

SUMMARY
Lung cancer remains the leading cause of cancer death. Genome sequencing of lung tumors from patients with squamous cell carcinoma has identified SMAD4 to be frequently mutated. Here, we use a mouse model to determine the molecular mechanisms by which Smad4 loss leads to lung cancer progression. Mice with ablation of Pten and Smad4 in airway epithelium develop metastatic adenosquamous tumors. Comparative transcriptomic and in vivo cistromic analyses determine that loss of PTEN and SMAD4 results in ELF3 and ErbB2 pathway activation due to decreased expression of ERRFI1, a negative regulator of ERBB2 in mouse and human cells. The combinatorial inhibition of ErbB2 and Akt signaling attenuate tumor progression and cell invasion, respectively. Expression profile analysis of human lung tumors substantiated the importance of the ErbB2/Akt/ELF3 signaling pathway as both a prognostic biomarker and a therapeutic drug target for treating lung cancer.
Graphical Abstract
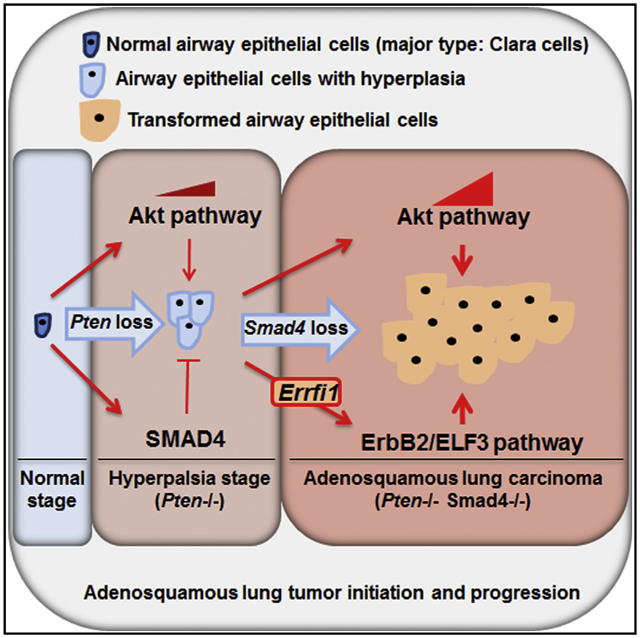
In Brief
Liu et al. now show that ablation of Smad4 and Pten in the pulmonary epithelium results in the development of metastatic adenosquamous lung tumors through activation of the ErbB2/ELF3/AKT pathway. ErbB2 activation in mice is due to downregulation of Errfi1 expression, a direct target of SMAD4.
INTRODUCTION
The dichotomous division of primary lung cancer into non-small cell lung carcinoma (NSCLC) and small cell lung carcinoma (SCLC) is based on histological appearance and clinical characteristics (Shames and Wistuba, 2014). NSCLC is the predominant type of primary lung cancer and is subdivided into adenocarcinoma (AD), squamous cell carcinoma (SCC), and large cell carcinoma (LCC), although, in over 50% of the cases, a mixed histological phenotype is detected, indicating a continuum of heterogeneity in lung cancer (Jemal et al., 2010; Walker, 2008). Despite the histological differences, the 5-year survival rate for all early-stage NSCLC lung cancers is estimated to be about 50%, but it is significantly reduced, to less than 5, in cancers that are diagnosed with distant metastasis (Howlader et al., 2014). Given the disproportionate number of lung cancer patients that present with distant metastasis or at a late disease stage (Howlader et al., 2014), lung cancer remains the leading cause of cancer-related death (Siegel et al., 2014). Therefore, there is an urgent need to identify biomarkers and drug targets for early diagnosis and treatment of human lung cancer.
Lung cancer initiation and progression is driven by the stepwise accumulation of genetic alterations (Sakashita et al., 2014), including inactivation of tumor suppressor genes, such as PTEN, p53, and SMAD4 (Cancer Genome Atlas Research Network, 2012), and mutation and/or amplification of oncogenic genes, such as Kras, EGFR, and ERBB2 that control cell proliferation (Cancer Genome Atlas Research Network, 2014). In particular, PTEN, a suppressor of the PI3K/Akt signaling pathway, is highly dysregulated (Cancer Genome Atlas Research Network, 2012) and is recognized as a prognostic marker in human lung cancer (Yanagawa et al., 2012). However, deletion of Pten in airway epithelial cells and its biological function in animal models of lung cancer has produced inconsistent results. While deletion of Pten in the airway epithelial cells using the Cre recombinase under the Club Cell Secretory Protein promoter (CCSPCre) resulted in no discernible effect on bronchial epithelial cell morphology in mice (Li et al., 2008; Iwanaga et al., 2008), deletion of Pten in the distal airways and alveolar cells resulted in hyperplasia and lung AD in mice (Perl et al., 2002; Davé et al., 2008; Yanagi et al., 2007). Recently, Pten deletion alone in airway basal cells was shown to initiate lung tumor formation, but with low incidence and long latency (Malkoski et al., 2014), and inactivation of Lkb1 and Pten using an Ad-Cre inhalation system resulted in development of lung cancer that closely resemble human lung SCC (Xu et al., 2014). Given these contradictory results, we expressed a codon optimized Cre recombinase under the control of the CCSP promoter (CCSPicre) to examine the role of Pten in lung cancer development.
In this report, we show that mice with efficient expression of Cre in their proximal airways (CCSPicre) crossed with floxed Pten allele mice develop airway epithelial hyperplasia without progression to tumor. In an attempt to identify the mechanism responsible for the failure of hyperplastic epithelial cells lacking Pten to progress to lung tumors, we discovered alterations in transforming growth factor β (TGF-β)-SMAD4 signaling axis in the hyperplastic airway epithelial cells lacking Pten. Concurrent ablation of Smad4 and Pten in the airways of mice resulted in the spontaneous development of proximal airway tumors with metastasis to distal organs. Transcriptomic and in vivo cistromic analyses of lung tumors demonstrated that ablation of Smad4 resulted in the activation of the ErbB2 signaling pathway. We then demonstrate that the activation of ErbB2 signaling is due to alteration in the expression of Errfi1 (ERBB Receptor Feedback Inhibitor 1), a negative regulator of ErbB2 activation and direct target of SMAD4 transcriptional regulation. Furthermore, we identified Elf3 as an essential target of the signaling pathways that control lung tumor cell proliferation and invasion. SMAD4 directly inhibits and ErbB2 signaling induces the expression and activity of Elf3. Thus, the ERRFI1/ErbB2/ELF3 signaling axis may be targeted for the treatment of lung tumors that have an attenuated expression of PTEN and SMAD4.
RESULTS
Deletion of Pten in Mouse Bronchial Epithelial Cells Results in Hyperplasia and Alters TGF-β Signaling
Our previous study showed that CCSPCre-mediated lung-specific deletion of Pten in bronchial epithelial cells had no discernible phenotype unless combined with Kras activation (Iwanaga et al., 2008). The lack of histological change in the airway epithelial cell in mice with deletion of Pten may have been in part due to the low efficiency of Cre-induced recombination. To address this possibility, we generated a mouse line that expresses an improved Cre (iCre) (Shimshek et al., 2002) inserted into the CCSP locus (CCSPiCre model) (Figure S1A). The Rosa 26 reporter mice crossed with CCSPiCre showed efficient recombination in epithelial cells lining the trachea, bronchi, and distal airways with some recombination in alveolar type II cells (Figure 1A). The recombination efficiency of CCSPiCre was higher than that of CCSPCre, as demonstrated by the increased X-gal staining of the lung (Figure 1A) (Li et al., 2008; Soriano, 1999). The recombination observed in mice expressing CCSPiCre is confined to the lungs with no recombination observed in other tissues examined (Figure S1B). Mice with the floxed Pten allele (Ptenf/f) (Lesche et al., 2002) were crossed with CCSPicre mice to achieve airway-specific deletion of Pten (Ptend/d). Immunohistochemical (IHC) staining for PTEN showed loss of the PTEN protein in the airway epithelium of Ptend/d mice with prominent hyperplasia at 9 months of age (Figure 1B). Western blot (WB) analysis of lung extract showed a significant reduction of PTEN protein and the expected increase in the p-AKT level (Figure 1C). Interestingly, even at 15 months of age hyperplasia did not progress to carcinoma indicating that in the absence of Pten other tumor suppressor genes may be altered and prevent lung tumor development.
Figure 1. Deletion of Pten in Mouse Bronchial Epithelial Cells Results in Hyperplasia and Alters TGF-β Signaling.
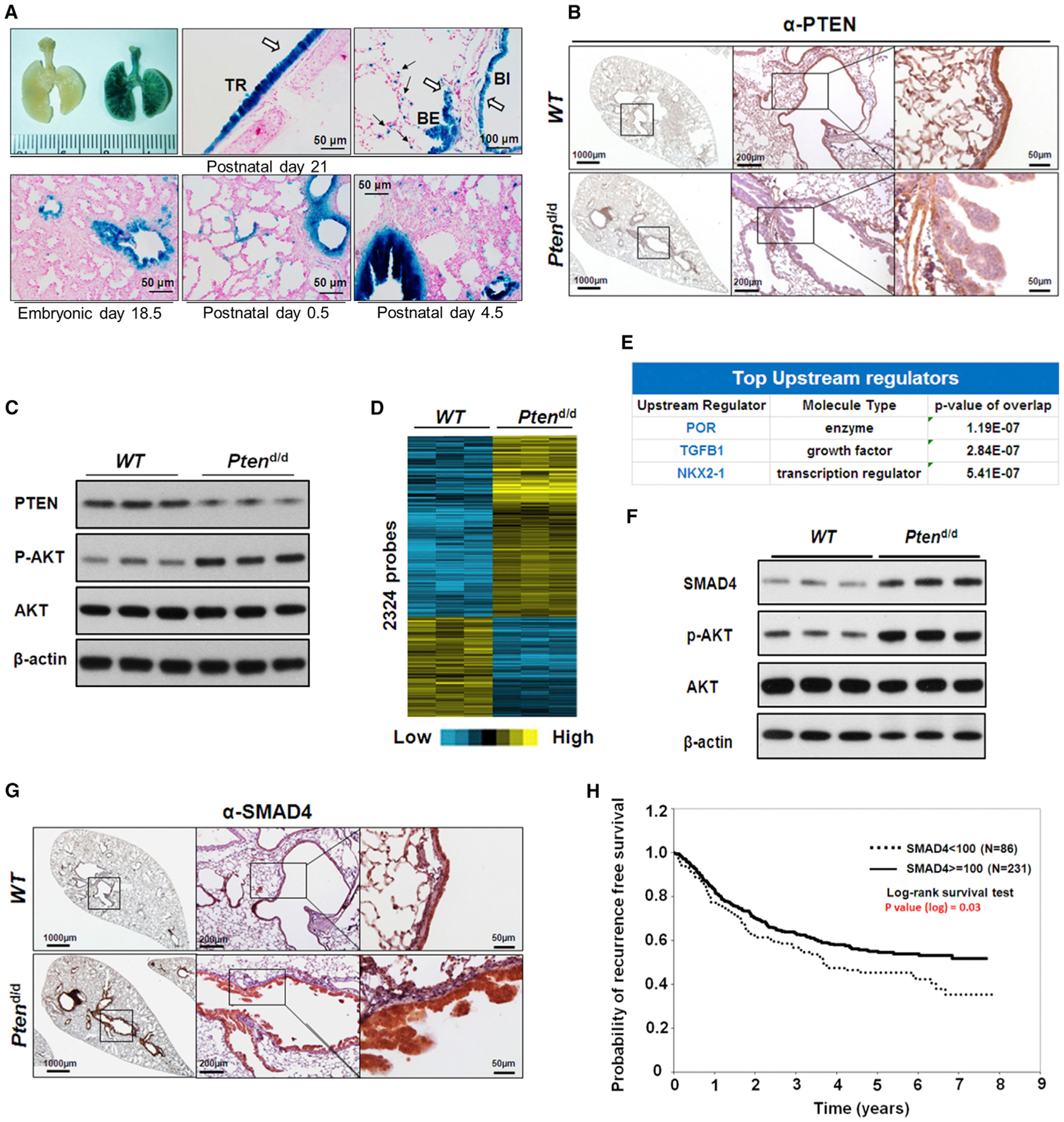
(A) Representative whole-mount X-gal staining of mouse lungs in different stages. The blue X-gal staining showed CCSPiCre expressed in tracheal and bronchial airway epithelial cells in R26R (left) and CCSPiCre/R26R (right) mice (upper panel). The white arrows indicate the blue staining of X-gal in tracheal and bronchial airway epithelial cells and the black arrows indicate the staining of X-gal in the alveolar type II cells; trachea (TR), bronchi (BI), and bronchiole (BE).
(B) IHC staining of PTEN in lungs of 7-month-old WT and Ptend/d mice. PTEN staining is strongly present in the epithelium (brown staining) of WT mice and not in the epithelial cells of the Ptend/d mouse lung.
(C) WB analysis of PTEN and AKT expression and Akt phosphorylation (p-AKT) in lungs of 7-month-old mice. Increased p-AKT was observed in Ptend/d mouse lungs.
(D) The heatmap for the 1,847 differentially regulated genes (2,324 probes) in the microarray analysis of lungs from 7-month-old Ptend/d mice (n = 6) when compared to 7-month-old WT mice (n = 6).
(E) IPA of gene microarray data revealed top changed upstream regulators in Ptend/d mice, as compared to WT mice.
(F) WB analysis of SMAD4 protein expression in 7-month-old mouse lungs.
(G) IHC staining of SMAD4 (brown staining) in 7-month-old WT and Ptend/d mouse lungs.
(H) Kaplan-Meier plot of probability of recurrence-free survival based on the cytoplasmic SMAD4 protein expression index in lung cancer patients from SMAD4 tissue array data set.
To identify which genes are significantly altered in the absence of Pten, microarray analysis comparing lung RNAs isolated from control and Ptend/d mice was performed and identified 1,847 unique genes that were significantly altered when Pten was ablated (Figure 1D). Ingenuity knowledge-based analysis (IPA) of the altered genes identified TGF-β1 as one of the top upstream regulators, indicating its potential role in preventing Ptend/d airway epithelia progression to tumor (Figure 1E). The TGF-β signaling pathway has been shown to be critical in many types of cancer (de Caestecker et al., 2000), including lung cancer (Jeon and Jen, 2010). Indeed, dramatic changes in the components of the TGF-β/BMP pathways were observed (Figures S1C and 1F). We validated the alteration of several genes in the TGF-β1 pathway in the lungs from Ptend/d mice (Table S1; Figure S1D) and focused on SMAD4, a tumor suppressor gene that is activated in response to TGF-β signaling (Shi et al., 1997) and has been recently found to be one of the highly mutated genes in human lung SCC (Cancer Genome Atlas Research Network, 2012). IHC and WB analyses of lung tissues showed a significant increase in expression of SMAD4 in the lungs of Ptend/d mouse, as compared with control mice (Figures 1F and 1G), supporting a possible protective role for TGF-β signaling in the prevention of lung tumors in Pten deficiency.
Higher Level of SMAD4 Is Associated with Improved Overall Survival in Human Lung Cancer
The tumor suppressor function of SMAD4 has been well established in a number of human cancers (Ding et al., 2011; Tascilar et al., 2001; Liu, 2001; Miyaki and Kuroki, 2003; Teng et al., 2006), but its role in lung cancer development remains less clear (Ke et al., 2008; Nagatake et al., 1996). In line with reported downregulated expression of SMAD4 in a subset of human primary lung tumors (Ke et al., 2008), TCGA expression analysis from a clinical database revealed the reverse correlation of SMAD4 copy number with tumor grades and patients’ 5-year survival rate (Figure S1E). In addition to the bioinformatics analysis of mRNA expression, we also examined the expression of the SMAD4 protein in 479 cases of AD and SCC lung cancer samples using tissue microarray (Table S2). Kaplan-Meier survival analysis of the tissue microarray results revealed that patients with higher retention of cytoplasmic SMAD4 expression showed significant improvement in recurrence-free survival (p = 0.03, log-rank test) (Figures 1H and S1F). Taken together, these data demonstrate that SMAD4 is a potential suppressor for lung cancer progression.
Deletion of Smad4 in Pten-Null Background Promotes Tumor Growth and Metastasis
To determine if increased expression of SMAD4 plays an inhibitory role in lung tumor development in Ptend/d mice, we crossed Smad4f/f mice (Yang et al., 2002) with CCSPicrePtenf/f mice to generate mice with a double deletion of Pten and Smad4 in the airway epithelium (Ptend/dSmad4d/d). Smad4d/d, although showing high deletion efficiency (Figure S2A), failed to show any discernable changes in the epithelium. Additional loss of Smad4 in the Ptend/d mice resulted in primary lung carcinoma in the proximal airways at 9 months of age (Figures 2A and 2B). H&E staining examination of the lung histology revealed a high rate of perineural invasion with architectural distortion (Figure 2B). Peroxidase acid Schiff (PAS) staining of the lungs identified glycogen and mucin-rich deposits in the tumors (Figure 2B). Furthermore, lung tumors expressed TTF1 and KRT7 biomarkers present in human lung AD (Su et al., 2006; Dennis et al., 2005), as well as p63, SOX2, and KRT5 markers for squamous cell lung cancers (Xu et al., 2014), suggesting mixed features of adeno and squamous carcinoma phenotype (Figures 2C and S2B). Double staining of KRT5 and KRT7 or TTF1, respectively, showed no overlapping between the markers of AD and SCC in the tumors. This further indicates that the Ptend/dSmad4d/d mouse tumors belong to adenosquamous cell carcinoma (Figures S2C and S2D). Meanwhile, IHC analysis of tumors revealed positive CCSP, but not surfactant protein C (SPC), indicating the lack of type II cell marker expression in the malignant cell (Figure 2D).
Figure 2. Deletion of Smad4 in the Pten-Null Background Promotes Tumor Growth and Metastasis.
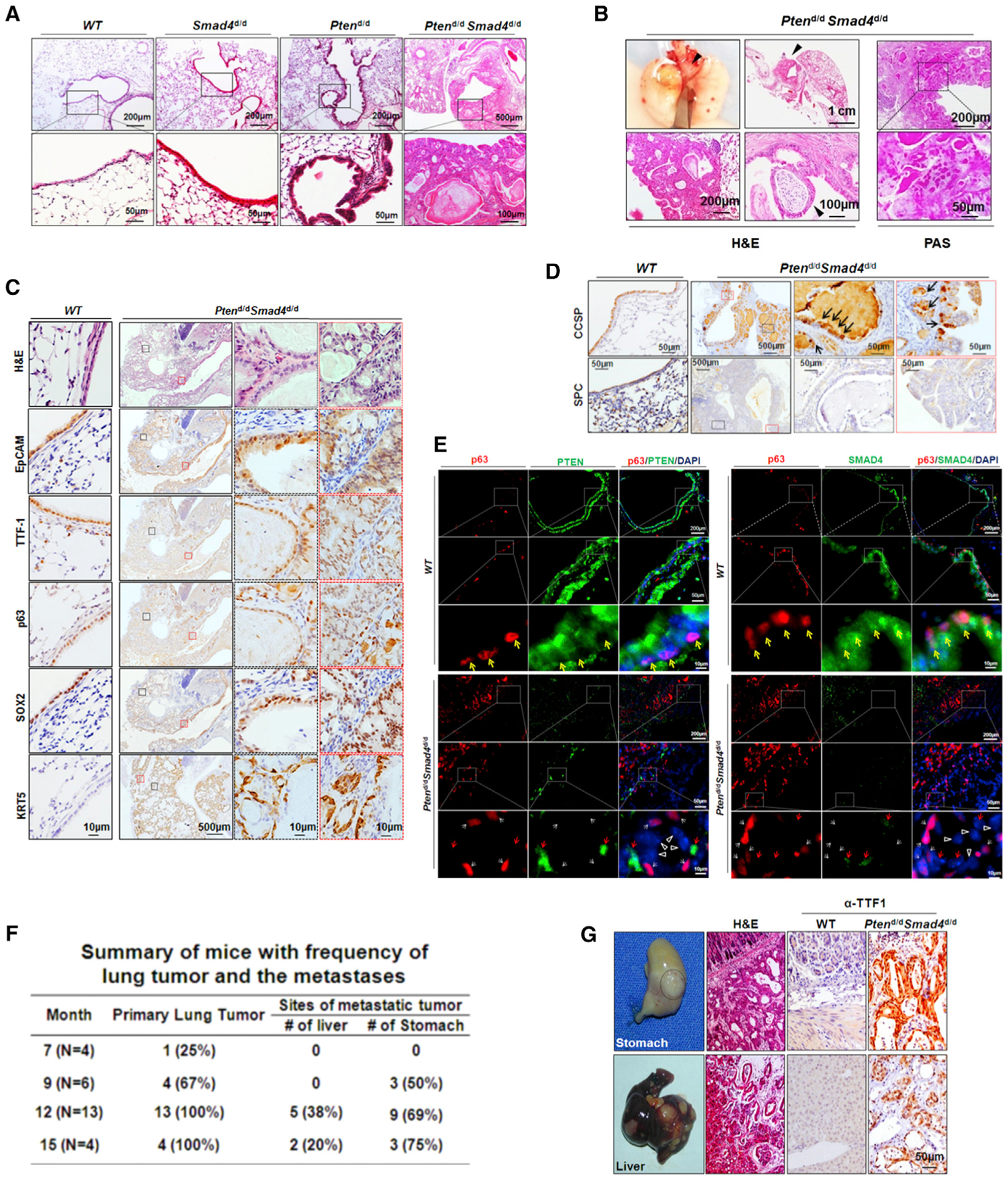
(A) H&E staining analysis of 9-month-old mouse lungs. Boxed areas are magnified in the panels directly below.
(B) H&E and PAS staining analysis of primary lung tumors from 9-month-old Ptend/dSmad4d/d mice. Primary lung tumors are indicated by black arrows.
(C) IHC staining of WT and Ptend/dSmad4d/d mouse lung tumors for p63, SOX2, KRT5, TTF1, and EpCAM. The regions labeled by black or red dash boxes are representative tumor epithelium magnified in adjacent panels.
(D) IHC staining of CCSP, the marker for Club cells, and of SPC, the marker for type II alveolar cells, on WT and Ptend/dSmad4d/d mouse lungs. The regions labeled by black or red dash boxes are representative tumor epithelium magnified in adjacent panels.
(E) Immunofluorescence staining of p63 and PTEN (left panel) or SMAD4 (right panel) in WT mouse lungs and Ptend/dSmad4d/d mouse tumors. Yellow arrows indicate the cells coexpressing p63 and PTEN/SMAD4; white arrows indicate the cells only expressing p63; red arrows indicate the cells only expressing PTEN/SMAD4; white triangles indicate the cells expressing neither p63 nor PTEN/SMAD4.
(F) Summary of Ptend/dSmad4d/d mice with frequency of lung tumor and metastases as it relates to age.
(G) Metastases in the stomach and liver from primary lung tumors. TTF1, a marker for the diagnosis of metastatic tumor from the lung, was positive in the stomach and liver.
The observed mouse tumors initiated from the proximal airway epithelial cells where CCSPiCre showed stronger expression compared with that of CCSPCre (Figure 1A) (Li et al., 2008). In order to determine if the CCSPiCre mouse targets recombination exclusively to the Club cells or is active in other cell types, such as p63-expressing basal cells in upper airways, we determined the specificity of Cre activity in the upper airways. First, we determined if there was Cre activity in basal cells of normal lung. Double immunofluorescence in the lungs of CCSPiCre mice crossed to the R26R Lacz reporter mice showed that there was overlapping staining of p63 and Lacz in a subpopulation of basal cells in the tracheal and bronchial epithelium (Figure S2E), indicating that Cre activity was not limited to Club cells of the lungs. Double staining of lungs with p63 and PTEN or SMAD4 in the tumors of the Ptend/dSmad4d/d mice determined if the increase in p63 positive cells had loss of PTEN and SMAD4 or if the increase in p63 was due to paracrine regulation of p63 cells that were then interspersed throughout the tumor mass. This analysis that lungs from Ptend/dSmad4d/d mice showed no expression of PTEN and SMAD4 in p63 positive cells in the tumors, while lungs from wild-type (WT) mice show expression of PTEN and SMAD4 in p63-expressing cells (Figure 2E), indicating some tumor cells might originate from transformed basal cells expressing CCSPiCre. Meanwhile, it was determined that some Ptend/dSmad4d/d mouse lung cells expressed neither p63 and PTEN or SMAD4 (Figure 2E). This may be due to the transformation of Ptend/dSmad4d/d mouse tumor cells other than basal cells or may indicate a complex lineage of cell differentiation during tumor progression. Taken together, the adenosquamous phenotype observed in our mouse model may not only be the result of transformation of differentiated Club cells, but also could be the result of transformation of a more yet to be determined cell type, at least including some basal cells having the CCSPiCre activity.
Over 60% of Ptend/dSmad4d/d mice developed primary lung tumors at 9 months of age, while 100% showed primary tumors at 12 months of age (Figure 2F). In addition to primary lung tumors, and in keeping with highly metastatic human lung tumors, approximately 70% and 40% of Ptend/dSmad4d/d mice at 12 months of age spontaneously developed metastasis to the stomach and liver, respectively (Nagashima et al., 2004) (Figure 2F). Detection of TTF1 in all liver tumors and a subset of stomach tumors indicated that they originated from the lung (Su et al., 2006; Dennis et al., 2005) (Figure 2G). Ki67 staining demonstrated that cell proliferation significantly increased in Ptend/dSmad4d/d tumors when compared to the WT, Smad4d/d, or Ptend/d mice, whereas no statistically significant changes in apoptosis were found among the WT, Smad4d/d, Ptend/d, and Ptend/dSmad4d/d mice, as measured by TUNEL assay (Figures S2F and S2G). Consistent with a previous finding that Pten deletion induces senescence in a prostate tumor mouse model (Ahmad et al., 2011), Ptend/d mice had high levels of senescence in lung epithelium (Figure S2H). Importantly, the senescence induced by Pten deletion was abolished upon Smad4 deletion in Ptend/dSmad4d/d mice (Figure S2H).
Small interfering RNA (siRNA)-mediated knockdown of PTEN and SMAD4 in immortalized human bronchial epithelial cell lines BEAS-2B and NL20 was conducted to determine the role of these regulators in human lung. Compared to controls, increased cell migration and invasion was observed in cells with knockdown of PTEN and SMAD4 (Figures S2I–S2K, S2M, and S2N). Interestingly, while knockdown of PTEN alone increased cell proliferation, migration, and invasion, knockdown of SMAD4 alone did not significantly alter any of these cellular activities (Figures S2L and S2O), Together, these findings demonstrate a prominent role for SMAD4 that prevents lung cancer development when the expression of Pten is altered in the airway epithelium.
Lung Tumors from Ptend/dSmad4d/d Mice Correlate Strongly with Human Lung Cancer
In order to investigate the molecular changes in the tumors of Ptend/dSmad4d/d, we performed comparative genomic expression profiles in lung tumors isolated from Ptend/dSmad4d/d mice and normal lung tissue from WT mice. A total of 2,128 unique genes were differentially regulated (p < 0.01, fold change >1.4) (Figure 3A). IPA of the differentially regulated genes identified cell movement as the top functional annotation, indicating a role for PTEN and SMAD4 in regulating cell migration and invasion Ptend/dSmad4d/d mice (Figure 3B). We also validated a number of the differentially regulated genes (e.g., Agr2, and Erbb2) with known function in cell motility (Figure 3C; Table S3). Comparative genomics using an expression data set of human lung tumors with distinct histological subtypes (Takeuchi et al., 2006) with the gene expression data set in the Ptend/dSmad4d/d mice found the highest correlation of Ptend/dSmad4d/d murine lung tumor gene signatures with that of the human LCNEC, SCLC, and SCC subtypes (Figure 3D), confirming the notion that genetic variations are common even among histologically diverse lung tumors.
Figure 3. Lung Tumor from Ptend/dSmad4d/d Mice Correlates Strongly with Human Lung Cancers.
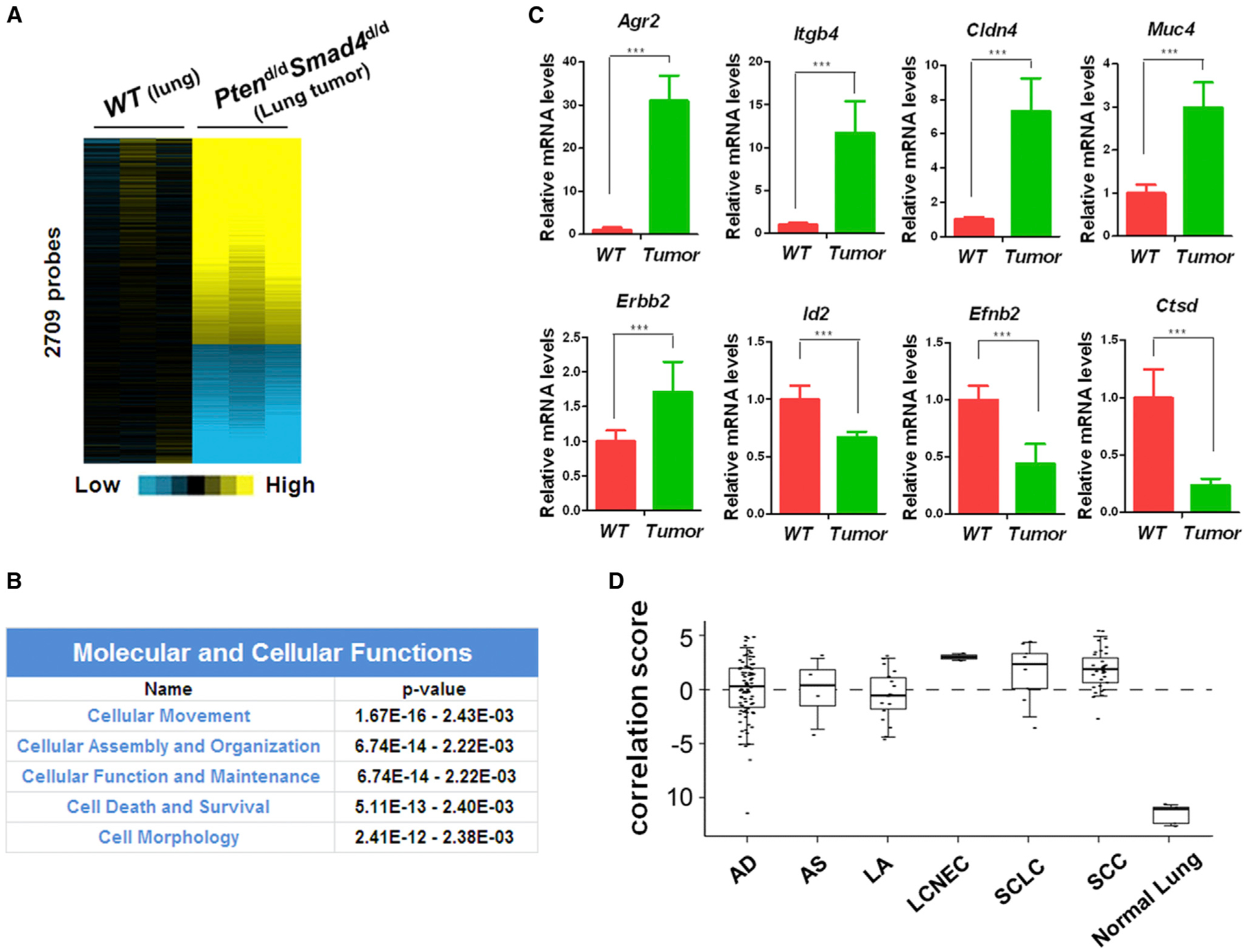
(A) The heatmap for the 2,128 differentially regulated genes (2,709 probes) in the microarray analysis of lung tumors from 12-month-old Ptend/dSmad4d/d mice (n = 3) when compared to lungs from 12-month-old WT mice (n = 3).
(B) Top molecular and cellular functions identified by IPA of the gene microarray data from lung tumors of Ptend/dSmad4d/d mice, in comparison with that of WT mouse lungs.
(C) Quantitative real-time PCR analysis of some of these significantly altered genes related to cell movement in the microarray (Student’s t test, p value is ***p < 0.001). Three lungs of WT mice and three lung tumors of Ptend/dSmad4d/d mice were analyzed.
(D) Alignment of the gene signature of our Ptend/dSmad4d/d mouse tumors with the Takeuchi lung cancer database (GSE11969). A score greater than zero represents a positive correlation with the murine gene signature. This data set encompasses expression profiles in 149 patients with NSCLC, nine patients with SCLC, and five patients with normal lung tissue. AD (n = 90, adenocarcinoma), AS (n = 4, adenosquamous carcinoma, LA (n = 18, large-cell carcinoma), and LCNEC (n = 2, large-cell neuroendocrine carcinoma). SCLC (n = 9, small cell lung cancer), SCC (n = 35, squamous cell lung cancer), and normal lung tissue (n = 5). Boxplot represents 5%, 25%, 75%, median, and 95%.
Deletion of Smad4 in Pten-Null Background Activates the ErbB2/Akt Signaling
In order to identify the early molecular targets that drive lung tumor development that are controlled by Smad4, gene microarray analysis was performed with RNA samples from the lung tissues of Ptend/d mice and Ptend/dSmad4d/d mice at the age of 7 months. At this age, mice had hyperplasia but no cancer so that this stage was considered to be the early/initiating stage of lung cancer in our mouse models. 744 genes were found to be significantly altered in lung tissues of Ptend/dSmad4d/d mice in comparison with those of Ptend/d mice, which reflected the early molecular signature of the tumors exerted by Smad4. We validated the expression of a representative of altered genes discovered in the array (Figures S4A and S4B).
Given that SMAD4 is a transcriptional factor (Massagué et al., 2005), we employed SMAD4 chromatin immunoprecipitation (ChIP) followed by high-throughput DNA sequencing (ChIP-seq) on whole-lung tissue from 7-month-old Ptend/d mice to identify genes that were directly bound by SMAD4. The SMAD4 binding sites were highly enriched in the proximal promoter regions of 13,103 genes (Table S4) as compared to the reference genome locus (Figures S4C and S4D). We identified 235 potential direct gene targets of SMAD4 by comparing genes with altered expression in microarray data (Ptend/dSmad4d/d versus Ptend/d) and genes identified in SMAD4 ChIP-seq data sets. Utilizing IPA, we found that ERBB2 regulated 24 of the 235 direct gene targets ranking top among upstream regulators (Figure 4A). Consistently, both ERBB2 protein expression and phosphorylation were increased in the lung epithelium of 7-month Ptend/dSmad4d/d mice, as compared to those of WT, Smad4d/d, and Ptend/d mice. As expected, phosphorylation of AKT (p-AKT) was increased in Ptend/d mice. Intriguingly, p-AKT was further increased upon deletion of Smad4 (Ptend/dSmad4d/d versus Ptend/d) (Figures 4B and 4C), suggesting SMAD4 may prevent the full activation of AKT upon the deletion of Pten. Upregulation of ErbB2/Akt signaling remained in the lung tumors of Ptend/dSmad4d/d mice at 12 months of age, as demonstrated by the increase of ERBB2 expression and ERBB2 and Akt phosphorylations (Figures 4D and 4E), suggesting a continuous activation of the ErbB2/Akt pathway in the process of Ptend/d Smad4d/d mouse tumor development. Similar to ERBB2, other members of the ERBB family, including EGFR, ERBB3, and ERBB4, also had increased phosphorylation (albeit at variable levels) upon the deletion of Pten and Smad4, especially in the lung tumors (Figure S4E). In contrast with the continuous hyperactivation of Akt, both MAPK and mTOR phosphorylations were only modestly altered in Ptend/dSmad4d/d mice, as compared to WT, Smad4d/d, or Ptend/d mice (Figure S4F).
Figure 4. Deletion of Smad4 in Pten-Null Background Activates the ErbB2/Akt Signaling.
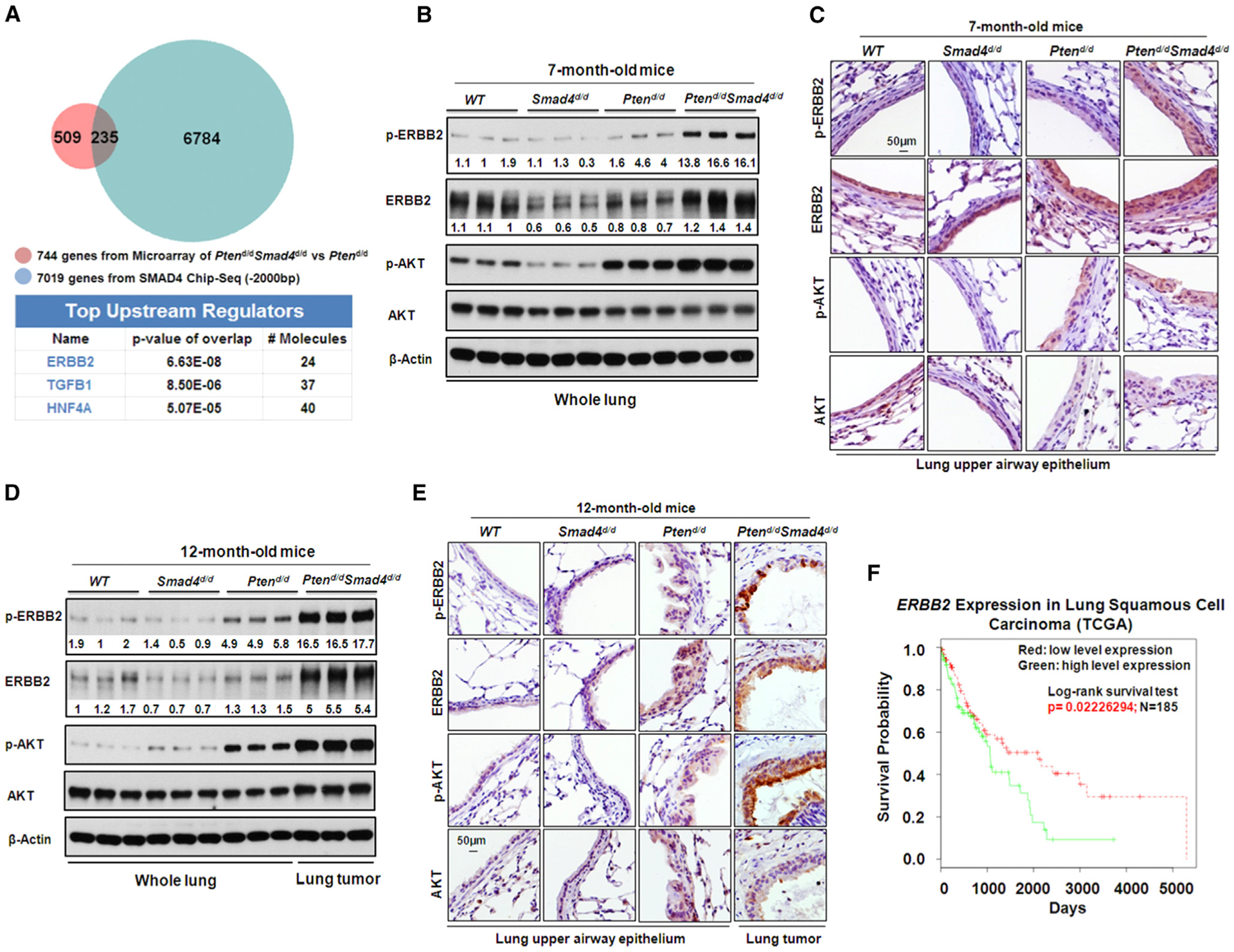
(A) Top changed upstream regulators identified by IPA of the intersection genes between gene microarray data set and SMAD4 ChIP-seq data set.
(B and C) WB (B) and IHC (C) analyses of protein expression and phosphorylation of the ERBB2/AKT signaling pathway in 7-month-old mouse lungs. p-ERBB2, ERBB2 phosphorylation. p-AKT, AKT phosphorylation. Number below each protein band indicates the relative band intensity (signal) measured by ImageJ software (NIH).
(D and E) IHC (D) and WB analyses (E) of protein expression and phosphorylation of ERBB2/AKT signaling pathway in 12-month-old mouse lungs and lung tumors. Number below each protein band indicates the relative band intensity (signal) measured by ImageJ software (NIH).
(F) Kaplan-Meier plot of survival probability based on the ERBB2 mRNA expression index in squamous cell lung carcinoma patients from TCGA data set. The mean expression value of ERBB2 across all investigated patients was calculated. Patients with ERBB2 expression higher than mean ERBB2 expression were classified into the ERBB2 high group, while patients with ERBB2 expression lower than mean ERBB2 expression were classified into the ERBB2 low group.
In order to investigate whether there was a similar phenotype in human cells, knockdown of PTEN and SMAD4 in BEAS-2B cells was performed and increased both ErbB2 and Akt phosphorylations were observed, confirming a cooperative regulatory function of PTEN and SMAD4 in human lung epithelial cells (Figure S4G). We next evaluated the clinical relevance of increased ERBB2 signaling using the lung cancer TGCA data sets. Consistent with our in vivo and in vitro studies, dividing lung SCC patients into two groups based on the expression of ERBB2, we found that higher expression of ERBB2 was associated with poor patient survival in lung SCC patients in the TCGA data set (Figure 4F). Together, these findings suggest that ErbB2 signaling plays an important role in Ptend/dSmad4d/d lung tumors and human lung cancers.
Blockade of ErbB2/Akt Signaling Pathway Inhibits Tumor Growth Induced by Pten/Smad4 Deficiency
Given the important oncogenic role of ErbB2/Akt signaling pathway in human cancers and its activation in Ptend/dSmad4d/d lung tumors, we investigated its role in enhanced cell growth and invasion. We used specific inhibitors that targeted the essential molecules within the ErbB2/Akt signaling pathways: GDC-0941, the specific inhibitor of PI3K; Herceptin, a mono-antibody against ERBB2; and Lapatinib, the dual kinase inhibitor of ERBB2 and EGFR (Folkes et al., 2008; Langer et al., 2004; Gril et al., 2008). While treatment with these inhibitors had no significant effect on invasion of BEAS-2B cells transfected with control siRNAs (siControl), single or combinatorial treatment with these inhibitors greatly inhibited the increase of cell invasion induced by the double knockdown of PTEN and SMAD4 (Figures 5A and 5B). Next, we tested the importance of ErbB2/Akt signaling in tumor progression of Ptend/dSmad4d/d mice. Given the combinatorial inhibitory effect of Lapatinib and GDC-0941 on cell invasion, we treated Ptend/dSmad4d/d mice (9-month-old) for 30 days using oral gavage. As compared to vehicle (DMSO) control, the combinatorial treatment with Lapatinib and GDC-0941 significantly inhibited phosphorylations of ERBB2 and AKT (Figure S5) and lung tumor growth and progression, as demonstrated by the significant decrease in the weight ratio of whole lung to whole body, the number of mice with surface tumors, the average number of surface tumors, and the size (diameter) of maximal surface tumors (Figures 5C–5G).
Figure 5. Blockade of ErbB2/Akt Signaling Pathway Inhibits Tumor Growth Induced by Pten/Smad4 Deficiency.

(A) Two chamber invasion assays of Beas-2B cells with knockdown of PTEN and SMAD4 (siSMAD4/PTEN) and the treatment of kinase inhibitors as indicated.
(B) Quantification of the results (A) using one-way ANOVA (p value is *p < 0.05, **p < 0.01, ***p < 0.01).
(C–G) Inhibition of tumor formation in 9-month-old Ptend/dSmad4d/d mice the treatment of kinase inhibitors. Representative whole-lung images and H&E staining of lung tissues in WT and Ptend/dSmad4d/d mice are shown in (C). Quantitative analysis of tumor growth in Ptend/dSmad4d/d mice with or without the treatment of kinase inhibitors is shown in (D)–(G). Statistical analysis was performed using one-way ANOVA (p value is *p < 0.05, **p < 0.01, ***p < 0.01) on the ratio of whole lung to whole body and using Student’s t test (p value is **p < 0.01) on the maximal diameter and the number of surface tumors. Black arrows indicate lung tumors.
Errfi1, a Negative Regulator of ErbB2 Signaling, Plays Critical Roles in Tumor Progression in the Pten-Null Background
Having defined the critical role of ErbB2 signaling in promoting tumor growth and progression in Ptend/dSmad4d/d mice, we determined the cause of the increase in ERBB2 phosphorylation (activity) in these mice. Combinatorial intersection analysis of our microarray data and SMAD4 ChIP-seq data revealed that Errfi1 (ERBB receptor feedback inhibitor 1), a known negative regulator of ERBB signaling (Hackel et al., 2001; Zhang et al., 2012; Zhang et al., 2007), was significantly downregulated in Ptend/dSmad4d/d lung tumors (Figure S6A) and was a potential target of SMAD4 in Ptend/d mice (Figures S6B and S6C). The downregulation of Errfi1 expression in Ptend/dSmad4d/d lungs was verified at both the mRNA level (Figure 6A) and the protein level (Figure 6B). As ERRFI1 was shown to inhibit ErbB2 signaling in thyroid and breast cancer (Lin et al., 2011; Anastasi et al., 2005) and to act as a tumor suppressor in lung cancer (Zhang et al., 2007; Tseng et al., 2005), our results suggested that downregulation of Errfi1 might contribute to increased ERBB2 phosphorylation and lung tumor growth and metastasis in mouse models. To investigate the role of Errfi1 in lung tumorigenesis, using the CCSPiCre, Errfi1 was conditionally ablated alone, Errfi1d/d, or in combination with Pten. Deletion of Errfi1 led to a great increase of ERBB2 phosphorylation in mouse lung upper airways (Figures 6C and 6D). While only 1 in 15 Errfi1d/d mice had lung tumor growth at 9 months of age, 71% of Errfi1d/dPtend/d mice formed lung tumors at the same age (Figures 6E and 6F). Interestingly, a similar tumor incidence was observed for Errfi1d/dPtend/d mice (71.4%) and Ptend/dSmad4d/d mice (67%). Markers staining indicated tumors from Errfi1d/dPtend/d mice belonged to AD (Figures S6D–S6F). Taken together, these results suggest that Errfi1 acts as a suppressor of ErbB2 signaling and prevents lung tumor growth in the Pten-null background; downregulation of Errfi1 upon the deletion of Smad4 in Ptend/dSmad4d/d mice led to increased ErbB2 signaling and lung tumorigenesis.
Figure 6. Errfi1, a Negative Regulator of ErbB2 Signaling, Plays Critical Roles in Tumor Progression in the Pten-Null Background.
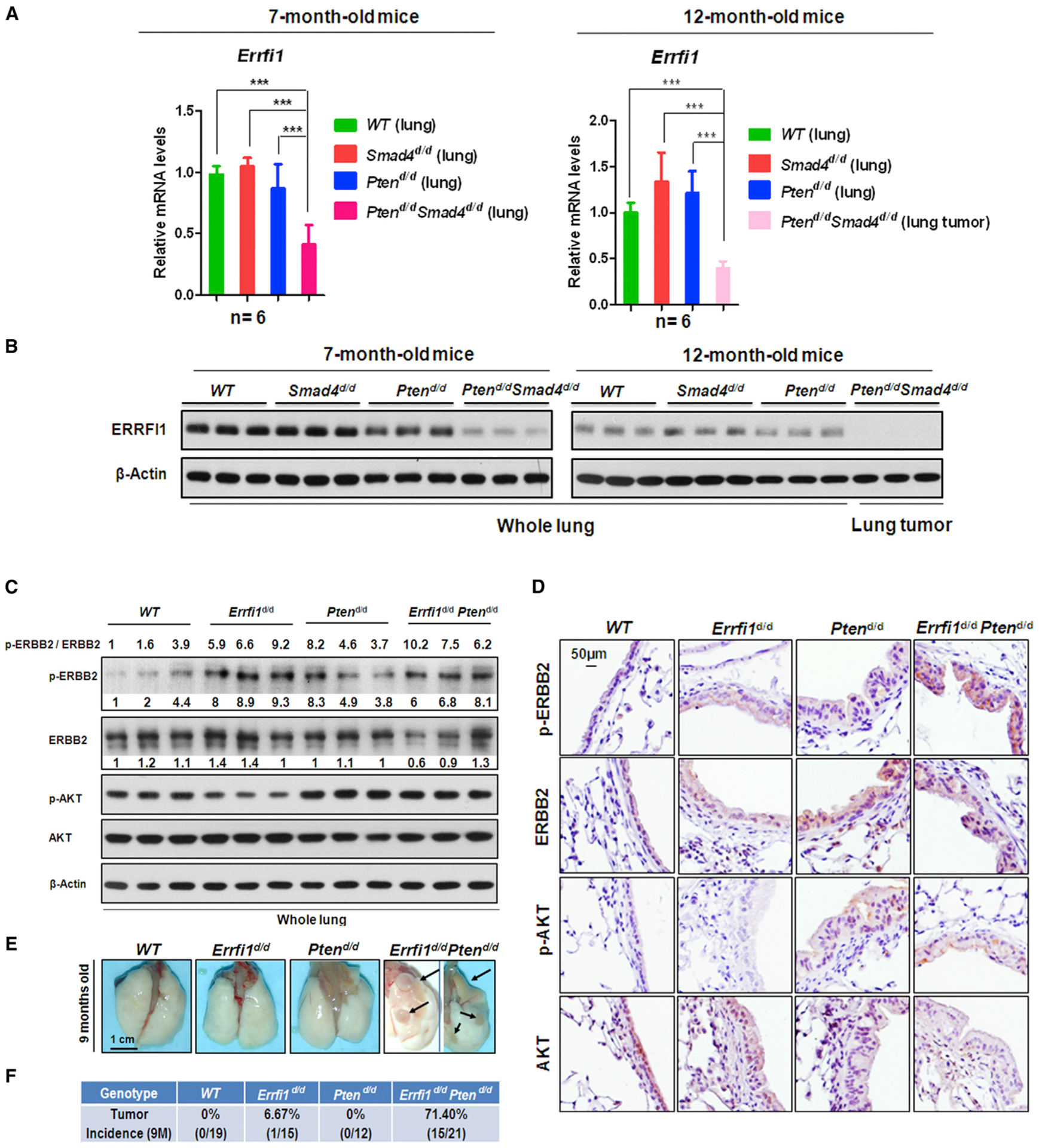
(A and B) Quantitative real-time PCR (A) and WB (B) analyses of Errfi1 expression level in mouse lungs and lung tumors.
(C and D) WB (C) and IHC (D) analyses of p-ERBB2/ERBB2 and p-AKT/AKT signaling in 9-month-old mouse lungs.
(E and F) Primary lung tumors were observed in the Errfi1d/dPtend/d mouse lungs. (E) The primary lung tumors are indicated by black arrows. (F) Summary of Errfi1d/dPtend/d mice with frequency of lung tumor.
ELF3 Is a Novel Target Gene in the ErbB2 Pathway Directly Regulated by SMAD4 in PTEN-Null or -Low Background
Given our findings on the essential role of activated ErbB2/Akt signaling in promoting Ptend/dSmad4d/d lung tumors, we next explored downstream target(s) of this pathway that may play a significant role in tumor progression. Analyzing the genes that showed a consistent change in the early- and late-stage lung tumors, we found ELF3, a transcriptional factor known to have oncogenic activity in breast cancer (Coppe et al., 2010), ranked on the top position in genes regulated by ErbB2 signaling (Figure S7A). Mining the SMAD4 ChIP-seq analysis demonstrated SMAD4 bound the proximal promoter region the ELF3 gene (Figure S7B), which was validated by ChIP-qPCR (Figure 7A). The upregulation of Elf3 was validated at both mRNA (Figure 7B) and protein levels (Figures 7C and 7D) in the lungs of 7-month-old Ptend/dSmad4d/d mice and in lung tumors isolated from 12-month-old Ptend/dSmad4d/d mice (Figure S7C). Interestingly, Elf3 was also found to be significantly upregulated in the lungs of Ptend/d mice, albeit at a lesser level than that of Ptend/dSmad4d/d mice, suggesting that Elf3 is regulated by both PTEN and SMAD4 tumor suppressors. Consistent with this in vivo finding, knockdown of PTEN and SMAD4 increased ELF3 mRNA expression in BEAS-2B cells (Figure 7E). In addition, ELF3 was downregulated by TGF-β1, an upstream stimulus of SMAD4, and depletion of SMAD4 abolished this inhibitory effect (Figure 7F). These results demonstrated that ELF3 was a gene directly and negatively regulated by SMAD4. We then determined the importance of ELF3 on cell proliferation and invasion. Knockdown of ELF3 significantly decreased both proliferation and invasion of BEAS-2B cells, most prominently in the context of both PTEN and SMAD4 double deletion (Figures 7G–7I). Analysis of Oncomine data sets revealed that ELF3 was highly upregulated in lung cancer (Figures S7E and S7F), and its upregulation is positively associated with poor survival and with early recurrence (Figures S7G and S7H).
Figure 7. ELF3 Is a Novel Target Gene of the ErbB2 Pathway and Is Directly Regulated by SMAD4 in PTEN-Null or -Low Background.
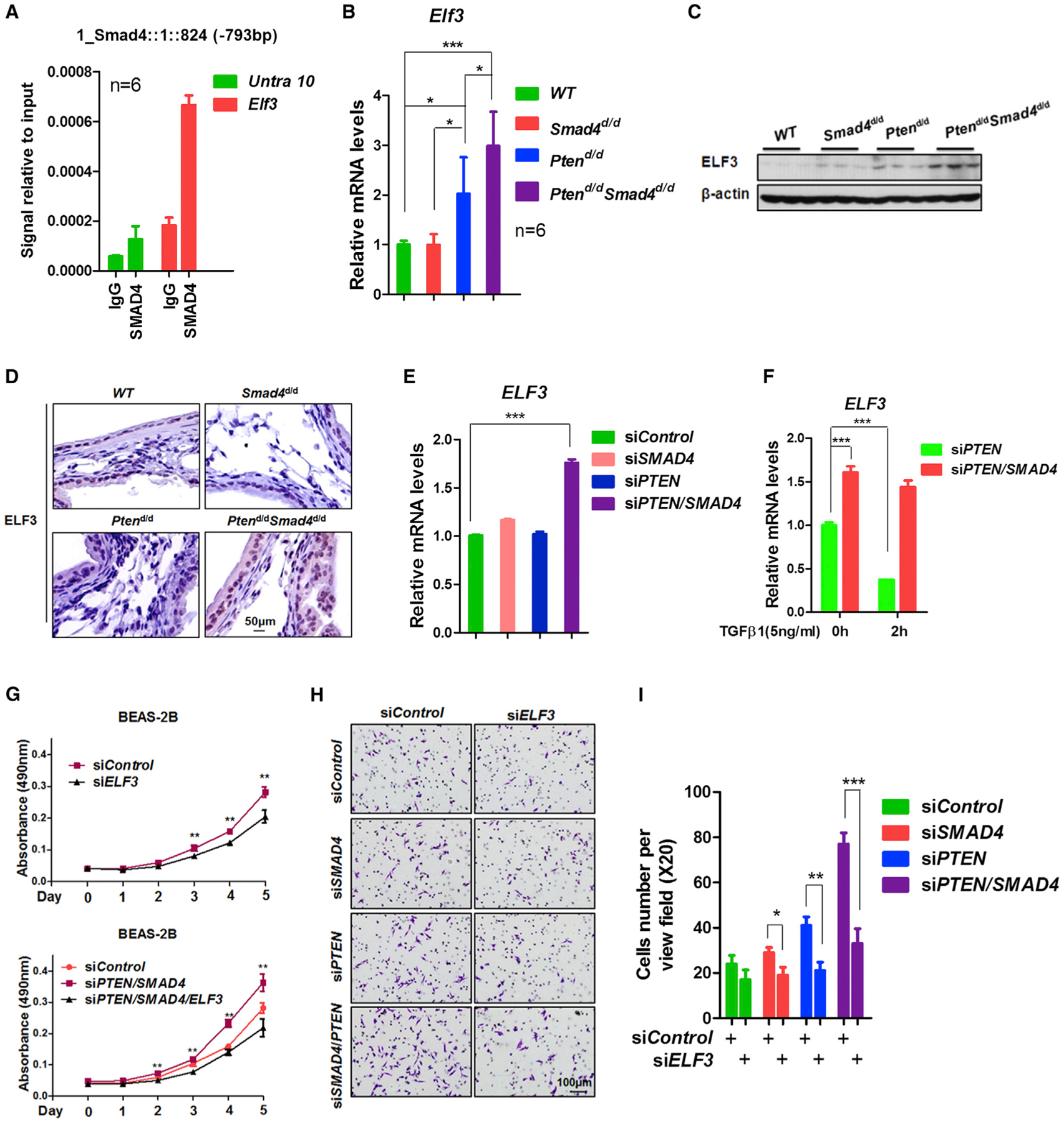
(A) ChIP-qPCR analysis of SMAD4 binding on the Elf3 promoter region in 7-month-old Ptend/d mouse lungs. The primers for Untra 10 are targeted on the untranslated region 10.
(B–D) Quantitative real-time PCR (B), WB (C), and IHC (D) analyses of Elf3 expression in 7-month-old mouse lungs.
(E) Quantitative real-time PCR analysis of ELF3 mRNA expression in BEAS-2B cells after knockdown of PTEN and/or SMAD4.
(F) Quantitative real-time PCR analysis of the ELF3 mRNA expression in BEAS-2B cells with the knockdown of PTEN and SMAD4 and with or without the treatment of TGF-β1. Statistical analysis was performed using one-way ANOVA (p value is *p < 0.05, **p < 0.01, and ***p < 0.001).
(G) MTT assay of cell viability in Beas-2B cells after the knockdown of PTEN, SMAD4, and ELF3. Statistical analysis was performed using Student’s t test (p value is **p < 0.01).
(H and I) Two-chamber invasion assay of Beas-2B cells after knockdown of PTEN, SMAD4, and/or Elf3. Representative images of cell invasion are shown in (H), and quantitation of cell invasion is shown in (I). Statistical analysis was performed using Student’s t test (p value is *p < 0.05, **p < 0.01, ***p < 0.01).
These results highlight the clinical relevance of the Ptend/d Smad4d/d lung tumor model: Pten deletion in airway epithelial cells induces airway epithelia cell hyperplasia probably secondary to the compensatory activation of SMAD4. Further depletion of Smad4 in Ptend/d mice results in activation of ErbB2, probably through the downregulation of ERRFI1, and further activates Akt signaling pathway and induces expression of ERBB2 target genes including Elf3, which promoted lung tumor growth and progression.
DISCUSSION
In this report, we have developed a new mouse model to identify critical molecules and signaling pathways that play key roles in driving tumor initiation and progression in hyperplastic (e.g., Pten-null) airway epithelial cells. Pten loss has been shown to be highly relevant in lung cancer development (Malkoski et al., 2014; Xu et al., 2014). However, initial investigations using the CCSPCre model to delete the Pten gene in mouse airway epithelial cells showed no discernible phenotype (Iwanaga et al., 2008). Here, we found that Pten ablation using the improved CCSPicre showed consistent airway epithelial hyperplasia without any progression to cancer (up to 15 months); these findings are in contrast to the reports that AD develop in the lungs of mice with SP-Ccre-mediated deletion of Pten (Yanagi et al., 2007). Further, a mixed ADC and SCC phenotype has been reported in the mice lacking Pten in the airway basal cells (Malkoski et al., 2014). These findings provide a cell-specific and geographically distinct mechanism of tumor suppressor activation for Pten and support the hypothesis that progression of hyperplastic epithelial cells in the proximal airways requires additional alterations in tumor suppressor genes and/or signaling pathways. In support of this hypothesis, we discovered the activated tumor suppressor TGF-β/SMAD4 signaling pathway in Ptend/d mice, a plausible mechanism protecting the hyperplastic airway epithelial cells to lung tumors. In support of our findings, a protective role for SMAD4 has been described in other epithelial derived tumors, such as prostate cancer (Ding et al., 2011); our findings demonstrate for the first time that concurrent loss of Pten and Smad4 induces tumor growth and metastasis in lung cancer and shows how similar cancer driver genes behave in epithelial tumors. Further, our novel lung tumor model also shares similar characteristics with human SCC lung cancer, including their initiation site in the proximal airways and lack type II alveolar airway phenotypic markers (Figures 2B–2D) (Sutherland and Berns, 2010). Additionally, much like human lung cancers, Ptend/dSmad4d/d lung tumors spontaneously metasta-size to other organs, including the liver and stomach; albeit, in humans, metastasis to the gastric mucosa had been reported at low frequency (Figures 2F and 2G) (Lee et al., 2011). In support of the relevance of the Ptend/dSmad4d/d lung cancer model, we show that the gene signature identified in this tumor model is highly consistent with that of human lung cancers (Figure 3D).
In our current study, we identified the activation of the ErbB2 pathway with concurrent loss of Pten and Smad4 prior to primary tumor formation. The hyperactivation of ErbB2 signaling is in parallel with remarkable reduction of Errfi1, a known inhibitor of ERBB2 activity, suggesting a molecular mechanism for the activation of ErbB2 signaling upon the loss of both Pten and Smad4. In support of this, concurrent loss of Errfi1 and Pten in lungs also led to upregulation of ERBB2 signaling and high tumor incidence. This important finding not only provides a mechanism for SMAD4-mediated suppression of lung tumor in hyperplastic airway epithelia cells in Pten-null mice, but also suggests that the ErbB2 signaling pathway could be used as a diagnostic biomarker and targeted to prevent lung tumor progression. Indeed, the ErbB2 signaling pathway is highly upregulated in human lung cancer (Minami et al., 2007; Stephens et al., 2004; Arcila et al., 2012; Greulich et al., 2012; Mazières et al., 2013), and ERBB2 has been recently found to be one of the driver genes in human lung AD (Cancer Genome Atlas Research Network, 2014). More importantly, we show that inhibition of the ErbB2 signaling pathway by combinatorial treatment with Herceptin and GDC-0941 inhibited cell invasion and tumor progression.
We have also identified ELF3 as a novel direct inhibitory target of SMAD4 that plays an important role in lung cancer, and ELF3 showed significant positive correlation with ERBB2 in human lung adenocarcinoma, indicating that it may be a very important downstream gene in ErbB2 signaling in lung cancer (Figure S7K). To our knowledge, our study is the first to demonstrate that Elf3 is highly upregulated upon deletion of Smad4 and plays an important role in lung cell proliferation and invasion. Interestingly, ELF3 was shown to be an upstream regulator of SPP1 (Coppe et al., 2010), which was identified as a key gene regulated by PTEN and SMAD4 in a murine model of metastatic prostate cancer (Ding et al., 2011). Although the overlap in altered genes observed in the prostate cancer tumors was minimal when compared to the lung Ptend/dSmad4d/d mouse model (data not shown), we found high expression of SPP1 and Elf3 in Ptend/d Smad4d/d lung tumors (Figures S7C and S7L), indicating that ELF3 might play the conserved role to drive cancer development and metastasis. In addition, we show that ELF3 is overexpressed in human lung cancer (Figures S7E and S7F) and its upregulation in human lung cancer cells with PI3KCA and SMAD4 mutations (Figures S7I and S7J). All the evidence indicates that ELF3 could be new potential target for lung cancer treatment.
Although the tumor types between Ptend/dSmad4d/d and Errfi1d/dPtend/d mouse models were different, both of them showed activated ERBB2 and AKT signaling. This indicated that other genes regulated by SMAD4 are required to work cooperatively with ERBB2/Akt signaling for the development of lung SCC in Ptend/dSmad4d/d mice. Comparing our Ptend/dSmad4d/d tumor gene array with the pure lung SCC mouse tumor array (Ad-Cre-Ptenf/fLkb1f/f) and the pure AD mouse tumor array (Ad-Cre-KrasG12D) (Xu et al., 2014), we found 341 genes only overlapping between the Ptend/dSmad4d/d tumor array and the Ad-Cre-Ptenf/fLkb1f/f mouse array (Figure S3), including the marker genes for SCC, such as p63, Sox2, and Krt5 (Table S5). Importantly, p63 and Sox2 have recently been proved to regulate the development of SCC in mice (Han et al., 2014; Mukhopadhyay et al., 2014). Consistent with the AD tumor type observed in Errfi1d/dPtend/d mouse models, p63, SOX2, and KRT5 are not expressed in these tumors (Figure S6D).
In summary, we successfully generated a metastatic lung tumor model with airway-specific deletion of Pten and Smad4 and showed that their sequential alterations induce oncogenic pathways including ErbB2/Akt/ELF3 and represses tumor suppressors, such as Errfi1, that are critical for lung tumor initiation and progression. Given the long latency of tumor progression in this model, it can be used to determine what other factors are critical for promoting tumor progression and metastasis. These studies include determining the impact of environmental exposures to airborne carcinogens as well as functional genomic screening for modifiers of tumor progression and metastasis in this model. Since the in vivo analysis in this mouse model can be recapitulated in human airway cells in vitro, further mechanistic studies can be conducted. These studies will be able to define the mechanism by which altering the expression of PTEN causes the increase in TGF-β signaling. Cistromic analysis in human cells will identify the genes directly regulated by SMAD4 in preventing tumor progression. These approaches will also identify other pathways that are ideal for treating lung cancer at early stages and for preventing cancer progression.
EXPERIMENTAL PROCEDURES
Generation of Ptend/d, Smad4d/d, Ptend/d Smad4d/d, Errfi1d/d, and Errfi1d/dPtend/d Mice
All animal experiments were performed in accordance with an IACUC approved protocol (Baylor College of Medicine, Houston, TX). Details are in the Supplemental Information.
Histopathology, Immunohistochemistry, Immunofluorescence, and Western Blotting
Mouse lungs were fixed in 4% paraformaldehyde and paraffin embedded according to standard protocols. Details are in the Supplemental Information.
Cell Derivation and Culture
Beas-2B (ATCC CRL-9609) and NL20 (ATCC CRL-2503) cells were purchased from ATCC and cultured by following the ATCC culture condition. 293T cells were purchased from Tissue Culture Core in Baylor College of Medicine. Details are in the Supplemental Information.
Senescence Experiment
Frozen lung sections were prepared from 10-month-old mice, and the standard protocol of Senescence Detection Kit (Abcam, cat. Ab65351) was utilized.
Microarray, Quantitative Real-Time PCR
Total RNAs were isolated from mouse lungs with TRIzol reagent (Invitrogen) and reversely transcribed into cDNA with a M-MLV kit (Invitrogen). The mRNA expression levels of human and mouse were determined by quantitative real-time PCR by using SYBR Green and TaqMan probes (Applied Biosystems). Statistical analysis was performed using one-way ANOVA and Student’s t test (p value is *p < 0.05, **p < 0.01, and ***p < 0.001).
Mouse Lung Cancer Microarray
Details are in the Supplemental Information.
Human Lung Cancer Samples and Tissue Microarray
All human lung tissues were obtained from the Lung Cancer Specialized Program of Research Excellence (SPORE) Tissue Bank at the MD Anderson Cancer Center and an IRB was approved for expression studies from this Tissue Bank. Details are in the Supplemental Information.
siRNA and Small Hairpin RNA Experiments
For siRNA experiments, ON-TARGET plus siRNAs SMARTpools (Thermo) were used. For small hairpin RNA (shRNA) experiments, all the pGIPZ lentivirus shRNA plasmids were purchased from the C-BASS Core at Baylor College of Medicine. Details are in the Supplemental Information.
CHIP-Seq and Chip-qPCR
Details are in the Supplemental Information.
Transwell Migration Assay, Invasion Assay, and MTS Assay
Standard 24-well Boyden control and invasion chambers (BD Biosciences) were used to assess cell migration and invasion following manufacturer suggestions. Details are in the Supplemental Information.
Inhibitors Treatment
Inhibitors GDC-0941(LC Laboratories, G-9252), Lapatinib (LC Laboratories, L-4899), and Herceptin (BOC Sciences, 180288-69-1) were dissolved in DMSO. DMSO was used as the control. Details are in the Supplemental Information.
Human Lung Tumor Expression Data Sets
The gene expression array data set GSE11969 (Takeuchi et al., 2006) was probed using the mouse lung cancer gene signature, using previously described approaches (Qin et al., 2013). Data sets in Oncomine (https://www.oncomine.org) were also examined. RNA sequencing data of Lung Squamous Cell Carcinoma (LUSC) and Lung Adenocarcinoma (LUAD) were obtained through TCGA pan-cancer synapse (https://www.synapse.org/#!Synapse:syn300013). Details are provided in Supplemental Experimental Procedures.
Supplementary Material
Highlights.
Airway-specific Pten loss causes epithelial hyperplasia and alters TGF-β signaling
Loss of Smad4 in Ptend/d mice results in metastatic adenosquamous carcinomas
Ablation of Smad4 and Pten causes repression of ERRFI1 and activation of ERBB2/ELF3
Loss of Errfi1 in Ptend/d mice promotes lung tumors
ACKNOWLEDGMENTS
We appreciate the help from our colleagues: Janet L. DeMayo, M.S., for technical assistance and lab management; Xueping Xu and S.N. Cho for the work on genotyping; Sophia Y. Tsai, Ph.D., Ming-Jer Tsai, Ph.D., Xiaotao Li, Ph.D., Jun Qin, Ph.D., John P. Lydon, Ph.D., and Sang Jun Han, Ph.D., for the discussion on the whole projects, Tunde Smith for collecting some tissue sections. We also thank the following Dan L. Duncan Cancer Center Shared Resources (NIH grant P30 CA125123) at Baylor College of Medicine: the Genetically Engineered Mouse Shared Resource, the Genomic and RNA Profiling Shared Resource, the Biostatistics and Informatics Shared Resource, and Cell Processing and Vector Production Shared Resource. This project was done in collaboration with the Adrienne Helis Malvin Medical Research Foundation through its direct engagement in the continuous active conduct of medical research in conjunction with Baylor College of Medicine and the Immune Therapy in a Novel Humanized Model of Lung Cancer Program (to F.J.D. and F.K.). Additional support included CPRIT grant RP120713 (to C.J.C.).
Footnotes
ACCESSION NUMBERS
The GEO accession number for the microarray data reported in this paper is GSE57133. The GEO accession number for the ChIP-seq data reported in this paper is GSE57177.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and seven tables and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2015.02.014.
REFERENCES
- Ahmad I, Patel R, Singh LB, Nixon C, Seywright M, Barnetson RJ, Brunton VG, Muller WJ, Edwards J, Sansom OJ, and Leung HY (2011). HER2 overcomes PTEN (loss)-induced senescence to cause aggressive prostate cancer. Proc. Natl. Acad. Sci. USA 108, 16392–16397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastasi S, Sala G, Huiping C, Caprini E, Russo G, Iacovelli S, Lucini F, Ingvarsson S, and Segatto O (2005). Loss of RALT/MIG-6 expression in ERBB2-amplified breast carcinomas enhances ErbB-2 oncogenic potency and favors resistance to Herceptin. Oncogene 24, 4540–4548. [DOI] [PubMed] [Google Scholar]
- Arcila ME, Chaft JE, Nafa K, Roy-Chowdhuri S, Lau C, Zaidinski M, Paik PK, Zakowski MF, Kris MG, and Ladanyi M (2012). Prevalence, clinicopathologic associations, and molecular spectrum of ERBB2 (HER2) tyrosine kinase mutations in lung adenocarcinomas. Clin. Cancer Res 18, 4910–4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2012). Comprehensive genomic characterization of squamous cell lung cancers. Nature 489, 519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2014). Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppe J-P, Amend C, Semeiks J, Baehner FL, Bayani N, Campisi J, Benz CC, Gray JW, and Neve RM (2010). ERBB receptor regulation of ESX/ELF3 promotes invasion in breast epithelial cells. Open Cancer J. 3, 89–100. [Google Scholar]
- Davé V, Wert SE, Tanner T, Thitoff AR, Loudy DE, and Whitsett JA (2008). Conditional deletion of Pten causes bronchiolar hyperplasia. Am. J. Respir. Cell Mol. Biol 38, 337–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Caestecker MP, Piek E, and Roberts AB (2000). Role of transforming growth factor-beta signaling in cancer. J. Natl. Cancer Inst 92, 1388–1402. [DOI] [PubMed] [Google Scholar]
- Dennis JL, Hvidsten TR, Wit EC, Komorowski J, Bell AK, Downie I, Mooney J, Verbeke C, Bellamy C, Keith WN, and Oien KA (2005). Markers of adenocarcinoma characteristic of the site of origin: development of a diagnostic algorithm. Clin. Cancer Res 11, 3766–3772. [DOI] [PubMed] [Google Scholar]
- Ding Z, Wu CJ, Chu GC, Xiao Y, Ho D, Zhang J, Perry SR, Labrot ES, Wu X, Lis R, et al. (2011). SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature 470, 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G, Chuckowree IS, Clarke PA, Depledge P, Eccles SA, et al. (2008). The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-yl-methyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J. Med. Chem 51, 5522–5532. [DOI] [PubMed] [Google Scholar]
- Greulich H, Kaplan B, Mertins P, Chen TH, Tanaka KE, Yun CH, Zhang X, Lee SH, Cho J, Ambrogio L, et al. (2012). Functional analysis of receptor tyrosine kinase mutations in lung cancer identifies oncogenic extracellular domain mutations of ERBB2. Proc. Natl. Acad. Sci. USA 109, 14476–14481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gril B, Palmieri D, Bronder JL, Herring JM, Vega-Valle E, Feigenbaum L, Liewehr DJ, Steinberg SM, Merino MJ, Rubin SD, and Steeg PS (2008). Effect of lapatinib on the outgrowth of metastatic breast cancer cells to the brain. J. Natl. Cancer Inst 100, 1092–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackel PO, Gishizky M, and Ullrich A (2001). Mig-6 is a negative regulator of the epidermal growth factor receptor signal. Biol. Chem 382, 1649–1662. [DOI] [PubMed] [Google Scholar]
- Han X, Li F, Fang Z, Gao Y, Li F, Fang R, Yao S, Sun Y, Li L, Zhang W, et al. (2014). Transdifferentiation of lung adenocarcinoma in mice with Lkb1 deficiency to squamous cell carcinoma. Nat. Commun 5, 3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlader N, Noone A, Krapcho M, Neyman N, Aminou R, Altekruse SF, Kosary CL, Ruhl J, Tatalovich Z, and Cho H, et al. , eds. (2014). SEER Cancer Statistics Review, 1975–2011 (Bethesda, MD: National Cancer Institute; ), based on November 2013 SEER data submission, posted to the SEER web site, April 2014. http://seer.cancer.gov/csr/1975_2011/. [Google Scholar]
- Iwanaga K, Yang Y, Raso MG, Ma L, Hanna AE, Thilaganathan N, Moghaddam S, Evans CM, Li H, Cai WW, et al. (2008). Pten inactivation accelerates oncogenic K-ras-initiated tumorigenesis in a mouse model of lung cancer. Cancer Res. 68, 1119–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Xu J, and Ward E (2010). Cancer statistics, 2010. CA Cancer J. Clin 60, 277–300. [DOI] [PubMed] [Google Scholar]
- Jeon HS, and Jen J (2010). TGF-beta signaling and the role of inhibitory Smads in non-small cell lung cancer. J. Thorac. Oncol 5, 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Z, Zhang X, Ma L, and Wang L (2008). Expression of DPC4/Smad4 in non-small-cell lung carcinoma and its relationship with angiogenesis. Neo-plasma 55, 323–329. [PubMed] [Google Scholar]
- Langer CJ, Stephenson P, Thor A, Vangel M, and Johnson DH; Eastern Cooperative Oncology Group Study 2598 (2004). Trastuzumab in the treatment of advanced non-small-cell lung cancer: is there a role? Focus on Eastern Cooperative Oncology Group study 2598. J. Clin. Oncol 22, 1180–1187. [DOI] [PubMed] [Google Scholar]
- Lee PC, Lo C, Lin MT, Liang JT, and Lin BR (2011). Role of surgical intervention in managing gastrointestinal metastases from lung cancer. World J. Gastroenterol 17, 4314–4320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesche R, Groszer M, Gao J, Wang Y, Messing A, Sun H, Liu X, and Wu H (2002). Cre/loxP-mediated inactivation of the murine Pten tumor suppressor gene. Genesis 32, 148–149. [DOI] [PubMed] [Google Scholar]
- Li H, Cho SN, Evans CM, Dickey BF, Jeong JW, and DeMayo FJ (2008). Cre-mediated recombination in mouse Clara cells. Genesis 46, 300–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CI, Du J, Shen WT, Whang EE, Donner DB, Griff N, He F, Moore FD Jr., Clark OH, and Ruan DT (2011). Mitogen-inducible gene-6 is a multifunctional adaptor protein with tumor suppressor-like activity in papillary thyroid cancer. J. Clin. Endocrinol. Metab 96, E554–E565. [DOI] [PubMed] [Google Scholar]
- Liu F (2001). SMAD4/DPC4 and pancreatic cancer survival. Commentary re: M. Tascilar et al. , The SMAD4 protein and prognosis of pancreatic ductal adenocarcinoma. Clin. Cancer Res, 7: 4115–4121, 2001. Clin. Cancer Res. 7, 3853–3856. [PubMed] [Google Scholar]
- Malkoski SP, Cleaver TG, Thompson JJ, Sutton WP, Haeger SM, Rodriguez KJ, Lu SL, Merrick D, and Wang XJ (2014). Role of PTEN in basal cell derived lung carcinogenesis. Mol. Carcinog 53, 841–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué J, Seoane J, and Wotton D (2005). Smad transcription factors. Genes Dev. 19, 2783–2810. [DOI] [PubMed] [Google Scholar]
- Mazières J, Peters S, Lepage B, Cortot AB, Barlesi F, Beau-Faller M, Besse B, Blons H, Mansuet-Lupo A, Urban T, et al. (2013). Lung cancer that harbors an HER2 mutation: epidemiologic characteristics and therapeutic perspectives. J. Clin. Oncol 31, 1997–2003. [DOI] [PubMed] [Google Scholar]
- Minami Y, Shimamura T, Shah K, LaFramboise T, Glatt KA, Liniker E, Borgman CL, Haringsma HJ, Feng W, Weir BA, et al. (2007). The major lung cancer-derived mutants of ERBB2 are oncogenic and are associated with sensitivity to the irreversible EGFR/ERBB2 inhibitor HKI-272. Oncogene 26, 5023–5027. [DOI] [PubMed] [Google Scholar]
- Miyaki M, and Kuroki T (2003). Role of Smad4 (DPC4) inactivation in human cancer. Biochem. Biophys. Res. Commun 306, 799–804. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay A, Berrett KC, Kc U, Clair PM, Pop SM, Carr SR, Witt BL, and Oliver TG (2014). Sox2 cooperates with Lkb1 loss in a mouse model of squamous cell lung cancer. Cell Rep. 8, 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagashima A, Abe Y, Yamada S, Nakagawa M, and Yoshimatsu T (2004). Long-term survival after surgical resection of liver metastasis from lung cancer. Jpn. J. Thorac. Cardiovasc. Surg 52, 311–313. [DOI] [PubMed] [Google Scholar]
- Nagatake M, Takagi Y, Osada H, Uchida K, Mitsudomi T, Saji S, Shimokata K, Takahashi T, and Takahashi T (1996). Somatic in vivo alterations of the DPC4 gene at 18q21 in human lung cancers. Cancer Res. 56, 2718–2720. [PubMed] [Google Scholar]
- Perl AK, Wert SE, Nagy A, Lobe CG, and Whitsett JA (2002). Early restriction of peripheral and proximal cell lineages during formation of the lung. Proc. Natl. Acad. Sci. USA 99, 10482–10487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Wu SP, Creighton CJ, Dai F, Xie X, Cheng CM, Frolov A, Ayala G, Lin X, Feng XH, et al. (2013). COUP-TFII inhibits TGF-β-induced growth barrier to promote prostate tumorigenesis. Nature 493, 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakashita S, Sakashita M, and Sound Tsao M (2014). Genes and pathology of non-small cell lung carcinoma. Semin. Oncol 41, 28–39. [DOI] [PubMed] [Google Scholar]
- Shames DS, and Wistuba II (2014). The evolving genomic classification of lung cancer. J. Pathol 232, 121–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Hata A, Lo RS, Massagué J, and Pavletich NP (1997). A structural basis for mutational inactivation of the tumour suppressor Smad4. Nature 388, 87–93. [DOI] [PubMed] [Google Scholar]
- Shimshek DR, Kim J, Hübner MR, Spergel DJ, Buchholz F, Casanova E, Stewart AF, Seeburg PH, and Sprengel R (2002). Codon-improved Cre recombinase (iCre) expression in the mouse. Genesis 32, 19–26. [DOI] [PubMed] [Google Scholar]
- Siegel R, Ma J, Zou Z, and Jemal A (2014). Cancer statistics, 2014. CA Cancer J. Clin 64, 9–29. [DOI] [PubMed] [Google Scholar]
- Soriano P (1999). Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet 21, 70–71. [DOI] [PubMed] [Google Scholar]
- Stephens P, Hunter C, Bignell G, Edkins S, Davies H, Teague J, Stevens C, O’Meara S, Smith R, Parker A, et al. (2004). Lung cancer: intragenic ERBB2 kinase mutations in tumours. Nature 431, 525–526. [DOI] [PubMed] [Google Scholar]
- Su YC, Hsu YC, and Chai CY (2006). Role of TTF-1, CK20, and CK7 immunohistochemistry for diagnosis of primary and secondary lung adenocarcinoma. Kaohsiung J. Med. Sci 22, 14–19. [DOI] [PubMed] [Google Scholar]
- Sutherland KD, and Berns A (2010). Cell of origin of lung cancer. Mol. Oncol 4, 397–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi T, Tomida S, Yatabe Y, Kosaka T, Osada H, Yanagisawa K, Mitsudomi T, and Takahashi T (2006). Expression profile-defined classification of lung adenocarcinoma shows close relationship with underlying major genetic changes and clinicopathologic behaviors. J. Clin. Oncol 24, 1679–1688. [DOI] [PubMed] [Google Scholar]
- Tascilar M, Skinner HG, Rosty C, Sohn T, Wilentz RE, Offerhaus GJ, Adsay V, Abrams RA, Cameron JL, Kern SE, et al. (2001). The SMAD4 protein and prognosis of pancreatic ductal adenocarcinoma. Clin. Cancer Res 7, 4115–4121. [PubMed] [Google Scholar]
- Teng Y, Sun AN, Pan XC, Yang G, Yang LL, Wang MR, and Yang X (2006). Synergistic function of Smad4 and PTEN in suppressing forestomach squamous cell carcinoma in the mouse. Cancer Res. 66, 6972–6981. [DOI] [PubMed] [Google Scholar]
- Tseng RC, Chang JW, Hsien FJ, Chang YH, Hsiao CF, Chen JT, Chen CY, Jou YS, and Wang YC (2005). Genomewide loss of heterozygosity and its clinical associations in non small cell lung cancer. Int. J. Cancer 117, 241–247. [DOI] [PubMed] [Google Scholar]
- Walker S (2008). Updates in non-small cell lung cancer. Clin. J. Oncol. Nurs 12, 587–596. [DOI] [PubMed] [Google Scholar]
- Xu C, Fillmore CM, Koyama S, Wu H, Zhao Y, Chen Z, Herter-Sprie GS, Akbay EA, Tchaicha JH, Altabef A, et al. (2014). Loss of Lkb1 and Pten leads to lung squamous cell carcinoma with elevated PD-L1 expression. Cancer Cell 25, 590–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagawa N, Leduc C, Kohler D, Saieg MA, John T, Sykes J, Yoshimoto M, Pintilie M, Squire J, Shepherd FA, and Tsao MS (2012). Loss of phosphatase and tensin homolog protein expression is an independent poor prognostic marker in lung adenocarcinoma. J. Thorac. Oncol 7, 1513–1521. [DOI] [PubMed] [Google Scholar]
- Yanagi S, Kishimoto H, Kawahara K, Sasaki T, Sasaki M, Nishio M, Yajima N, Hamada K, Horie Y, Kubo H, et al. (2007). Pten controls lung morphogenesis, bronchioalveolar stem cells, and onset of lung adenocarcinomas in mice. J. Clin. Invest 117, 2929–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Li C, Herrera PL, and Deng CX (2002). Generation of Smad4/Dpc4 conditional knockout mice. Genesis 32, 80–81. [DOI] [PubMed] [Google Scholar]
- Zhang YW, Staal B, Su Y, Swiatek P, Zhao P, Cao B, Resau J, Sigler R, Bronson R, and Vande Woude GF (2007). Evidence that MIG-6 is a tumor-suppressor gene. Oncogene 26, 269–276. [DOI] [PubMed] [Google Scholar]
- Zhang YW, Staal B, Dykema KJ, Furge KA, and Vande Woude GF (2012). Cancer-type regulation of MIG-6 expression by inhibitors of methylation and histone deacetylation. PLoS ONE 7, e38955. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.