

Abstract
Since January 2020, the world is facing the COVID-19 pandemic caused by SARS-CoV-2. In a big effort to cope with this outbreak, two Uruguayan institutions, Institut Pasteur de Montevideo and Universidad de la República, have developed and implemented a diagnosis pipeline based on qRT-PCR using entirely local resources. In this context, we performed comparative quantitative proteomic analysis from oro- and naso-pharyngeal swabs used for diagnosis. Tryptic peptides obtained from five positive and five negative samples were analysed by nano-LC-MS/MS using a Q-Exactive Plus mass spectrometer. Data analysis was performed using PatternLab for Proteomics software. From all SARS-CoV-2 positive swabs we were able to detect peptides of the SARS-CoV-2 nucleoprotein that encapsulates and protect the RNA genome. Additionally, we detected an average of 1100 human proteins from each sample. The most abundant proteins exclusively detected in positive swabs were “Guanylate-binding protein 1”, “Tapasin” and “HLA class II histocompatibility antigen DR beta chain”. The biological processes overrepresented in infected host cells were “SRP-dependent cotranslational protein targeting to membrane”, “nuclear-transcribed mRNA catabolic process, nonsense-mediated decay”, “viral transcription” and “translational initiation”. Data is available via ProteomeXchange with identifier PXD020394. We expect that this data can contribute to the future development of mass spectrometry based approaches for COVID-19 diagnosis. Also, we share this preliminary proteomic characterization concerning the host response to infection for its reuse in basic investigation.
Keywords: SARS-CoV-2, COVID-19, Quantitative proteomics, Shotgun proteomics, Nucleoprotein
Specifications Table
| Subject | Biochemistry, Genetics and Molecular Biology (General). |
| Specific subject area | Label-free quantitative proteomics, virology, SARS-CoV-2. |
| Type of data | Tables. Figures. Supplementary data as excel output files. Supplementary figure. Supplementary tables. Raw data in public repository. |
| How data were acquired | Swabs for clinical diagnosis of COVID-19 were biological inactivated with 2% SDS. Protein samples were run on a SDS-PAGE and processed for mass spectrometry analysis. Data was acquired using a nano-HPLC (UltiMate 3000, Thermo) coupled to a Q-Orbitrap mass spectrometer (Q-Exactive Plus, Thermo). Protein identification and relative quantification were performed with PatternLab for Proteomics v4.0 software. Data was further analysed with Panther Server Classification System (http://pantherdb.org). |
| Data format | RAW data (ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD020394) and supplementary excel files with data analysis output, figures and tables. |
| Parameters for data collection | Five SARS-CoV-2 positive and five negatives oro- and naso-pharyngeal swabs were obtained from the COVID-19 diagnostic laboratory installed at Institut Pasteur de Montevideo. They were selected in a random way. Data from swabs samples origin was confidential and thus totally unknown. |
| Description of data collection | Sample was collected by health professionals from local public hospitals. |
| Data source location | Institut Pasteur de Montevideo Montevideo Uruguay |
| Data accessibility | Repository name: ProteomeXchange Consortium via the PRIDE partner Data identification number: PXD020394 Direct URL to data: http://www.ebi.ac.uk/pride/archive/projects/PXD020394 ftp://ftp.pride.ebi.ac.uk/pride/data/archive/2020/07/PXD020394 |
Value of the Data
-
•
This dataset provides evidence of mass spectrometry detection of SARS-CoV-2 proteins directly from clinical samples. Also offers insights into the human biological processes altered by SARS-CoV-2 infection.
-
•
This dataset is intended to be used by researches that have the aim to develop an alternative diagnosis tool based on mass spectrometry using clinical samples (oro/naso-pharyngeal swabs). Also, it would be useful for researchers in the areas of virology and immunology since it provides unbiased insights into host proteome response to SARS-CoV-2 infection.
-
•
The dataset set offers information of SARS-CoV-2 peptides detected from clinical samples and could be used for further development of alternative diagnostic strategies. Additionally, it gives preliminary information on biological processes and pathways turned on in oro- and naso-pharyngeal mucosa in response to this viral infection.
-
•
To deal with this pandemic, produced by a totally unknown infectious agent, the scientific community is carrying forward an unprecedented effort to rapidly generate knowledge and diagnostic methods with impact in the health of the people currently infected. In that sense, sharing this data could help to accelerate different human health related scientific developments.
1. Data description
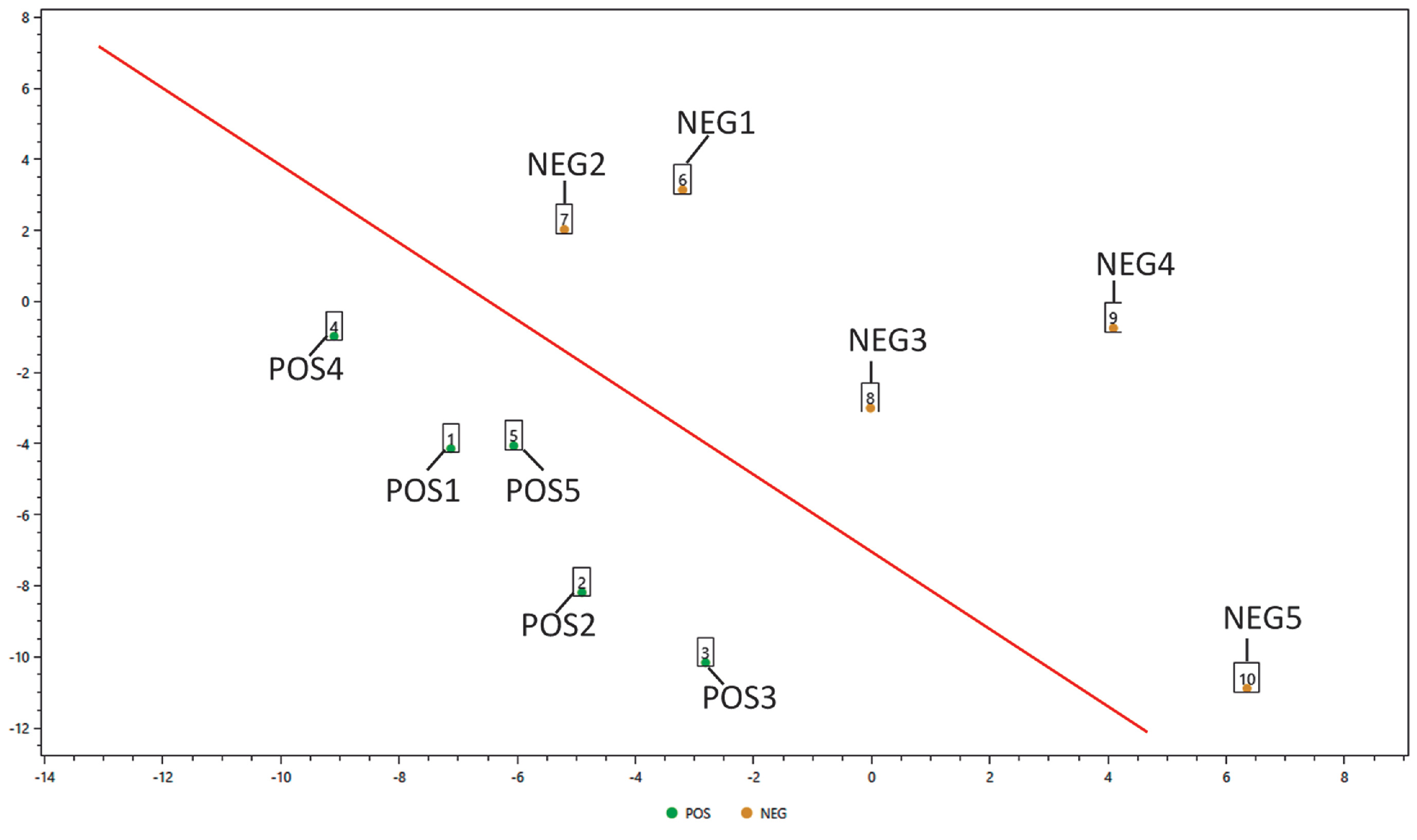
The proteomic dataset presented here was generated from oro- and naso-pharyngeal swabs obtained from the diagnostic pipeline implemented at Institut Pasteur de Montevideo. Five positive and 5 negative swabs were first inactivated and the proteins were extracted. Proteins were separated shortly (1 cm) in a SDS-PAGE and processed for mass spectrometry analysis as described in Materials and Methods section. We performed a Principal Component Analysis (PCA) and we observed that negative and positive samples grouped separately (supplementary figure 1). Also, we were able to identify an average of 1100 proteins from each sample. Details regarding the identified proteins are depicted in Supplementary table 1 (molecular weight, protein coverage, spectrum count, proteins score and other statistical information is given). The nucleoprotein SARS-CoV-2 (Uniprot accession number: P0DTC9) was identified in all sample previously assigned as positive for SARS-CoV-2 by qRT-PCR standard diagnostic tool. The tryptic peptide RGPEQTQGNFGDQELIR (MH+ = 1944.95) was systematically detected in all positive samples, even in those where the viral protein was identified with lower spectral counts.
We further performed comparative and quantitative analyses of the datasets using PatternLab for Proteomics v4.0 software. The number of proteins uniquely detected in each sample group is shown in Fig. 1 (Venn diagram). The most abundant proteins uniquely detected in positive swabs were the human proteins “Guanylate-binding protein 1” (Uniprot accession numbers P32455), “HLA class II histocompatibility antigen DR beta chain” (Unirpot accession number D7RIG5) and the nucleoprotein from SARS-CoV-2 (Uniprot accession number P0DTC9). Details regarding these and the other differentially detected proteins are depicted in Supplementary Table 2 (first and second tabs in the excel file show the list of proteins uniquely detected in positive and negative samples, respectively; information related to the number of replicates, spectrum count and description is given).
Fig. 1.

Venn diagram showing the overlapping of proteins identified in positive and negative samples (proteins identified in at least 3 replicates of each condition were considered). The number of proteins statistically detected as uniquely present in positive samples (light gray) and negative samples (dark gray) are shown. The number of proteins in the intersection represents proteins common to both conditions.
Considering the proteins present in both conditions, we analysed the significant difference in relative abundance between the two sample sets by means of spectrum counts. In this way, we found 57 proteins with increased relative abundance in positive samples and 24 proteins with increased relative abundance in negative samples. The results are shown as a Volcano plot where the x-axis represents the log2(p-value) and the y-axis the log2(fold change) (Fig. 2).
Fig. 2.

Volcano plot indicating proteins present in both sample sets but with statistical difference in relative abundance according to spectrum counts. Each spot represents one protein. In the y-axis Log2(p-value) is indicated. In the x-axis Log2(fold-change) is shown (negative values indicate proteins overrepresented in negative samples and positive values show proteins overrepresented in positive samples). Blue dots correspond to proteins that show a statistical difference in relative abundance between conditions.
Human proteins exclusively detected in positive samples together with those statistically increased in the same condition were considered to perform a biological processes overrepresentation test and a Reactome pathways analysis using Panther db (http://www.pantherdb.org/). The most significant overrepresented biological processes were “SRP-dependent cotranslational protein targeting to membrane”, “nuclear-transcribed mRNA catabolic process, nonsense-mediated decay”, “viral transcription” and “translational initiation”. The most significant overrepresented Reactome pathways were “SRP-dependent cotranslational protein targeting to membrane”, “Viral mRNA Translation”, “Peptide chain elongation” and “Nonsense Mediated Decay (NMD) enhanced by the Exon Junction Complex (ECJ)”. All overrepresented biological process and pathways are depicted in Tables 1 and 2 (details regarding biologic processes and Reactome pathways, respectively; fold-changes and statistical values are provided).
Table 1.
Biological processes overrepresented in positive samples.
| GO Biological Process complete | ||||||
|---|---|---|---|---|---|---|
| Homo sapiens (REF) | Input (proteins overrepresented in positive samples) |
|||||
| #* | #⁎⁎ | Expected⁎⁎⁎ | Fold enrichment | +/- | Raw p-value |
|
| SRP-dependent cotranslational protein targeting to membrane | 96 | 17 | 0.46 | 37.3 | + | 2.75E-17 |
| nuclear-transcribed mRNA catabolic process, nonsense-mediated decay | 120 | 17 | 0.57 | 29.84 | + | 8.36E-16 |
| viral transcription | 115 | 16 | 0.55 | 29.30 | + | 1.41E-14 |
| translation initiation | 143 | 16 | 0.68 | 23.57 | + | 3.43E-13 |
| aerobic respiration | 78 | 8 | 0.37 | 21.60 | + | 6.61E-05 |
| antigen processing and presentation of exogenous peptide antigen | 176 | 10 | 0.84 | 11.97 | + | 1.70E-04 |
| rRNA processing | 253 | 11 | 1.20 | 9.16 | + | 4.43E-04 |
| electron transport chain | 175 | 9 | 0.83 | 10.83 | + | 2.03E-03 |
| interferon-gamma-mediated signaling pathway | 72 | 6 | 0.34 | 17.55 | + | 1.69E-02 |
| oxidative phosphorylation | 119 | 7 | 0.57 | 12.39 | + | 2.05E-02 |
All genes from Homo sapiens were used as comparative dataset.
The number of proteins found in the input dataset that were assigned to the corresponding biological process.
This column represents the expected number of H. sapiens genes according to the size of dataset used as an input. The input dataset was all proteins incremented in positive samples when compared to negative samples. Fisher exact was used as statistical analysis with Bonferroni correction for multiple testing. Biological processes are ordered according to the p-value.
Table 2.
Reactome pathway overrepresented in positive samples.
| Reactome pathway | ||||||
|---|---|---|---|---|---|---|
| Homo sapiens (REF) | Input (proteins overrepresented in positive samples) |
|||||
| #* | #⁎⁎ | Expected⁎⁎⁎ | Fold enrichment | +/- | Raw p-value |
|
| SRP-dependent cotranslational protein targeting to membrane | 112 | 17 | 0.53 | 31.97 | + | 6.62E-17 |
| Viral mRNA Translation | 89 | 15 | 0.42 | 35.50 | + | 3.04E-15 |
| Peptide chain elongation | 89 | 15 | 0.42 | 35.50 | + | 3.04E-15 |
| Nonsense Mediated Decay (NMD) enhanced by the Exon Junction Complex (EJC) | 115 | 16 | 0.55 | 29.30 | + | 3.21E-15 |
| Selenocysteine synthesis | 93 | 15 | 0.44 | 33.97 | + | 5.94E-15 |
| Eukaryotic Translation Termination | 93 | 15 | 0.44 | 33.97 | + | 5.94E-15 |
| Nonsense Mediated Decay (NMD) independent of the Exon Junction Complex (EJC) | 95 | 15 | 0.45 | 33.97 | + | 7.31E-15 |
| Formation of a pool of free 40S subunits | 101 | 15 | 0.48 | 31.28 | + | 1.67E-14 |
| Response of EIF2AK4 (GCN2) to amino acid deficiency | 101 | 15 | 0.48 | 31.28 | + | 1.67E-14 |
| Regulation of expression of SLITs and ROBOs | 169 | 17 | 0.80 | 21.19 | + | 3.82E-14 |
| L13a-mediated translational silencing of Ceruloplasmin expression | 111 | 15 | 0.53 | 28.46 | + | 6.03E-14 |
| GTP hydrolysis and joining of the 60S ribosomal subunit | 112 | 15 | 0.53 | 28.21 | + | 6.81E-14 |
| Major pathway of rRNA processing in the nucleolus and cytosol | 182 | 15 | 0.86 | 17.36 | + | 5.18E-11 |
| Formation of the ternary complex, and subsequently, the 43S complex | 51 | 7 | 0.24 | 28.91 | + | 2.17E-05 |
| Ribosomal scanning and start codon recognition | 58 | 7 | 0.28 | 25.42 | + | 4.88E-05 |
| Translation initiation complex formation | 58 | 7 | 0.28 | 25.42 | + | 4.88E-05 |
| Mitochondrialproteinimport | 65 | 6 | 0.31 | 19.44 | + | 2.21E-03 |
| Interferon gamma signaling | 91 | 6 | 0.43 | 13.89 | + | 1.38E-02 |
| Downstream TCR signaling | 102 | 6 | 0.48 | 12.39 | + | 2.56E-02 |
All genes from H. sapiens were used as comparative dataset.
The number of proteins found in the input dataset that were assigned to the corresponding biological process.
This column represents the expected number of H. sapiens genes according to the size of dataset used as an input. The input dataset was all proteins incremented in positive samples when compared to negative samples. Fisher exact was used as statistical analysis with Bonferroni correction for multiple testing. Biological processes are ordered according to the p-value. Pathways with less than 5 proteins found in the input dataset were not considered.
Raw data is available via ProteomeXchange with identifier PXD020394.
2. Experimental design, materials and methods
2.1. Sample collection and inactivation
Samples were obtained by health professional at local public hospitals and delivered to the diagnostic laboratory located at Institut Pasteur de Montevideo. As it is a preliminary and exploratory research, information regarding any aspect of patients was confidential and totally unknown. Samples containing both, oro- and naso-pharyngeal swabs, in the same sample tube and without addition of transport media (to avoid contamination with medium proteins) were randomly chosen. Samples were previously analysed by standard qRT-PCR diagnostic method. We collected five SARS-CoV-2 positive and five negative samples. For viral inactivation, 2 mL of 2% SDS was added and incubated overnight at room temperature. Inactivation of SARS virus using SDS was reported in [1]. These procedures were performed in a Biosafety laboratory level 2.
2.2. Protein extraction and sample preparation for mass spectrometry analysis
Protein extraction from swabs containing SDS solution was performed by vigorous vortexing. Supernatant was recovered and stored at −20 °C. Protein quantification was carried out by densitometry analysis in gels. For that purpose, 6 μl of 4X loading SDS-PAGE sample buffer was added to 18 μl of each sample and heated for 10 min at 100 °C. Samples were loaded into pre-cast gels (NuPAGE™ 4–12%, Bis-Tris, 1.0 mm, Mini Protein Gel, 10-well, Invitrogen). Five and 2.5 μl of LMW-SDS Marker Kit (GE Healthcare) were also loaded to be used as standard. Electrophoresis was run at 150 V. Gels were fixed with 50% ethanol / 10% acetic acid, for 30 min at room temperature with gentle agitation and stained overnight with colloidal Coomassie blue in the same conditions. After destaining by ultrapure water washing, gel images were digitalized with UMAX Power-Look 1120 scanner and LabScan 5.0 software (GE Healthcare). Quantification was performed using ImageQuant TL software (v8.1), 1D analysis module, and the LMW-SDS Marker Kit (GE Healthcare) as standard.
Then, 12.5 μg of each sample were loaded into a 12.5% acrylamide gel, run at 10 mA per gel until samples enter 1 cm into the resolving gel. The gel was fixed and stained as described above. Gel fragment containing the samples were sliced with a scalpel and transferred to an eppendorf tube. Sample processing for mass spectrometry analysis was performed as described in [2] which includes sample reduction with 10 mM DTT at 56 °C for 1 h with vigorous agitation; cysteine alkylation with 50 mM iodoacetamide at room temperature for 45 min with vigorous agitation and protected from light; in gel protein digestion with 2 μg trypsin (sequence grade, Promega) in 50 mM ammonium bicarbonate, overnight at 37 °C; peptide extraction by the addition of 60% acetonitrile/ 0.1% trifluoroacetic acid with vigorous agitation; vacuum drying concentration and resuspension in 0.1% trifluoroacetic acid.
2.3. LC-MS/MS analysis
Samples were analysed by nano-LC MS/MS using a shotgun strategy on a nano-HPLC, UltiMate 3000 (Thermo) coupled on line to a Q-Exactive Plus (Q-Orbitrap) (Thermo) mass spectrometer through an Easy-Spray source (Thermo). Five μg of tryptic peptides was injected into an Acclaim PepMapTM 100 C18 nano-trap column (75 μm x 2 cm, 3 μm particle size, Thermo) and separated using a 75 μm x 50 cm, PepMapTMRSLC C18 analytical column (2 μm particle size, 100 Å, Thermo) at a constant flow rate of 250 nL/min and 40 °C. Column was equilibrated at 1% of mobile phase B (A: 0.1% formic acid; B: 0.1% formic acid in acetonitrile) followed by an elution gradient from 1% to 35% B over 150 min and 35–99% B over 20 min. Two technical replicates were run for each sample.
The mass spectrometer was operated in the positive mode. Ion spray voltage was set at 1.7 kV; capillary temperature at 250 °C and S-lens RF level at 50. Mass analysis was carried out with a data dependent mode in two steps: acquisition of full MS scans in a range of m/z from 200 to 2000; followed by HCD fragmentation of the 12 most intense ions in each segment using a stepped normalized collision energy of 25, 30 and 35. Full MS scans were acquired at a resolution of 70,000 at 200 m/z, an AGC target value of 1E06 and a maximum ion injection time of 100 ms. For MS/MS acquisition the resolution was 17,500 at 200 m/z, AGC target value: 1E05 and maximum ion injection time: 50 ms. Precursor ions with unassigned, single, five and higher charge states were excluded for fragmentation. A dynamic exclusion time was set at 30 s.
2.4. Protein identification and analysis
PatternLab for Proteomics (version 4.0) software (http://www.patternlabforproteomics.org) [3] was employed to generate a target-reverse database using sequences from Homo sapiens and Severe acute respiratory syndrome coronavirus 2, both downloaded from Uniprot consortium in June, 2020 (http://www.uniprot.org). In addition, 127 common mass spectrometry contaminants were incorporated. Thermo raw files were searched against the database using the software integrated Comet search engine applying the following parameters: trypsin as proteolytic enzyme with full specificity and 1 missed cleavage; oxidation of methionine as variable modification, carbamidomethylation of cysteines as fixed modification; 40 ppm of tolerance from the measured precursor m/z. XCorr and Z-Score were used as the primary and secondary search engine scores, respectively.
Peptide spectrum matches were filtered using the Search Engine Processor (SEPro) and acceptable FDR criteria was set on 1% at the protein level. PatternLab's Buzios module was employed to perform a principal component Analysis (supplementary figure 1). PatternLab's Approximately Area Proportional Venn Diagram module was used to compare the proteins identified in positive and negative swabs and to determine which proteins were uniquely detected in each sample set using a probability value of 0.05 as a filtering option. PatternLab's T-Fold module was used to detect proteins present in both conditions but at significantly different relative abundance by spectral count analysis.
2.5. Bioinformatics analysis
A statistical overrepresentation test was performed using the Panther Server Classification System (http://pantherdb.org) released 2020-04-07 [4]. The annotation data set “GO biological processes complete” and “Reactome pathway” were used for analysis. The release date of GO ontology dataset was 2020-03-23. The protein showing significant increased relative abundances in positive swabs were used for analysis. Homo sapiens database was used as reference List.
2.6. Mass spectrometry data repository
All proteomic data have been deposited into the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD020394 and is available [5].
Ethics statement
Oro- and naso-pharyngeal samples were remitted to Institut Pasteur de Montevideo, that has been validated by the Ministry of Health of Uruguay as an approved center providing diagnostic testing for COVID-19. Samples were deidentified and anonymised before receipt by the study investigators. Samples were selected in a random way. Data from swabs samples origin was confidential and thus totally unknown.
CRediT authorship contribution statement
Bernardina Rivera: Methodology, Investigation, Writing - review & editing. Alejandro Leyva: Methodology, Investigation, Writing - review & editing. María Magdalena Portela: Methodology, Investigation, Writing - review & editing. Gonzalo Moratorio: Supervision, Resources, Writing - review & editing. Pilar Moreno: Supervision, Resources, Writing - review & editing. Rosario Durán: Project administration, Conceptualization, Methodology, Investigation, Writing - review & editing. Analía Lima: Project administration, Conceptualization, Methodology, Investigation, Writing - review & editing, Writing - original draft, Visualization.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships which have, or could be perceived to have, influenced the work reported in this article.
Acknowledgments
The authors acknowledge to all the scientists and technicians from Insitut Pasteur de Montevideo and Universidad de la República del Uruguay that participate in the design, installation, implementation of COVID-19 diagnostic center.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.dib.2020.106121.
Appendix. Supplementary materials
{kind=link}
References
- 1.Darnell M.E.R., Subbarao K., Feinstone S.M., Taylor D.R. Inactivation of the coronavirus that induces severe acute respiratory syndrome, SARS-CoV. J. Virol. Methods. 2004;121:85–91. doi: 10.1016/j.jviromet.2004.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rossello J., Lima A., Gil M., Duarte J.R., Correa A., Carvalho P.C., Kierbel A., Durán R. The EAL-domain protein FcsR regulates flagella, chemotaxis and type III secretion system in Pseudomonas aeruginosa by a phosphodiesterase independent mechanism. Sci. Rep. 2017 doi: 10.1038/s41598-017-09926-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carvalho P.C., Lima D.B., Leprevost F.V, Santos M.D.M., Fischer J.S.G., Aquino P.F., Moresco J.J., Yates J.R., Barbosa V.C., Iii J.R.Y., Barbosa V.C. Integrated analysis of shotgun proteomic data with PatternLab for proteomics 4.0. Nat. Protoc. 2016;11:102–117. doi: 10.1038/nprot.2015.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mi H., Poudel S., Muruganujan A., Casagrande J.T., Thomas P.D. PANTHER version 10: expanded protein families and functions, and analysis tools. Nucleic Acids Res. 2016;44:D336–D342. doi: 10.1093/nar/gkv1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D.J., Inuganti A., Griss J., Mayer G., Eisenacher M., Pérez E., Uszkoreit J., Pfeuffer J., Sachsenberg T., Yilmaz Ş., Tiwary S., Cox J., Audain E., Walzer M., Jarnuczak A.F., Ternent T., Brazma A., Vizcaíno J.A. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019;47:D442–D450. doi: 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.