. This work was supported by the German Research Foundation (DFG) (SFB-TR84 114933180 to L.E.S., S.H., A. Hocke, N.S., M.W., and C.D.; INST 37/1049-1, INST 216/981-1, INST 257/605-1, INST 269/768-1, INST 217/988-1, INST 217/577-1, and EXC2151/1 to J.L.S.; SFB TR57 and SPP1937 to J.N.; GRK2157 to A.-E.S.; and ME 3644/5-1 to H.E.M.); the Berlin University Alliance (BUA) (PreEP-Corona grant to L.E.S., V.C., and C.D.), the Berlin Institute of Health (BIH) (to L.E.S., A. Hocke, and C.D.); Helmholtz-Gemeinschaft Deutscher Forschungszentren, Germany (sparse2big to J.L.S.), EU projects SYSCID (733100 to J.L.S.), ERA CVD (00160389 to J.L.S.); the DZIF, Germany (TTU 04.816 and 04.817 to J.N.); the Hector Foundation (M89 to J.N.); the Helmholtz Association of German Research Centres (to A.-E.S.); the EU projects ONE STUDY (260687 to B.S.), BIO-DrIM (305147 to B.S.), and INsTRuCT (860003 to B.S.); the German Federal Ministry of Education and Research, Germany (BMBF) (project RAPID to M.W., C.D., S.H., and A. Hocke, project SYMPATH to M.W. and N.S.); a Charité 3R project (to B.S. and S.H.); and Radboud University Medical Centre Hypatia Grant (2018 to Y.L.).
Author Contributions
Conceptualization, J.S.-S., N.R., K.B., B.K., L.B., E.D.D., F.K., J.L.S., A.C.A., Y.L., J.N., B.S., A.-E.S., and L.E.S.; Methodology, J.S.-S., D.P., T.K., S.B., L.B., E.D.D., M.G., D.W., M. Beckstette., T.S.K., A.S., O.D., H.M., A.R.S., C.C., D.K., E.V., C.J.X., A.D., C.T., S.H., C.R.G., M.L., E.W., T.U., M. Becker., R.G., C.D.-H., C.v.K., and K.H.; Software/Data Analysis, J.S.-S., N.R., K.B., S.S., B.Z., T.K., L.B., A.S., T.U., and M. Becker.; Investigation, J.S.-S., K.B., T.P., A. Horne., M.H., J.L.S., A.C.A., M.W., Y.L., J.N., B.S., A.-E.S., and L.E.S.; Biospecimen/Enzyme Resources, B.K., S.B., M.P., S.H., H.M.-R., F.M., A.U., L.B.d.J., L.J., C.R.G., P.G., M.S., C.D.-H., N.N., K.K., R.T., V.C., J.R., K.M.K., M.T.V., G.R., F.K., J.N., and M.W.; Writing – Original Draft, J.S.-S., N.R., K.B., L.B., E.D.D., C.M., J.L.S., A.C.A., Y.L., J.N., B.S., A.-E.S., and L.E.S.; Writing – Review & Editing, J.S.-S., N.R., M.W., K.B., L.B., E.D.D., T.P., M. Becker., T.S.K., S.H., A. Hocke., M.S., H.-D.V., C.D.-H., N.S., C.v.K., F.K., J.L.S., A.C.A., Y.L., J.N., B.S., A.-E.S., and L.E.S.
Declaration of Interests
The authors declare no competing interests.
Published: August 5, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2020.08.001.
Contributor Information
Deutsche COVID-19 OMICS Initiative (DeCOI):
Angel Angelov, Robert Bals, Alexander Bartholomäus, Anke Becker, Daniela Bezdan, Ezio Bonifacio, Peer Bork, Thomas Clavel, Maria Colome-Tatche, Andreas Diefenbach, Alexander Dilthey, Nicole Fischer, Konrad Förstner, Julia-Stefanie Frick, Julien Gagneur, Alexander Goesmann, Torsten Hain, Michael Hummel, Stefan Janssen, Jörn Kalinowski, René Kallies, Birte Kehr, Andreas Keller, Sarah Kim-Hellmuth, Christoph Klein, Oliver Kohlbacher, Jan O. Korbel, Ingo Kurth, Markus Landthaler, Yang Li, Kerstin Ludwig, Oliwia Makarewicz, Manja Marz, Alice McHardy, Christian Mertes, Markus Nöthen, Peter Nürnberg, Uwe Ohler, Stephan Ossowski, Jörg Overmann, Silke Peter, Klaus Pfeffer, Anna R. Poetsch, Alfred Pühler, Nikolaus Rajewsky, Markus Ralser, Olaf Rieß, Stephan Ripke, Ulisses Nunes da Rocha, Philip Rosenstiel, Antoine-Emmanuel Saliba, Leif Erik Sander, Birgit Sawitzki, Philipp Schiffer, Eva-Christina Schulte, Joachim L. Schultze, Alexander Sczyrba, Oliver Stegle, Jens Stoye, Fabian Theis, Janne Vehreschild, Jörg Vogel, Max von Kleist, Andreas Walker, Jörn Walter, Dagmar Wieczorek, and John Ziebuhr
Supplemental Information
Cohort Details, Related to Figures 1, 2, 3, 4, 5, 6, 7, and S1–S6
Detailed Information on Antibody Panels Used for Mass Cytometry Analysis, Related to Figures 1, 3, and 6
List of Antibodies Used for Multi-color Flow Cytometry, Related to Figures 1, 3, and 6
Cluster-Specific Marker Gene Lists from the scRNA-Seq Analyses, Related to Figures 2, 4, 5, 7, S2, S3, S4, and S6
References
- Ahmad S., Singh P., Sharma A., Arora S., Shriwash N., Rahmani A.H., Almatroodi S.A., Manda K., Dohare R., Syed M.A. Transcriptome meta-analysis deciphers a dysregulation in immune response-associated gene signatures during sepsis. Genes (Basel) 2019;10:1005. doi: 10.3390/genes10121005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.C., Geurts P., Aerts J. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017;14:1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alder M.N., Opoka A.M., Lahni P., Hildeman D.A., Wong H.R. Olfactomedin-4 Is a Candidate Marker for a Pathogenic Neutrophil Subset in Septic Shock. Crit. Care Med. 2017;45:e426–e432. doi: 10.1097/CCM.0000000000002102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angerer P., Haghverdi L., Büttner M., Theis F.J., Marr C., Buettner F. destiny: diffusion maps for large-scale single-cell data in R. Bioinformatics. 2016;32:1241–1243. doi: 10.1093/bioinformatics/btv715. [DOI] [PubMed] [Google Scholar]
- Aran D., Looney A.P., Liu L., Wu E., Fong V., Hsu A., Chak S., Naikawadi R.P., Wolters P.J., Abate A.R. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019;20:163–172. doi: 10.1038/s41590-018-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., The Gene Ontology Consortium Gene ontology: tool for the unification of biology. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao L., Deng W., Huang B., Gao H., Liu J., Ren L., Wei Q., Yu P., Xu Y., Qi F. The pathogenicity of SARS-CoV-2 in hACE2 transgenic mice. Nature. 2020;583:830–833. doi: 10.1038/s41586-020-2312-y. [DOI] [PubMed] [Google Scholar]
- Barnes B.J., Adrover J.M., Baxter-Stoltzfus A., Borczuk A., Cools-Lartigue J., Crawford J.M., Daßler-Plenker J., Guerci P., Huynh C., Knight J.S. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020;217:e20200652. doi: 10.1084/jem.20200652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayik D., Zhou Y., Park C., Hong C., Vail D., Silver D.J., Lauko A., Roversi G., Watson D.C., Lo A. Myeloid-derived suppressor cell subsets drive glioblastoma growth in a sex-specific manner. Cancer Discov. 2020;10:1210–1225. doi: 10.1158/2159-8290.CD-19-1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardo D., Marin A.C., Fernández-Tomé S., Montalban-Arques A., Carrasco A., Tristán E., Ortega-Moreno L., Mora-Gutiérrez I., Díaz-Guerra A., Caminero-Fernández R. Human intestinal pro-inflammatory CD11chighCCR2+CX3CR1+ macrophages, but not their tolerogenic CD11c-CCR2-CX3CR1- counterparts, are expanded in inflammatory bowel disease. Mucosal Immunol. 2018;11:1114–1126. doi: 10.1038/s41385-018-0030-7. [DOI] [PubMed] [Google Scholar]
- Bowers N.L., Helton E.S., Huijbregts R.P.H., Goepfert P.A., Heath S.L., Hel Z. Immune suppression by neutrophils in HIV-1 infection: role of PD-L1/PD-1 pathway. PLoS Pathog. 2014;10:e1003993. doi: 10.1371/journal.ppat.1003993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun J., Loyal L., Frentsch M., Wendisch D., Georg P., Kurth F., Hippenstiel S., Dingeldey M., Kruse B., Fauchere F., et al. (2020). SARS-CoV-2-reactive T cells in healthy donors and patients with COVID-19. Nature. Published online July 29, 2020. 10.1038/s41586-020-2598-9. [DOI] [PubMed]
- Bronte V., Serafini P., Mazzoni A., Segal D.M., Zanovello P. L-arginine metabolism in myeloid cells controls T-lymphocyte functions. Trends Immunol. 2003;24:302–306. doi: 10.1016/s1471-4906(03)00132-7. [DOI] [PubMed] [Google Scholar]
- Bronte V., Brandau S., Chen S.H., Colombo M.P., Frey A.B., Greten T.F., Mandruzzato S., Murray P.J., Ochoa A., Ostrand-Rosenberg S. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016;7:12150. doi: 10.1038/ncomms12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbon S., Douglass E., Dunn N., Good B., Harris N.L., Lewis S.E., Mungall C.J., Basu S., Chisholm R.L., Dodson R.J., The Gene Ontology Consortium The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019;47(D1):D330–D338. doi: 10.1093/nar/gky1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassetta L., Baekkevold E.S., Brandau S., Bujko A., Cassatella M.A., Dorhoi A., Krieg C., Lin A., Loré K., Marini O. Deciphering myeloid-derived suppressor cells: isolation and markers in humans, mice and non-human primates. Cancer Immunol. Immunother. 2019;68:687–697. doi: 10.1007/s00262-019-02302-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castell S.D., Harman M.F., Morón G., Maletto B.A., Pistoresi-Palencia M.C. Neutrophils which migrate to lymph nodes modulate CD4+ T cell response by a PD-L1 dependent mechanism. Front. Immunol. 2019;10:105. doi: 10.3389/fimmu.2019.00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G., Wu D., Guo W., Cao Y., Huang D., Wang H., Wang T., Zhang X., Chen H., Yu H. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Invest. 2020;130:2620–2629. doi: 10.1172/JCI137244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua R.L., Lukassen S., Trump S., Hennig B.P., Wendisch D., Pott F., Debnath O., Thürmann L., Kurth F., Völker M.T. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 2020 doi: 10.1038/s41587-020-0602-4. Published online June 26, 2020. [DOI] [PubMed] [Google Scholar]
- Chun E., Lavoie S., Michaud M., Gallini C.A., Kim J., Soucy G., Odze R., Glickman J.N., Garrett W.S. CCL2 Promotes Colorectal Carcinogenesis by Enhancing Polymorphonuclear Myeloid-Derived Suppressor Cell Population and Function. Cell Rep. 2015;12:244–257. doi: 10.1016/j.celrep.2015.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibrián D., Sánchez-Madrid F. CD69: from activation marker to metabolic gatekeeper. Eur. J. Immunol. 2017;47:946–953. doi: 10.1002/eji.201646837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J. From mice to monkeys, animals studied for coronavirus answers. Science. 2020;368:221–222. doi: 10.1126/science.368.6488.221. [DOI] [PubMed] [Google Scholar]
- Cuevas V.D., Anta L., Samaniego R., Orta-Zavalza E., Vladimir de la Rosa J., Baujat G., Domínguez-Soto Á., Sánchez-Mateos P., Escribese M.M., Castrillo A. MAFB Determines Human Macrophage Anti-Inflammatory Polarization: Relevance for the Pathogenic Mechanisms Operating in Multicentric Carpotarsal Osteolysis. J. Immunol. 2017;198:2070–2081. doi: 10.4049/jimmunol.1601667. [DOI] [PubMed] [Google Scholar]
- Darcy C.J., Minigo G., Piera K.A., Davis J.S., McNeil Y.R., Chen Y., Volkheimer A.D., Weinberg J.B., Anstey N.M., Woodberry T. Neutrophils with myeloid derived suppressor function deplete arginine and constrain T cell function in septic shock patients. Crit. Care. 2014;18:R163. doi: 10.1186/cc14003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davison G.M., Nkambule B.B., Mkandla Z., Hon G.M., Kengne A.P., Erasmus R.T., Matsha T.E. Platelet, monocyte and neutrophil activation and glucose tolerance in South African Mixed Ancestry individuals. Sci. Rep. 2017;7:40329. doi: 10.1038/srep40329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kleijn S., Langereis J.D., Leentjens J., Kox M., Netea M.G., Koenderman L., Ferwerda G., Pickkers P., Hermans P.W.M. IFN-γ-stimulated neutrophils suppress lymphocyte proliferation through expression of PD-L1. PLoS ONE. 2013;8:e72249. doi: 10.1371/journal.pone.0072249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Santo C., Salio M., Masri S.H., Lee L.Y.H., Dong T., Speak A.O., Porubsky S., Booth S., Veerapen N., Besra G.S. Invariant NKT cells reduce the immunosuppressive activity of influenza A virus-induced myeloid-derived suppressor cells in mice and humans. J. Clin. Invest. 2008;118:4036–4048. doi: 10.1172/JCI36264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimopoulos G., de Mast Q., Markou N., Theodorakopoulou M., Komnos A., Mouktaroudi M., Netea M.G., Spyridopoulos T., Verheggen R.J., Hoogerwerf J. Favorable Anakinra Responses in Severe Covid-19 Patients with Secondary Hemophagocytic Lymphohistiocytosis. Cell Host Microbe. 2020;28:117–123. doi: 10.1016/j.chom.2020.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellinghaus D., Degenhardt F., Bujanda L., Buti M., Albillos A., Invernizzi P., Fernández J., Prati D., Baselli G., Asselta R. Genomewide Association Study of Severe Covid-19 with Respiratory Failure. N. Engl. J. Med. 2020 doi: 10.1056/NEJMoa2020283. Published online June 17, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer-Riepe L., Daber N., Schulte-Schrepping J., Véras De Carvalho B.C., Russo A., Pohlen M., Fischer J., Chasan A.I., Wolf M., Ulas T. CD163 expression defines specific, IRF8-dependent, immune-modulatory macrophages in the bone marrow. J. Allergy Clin. Immunol. 2020 doi: 10.1016/j.jaci.2020.02.034. Published online March 19, 2020. [DOI] [PubMed] [Google Scholar]
- Giamarellos-Bourboulis E.J., Netea M.G., Rovina N., Akinosoglou K., Antoniadou A., Antonakos N., Damoraki G., Gkavogianni T., Adami M.E., Katsaounou P. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe. 2020;27:992–1000. doi: 10.1016/j.chom.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grifoni A., Weiskopf D., Ramirez S.I., Mateus J., Dan J.M., Moderbacher C.R., Rawlings S.A., Sutherland A., Premkumar L., Jadi R.S. Targets of T cell responses to SARS-CoV-2 coronavirus in humans with COVID-19 disease and unexposed individuals. Cell. 2020;181:1489–1501. doi: 10.1016/j.cell.2020.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- Hadjadj J., Yatim N., Barnabei L., Corneau A., Boussier J., Smith N., Pere H., Charbit B., Bondet V., Chenevier-Gobeaux C., Breillat P. Impaired type I interferon activity and exacerbated inflammatory responses in severe COVID-19 patients. Science. 2020;369:718–724. doi: 10.1126/science.abc6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafemeister C., Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20:296. doi: 10.1186/s13059-019-1874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesselink L., Spijkerman R., van Wessem K.J.P., Koenderman L., Leenen L.P.H., Huber-Lang M., Hietbrink F. Neutrophil heterogeneity and its role in infectious complications after severe trauma. World J. Emerg. Surg. 2019;14:24. doi: 10.1186/s13017-019-0244-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano N., Butler M.O., Xia Z., Ansén S., von Bergwelt-Baildon M.S., Neuberg D., Freeman G.J., Nadler L.M. Engagement of CD83 ligand induces prolonged expansion of CD8+ T cells and preferential enrichment for antigen specificity. Blood. 2006;107:1528–1536. doi: 10.1182/blood-2005-05-2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann S., Harms H., Ulm L., Nabavi D.G., Mackert B.M., Schmehl I., Jungehulsing G.J., Montaner J., Bustamante A., Hermans M., PREDICT Investigators Stroke-induced immunodepression and dysphagia independently predict stroke-associated pneumonia - The PREDICT study. J. Cereb. Blood Flow Metab. 2017;37:3671–3682. doi: 10.1177/0271678X16671964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horby P., Lim W.S., Emberson J.R., Mafham M., Bell J.L., Linsell L., Phil D., Sta-Plin N., Brightling C., Med F. Dexamethasone in Hospitalized Patients with Covid-19 - Preliminary Report. N. Engl. J. Med. 2020 doi: 10.1056/NEJMoa2021436. Published online July 17, 2020. [DOI] [Google Scholar]
- Hothorn T., Bretz F., Westfall P. Simultaneous inference in general parametric models. Biom. J. 2008;50:346–363. doi: 10.1002/bimj.200810425. [DOI] [PubMed] [Google Scholar]
- Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X., He C., Lin G., Lu L., Xing K., Hua X., Sun S., Mao Y., Song Y., Wang J., Li S. Induced CD10 expression during monocyte-to-macrophage differentiation identifies a unique subset of macrophages in pancreatic ductal adenocarcinoma. Biochem. Biophys. Res. Commun. 2020;524:1064–1071. doi: 10.1016/j.bbrc.2020.02.042. [DOI] [PubMed] [Google Scholar]
- Jamilloux Y., Henry T., Belot A., Viel S., Fauter M., El Jammal T., Walzer T., François B., Sève P. Should we stimulate or suppress immune responses in COVID-19? Cytokine and anti-cytokine interventions. Autoimmun. Rev. 2020;19:102567. doi: 10.1016/j.autrev.2020.102567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janky R., Verfaillie A., Imrichová H., Van de Sande B., Standaert L., Christiaens V., Hulselmans G., Herten K., Naval Sanchez M., Potier D. iRegulon: from a gene list to a gene regulatory network using large motif and track collections. PLoS Comput. Biol. 2014;10 doi: 10.1371/journal.pcbi.1003731. e1003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janols H., Wullt M., Bergenfelz C., Björnsson S., Lickei H., Janciauskiene S., Leandersson K., Bredberg A. Heterogeneity among septic shock patients in a set of immunoregulatory markers. Eur. J. Clin. Microbiol. Infect. Dis. 2014;33:313–324. doi: 10.1007/s10096-013-1957-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamp V.M., Pillay J., Lammers J.-W.J., Pickkers P., Ulfman L.H., Koenderman L. Human suppressive neutrophils CD16bright/CD62Ldim exhibit decreased adhesion. J. Leukoc. Biol. 2012;92:1011–1020. doi: 10.1189/jlb.0612273. [DOI] [PubMed] [Google Scholar]
- Kanehisa M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019;28:1947–1951. doi: 10.1002/pro.3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kangelaris K.N., Prakash A., Liu K.D., Aouizerat B., Woodruff P.G., Erle D.J., Rogers A., Seeley E.J., Chu J., Liu T. Increased expression of neutrophil-related genes in patients with early sepsis-induced ARDS. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015;308:L1102–L1113. doi: 10.1152/ajplung.00380.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klok F.A., Kruip M.J.H.A., van der Meer N.J.M., Arbous M.S., Gommers D.A.M.P.J., Kant K.M., Kaptein F.H.J., van Paassen J., Stals M.A.M., Huisman M.V., Endeman H. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 2020;191:145–147. doi: 10.1016/j.thromres.2020.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.R., Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotecha N., Krutzik P.O., Irish J.M. Web-based analysis and publication of flow cytometry experiments. Curr. Protoc. Cytom. 2010;Chapter 10:Unit10.17. doi: 10.1002/0471142956.cy1017s53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuri-Cervantes L., Pampena M.B., Meng W., Rosenfeld A.M., Ittner C.A.G., Weisman A.R., Agyekum R., Mathew D., Baxter A.E., Vella L. Immunologic perturbations in severe COVID-19/SARS-CoV-2 infection. bioRxiv. 2020 doi: 10.1101/2020.05.18.101717. [DOI] [Google Scholar]
- Kurth F., Roennefarth M., Thibeault C., Corman V.M., Müller-Redetzky H., Mittermaier M., Ruwwe-Glösenkamp C., Heim K.M., Krannich A., Zvorc S. Studying the pathophysiology of coronavirus disease 2019: a protocol for the Berlin prospective COVID-19 patient cohort (Pa-COVID-19) Infection. 2020;48:619–626. doi: 10.1007/s15010-020-01464-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kverneland A.H., Streitz M., Geissler E., Hutchinson J., Vogt K., Boës D., Niemann N., Pedersen A.E., Schlickeiser S., Sawitzki B. Age and gender leucocytes variances and references values generated using the standardized ONE-Study protocol. Cytometry A. 2016;89:543–564. doi: 10.1002/cyto.a.22855. [DOI] [PubMed] [Google Scholar]
- Kwok I., Becht E., Xia Y., Ng M., Teh Y.C., Tan L., Evrard M., Li J.L.Y., Tran H.T.N., Tan Y. Combinatorial Single-Cell Analyses of Granulocyte-Monocyte Progenitor Heterogeneity Reveals an Early Uni-potent Neutrophil Progenitor. Immunity. 2020;S1074-7613:30235-1. doi: 10.1016/j.immuni.2020.06.005. [DOI] [PubMed] [Google Scholar]
- Langereis J.D., Pickkers P., de Kleijn S., Gerretsen J., de Jonge M.I., Kox M. Spleen-derived IFN-γ induces generation of PD-L1+-suppressive neutrophils during endotoxemia. J. Leukoc. Biol. 2017;102:1401–1409. doi: 10.1189/jlb.3A0217-051RR. [DOI] [PubMed] [Google Scholar]
- Lee J.S., Park S., Jeong H.W., Ahn J.Y., Choi S.J., Lee H., Choi B., Nam S.K., Sa M., Kwon J.-S. Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Sci. Immunol. 2020;5 doi: 10.1126/sciimmunol.abd1554. eabd1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenth R.V. Least-squares means: The R package lsmeans. J. Stat. Softw. 2016;69:1–33. [Google Scholar]
- Leshner M., Wang S., Lewis C., Zheng H., Chen X.A., Santy L., Wang Y. PAD4 mediated histone hypercitrullination induces heterochromatin decondensation and chromatin unfolding to form neutrophil extracellular trap-like structures. Front. Immunol. 2012;3:307. doi: 10.3389/fimmu.2012.00307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.-K., Lu Q.-B., Chen W.-W., Xu W., Liu R., Zhang S.-F., Du J., Li H., Yao K., Zhai D. Arginine deficiency is involved in thrombocytopenia and immunosuppression in severe fever with thrombocytopenia syndrome. Sci. Transl. Med. 2018;10 doi: 10.1126/scitranslmed.aat4162. eaat4162. [DOI] [PubMed] [Google Scholar]
- Liao M., Liu Y., Yuan J., Wen Y., Xu G., Zhao J., Cheng L., Li J., Wang X., Wang F. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020;26:842–844. doi: 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]
- Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J.P., Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R.-J., Chu J.-S., Chien H.-L., Tseng C.-H., Ko P.-C., Mei Y.-Y., Tang W.-C., Kao Y.-T., Cheng H.-Y., Liang Y.-C., Lin S.Y. MCPIP1 suppresses hepatitis C virus replication and negatively regulates virus-induced proinflammatory cytokine responses. J. Immunol. 2014;193:4159–4168. doi: 10.4049/jimmunol.1400337. [DOI] [PubMed] [Google Scholar]
- Loftus T.J., Mohr A.M., Moldawer L.L. Dysregulated myelopoiesis and hematopoietic function following acute physiologic insult. Curr. Opin. Hematol. 2018;25:37–43. doi: 10.1097/MOH.0000000000000395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long Q.-X., Liu B.-Z., Deng H.-J., Wu G.-C., Deng K., Chen Y.-K., Liao P., Qiu J.-F., Lin Y., Cai X.-F. Antibody responses to SARS-CoV-2 in patients with COVID-19. Nat. Med. 2020;26:845–848. doi: 10.1038/s41591-020-0897-1. [DOI] [PubMed] [Google Scholar]
- Lucas C., Wong P., Klein J., Castro T.B.R., Silva J., Sundaram M., Ellingson M.K., Mao T., Oh J.E., Israelow B., Yale IMPACT Team Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature. 2020 doi: 10.1038/s41586-020-2588-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüdtke A., Ruibal P., Becker-Ziaja B., Rottstegge M., Wozniak D.M., Cabeza-Cabrerizo M., Thorenz A., Weller R., Kerber R., Idoyaga J. Ebola Virus Disease Is Characterized by Poor Activation and Reduced Levels of Circulating CD16+ Monocytes. J. Infect. Dis. 2016;214(Suppl 3):S275–S280. doi: 10.1093/infdis/jiw260. [DOI] [PubMed] [Google Scholar]
- MacParland S.A., Liu J.C., Ma X.Z., Innes B.T., Bartczak A.M., Gage B.K., Manuel J., Khuu N., Echeverri J., Linares I. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun. 2018;9:4383. doi: 10.1038/s41467-018-06318-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier M.J. DirichletReg: Dirichlet Regression for Compositional Data in R. 2014. https://epub.wu.ac.at/4077/
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011;17:10. [Google Scholar]
- Maslove D.M., Shapira T., Tyryshkin K., Veldhoen R.A., Marshall J.C., Muscedere J. Validation of diagnostic gene sets to identify critically ill patients with sepsis. J. Crit. Care. 2019;49:92–98. doi: 10.1016/j.jcrc.2018.10.028. [DOI] [PubMed] [Google Scholar]
- Mathew D., Giles J.R., Baxter A.E., Greenplate A.R., Wu J.E., Alanio C., Oldridge D.A., Kuri-Cervantes L., Pampena M.B., D’Andrea K. Deep immune profiling of COVID-19 patients reveals patient heterogeneity and distinct immunotypes with implications for therapeutic interventions. bioRxiv. 2020 doi: 10.1101/2020.05.20.106401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKechnie J.L., Blish C.A. The Innate Immune System: Fighting on the Front Lines or Fanning the Flames of COVID-19? Cell Host Microbe. 2020;27:863–869. doi: 10.1016/j.chom.2020.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meghraoui-Kheddar A., Chousterman B.G., Guillou N., Barone S.M., Granjeaud S., Vallet H., Corneau A., Guessous K., Boissonnas A., Irish J.M. Two new immature and dysfunctional neutrophil cell subsets define a predictive signature of sepsis useable in clinical practice. bioRxiv. 2020 doi: 10.1101/2020.05.29.123992. [DOI] [Google Scholar]
- Mei H.E., Leipold M.D., Schulz A.R., Chester C., Maecker H.T. Barcoding of live human peripheral blood mononuclear cells for multiplexed mass cytometry. J. Immunol. 2015;194:2022–2031. doi: 10.4049/jimmunol.1402661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei H.E., Leipold M.D., Maecker H.T. Platinum-conjugated antibodies for application in mass cytometry. Cytometry A. 2016;89:292–300. doi: 10.1002/cyto.a.22778. [DOI] [PubMed] [Google Scholar]
- Mengos A.E., Gastineau D.A., Gustafson M.P. The CD14(+) HLA-DR(lo/neg) Monocyte: An immunosuppressive phenotype that restrains responses to cancer immunotherapy. Front. Immunol. 2019;10:1147. doi: 10.3389/fimmu.2019.01147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merad M., Martin J.C. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat. Rev. Immunol. 2020;20:355–362. doi: 10.1038/s41577-020-0331-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messner C.B., Demichev V., Wendisch D., Michalick L., White M., Freiwald A., Textoris-Taube K., Vernardis S.I., Egger A.-S., Kreidl M. Ultra-high-throughput clinical proteomics reveals classifiers of COVID-19 infection. Cell Syst. 2020;11:11–24. doi: 10.1016/j.cels.2020.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer C., Cagnon L., Costa-Nunes C.M., Baumgaertner P., Montandon N., Leyvraz L., Michielin O., Romano E., Speiser D.E. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol. Immunother. 2014;63:247–257. doi: 10.1007/s00262-013-1508-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton E.A., He X.-Y., Denorme F., Campbell R.A., Ng D., Salvatore S.P., Mostyka M., Baxter-Stoltzfus A., Borczuk A.C., Loda M. Neutrophil Extracellular Traps (NETs) Contribute to Immunothrombosis in COVID-19 Acute Respiratory Distress Syndrome. Blood. 2020 doi: 10.1182/blood.2020007008. Published online June 29, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monneret G., Lepape A., Voirin N., Bohé J., Venet F., Debard A.L., Thizy H., Bienvenu J., Gueyffier F., Vanhems P. Persisting low monocyte human leukocyte antigen-DR expression predicts mortality in septic shock. Intensive Care Med. 2006;32:1175–1183. doi: 10.1007/s00134-006-0204-8. [DOI] [PubMed] [Google Scholar]
- Mortaz E., Alipoor S.D., Adcock I.M., Mumby S., Koenderman L. Update on neutrophil function in severe inflammation. Front. Immunol. 2018;9:2171. doi: 10.3389/fimmu.2018.02171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naranjo-Gómez J.S., Castillo J.A., Rojas M., Restrepo B.N., Diaz F.J., Velilla P.A., Castaño D. Different phenotypes of non-classical monocytes associated with systemic inflammation, endothelial alteration and hepatic compromise in patients with dengue. Immunology. 2019;156:147–163. doi: 10.1111/imm.13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng L.G., Ostuni R., Hidalgo A. Heterogeneity of neutrophils. Nat. Rev. Immunol. 2019;19:255–265. doi: 10.1038/s41577-019-0141-8. [DOI] [PubMed] [Google Scholar]
- Ni L., Ye F., Cheng M.L., Feng Y., Deng Y.Q., Zhao H., Wei P., Ge J., Gou M., Li X. Detection of SARS-CoV-2-Specific Humoral and Cellular Immunity in COVID-19 Convalescent Individuals. Immunity. 2020;52:971–977. doi: 10.1016/j.immuni.2020.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowicka M., Krieg C., Crowell H.L., Weber L.M., Hartmann F.J., Guglietta S., Becher B., Levesque M.P., Robinson M.D. CyTOF workflow: differential discovery in high-throughput high-dimensional cytometry datasets. F1000Res. 2017;6:748. doi: 10.12688/f1000research.11622.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong E.Z., Chan Y.F.Z., Leong W.Y., Lee N.M.Y., Kalimuddin S., Haja Mohideen S.M., Chan K.S., Tan A.T., Bertoletti A., Ooi E.E., Low J.G.H. A dynamic immune response shapes COVID-19 progression. Cell Host Microbe. 2020;27:879–882. doi: 10.1016/j.chom.2020.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellin D., Loperfido M., Baricordi C., Wolock S.L., Montepeloso A., Weinberg O.K., Biffi A., Klein A.M., Biasco L. A comprehensive single cell transcriptional landscape of human hematopoietic progenitors. Nat. Commun. 2019;10:2395. doi: 10.1038/s41467-019-10291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlman S., Dandekar A.A. Immunopathogenesis of coronavirus infections: implications for SARS. Nat. Rev. Immunol. 2005;5:917–927. doi: 10.1038/nri1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiler S., Massberg S., Engelmann B. Biological basis and pathological relevance of microvascular thrombosis. Thromb. Res. 2014;133(Suppl 1):S35–S37. doi: 10.1016/j.thromres.2014.03.016. [DOI] [PubMed] [Google Scholar]
- Pillay J., Kamp V.M., van Hoffen E., Visser T., Tak T., Lammers J.W., Ulfman L.H., Leenen L.P., Pickkers P., Koenderman L. A subset of neutrophils in human systemic inflammation inhibits T cell responses through Mac-1. J. Clin. Invest. 2012;122:327–336. doi: 10.1172/JCI57990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popescu D.M., Botting R.A., Stephenson E., Green K., Webb S., Jardine L., Calderbank E.F., Polanski K., Goh I., Efremova M. Decoding human fetal liver haematopoiesis. Nature. 2019;574:365–371. doi: 10.1038/s41586-019-1652-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu P., Simonds E.F., Bendall S.C., Gibbs K.D., Jr., Bruggner R.V., Linderman M.D., Sachs K., Nolan G.P., Plevritis S.K. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat. Biotechnol. 2011;29:886–891. doi: 10.1038/nbt.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quah P., Li A., Phua J. Mortality rates of patients with COVID-19 in the intensive care unit: a systematic review of the emerging literature. Crit. Care. 2020;24:285. doi: 10.1186/s13054-020-03006-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remy K.E., Brakenridge S.C., Francois B., Daix T., Deutschman C.S., Monneret G., Jeannet R., Laterre P.-F., Hotchkiss R.S., Moldawer L.L. Immunotherapies for COVID-19: lessons learned from sepsis. Lancet Respir. Med. 2020;2:2–5. doi: 10.1016/S2213-2600(20)30217-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes M., Filbin M.R., Bhattacharyya R.P., Billman K., Eisenhaure T., Hung D.T., Levy B.D., Baron R.M., Blainey P.C., Goldberg M.B., Hacohen N. An immune-cell signature of bacterial sepsis. Nat. Med. 2020;26:333–340. doi: 10.1038/s41591-020-0752-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reymond N., Imbert A.M., Devilard E., Fabre S., Chabannon C., Xerri L., Farnarier C., Cantoni C., Bottino C., Moretta A. DNAM-1 and PVR regulate monocyte migration through endothelial junctions. J. Exp. Med. 2004;199:1331–1341. doi: 10.1084/jem.20032206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riegel A., Maurer T., Prior B., Stegmaier S., Heppert V., Wagner C., Hänsch G.M. Human polymorphonuclear neutrophils express RANK and are activated by its ligand, RANKL. Eur. J. Immunol. 2012;42:975–981. doi: 10.1002/eji.201141786. [DOI] [PubMed] [Google Scholar]
- Ritchie A.I., Singanayagam A. Immunosuppression for hyperinflammation in COVID-19: a double-edged sword? Lancet. 2020;395:1111. doi: 10.1016/S0140-6736(20)30691-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbiani D.F., Gaebler C., Muecksch F., Lorenzi J.C.C., Wang Z., Cho A., Agudelo M., Barnes C.O., Gazumyan A., Finkin S. Convergent antibody responses to SARS-CoV-2 in convalescent individuals. Nature. 2020 doi: 10.1038/s41586-020-2456-9. Published online June 18, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Cerrillo I., Landete P., Aldave B., Sanchez-Alonso S., Sanchez-Azofra A., Marcos-Jimenez A., Avalos E., Alcaraz-Serna A., de los Santos I., Mateu-Albero T. Differential Redistribution of Activated Monocyte and Dendritic Cell Subsets to the Lung Associates with Severity of COVID-19. medRxiv. 2020 doi: 10.1101/2020.05.13.20100925. [DOI] [Google Scholar]
- Sander L.E., Sackett S.D., Dierssen U., Beraza N., Linke R.P., Müller M., Blander J.M., Tacke F., Trautwein C. Hepatic acute-phase proteins control innate immune responses during infection by promoting myeloid-derived suppressor cell function. J. Exp. Med. 2010;207:1453–1464. doi: 10.1084/jem.20091474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawitzki B., Harden P.N., Reinke P., Moreau A., Hutchinson J.A., Game D.S., Tang Q., Guinan E.C., Battaglia M., Burlingham W.J. Regulatory cell therapy in kidney transplantation (The ONE Study): a harmonised design and analysis of seven non-randomised, single-arm, phase 1/2A trials. Lancet. 2020;395:1627–1639. doi: 10.1016/S0140-6736(20)30167-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scapini P., Marini O., Tecchio C., Cassatella M.A. Human neutrophils in the saga of cellular heterogeneity: insights and open questions. Immunol. Rev. 2016;273:48–60. doi: 10.1111/imr.12448. [DOI] [PubMed] [Google Scholar]
- Schrijver I.T., Kemperman H., Roest M., Kesecioglu J., de Lange D.W. Myeloperoxidase can differentiate between sepsis and non-infectious SIRS and predicts mortality in intensive care patients with SIRS. Intensive Care Med. Exp. 2017;5:43. doi: 10.1186/s40635-017-0157-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultze J.L., Mass E., Schlitzer A. Emerging Principles in Myelopoiesis at Homeostasis and during Infection and Inflammation. Immunity. 2019;50:288–301. doi: 10.1016/j.immuni.2019.01.019. [DOI] [PubMed] [Google Scholar]
- Schulz A.R., Mei H.E. Methods in Molecular Biology. Humana Press Inc.; 2019. Surface barcoding of live PBMC for multiplexed mass cytometry; pp. 93–108. [DOI] [PubMed] [Google Scholar]
- Schuyler R.P., Jackson C., Garcia-Perez J.E., Baxter R.M., Ogolla S., Rochford R., Ghosh D., Rudra P., Hsieh E.W.Y. Minimizing Batch Effects in Mass Cytometry Data. Front. Immunol. 2019;10:2367. doi: 10.3389/fimmu.2019.02367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segawa S., Kondo Y., Nakai Y., Iizuka A., Kaneko S., Yokosawa M., Furuyama K., Tsuboi H., Goto D., Matsumoto I., Sumida T. Placenta Specific 8 Suppresses IL-18 Production through Regulation of Autophagy and Is Associated with Adult Still Disease. J. Immunol. 2018;201:3534–3545. doi: 10.4049/jimmunol.1800667. [DOI] [PubMed] [Google Scholar]
- Silvestre-Roig C., Fridlender Z.G., Glogauer M., Scapini P. Neutrophil Diversity in Health and Disease. Trends Immunol. 2019;40:565–583. doi: 10.1016/j.it.2019.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiel L., Delabranche X., Galoisy A.C., Severac F., Toti F., Mauvieux L., Meziani F., Boisramé-Helms J. Neutrophil Fluorescence: A New Indicator of Cell Activation During Septic Shock-Induced Disseminated Intravascular Coagulation. Crit. Care Med. 2016;44:e1132–e1136. doi: 10.1097/CCM.0000000000001851. [DOI] [PubMed] [Google Scholar]
- Stiel L., Meziani F., Helms J. Neutrophil Activation During Septic Shock. Shock. 2018;49:371–384. doi: 10.1097/SHK.0000000000000980. [DOI] [PubMed] [Google Scholar]
- Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tak T., Wijten P., Heeres M., Pickkers P., Scholten A., Heck A.J.R., Vrisekoop N., Leenen L.P., Borghans J.A.M., Tesselaar K., Koenderman L. Human CD62Ldim neutrophils identified as a separate subset by proteome profiling and in vivo pulse-chase labeling. Blood. 2017;129:3476–3485. doi: 10.1182/blood-2016-07-727669. [DOI] [PubMed] [Google Scholar]
- Testa U., Riccioni R., Diverio D., Rossini A., Lo Coco F., Peschle C. Interleukin-3 receptor in acute leukemia. Leukemia. 2004;18:219–226. doi: 10.1038/sj.leu.2403224. [DOI] [PubMed] [Google Scholar]
- Thomas M.P., Whangbo J., McCrossan G., Deutsch A.J., Martinod K., Walch M., Lieberman J. Leukocyte protease binding to nucleic acids promotes nuclear localization and cleavage of nucleic acid binding proteins. J. Immunol. 2014;192:5390–5397. doi: 10.4049/jimmunol.1303296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trouillet-Assant S., Viel S., Gaymard A., Pons S., Richard J.C., Perret M., Villard M., Brengel-Pesce K., Lina B., Mezidi M., COVID HCL Study group Type I IFN immunoprofiling in COVID-19 patients. J. Allergy Clin. Immunol. 2020;146:206–208. doi: 10.1016/j.jaci.2020.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhel F., Azzaoui I., Grégoire M., Pangault C., Dulong J., Tadié J.M., Gacouin A., Camus C., Cynober L., Fest T. Early expansion of circulating granulocytic myeloid-derived suppressor cells predicts development of nosocomial infections in patients with sepsis. Am. J. Respir. Crit. Care Med. 2017;196:315–327. doi: 10.1164/rccm.201606-1143OC. [DOI] [PubMed] [Google Scholar]
- Veglia F., Perego M., Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018;19:108–119. doi: 10.1038/s41590-017-0022-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venet F., Demaret J., Gossez M., Monneret G. Myeloid cells in sepsis-acquired immunodeficiency. Ann. N Y Acad. Sci. 2020 doi: 10.1111/nyas.14333. Published online March 23, 2020. [DOI] [PubMed] [Google Scholar]
- Vo A.V., Takenaka E., Shibuya A., Shibuya K. Expression of DNAM-1 (CD226) on inflammatory monocytes. Mol. Immunol. 2016;69:70–76. doi: 10.1016/j.molimm.2015.11.009. [DOI] [PubMed] [Google Scholar]
- Volkmann J., Schmitz J., Nordlohne J., Dong L., Helmke A., Sen P., Immenschuh S., Bernhardt W.M., Gwinner W., Bräsen J.H. Kidney injury enhances renal G-CSF expression and modulates granulopoiesis and human neutrophil CD177 in vivo. Clin. Exp. Immunol. 2020;199:97–108. doi: 10.1111/cei.13372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D., Hu B., Hu C., Zhu F., Liu X., Zhang J., Wang B., Xiang H., Cheng Z., Xiong Y. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA. 2020;323:1061–1069. doi: 10.1001/jama.2020.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L.L., Wang W.J., Chen D.X., Xu B. Dysregulation of the immune response affects the outcome of critical COVID-19 patients. J. Med. Virol. 2020 doi: 10.1002/jmv.26181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham, H. (2016). ggplot2: Elegant Graphics for Data Analysis. (Springer-Verlag, New York).
- Wilk A.J., Rustagi A., Zhao N.Q., Roque J., Martínez-Colón G.J., McKechnie J.L., Ivison G.T., Ranganath T., Vergara R., Hollis T. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 2020;26:1070–1076. doi: 10.1038/s41591-020-0944-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X., Wen M., Zhan S., He J., Chen W. [An increased neutrophil/lymphocyte ratio is an early warning signal of severe COVID-19] Nan Fang Yi Ke Da Xue Xue Bao. 2020;40:333–336. doi: 10.12122/j.issn.1673-4254.2020.03.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z., Shi L., Wang Y., Zhang J., Huang L., Zhang C., Liu S., Zhao P., Liu H., Zhu L. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020;8:420–422. doi: 10.1016/S2213-2600(20)30076-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Q., He D.M., Shu G.F., Cao B., Xia Y.Q., Xing Y., Ni M., Chen J.F., Shi S.L., Gu H.F. Increased formation of neutrophil extracellular traps is associated with gut leakage in patients with type 1 but not type 2 diabetes. J. Diabetes. 2019;11:665–673. doi: 10.1111/1753-0407.12892. [DOI] [PubMed] [Google Scholar]
- Younos I.H., Abe F., Talmadge J.E. Myeloid-derived suppressor cells: their role in the pathophysiology of hematologic malignancies and potential as therapeutic targets. Leuk. Lymphoma. 2015;56:2251–2263. doi: 10.3109/10428194.2014.987141. [DOI] [PubMed] [Google Scholar]
- Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F., Yu T., Du R., Fan G., Liu Y., Liu Z., Xiang J., Wang Y., Song B., Gu X. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054–1062. doi: 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z., Ren L., Zhang L., Zhong J., Xiao Y., Jia Z., Guo L., Yang J., Wang C., Jiang S. Overly Exuberant Innate Immune Response to SARS-CoV-2 Infection. SSRN Electron. J. 2020 doi: 10.2139/ssrn.3551623. [DOI] [Google Scholar]
- Zuo Y., Yalavarthi S., Shi H., Gockman K., Zuo M., Madison J.A., Blair C., Weber A., Barnes B.J., Egeblad M. Neutrophil extracellular traps in COVID-19. JCI Insight. 2020;5:e138999. doi: 10.1172/jci.insight.138999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cohort Details, Related to Figures 1, 2, 3, 4, 5, 6, 7, and S1–S6
Detailed Information on Antibody Panels Used for Mass Cytometry Analysis, Related to Figures 1, 3, and 6
List of Antibodies Used for Multi-color Flow Cytometry, Related to Figures 1, 3, and 6
Cluster-Specific Marker Gene Lists from the scRNA-Seq Analyses, Related to Figures 2, 4, 5, 7, S2, S3, S4, and S6
Data Availability Statement
ScRNA-seq data generated during this study are deposited at the European Genome-phenome Archive (EGA) under access number EGAS00001004571, which is hosted by the EBI and the CRG.