

Abstract
White-rot (WR) fungi are pivotal decomposers of dead organic matter in forest ecosystems and typically use a large array of hydrolytic and oxidative enzymes to deconstruct lignocellulose. However, the extent of lignin and cellulose degradation may vary between species and wood type. Here, we combined comparative genomics, transcriptomics and secretome proteomics to identify conserved enzymatic signatures at the onset of wood-decaying activity within the Basidiomycota genus Pycnoporus. We observed a strong conservation in the genome structures and the repertoires of protein-coding genes across the four Pycnoporus species described to date, despite the species having distinct geographic distributions. We further analysed the early response of P. cinnabarinus, P. coccineus and P. sanguineus to diverse (ligno)-cellulosic substrates. We identified a conserved set of enzymes mobilized by the three species for breaking down cellulose, hemicellulose and pectin. The co-occurrence in the exo-proteomes of H2O2-producing enzymes with H2O2-consuming enzymes was a common feature of the three species, although each enzymatic partner displayed independent transcriptional regulation. Finally, cellobiose dehydrogenase-coding genes were systematically co-regulated with at least one AA9 lytic polysaccharide monooxygenase gene, indicative of enzymatic synergy in vivo. This study highlights a conserved core white-rot fungal enzymatic mechanism behind the wood-decaying process.
Keywords: wood decay, lignocellulose, CAZyme, lytic polysaccharide monooxygenase, Class II Peroxidase
1. Introduction
Saprotrophic fungi of Northern Hemisphere and tropical forests impact the carbon cycling through mineralization and alteration of C storage in wood and litter dead organic matter.1,2 White-rot (WR) fungi are wood decayers with the capacity to mineralize lignin with ultimate formation of CO2 and H2O.3. WR fungi deploy a wide arsenal of hydrolytic and oxidative enzymes to degrade wood and their genomes typically contain genes coding for glycoside hydrolases, carbohydrate esterases and polysaccharide lyases that collectively cleave cellulose, hemicellulose and pectin backbones and lateral chains, and oxidative enzymes that target the highly recalcitrant lignin, crystalline cellulose or cellulose-bound xylan.4,5 Beyond these shared features, several studies have highlighted significant polymorphism between WR fungi regarding their ability to selectively degrade lignin over cellulose6–8 and in the gene portfolios involved in lignocellulose breakdown.9–11 Scarce studies at the intra-genus level have shown that functional diversity between species may arise from diversity in gene content.12,13
Among WR fungi, the genus Pycnoporus (Basidiomycota, Agaricomycetes) has been studied for the efficiency of lignin degradation, the capacity to secrete laccases and biotechnological applications related to aromatic compound functionalization, biopolymer synthesis and biomass pre-treatment in the pulp and paper industry.14 Four Pycnoporus species have been differentiated,15,16 which form a monophyletic group within the Trametes clade.17 The four species are found in different geo-climatic areas with limited geographical overlap; P. cinnabarinus is widely distributed in the Northern hemisphere, P. coccineus is found in countries bordering the Indian and Pacific Oceans, P. sanguineus is found in the tropics and subtropics of both hemispheres and P. puniceus is found in paleotropical areas.15,16 The four species are found on stumps and either standing or fallen trunks of deciduous trees.
Our aim was to investigate the Pycnoporus intra-genus genomic and functional diversity focusing on lignocellulose breakdown. We examined whether divergence in distinct geographic areas had led to genomic diversity, and if there was a signature of conserved enzymatic mechanics in terms of transcriptome and exo-proteome responses to lignocellulosic substrates. We sequenced the genomes of P. coccineus, P. sanguineus and P. puniceus monokaryotic strains. We overviewed the genomic features among the three species, in comparison to the previously sequenced genome of P. cinnabarinus9 and to other evolutionarily related wood-decay fungi. Then, we captured the transcriptomic and exoproteomic responses of three Pycnoporus species to a panel of cellulosic and lignocellulosic substrates representative of Gramineae and hardwoods. The focus was the early responses to the substrates in order to minimize inter-species differences influenced by varied growth abilities on the substrates. Our omics integrative approach enabled to identify a common set of lignocellulose-degrading enzymes mobilized by the fungi at the initial stage of lignocellulose degradation, leading to discoveries of genus-wide conserved expression patterns indicative of conserved enzymatic synergies.
2. Materials and methods
2.1. Genome sequencing and assembly
The monokaryotic strains P. coccineus BRFM 310, P. sanguineus BRFM 1264 and P. puniceus BRFM 1868 were generated after fruiting of the parental strains BRFM 66 (IMB WOO6-2), BRFM 902 and BRFM 1856, respectively, as described previously14 (Supplementary Information). All strains were maintained at the International Centre of Microbial Resources (CIRM; https://www6.inra.fr/cirm/). The P. coccineus BRFM 310 genome was sequenced using the Illumina platform (99.4×) and assembled with AllPathsLG version R4665218 (GenBank accession number: NCSW00000000). The P. sanguineus BRFM 1264 genome was sequenced using 454 (16.8×) and Solexa (87×) technologies and assembled with CABOG19 (GenBank accession number: VOCM00000000). The P. puniceus BRFM 1868 genome was sequenced using PacBio technology (97×) and assembled with FALCON, improved with finisherSC,20 polished with Quiver (GenBank accession number: VICQ00000000). The three genomes were annotated using the JGI annotation pipeline,21 which takes multiple inputs (scaffolds, ESTs and known genes) and runs several analytical tools for gene prediction and annotation, and deposits the results in the JGI Genome Portal MycoCosm (http://genome.jgi.doe.gov/fungi). The previously sequenced and annotated genome of P. cinnabarinus BRFM 1379 was also deposited in MycoCosm.
2.2. Construction of phylogenetic tree
We constructed a phylogeny based on orthologous genes among the selected fungi using FastOrtho with the parameters set to 50% identity, 50% coverage and inflation 3.0.22 The protein sequences used for the process were downloaded from the JGI fungal portal MycoCosm. We identified the clusters of orthologous genes with single copy genes, aligned the sequences of each cluster with MAFFT 7.221,23 eliminated ambiguous regions (containing gaps and poorly aligned regions) and concatenated the alignments with Gblocks 0.91b.24 We constructed a phylogenetic tree with RAxML 7.7.225 using the standard algorithm, the PROTGAMMAWAG model of sequence evolution and 500 bootstrap replicates.
2.3. Comparative genomic analysis
Genome completeness with single copy orthologues was calculated using BUSCO v3.0.2 with default parameters.26 The coverage of transposable elements (TEs) in genomes was calculated using a custom pipeline transposon identification nominative genome overview.27 The counts for plant cell wall (PCW)-degrading enzymes, predicted secreted auxiliary activity (AA) enzymes and fungal cell wall (FCW)-degrading enzymes were combined and visualized with custom R scripts, proteomic information navigated genomic outlook28 incorporating R packages ggplot2, ggtree and egg.29–31
2.4. Expert functional annotations
Genes from the A- and B-mating type loci were identified and manually curated as described in Kues et al.32 CAZymes and AA were annotated as in Lombard et al.33 Gene models from AA2 Class II peroxidases, AA3_2 glucose-methanol-choline (GMC)-oxidoreductases and AA5 copper radical oxidases were further inspected by sequence-by-sequence exhaustive analysis and phylogenetic analysis. Peptidases were annotated using Blastx searches of gene models against InterPro and MEROPS databases34,35 followed by manual curation. Glutathione transferases (GST)-coding genes were annotated with a combination of automated blastp using functionally characterized GST sequences from P. chrysosporium,36–38 phylogenetic analysis and active site comparison. Genes coding for hydrophobins, laccases, the secretory pathway and carbon catabolism were also manually inspected. Proteins were predicted secreted if they fulfilled three conditions: (i) presence of a secretion signal peptide, (ii) absence of endoplasmic reticulum retention motif and (iii) absence of transmembrane helix outside the signal peptide, as previously described.39 Predicted secreted proteins shorter than 300 amino acids were annotated as small secreted proteins (SSPs). Detailed analyses of expert annotations are provided in the Supporting Information file.
2.5. Cultures
Media for cultures on agar plates contained diammonium tartrate (1.84 g/l), yeast nitrogen base (0.17 g/l), agar (15 g/l) and were supplemented with either maltose (20 g/l), Avicel (AVI) PH 101 (Fluka) (15 g/l), ground and sifted wheat straw (WS) fragments <2 mm (15 g/l), Pinus halepensis pine wood fragments <2 mm (15 g/l) or Populus tremuloides Wiley-milled aspen (Asp) fragments (180 µm < fragment size < 2 mm; 15 g/l). The plates were inoculated with one fungal disk (4 mm diameter) of 7-day-old mycelia and incubated at 30 °C. Liquid cultures were maintained at 30 °C in a rotary shaker at 120 rpm in 250-ml Erlenmeyer flasks containing 100 ml of culture medium (Supplementary Information) supplemented with either maltose (20 g/l), AVI (15 g/l), WS fragments (15 g/l), P. halepensis wood fragments (15 g/l) or P. tremuloides fragments (15 g/l). Each culture was done in triplicate. Inoculums of the liquid cultivations were prepared as described in Herpoël et al.40
2.6. Integration of transcriptome and exo-proteome profiles
LC-MS/MS analysis of the secreted proteins was performed as described in Navarro et al.41 Briefly, 10 µg of diafiltered proteins were loaded on SDS-PAGE gels and allowed to migrate on a 0.5 cm length. Each lane was cut into two slices for in-gel digestion according to a standard trypsinolysis protocol. On-line analysis of the peptides was performed with a Q-exactive mass spectrometer (Thermo Fisher Scientific), using a nanoelectrospray ion source. Protein identification was performed by querying MS/MS data against the genome of P. cinnabarinus BRFM 137, P. coccineus BRFM 310 or P. sanguineus BRFM 1264, together with an in-house contaminant database, using the X! Tandem Cyclone software. All peptides that matched with an E value lower than 0.05 were parsed with X! Tandem pipeline software. Proteins identified with at least two unique peptides and a log (E value) lower than −2.6 were validated.
For total RNA extractions, mycelia were ground in liquid nitrogen using a Freezer/Mill Cryogenic Grinder (SPEX Sample Prep, UK). Total RNA was extracted from 100 mg ground tissue in 1 ml TRIZOL (Ambion). Nucleic acids were precipitated with isopropanol, resuspended in water and treated with RNase-Free DNase I (QIAGEN). Total RNA was precipitated with LiCl and resuspended in DEPC-treated water. RNA quantity and quality were determined using the Experion RNA StdSens kit (QIAGEN). The transcriptome response of P. sanguineus to pine could not be analysed because of poor quality of the extracted RNAs. Double stranded cDNAs were synthesized from PolyA RNA and fragmented (200–300 bp) before construction of the sequencing libraries (Kapa Library Amplification Kit; Kapa Biosystems). Sequencing was done on the Illumina HighSeq-2500 JGI platform generating paired end reads of 150 bp each. Paired end 150 bp Illumina reads were trimmed for quality and aligned to the corresponding genome using TopHat 2 with only unique mapping allowed.42 Gene models for which the mean raw read counts were inferior to 5 were considered as not transcribed and their read counts were changed to 0.
The counts of mapped Illumina reads from biological triplicates of each growth condition (GEO accession number GSE82486) were normalized with the DESeq2 package and log2 transformed.43 The normalized read counts of genes coding for CAZymes, peptidases, hydrophobins and SSPs from P. cinnabarinus BRFM 137, P. coccineus BRFM 310 and P. sanguineus BRFM 1264 were retrieved and combined by conducting; (i) removal of batch effects with Combat function in SVA package44 and (ii) quantile normalization with the preprocessCore package.45 We used self-organizing map (SOM) to group genes into nodes according to similar transcript levels obtained from the different substrate conditions. Self-organising maps were constructed with the R package kohonen.46 The genes showing similar transcription levels were sorted and grouped into nodes of SOMs. It was empirically found that about 35 genes in a single node of the SOM gave the best resolution of the gene clusters. In terms of the standard formula ‘X × sqrt (N)’ to calculate the number of map units, where N was the number of the rows/genes of the data, X was 1.5. The number of iterations (epochs) was 100 times more than the map units to minimize the mean distance between the weights of the neighbouring nodes. The default initialization, learning rate and radius were used. Hexagonal SOM models were constructed. The mean reads (>12 log2) of the nodes (grouped genes) with the replicates combined were calculated for each substrate.
We integrated SOM with the protein secretome information analysed by peptide-LC-MS/MS, using SOM Harboring Informative Nodes with Gene Ontology.10,47,48
2.7. Transcription regulation of co-orthologous genes
One-to-one orthologous genes from P. cinnabarinus BRFM 137, P. coccineus BRFM 310 and P. sanguineus BRFM 1264 were retrieved using OrthoFinder v. 2.3.8.49 Heatmaps were created on the log2-fold change of transcript read counts in each growth condition when compared with growth on maltose after DESeq2 normalization using the ‘Heatmap’ function from the package ‘ComplexHeatmap’ v1.10.1 in R. Pairwise comparisons of gene expression based on Pearson correlation coefficients among all replicates was performed on CAZyme, peptidase, hydrophobin and SSP co-orthologs after read count normalization by DESeq2, batch effect removal and quantile normalization ‘cor’ function in R and visualized as heatmap with R package, ggplot2.
3. Results and discussion
3.1. Four Pycnoporus species share similar genomic features and CAZomes
To assess the genomic diversity in the genus Pycnoporus, the genome of P. coccineus BRFM 310 (herein named Pycco), P. sanguineus BRFM 1264 (Pycsa) and P. puniceus BRFM 1868 (Pycpun) were sequenced and compared with that of P. cinnabarinus BRFM 137 (herein named Pycci).9 The genome size of P. coccineus, P. sanguineus and P. puniceus, ranging from 30 to 36 Mb were in line with that of P. cinnabarinus (33.67 Mb) and WR relatives (Table 1; Supplementary Fig. S1 and Table S1). We observed low amounts of repeat sequences (1.8–12.3%) and the absence of major rearrangements in the genomes (Supplementary Figs S2 and S3). The genes coding for mating type, Class II peroxidases, CAZymes, peptidases, GSTs, hydrophobins, proteins from the secretory pathway, the glycosylation pathway, the carbon catabolism pathway and SSPs were inspected by expert annotation (Supplementary Tables S2–S15 and Figs S4–S13). We observed a high proportion of conserved protein-coding genes across the genomes (82.3% of the P. cinnabarinus protein-coding genes) and a low proportion of species-specific genes (4–5%; Supplementary Fig. S14). Inspection of mating type genes showed high sequence identity between the alleles of the four Pycnoporus species (Supplementary Table S4). In particular, P. coccineus BRFM 310 and P. sanguineus BRFM 1264 alleles were much more similar to each other than to that of the two other Pycnoporus species, in support of the notion that these two species are closely related.15,50
Table 1.
Features of P. coccineus BRFM 310, P. puniceus BRFM 1868 and P. sanguineus BRFM 1264 genome assemblies and annotations
| P. cinnabarinus | P. coccineus | P. puniceus | P. sanguineus | |
|---|---|---|---|---|
| Genome size (Mbp) | 33.67 | 32.76 | 30.26 | 36.04 |
| Number of contigs | 2,036 | 469 | 105 | 2,046 |
| Number of scaffolds | 784 | 222 | 105 | 657 |
| Scaffold N50 | 54 | 20 | 12 | 35 |
| Scaffold L50 (Mbp) | 0.17 | 0.47 | 0.79 | 0.32 |
| Transposable element coverage (%) | 8.15 | 1.8 | 12.33 | 4.91 |
| Transposable element coverage (Mbp) | 2.74 | 0.59 | 3.73 | 1.77 |
| Number of predicted proteins | 10,442 | 12,690 | 10,050 | 14,165 |
| BUSCO complete protein sequences | 1,268 | 1,321 | 1,309 | 1,293 |
| BUSCO fragmented protein sequences | 32 | 7 | 14 | 21 |
| BUSCO missing protein sequences | 35 | 7 | 12 | 21 |
The reliability of gene structural annotations was assessed using universal single-copy orthologs (BUSCO). The genome of P. cinnabarinus BRFM 1379 is indicated for comparison.
The CAZyme gene repertoires (CAZome) in the three newly sequenced genomes were similar to that of P. cinnabarinus (mean 436 CAZymes classified into 108 CAZy families). The gene counts for FCW-degrading enzymes were similar to that of other Polyporales fungi (Supplementary Fig. S15). As a common feature of WR fungi,4,51Pycnoporus genomes contained a large number of genes coding for PCW-active CAZymes when compared with brown-rot fungi, which use Fenton chemistry in combination with a limited number of CAZymes for PCW breakdown (Fig. 1, Supplementary Figs S16–S18). The genomes were rich in lytic polysaccharide monooxygenases (LPMOs) active on crystalline cellulose and β-(1,4)-linked hemicellulose polysaccharides (CAZy family AA9; 13–17 genes). The Class II PODs (in total 9–11 genes) were identified as manganese peroxidases (MnP), versatile peroxidases (VP) and lignin peroxidases (LiP; Supplementary Fig. S19 and Table S5). We observed a high number of predicted secreted oxidoreductases that could act as AA enzymes for the oxidative degradation of PCWs. Among them, GMC-oxidoreductases from CAZy subfamily AA3_2 (20–22 genes) have pivotal roles in PCW degradation. Secreted AA3_2 are involved in the generation of H2O2, a co-substrate for class II peroxidases and LPMOs. AA3_2 also contribute to the oxidation of saccharides and to the redox cycling of aromatic alcohols and quinones. In addition, we identified three AA5_1 glyoxal oxidase genes in each of the genomes, which encode copper radical oxidases involved in extracellular H2O2 production (Supplementary Figs S9–S13 and Table S6).
Figure 1.

Gene counts for CAZyme domains of PCW and FCW-degrading enzymes. The bar plots show the total count of genes including PCW and FCW-degrading enzymes (left); and the ratio of PCW to FCW-degrading enzymes (right). The counts for AA enzymes that could contribute to PCW degradation include AA1_1 laccases and predicted secreted AA3, AA4 and AA5. The counts for PCW-active LPMOs include AA9, AA13, AA14 and AA16. Enzymes active on FCWs, cellulose, hemicellulose or pectin were classified according to Supplementary Figs S15–S18.
3.2. Pycnoporus species show diverse responses to lignocellulosic substrates
To assess the functional diversity within the genus Pycnoporus, we compared the ability of the phylogenetically most closely related species; P. cinnabarinus, P. coccineus and P. sanguineus to grow on a variety of plant-derived carbon sources on agar plate assays. In these conditions, we observed differences in fungal growth on complex lignocellulosic substrates (Fig. 2a and Supplementary Fig. S20). We next analysed the early response of the three species to cellulose, WS and woody substrates in agitated liquid culture media and compared their transcriptomes and the secreted proteins collected from the media. Maltose was used as a control, as it was shown not to induce carbon catabolic repression in ascomycetes.52 AVI was used as a cellulose-enriched substrate, and WS, pine and Asp were used as representatives of Gramineae, softwood and hardwood, respectively. At day 3, most cultures had initiated growth and consumed all maltose initially present in the medium. This time-point was therefore selected to analyse the early response of each species to the substrates (Fig. 3; Supplementary Figs S21 and S22).
Figure 2.
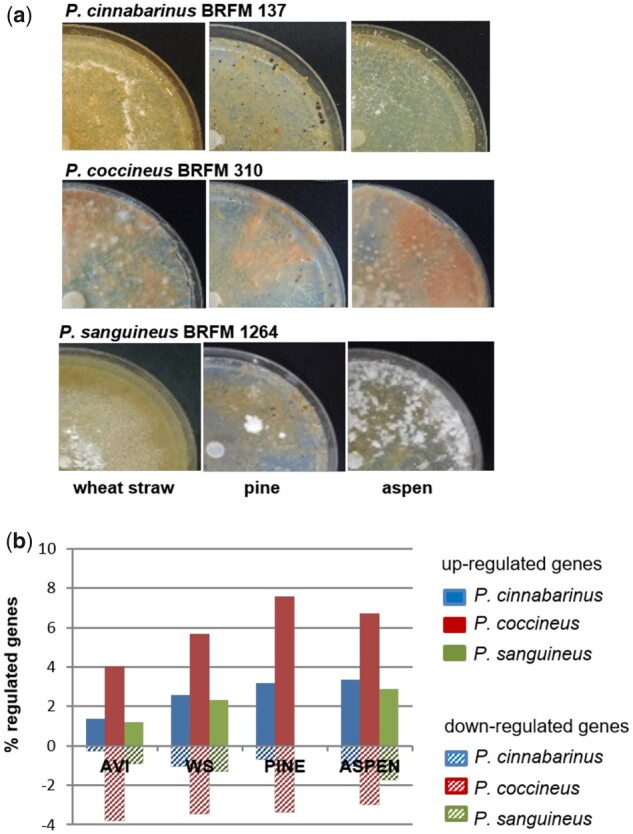
Phenotype polymorphism across three Pycnoporus species. (a) Agar plates after 6 weeks cultivation on ground WS, pine or Asp. P. cinnabarinus BRFM 137 did not develop mycelium on pine and Asp. The white dots formed by P. sanguineus on pine and Asp are arthrospores indicating that the fungus stopped growing to form dormant structures. (b) Percentage of regulated genes after 3-day growth in liquid cultures on AVI, WS, pine or Asp when compared with maltose (|fold change| ≥ 4) in the three Pycnoporus species. No RNASeq data was available for P. sanguineus grown on pine.
Figure 3.
Cross-species comparison of the early response of three Pycnoporus species to lignocellulosic carbon sources.
The global transcriptome responses varied among the species. Especially, P. coccineus showed the highest proportion of regulated genes with up to 7.6% of the genes up-regulated on pine (fold change in transcript abundance ≥4; Fig. 2b). Similarity between the transcriptomes of the three species was assessed by analysing the differential expression of one‐to‐one orthologous genes (co‐orthologs) after 3-day growth on cellulose, WS or Asp when compared with maltose. To identify co‐orthologs, in silico-deduced proteomes of the three species were clustered into 13,836 orthogroups using OrthoFinder, of which 6,524 represented co‐orthologs. Similar transcript regulations were frequent between co-orthologs of two species. Surprisingly, however, we observed poor conservation of transcript regulation of co-orthologs across the three species, including for genes with high transcription induction or repression on particular carbon sources (Fig. 4a). We examined the transcript read counts of CAZyme-coding genes. Transcriptome profiles were more similar within the species cultured under the different conditions than between the species cultured on the same substrates (Supplementary Fig. S23). Also, we observed in each species that approximately half of the regulated CAZymes (41–51%) were up-regulated in response to cellulose, WS and Asp, highlighting the presence of core regulations to (ligno)cellulosic substrates with diverse compositions (Supplementary Fig. S24).
Figure 4.

Global transcriptome similarity between co‐orthologous genes from P. cinnabarinus, P. coccineus and P. sanguineus. (a) Heatmap of changes in transcript read counts (log2 fold change) after 3-day growth on each carbon source when compared with maltose for 6,524 groups of 1-to-1 co‐ortholog genes. (b) Pearson correlation coefficient for the normalized transcript read counts in each growth condition for the 405 1-to-1 co‐ortholog CAZyme, peptidase, SSP and hydrophobin genes identified in the genomes. The comparisons of the response of each species to various substrates are highlighted in black boxes. Cross-species comparisons on a same substrate are highlighted in blue boxes. M: maltose; AVI: Avicel; WS: wheat straw; Asp: aspen.
3.3. Conserved gene regulations in response to lignocellulosic substrates
In search for conserved enzymatic mechanisms involved in the initiation of lignocellulose breakdown, we analysed the gene sets sharing similar transcription profiles at the onset of the response to cellulose, WS or Asp. We focused on genes coding for proteins typically found in fungal exo-proteomes, i.e. CAZymes, peptidases, SSPs and hydrophobins.53 A total of 2,227 manually curated genes from the genomes of P. cinnabarinus, P. coccineus and P. sanguineus were analysed. In order to combine all RNA-seq data in a single cross-species analysis, transcript read counts were normalized using the DESeq2 package,43 the normalized read counts were log2 transformed and subjected to removal of batch effects and to quantile normalization. We checked the impact of each normalization step on the distribution of the data (Supplementary Figs S25–S28). Inspecting the one-to-one co-orthologs from this set of genes (405 orthology groups), we observed low conservation of the normalized transcript read counts across the species, except for growth on maltose and cellulose, and for P. coccineus and P. sanguineus grown on Asp, indicating that complex lignocellulosic substrates induced more diverse responses across the species (Fig. 4b).
We grouped genes with similar transcript profiles into nodes (clusters of co-regulated genes) using the SOM unsupervised learning method. SOM is a data-driven clustering method constructing a topographic organization of nodes in which neighbouring nodes share similar transcriptome patterns, and thereby alleviates the requirement for arbitrary clustering thresholds. SOM allows the co-localization on the SOM map of genes with similar transcript levels. We used SOM on the normalized read counts from the three species in order to identify both the co-regulated genes within each species and the genes with similar transcript patterns across the species. We produced 72 nodes containing on average 31 genes per node (Fig. 5a and b). Exo-proteome data (Supplementary Tables S16–S18) were combined to the SOM map to integrate gene transcription and protein secretion information.
Figure 5.

Clustering of genes coding for predicted CAZymes, peptidases, SSPs and hydrophobins in three Pycnoporus species according to their transcription profile on maltose (M), AVI, WS and Asp. (a) SOM clustering resulted in 72 nodes with average 31 genes per node. Nodes containing genes highly transcribed or up-regulated on cellulose (blue), Asp (green) or WS (orange) when compared with maltose are highlighted.(b) Hierarchical clustering of the nodes according to the averaged normalized transcript read counts on each carbon source. (c) Gene content and transcript profiles of nodes 57, 39 and 58. AA8-AA3-1: cellobiose dehydrogenase.
We first examined the transcriptional response to cellulose. We identified 19 nodes containing 250 genes highly transcribed (mean normalized log2 read count ≥12) or up-regulated (mean fold change ≥4 compared with maltose; Supplementary Table S19). Inspecting the gene content for these nodes (e.g. Fig. 5c), we looked for shared gene differential expression across the three species. In the three species, we found up-regulation for CAZyme-coding genes involved in β-(1,4)-glucan linkage breakdown including endoglucanases from family 5 subfamily 5 (GH5_5), GH45 and GH131, cellobiohydrolases (GH6, GH7), cellobiose dehydrogenases (CDH) and AA9 LPMOs (Fig. 6a). Also, there was a wide panel of enzymes active on hemicelluloses from families GH10, GH74, GH5_7, GH12, GH115, CE1, CE15, CE16, enzymes active on pectin CE8, GH28 and enzymes with promiscuous activities on glycosidic bounds GH1 and GH3. Among the 20 CAZyme families identified, 14 were represented by co-orthologous genes from the 3 species, showing conservation of their regulation in response to cellulose across the genus. Globally, 50% of the enzymes encoded by the up-regulated genes were detected in the culture medium (Fig. 6a), strengthening a role for these enzymes as key players in lignocellulose breakdown by Pycnoporus fungi.
Figure 6.

Shared expression regulation of CAZyme genes across the three Pycnoporus species. (a) Numbers of genes with shared differential expression on cellulose and numbers of proteins secreted during growth on cellulose. (b) Numbers of genes with shared specific differential expression on lignocellulosic substrates, not on cellulose. The numbers of orthologous groups of genes with conserved transcription regulation and secretion across the three species are indicated.
To identify the genes specifically regulated for the complex lignocellulosic substrates, we selected genes up-regulated on WS or Asp, not on cellulose, when compared with maltose. There were 13 nodes including 225 genes that met at least one of the following criteria; (i) average normalized log2 read counts >12 on WS or Asp and <12 on maltose and cellulose, or (ii) mean fold change ≥4 on WS or Asp, and ≤4 on cellulose when compared with maltose. From these nodes, we identified the gene families with homologs among the three species. These included genes coding for enzymes that target hemicelluloses (CE16 and CE4 acetylesterases, GH2 β-mannosidases or β-glucuronidases, GH16 xyloglucan hydrolase, GH27 α-galactosidases, GH30 β-glucosidase/β-xylosidase, GH51 α-l-arabinofuranosidases and GH31 α-xylosidases/α-glucosidases), enzymes that target pectin (GH28 polygalacturonases, PL14_4 β-1,4-glucuronan lyases), AA1_1 laccases and AA3_2 GMC-oxidoreductases (Fig. 6b, Supplementary Table S20). Genes coding for glycoside hydrolases active on the FCW (GH18 chitinases, GH76 α-mannanases) and peptidases were also specifically up-regulated in the three species.
The genomes of the three species encoded the three types of class II peroxidases involved in lignin breakdown; MnP, LiP and VP (Supplementary Table S5). Orthologous MnP and LiP genes (defined by protein sequence phylogeny; Supplementary Fig. S6) did not show conserved regulation across the species in response to lignocellulosic substrates (Fig. 7). The highest induction factors were found in P. coccineus and a MnP (protein ID #1468611)- and a LiP (#1431101)-coding gene reached 800- and 1,500-fold induction on Asp, respectively. For the three species, we observed no up-regulation of the VP-coding genes.
Figure 7.

Regulation of Class II peroxidase genes in response to the substrates. Orthologous genes are grouped for comparison of their transcription profiles.
3.4. Co-regulated genes indicative of enzymatic synergies
We investigated potential enzymatic synergies conserved in the three Pycnoporus species through co-regulated gene transcription with co-secreted corresponding proteins. In each species, one single CDH gene was present in the genome, which shared a similar transcription pattern with AA9 LPMO-coding genes (nodes 39, 57 and 58; Figs 5c and 8). These AA9 LPMO and CDH genes were up-regulated on cellulose and the corresponding proteins were identified in the exo-proteomes. AA9 LPMOs oxidatively cleave glycosidic chains at the cellulose surface thereby creating new substrate-binding sites for hydrolytic cellulases.54 CDHs are composed of a flavin adenine dinucleotide-binding dehydrogenase domain (AA3_1) connected via a flexible linker to a haem b-binding cytochrome domain55 (Cytb; AA8). In vitro, CDHs behave as electron donors for AA9 LPMOs and boost LPMO activity (reviewed in Berrin et al.56). Our results suggest CDH as a biologically relevant electron donor for AA9 LPMOs.
Figure 8.
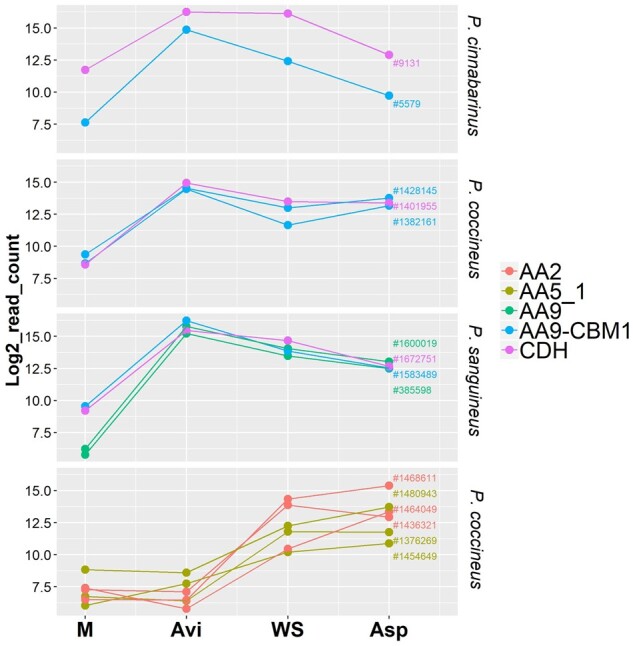
Conserved co-regulations of CDH and AA9 LPMO genes in P. cinnabarinus, P. coccineus and P. sanguineus and co-regulations of MnP- and GLOX-genes in P. coccineus. Transcript levels are expressed as log2-transformed read counts after 3-day-growth on maltose (M), AVI, WS or Asp. CBM: carbohydrate-binding module.
As indicated above, genes coding for predicted class II peroxidases (AA2s) were specifically up-regulated on WS or Asp, but not on cellulose (Fig. 5b). Node 40, 41 and 32 contained three mnp genes from P. coccineus co-regulated with the three predicted glyoxal oxidase genes identified in this genome, suggesting that glyoxal oxidases might provide these MnPs with the H2O2 required for their activity (Fig. 6). In contrast in P. cinnabarinus and P. sanguineus, we did not observe any co-regulations at the transcript level between class II peroxidases and glyoxal oxidases.
3.5. Differentially regulated genes indicative of detoxification
In natural environments, lignocellulose degradation may lead to the release of toxic degradation products and extractives (e.g. phenols, terpenoids and tannins).57,58 We investigated if the regulation of genes involved in detoxification was conserved across the three Pycnoporus species in our growth conditions. For this purpose, we examined genes coding for putative GST in the cross-species SOM analysis.
We identified a GST gene from class GTT2 in P. sanguineus that was clustered in node 31 filtered for high transcript level on WS and Asp, and one GTT2 gene from P. cinnabarinus that was clustered in node 22 filtered for up-regulation on WS (Supplementary Table S20). One isoform of this class from Phanerochaete chrysosporium has been functionally characterized and has been shown to reduce high-molecular weight peroxides.59,60 In contrast, a total of four GST genes of P. coccineus were regulated on the tested substrates; two GST class A (FuA) genes were up-regulated on all the tested substrates (Supplementary Table S19) whereas one S-glutathionyl hydroquinone reductase and one GST Omega were specifically up-regulated in response to WS or Asp (Supplementary Table S20). For the case of P. chrysosporium, these classes of GSTs have potential roles in the detoxification of wood-derived molecules acting as ligandins for wood extractives or as catalysts for deglutathionylation of various substrates, including hydroquinone conjugates and terpenes.36,59,61 These findings suggest that different GST-related detoxification systems may exist in P. coccineus.
Cytochrome P450 (CYP450) monooxygenases are commonly involved in the first step of eliminating toxic molecules including molecules released from lignocellulose. CYP450 can initiate the modification of these molecules via hydroxylation, epoxidation or monooxygenation.62,63 The genomes of P. cinnabarinus, P. coccineus and P. sanguineus contain 107, 132 and 113 predicted CYP450, respectively. Inspection of the transcript profiles for these genes showed that P. coccineus had the highest number of up-regulated CYP450 genes in response to the tested substrates (17 genes) compared with P. cinnabarinus and P. sanguineus (14 and 6 up-regulated genes, respectively). Some CYP450 genes were commonly up-regulated in at least two species, which were from families CYP63, CYP5035, CYP5139, CYP5144 and CYP5150 According to the Fungal cytochrome P450 database64 (Supplementary Table S13). Of these, CYP63, CYP5139 and CYP5144 were shown to be active on multiple xenobiotic compounds such as polycyclic aromatic hydrocarbons, alkylphenols and alkanes.63,65,66 CYP5035s oxidize plant chemicals (i.e. resin and flavonoids), and the number of genes for CYP5035 and CYP5150 expanded in basidiomycetes that grow on wood or litter.63 Our results suggest that these CYP450 families could be differently utilized by the species for detoxification of molecules released from the lignocellulosic substrates.
4. Discussion
4.1. Phenotype diversity outreaches diversity of the predicted proteome across the genus Pycnoporus
The four Pycnoporus species studied here are found in different geo-climatic areas15,16 covering the Northern hemisphere (P. cinnabarinus), countries bordering the Indian and Pacific Oceans (P. coccineus), the tropics and subtropics of both hemispheres (P. sanguineus) and paleotropical areas (P. puniceus), with overlap limited to Japan and Eastern China. Accordingly, the parental strains of the monokaryons studied here were collected in Europe, China, South America and Indonesia, respectively. Here, we show that the genomes sequenced from geographically distant strains did not show any major rearrangements in structure and protein-coding gene composition. Our findings suggest that geographical isolation of the species the genus Pycnoporus has not yet lead to any noticeable genome level diversity. In addition, the high sequence identity between mating type genes of the four species, particularly between P. coccineus and P. sanguineus suggests that the speciation events are recent in this genus. In the genus Laetiporus, another group of wood-decay fungi from the order Polyporales that originated about 20 mya,67 several regional speciation events have been estimated between 9.88 and 2.89 mya. Some of these speciation events have been related to vicariance that emerged due to tectonic activity and the disconnection of the continental plates or from wind and ocean current dispersion of basidiospores.67 It is currently estimated that the genus Pycnoporus originated about 40–50 mya (Nagy LG, personal communication). Further molecular studies are needed to understand the dispersal routes and the role of regional speciation in the evolutionary history of Pycnoporus.
Despite conserved CAZomes, the P. cinnabarinus, P. coccineus and P. sanguineus strains analysed here showed different abilities to grow on (ligno)cellulosic substrates. This finding demonstrates that gene repertoires obtained from genome sequencing are not sufficient to predict the ability of a strain to grow on recalcitrant raw biomass. The comparison of the early responses of the fungi to the substrates showed that gene transcription profiles better correlated with the strains rather than with the substrates. A similar trend was observed in the WR fungus Pleurotus ostreatus, between monokaryotic strains issued from a same parental dikaryotic strain. In P. ostreatus, these differences in gene regulation have been partly attributed to the presence of transposable elements near the differentially regulated genes.68 A closer analysis of transposable element localization or methylation status of each monokaryon haplotype would help understanding the transcription regulation we observed.
4.2. Conserved sets of enzymes produced at the onset of wood-decay activity
As a conserved response to cellulose across three Pycnoporus species, we identified here a set of CAZyme genes with shared transcriptional regulations. For a sub-set of these genes, the corresponding enzymes were commonly found in the secretomes of the three species at the analysed time point, further strengthening a role for these enzymes as key players in lignocellulose breakdown by these fungi. These included genes coding for enzymes active on β-1,4-glucan linkages (GH5_5 and GH131 endo-β-1,4-glucanases, GH6 and GH7 cellobiohydrolases and AA9 LPMOs) and genes targeting the hemicellulose backbone (GH10 endoxylanases, GH12 and GH74 xyloglucan hydrolases, GH5_7 β-mannosidases) or branched groups (CE1 feruloyl esterases) and enzymes active on pectin (GH28 polygalacturonases). In the presence of lignocellulosic substrates, additional genes were commonly induced and the corresponding CAZymes secreted, that target hemicelluloses (GH16 xyloglucan hydrolase, GH27 β-galactosidases, GH30 β-glucosidases/β-xylosidases, GH51 α-l-arabinofuranosidases), peptidases and laccases (AA1_1).
4.3. AA9 LPMOs and their enzymatic partners
AA9 LPMOs are known as key players in oxidative cellulose depolymerization by wood-decay fungi.56 Expansions in AA9 gene numbers is a common feature of the genomes of WR fungi,69 but whether members of the AA9 gene family play different roles during wood decay has not been elucidated. We identified 17, 16 and 11 genes coding for AA9 LPMOs in P. cinnabarinus, P. coccineus and P. sanguineus, from which 11 (64%), 13 (81%) and 10 (90%), respectively, were up-regulated during the early response to cellulose-containing substrates. In each strain, several AA9 LPMO genes shared similar transcription profiles, and several AA9 LPMOs were concomitantly secreted, indicating that several AA9 LPMOs co-secreted by the fungi might act simultaneously on the substrate.
The identification of enzymatic partners for LPMOs is a very active field of research.70–73 Looking at conserved co-regulations across the three strains, we identified CDHs as invariably co-regulated with at least one AA9 LPMO gene. CDHs have been shown to reduce O2 and generate H2O2 which in turn can fuel, via the Fenton reaction, the production of hydroxyl radicals that disrupt lignocellulose polymers.74 More recently, CDHs have been shown to activate AA9 LPMOs in vitro through electron transfer75 and it has been proposed that CDHs could also activate AA9 LPMOs through the generation of H2O2, a co-substrate for the enzyme.76 Our findings support the hypothesis of synergy between AA9 LPMOs and CDHs in vivo as a conserved mechanism for cellulose degradation in the genus Pycnoporus spp.
4.4. Co-secretion of extracellular H2O2-generating and -consuming enzymes
Extracellular H2O2 is a central factor in oxidative lignocellulose breakdown. It was proposed that WR fungi could avoid oxidative damages due to H2O2 accumulation in the vicinity of the hyphae by the co-secretion of H2O2-generating and H2O2-consuming enzymatic partners.72 Some secreted GMC-oxidoreductases such as glucose dehydrogenases and aryl-alcohol quinone oxidoreductases (AA3_2), which generate H2O2, are able to prime AA9 LPMO activity in vitro.70,73 Similarly, glyoxal oxidases can generate H2O2 and prime MnP and LiP activity in vitro.77 A biological relevance for these synergetic activities was previously suggested from the co-occurrence in fungal secretomes of class II peroxidases with GMC-oxidoreductases10,51,78–82 or glyoxal oxidases.10,51,79,81,83–85 Here, we confirmed the co-occurrence in the secretomes of GMC-oxidoreductases and glyoxal oxidases with class II peroxidases and AA9 LPMOs as a common feature of three Pycnoporus species. However, we detected no evidence for co-regulation of the corresponding genes at the transcript level, except between three MnPs and two glyoxal oxidase encoding genes in P. coccineus. We hypothesize that independent tight regulations of gene expression for H2O2-generating and H2O2-consuming enzymes at the transcription level could allow a more flexible system to rapidly adapt extracellular reactive oxygen species concentrations and avoid oxidative damage to the hyphae.
5. Conclusion
Although WR fungi are well-studied for the diversity of enzymatic systems for lignocellulose breakdown, their intra-genus diversity has not been extensively explored so far. Here, we investigated the functional diversity within the genus Pycnoporus, a group of fungi from the Trametes clade renowned for the ecological significance and a great potential for novel biocatalysts. We introduced a new method for cross-species comparisons of transcriptomes, using the genome sequences of the four species described in the genus and expert gene functional annotations. The transcriptomic aspect of observations was validated with experimentally identified secreted proteins, supporting our hypotheses. We identified a conserved set of genes responsive to lignocellulosic substrates that outlines key enzymatic mechanics for wood decomposition activity in these fungi. We observed that the mechanisms involved in oxidative lignin breakdown were more diverse than those in polysaccharide breakdown and the fungal enzymes detoxifying lignocellulose-released products were different among the species.
Supplementary Material
Acknowledgements
We are grateful to Valentin Loux from the INRA MIGALE bioinformatics platform (http://migale.jouy.inra.fr) and to Estelle Bonnin (INRA, France) for substrate composition analysis. Magali Lescot (MIO, France) and Oswaldo Forey (INRA) provided fruitful advises for comparative genomics and graphical output, respectively. Miaomiao Zhou (Westerdijk Fungal Biodiversity Institute) provided support for gene expert annotation.
Accession numbers
NCSW00000000, VOCM00000000, VICQ00000000, GSE82486.
Funding
This work was supported by The French National Agency for Research (ANR-14-CE06-0020). H.H. was granted by the Institut national de recherche pour l’agriculture, l’alimentation et l’environnement and Région SUD Provence-Alpes-Côte d’Azur. The work conducted by the U.S. Department of Energy (DOE) Joint Genome Institute, a DOE Office of Science User Facility, is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. The work by the Consejo Superior de Investigaciones Científicas was supported by the Ministerio de Ciencias, Innovación y Universidades de España (BIO2017-86559-R).
Conflict of interest
None declared.
References
- 1. Rajala T., Peltoniemi M., Pennanen T., Mäkipä R.. 2012, Fungal community dynamics in relation to substrate quality of decaying Norway spruce (Picea abies [L.] Karst.) logs in boreal forests, FEMS Microbiol. Ecol., 81, 494–505. [DOI] [PubMed] [Google Scholar]
- 2. Valentín L., Rajala T., Peltoniemi M., Heinonsalo J., Pennanen T., Mäkipää R.. 2014, Loss of diversity in wood-inhabiting fungal communities affects decomposition activity in Norway spruce wood, Front. Microbiol., 5, 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Blanchette R.A. 1991, Delignification by wood-decay fungi, Annu. Rev. Phytopathol., 29, 381–403. [Google Scholar]
- 4. Riley R., Salamov A.A., Brown D.W., et al. 2014, Extensive sampling of basidiomycete genomes demonstrates inadequacy of the white-rot/brown-rot paradigm for wood decay fungi, Proc. Natl. Acad. Sci. U. S. A., 111, 9923–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Couturier M., Ladevèze S., Sulzenbacher G., et al. 2018, Lytic xylan oxidases from wood-decay fungi unlock biomass degradation, Nat. Chem. Biol., 14, 306–10. [DOI] [PubMed] [Google Scholar]
- 6. Fernandez-Fueyo E., Ruiz-Dueñas F.J., Ferreira P., et al. 2012, Comparative genomics of Ceriporiopsis subvermispora and Phanerochaete chrysosporium provide insight into selective ligninolysis, Proc. Natl. Acad. Sci. U. S. A., 109, 5458–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kuuskeri J., Mäkelä M.R., Isotalo J., Oksanen I., Lundell T.. 2015, Lignocellulose-converting enzyme activity profiles correlate with molecular systematics and phylogeny grouping in the incoherent genus Phlebia (Polyporales, Basidiomycota), BMC Microbiol., 15, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hastrup A.C.S., Howell C., Larsen F.H., Sathitsuksanoh N., Goodell B., Jellison J.. 2012, Differences in crystalline cellulose modification due to degradation by brown and white rot fungi, Fungal Biol., 116, 1052–63. [DOI] [PubMed] [Google Scholar]
- 9. Levasseur A., Lomascolo A., Chabrol O., et al. 2014, The genome of the white-rot fungus Pycnoporus cinnabarinus: a basidiomycete model with a versatile arsenal for lignocellulosic biomass breakdown, BMC Genomics, 15, 486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Miyauchi S., Rancon A., Drula E., et al. 2018, Integrative visual omics of the white-rot fungus Polyporus brumalis exposes the biotechnological potential of its oxidative enzymes for delignifying raw plant biomass, Biotechnol. Biofuels, 11, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ohm R.A., Riley R., Salamov A., Min B., Choi I.-G., Grigoriev I.V.. 2014, Genomics of wood-degrading fungi, Fungal Genet. Biol., 72, 82–90. [DOI] [PubMed] [Google Scholar]
- 12. Suzuki H., MacDonald J., Syed K., et al. 2012, Comparative genomics of the white-rot fungi, Phanerochaete carnosa and P. chrysosporium, to elucidate the genetic basis of the distinct wood types they colonize, BMC Genomics, 13, 444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dai Y., Li X., Song B., et al. 2019, Genomic analyses provide insights into the evolutionary history and genetic diversity of Auricularia species, Front. Microbiol., 10, 2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lomascolo A., Cayol J.-L., Roche M., et al. 2002, Molecular clustering of Pycnoporus strains from various geographic origins and isolation of monokaryotic strains for laccase hyperproduction, Mycol. Res., 106, 1193–203. [Google Scholar]
- 15. Nobles M.K., Frew B.P.. 1962, Studies in wood-inhabiting Hymenomycetes V. The genus Pycnoporus Karst, Can. J. Bot., 40, 987–1016. [Google Scholar]
- 16. Ryvarden L., Johansen I.. 1980, A Preliminary Polypore Flora of East Africa. Oslo: Fungiflora. [Google Scholar]
- 17. Justo A., Hibbett D.S.. 2011, Phylogenetic classification of Trametes (Basidiomycota, Polyporales) based on a five-marker dataset, Taxon, 60, 1567–83. [Google Scholar]
- 18. Gnerre S., MacCallum I., Przybylski D., et al. 2011, High-quality draft assemblies of mammalian genomes from massively parallel sequence data, Proc. Natl. Acad. Sci. U. S. A., 108, 1513–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miller J.R., Delcher A.L., Koren S., et al. 2008, Aggressive assembly of pyrosequencing reads with mates, Bioinformatics, 24, 2818–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ka-Kit L., LaButti K., Khalak A., Tse D.. 2015, FinisherSC: a repeat-aware tool for upgrading de novo assembly using long reads, Bioinformatics, 31, 3207–9. [DOI] [PubMed] [Google Scholar]
- 21. Grigoriev I.V., Nikitin R., Haridas S., et al. 2014, MycoCosm portal: gearing up for 1000 fungal genomes, Nucleic Acids Res., 42, D699–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wattam A.R., Abraham D., Dalay O., et al. 2014, PATRIC, the bacterial bioinformatics database and analysis resource, Nucleic Acids Res., 42, D581–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Katoh K., Standley D.M.. 2013, MAFFT multiple sequence alignment software version 7: improvements in performance and usability, Mol. Biol. Evol., 30, 772–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Castresana J. 2000, Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis, Mol. Biol. Evol., 17, 540–52. [DOI] [PubMed] [Google Scholar]
- 25. Stamatakis A. 2014, RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies, Bioinformatics, 30, 1312–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sima F.A., Waterhouse R.M., Ioannidis P., Kriventseva E.V., Zdobnov E.M.. 2015, Genome analysis BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs, Bioinformatics, 31, 3210–2. [DOI] [PubMed] [Google Scholar]
- 27. Morin E., Miyauchi S., San Clemente H., et al. 2019, Comparative genomics of Rhizophagus irregularis, R. cerebriforme, R. diaphanus and Gigaspora rosea highlights specific genetic features in Glomeromycotina, New Phytol., 222, 1584–98. [DOI] [PubMed] [Google Scholar]
- 28. Miyauchi S. 2020, Visual data pipeline: Proteomic Information Navigated Genomic Outlook (PRINGO). https://github.com/ShingoMiyauchi/PRINGO.
- 29. Auguie B. 2017, Overview of the egg package. https://cran.r-project.org/web/packages/egg/index.html.
- 30. Yu G., Smith D.K., Zhu H., Guan Y., Lam T.T.-Y.. 2017, ggtree: an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data, Methods Ecol. Evol., 8, 28–36. [Google Scholar]
- 31. Wickham H. 2009, ggplot2: Elegant Graphics for Data Analysis. New York: Springer. [Google Scholar]
- 32. Kues U., Nelson D.R., Liu C., et al. 2015, Genome analysis of medicinal Ganoderma spp. with plant-pathogenic and saprotrophic life-styles, Phytochemistry, 114, 18–37. [DOI] [PubMed] [Google Scholar]
- 33. Lombard V., Golaconda Ramulu H., Drula E., Coutinho P.M., Henrissat B.. 2014, The carbohydrate-active enzymes database (CAZy) in 2013, Nucleic Acids Res., 42, D490–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mitchell A., Chang H.-Y., Daugherty L., et al. 2015, The InterPro protein families database: the classification resource after 15 years, Nucleic Acids Res., 43, D213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rawlings N.D., Barrett A.J., Finn R.. 2016, Twenty years of the MEROPS database of proteolytic enzymes, their substrates and inhibitors, Nucleic Acids Res., 44, D343–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Meux E., Morel M., Lamant T., et al. 2013, New substrates and activity of Phanerochaete chrysosporium Omega glutathione transferases, Biochimie, 95, 336–46. [DOI] [PubMed] [Google Scholar]
- 37. Mathieu Y., Prosper P., Favier F., et al. 2013, Diversification of fungal specific Class A glutathione transferases in saprotrophic fungi, PLoS One, 8, e80298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Roret T., Thuillier A., Favier F., Gelhaye E., Didierjean C., Morel-Rouhier M.. 2015, Evolutionary divergence of Ure2pA glutathione transferases in wood degrading fungi, Fungal Genet. Biol., 83, 103–12. [DOI] [PubMed] [Google Scholar]
- 39. Pellegrin C., Morin E., Martin F.M., Veneault-Fourrey C.. 2015, Comparative analysis of secretomes from ectomycorrhizal fungi with an emphasis on small-secreted proteins, Front. Microbiol., 6, 1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Herpoël I., Moukha S., Lesage-Meessen L., Sigoillot J.-C., Asther M.. 2000, Selection of Pycnoporus cinnabarinus strains for laccase production, FEMS Microbiol. Lett., 183, 301–6. [DOI] [PubMed] [Google Scholar]
- 41. Navarro D., Rosso M.-N., Haon M., et al. 2014, Fast solubilization of recalcitrant cellulosic biomass by the basidiomycete fungus Laetisaria arvalis involves successive secretion of oxidative and hydrolytic enzymes, Biotechnol. Biofuels, 7, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L.. 2013, TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions, Genome Biol., 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Love M.I., Huber W., Anders S.. 2014, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2, Genome Biol., 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Leek J.T., Johnson W.E., Parker H.S., Jaffe A.E., Storey J.D.. 2012, The SVA package for removing batch effects and other unwanted variation in high-throughput experiments, Bioinformatics, 28, 882–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bolstad B. 2019, preprocessCore: a collection of pre-processing functions. R package version 1.46.0. https://github.com/bmbolstad/preprocessCore.
- 46. Wehrens R., Buydens L.M.C.. 2007, Self- and super-organizing maps in R: the Kohonen Package, J. Stat. Softw., 21, 1–19. [Google Scholar]
- 47. Miyauchi S., Navarro D., Grigoriev I.V., et al. 2016, Visual comparative omics of fungi for plant biomass deconstruction, Front. Microbiol., 7, 1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Miyauchi S., Navarro D., Grisel S., Chevret D., Berrin J.-G., Rosso M.-N.. 2017, The integrative omics of white-rot fungus Pycnoporus coccineus reveals co-regulated CAZymes for orchestrated lignocellulose breakdown, PLoS One, 12, e0175528–e0175528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Emms D.M., Kelly S.. 2019, OrthoFinder: phylogenetic orthology inference for comparative genomics, Genome Biol., 20, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lesage-Meessen L., Haon M., Uzan E., et al. 2011, Phylogeographic relationships in the polypore fungus Pycnoporus inferred from molecular data, FEMS Microbiol. Lett., 325, 37–48. [DOI] [PubMed] [Google Scholar]
- 51. Hori C., Gaskell J., Igarashi K., et al. 2014, Temporal alterations in the secretome of the selective ligninolytic fungus Ceriporiopsis subvermispora during growth on aspen wood reveal this organism’s strategy for degrading lignocellulose, Appl. Environ. Microbiol., 80, 2062–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Brown N.A., Ries L.N.A., Goldman G.H.. 2014, How nutritional status signalling coordinates metabolism and lignocellulolytic enzyme secretion, Fungal Genet. Biol., 72, 48–63. [DOI] [PubMed] [Google Scholar]
- 53. Alfaro M., Oguiza J.A., Ramírez L., Pisabarro A.G.. 2014, Comparative analysis of secretomes in basidiomycete fungi, J. Proteomics, 102, 28–43. [DOI] [PubMed] [Google Scholar]
- 54. Tandrup T., Frandsen K.E.H., Johansen K.S., Berrin J.-G., Lo Leggio L.. 2018, Recent insights into lytic polysaccharide monooxygenases (LPMOs), Biochem. Soc. Trans., 46, 1431–47. [DOI] [PubMed] [Google Scholar]
- 55. Henriksson G., Johansson G., Pettersson G.. 2000, A critical review of cellobiose dehydrogenases, J. Biotechnol., 78, 93–113. [DOI] [PubMed] [Google Scholar]
- 56. Berrin J.-G., Rosso M.-N., Abou Hachem M.. 2017, Fungal secretomics to probe the biological functions of lytic polysaccharide monooxygenases, Carbohydr. Res., 448, 155–60. [DOI] [PubMed] [Google Scholar]
- 57. Mäkelä M.R., Marinović M., Nousiainen P., et al. 2014, Aromatic metabolism of filamentous fungi in relation to the presence of aromatic compounds in plant biomass, Adv. Appl. Microbiol .,91, 63–137. [DOI] [PubMed] [Google Scholar]
- 58. Fernández-González A.J., Valette N., Kohler A., et al. 2018, Oak extractive-induced stress reveals the involvement of new enzymes in the early detoxification response of Phanerochaete chrysosporium, Environ. Microbiol., 20, 3890–901. [DOI] [PubMed] [Google Scholar]
- 59. Morel M., Meux E., Mathieu Y., et al. 2013, Xenomic networks variability and adaptation traits in wood decaying fungi, Microb. Biotechnol., 6, 248–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Thuillier A., Chibani K., Belli G., et al. 2014, Transcriptomic responses of Phanerochaete chrysosporium to oak acetonic extracts: focus on a new glutathione transferase, Appl. Environ. Microbiol., 80, 6316–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mathieu Y., Prosper P., Buée M., et al. 2012, Characterization of a Phanerochaete chrysosporium glutathione transferase reveals a novel structural and functional class with ligandin properties, J. Biol. Chem., 287, 39001–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Doddapaneni H., Yadav J.S.. 2005, Microarray-based global differential expression profiling of P450 monooxygenases and regulatory proteins for signal transduction pathways in the white rot fungus Phanerochaete chrysosporium, Mol. Genet. Genomics, 274, 454–66. [DOI] [PubMed] [Google Scholar]
- 63. Syed K., Shale K., Pagadala N.S., Tuszynski J.. 2014, Systematic identification and evolutionary analysis of catalytically versatile cytochrome p450 monooxygenase families enriched in model basidiomycete fungi, PLoS One, 9, e86683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Moktali V., Park J., Fedorova-Abrams N.D., et al. 2012, Systematic and searchable classification of cytochrome P450 proteins encoded by fungal and oomycete genomes, BMC Genomics, 13, 525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Qhanya L.B., Matowane G., Chen W., et al. 2015, Genome-wide annotation and comparative analysis of cytochrome P450 monooxygenases in Basidiomycete biotrophic plant pathogens, PLoS One, 10, e0142100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ichinose H. 2013, Cytochrome P450 of wood-rotting basidiomycetes and biotechnological applications, Biotechnol. Appl. Biochem., 60, 71–81. [DOI] [PubMed] [Google Scholar]
- 67. Song J., Cui B.-K.. 2017, Phylogeny, divergence time and historical biogeography of Laetiporus (Basidiomycota, Polyporales), BMC Evol. Biol., 17, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Castanera R., López-Varas L., Borgognone A., et al. 2016, Transposable elements versus the fungal genome: impact on whole-genome architecture and transcriptional profiles, PLoS Genet., 12, e1006108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Nagy L.G., Riley R., Bergmann P.J., et al. 2017, Genetic bases of fungal white rot wood decay predicted by phylogenomic analysis of correlated gene-phenotype evolution, Mol. Biol. Evol., 34, 35–44. [DOI] [PubMed] [Google Scholar]
- 70. Garajova S., Mathieu Y., Beccia M.R., et al. 2016, Single-domain flavoenzymes trigger lytic polysaccharide monooxygenases for oxidative degradation of cellulose, Sci. Rep., 6, 28276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kracher D., Scheiblbrandner S., Felice A.K.G., et al. 2016, Extracellular electron transfer systems fuel cellulose oxidative degradation, Science, 352, 1098–101. [DOI] [PubMed] [Google Scholar]
- 72. Bissaro B., Várnai A., Røhr Å.K., Eijsink V.G.H.. 2018, Oxidoreductases and reactive oxygen species in conversion of lignocellulosic biomass, Microbiol. Mol. Biol. Rev., 82, e00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sützl L., Laurent C.V.F.P., Abrera A.T., Schütz G., Ludwig R., Haltrich D.. 2018, Multiplicity of enzymatic functions in the CAZy AA3 family, Appl. Microbiol. Biotechnol., 102, 2477–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hyde S.M., Wood P.M.. 1997, A Mechanism for production of hydroxyl radicals by the brown-rot fungus Coniophora puteana: Fe(III) reduction by cellobiose dehydrogenase and Fe(II) oxidation at a distance from the hyphae, Microbiology, 143, 259–66. [DOI] [PubMed] [Google Scholar]
- 75. Tan T.-C., Kracher D., Gandini R., et al. 2015, Structural basis for cellobiose dehydrogenase action during oxidative cellulose degradation, Nat. Commun., 6, 7542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bissaro B., Røhr Å.K., Müller G., et al. 2017, Oxidative cleavage of polysaccharides by monocopper enzymes depends on H2O2, Nat. Chem. Biol., 13, 1123–8. [DOI] [PubMed] [Google Scholar]
- 77. Kersten P., Cullen D.. 2014, Copper radical oxidases and related extracellular oxidoreductases of wood-decay Agaricomycetes, Fungal Genet. Biol., 72, 124–30. [DOI] [PubMed] [Google Scholar]
- 78. Fernández-Fueyo E., Ruiz-Dueñas F.J., Miki Y., Martínez M.J., Hammel K.E., Martínez A.T.. 2012, Lignin-degrading peroxidases from genome of selective ligninolytic fungus Ceriporiopsis subvermispora, J. Biol. Chem., 287, 16903–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Salvachúa D., Martínez A.T., Tien M., et al. 2013, Differential proteomic analysis of the secretome of Irpex lacteus and other white-rot fungi during wheat straw pretreatment, Biotechnol. Biofuels, 6, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Fernández-Fueyo E., Ruiz-Dueñas F.J., López-Lucendo M.F., et al. 2016, A secretomic view of woody and nonwoody lignocellulose degradation by Pleurotus ostreatus, Biotechnol. Biofuels, 9, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kuuskeri J., Häkkinen M., Laine P., et al. 2016, Time-scale dynamics of proteome and transcriptome of the white-rot fungus Phlebia radiata: growth on spruce wood and decay effect on lignocellulose, Biotechnol. Biofuels, 9, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Moody S.C., Dudley E., Hiscox J., Boddy L., Eastwood D.C.. 2018, Interdependence of primary metabolism and xenobiotic mitigation characterizes the proteome of Bjerkandera adusta during wood decomposition, Appl. Environ. Microbiol., 84, e01401–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kersten P.J. 1990, Glyoxal oxidase of Phanerochaete chrysosporium: its characterization and activation by lignin peroxidase, Proc. Natl. Acad. Sci. U. S. A., 87, 2936–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Martinez D., Challacombe J., Morgenstern I., et al. 2009, Genome, transcriptome, and secretome analysis of wood decay fungus Postia placenta supports unique mechanisms of lignocellulose conversion, Proc. Natl. Acad. Sci. U. S. A., 106, 1954–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Daou M., Faulds C.B.. 2017, Glyoxal oxidases: their nature and properties, World J. Microbiol. Biotechnol., 33, 87. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.