

Abstract
In the PNS, myelination occurs postnatally when Schwann cells (SCs) contact axons. Axonal factors, such as Neuregulin-1 Type III, trigger promyelinating signals that upregulate myelin genes. Neuregulin-1 Type III has been proposed to activate calcineurin signaling in immature SCs to initiate differentiation and myelination. However, little is known about the role of calcineurin in promyelinating SCs after birth. By creating a SC conditional KO of calcineurin B (CnBscko), we assessed the effects of CnB ablation on peripheral myelination after birth in both male and female mice. Surprisingly, CnBscko mice have minimal myelination defects, no alteration of myelin thickness, and normal KROX20 expression. In contrast, we did find a unique role for calcineurin in SCs after nerve injury. Following nerve crush, CnBscko mice have slower degeneration of myelin compared with WT mice. Furthermore, absence of CnB in primary SCs delays clearance of myelin debris. SCs clear myelin via autophagy and recent literature has demonstrated that calcineurin can regulate autophagy via dephosphorylation of transcription factor EB (TFEB), a master regulator of lysosomal biogenesis and autophagy. We demonstrate that loss of CnB reduces autophagic flux in primary SCs, indicating a possible mechanism for impaired myelin clearance. In addition, ablation of CnB impairs TFEB translocation to the nucleus 3 d after crush, suggesting that calcineurin may regulate autophagy in SCs via TFEB activation. Together, our data indicate that calcineurin is not essential for myelination but has a novel role in myelin clearance after injury.
SIGNIFICANCE STATEMENT Our data offer a novel mechanism for activation of autophagy after peripheral nerve injury. Efficient clearance of myelin after injury by Schwann cells is important for axonal regrowth and remyelination, which is one reason why the PNS is significantly better at recovery compared with the CNS. Improved understanding of myelin clearance allows for the identification of pathways that are potentially accessible to increase myelin clearance and improve remyelination and recovery. Finally, this paper clarifies the role of calcineurin in Schwann cells and myelination.
Keywords: autophagy, calcineurin, myelin, nerve injury, nerve regeneration, Schwann cell
Introduction
In the PNS, neural crest derivatives called Schwann cells (SCs) wrap axons in concentric lipid membrane bilayers to form myelin. Myelin acts as insulation for propagation of the electrical signal along the axon. Development of peripheral nerves is dependent on signaling between SCs and axons. Early SCs, called SC precursors (SCPs), provide trophic support to axons while SCPs require signaling from axons, most importantly neuregulin-1 Type III (NRG1), to survive, differentiate into promyelinating SCs, and later produce myelin (Jessen et al., 1994; Morris et al., 1999; Taveggia et al., 2005). NRG1 binds the ErbB2/3 receptors on SCs and activates downstream promyelinating pathways, including the Ras/extracellular signal regulated kinase 1/2 (ERK1/2) and phosphotidylinositol-3-kinase (PI3K)/AKT pathways. Manipulation of these pathways can alter myelin thickness both in vivo and in vitro (Ogata et al., 2004; Goebbels et al., 2010; Sheean et al., 2014; Domenech-Estevez et al., 2016). For example, conditional deletion of Erk1/2 in SCs causes a significant decrease in the number of myelinated axons and in the number of KROX20-positive cells, a marker of mature SCs (Newbern et al., 2011).
Previous literature has suggested that NRG1 also activates the phosphatase calcineurin (Kao et al., 2009). Calcineurin, a serine/threonine protein phosphatase activated by calcium, is composed of two subunits, calcineurin A (CnA) and calcineurin B (CnB), and frequently studied for its role in dephosphorylation and activation of the NFAT transcription factors (nuclear factor of activated T cells). In vivo deletion of the regulatory subunit, CnB, in neural crest cells inhibits calcineurin activity and causes a radial sorting delay and hypomyelination at embryonic and perinatal stages (Kao et al., 2009). However, these mice died shortly after birth, so it was unclear whether these defects were permanent or transient (Jessen and Mirsky, 2010). Therefore, to assess the role of calcineurin exclusively in SCs, we generated a SC conditional CnB KO mouse using CnB-floxed and P0-Cre mice (CnBscko) and assessed peripheral myelination after birth. Surprisingly, CnBscko mice have minimal myelination defects and no reduction in myelin protein expression, suggesting a less important role for calcineurin in developmental myelination than previously believed.
Despite having little effect on myelination in development, we discovered that calcineurin has a role in SCs after injury. Unlike the CNS, the PNS has a phenomenal ability to regenerate following injury. Within minutes of insult, axons begin to degenerate distal to the site of injury in a process known as Wallerian degeneration. This step is initiated by axons and triggers a trans-differentiation process in SCs, shifting from promyelinating SCs into “demyelinating” and then “repair” SCs (Jessen and Mirsky, 2010; Park et al., 2019). Demyelinating SCs first fragment their myelin sheath through changes in actin polymerization. This fragmentation of myelin then pinches off into the cytoplasm as myelin ovoids (Jung et al., 2011). SCs can break down and digest their own myelin via multiple parallel pathways, allowing for the regrowth of the axonal neurite. Data have demonstrated that SCs can clear myelin debris via autophagic destruction called “myelinophagy” (Gomez-Sanchez et al., 2015; Jang et al., 2016; Park et al., 2019), receptor-mediated phagocytosis (Lutz et al., 2017), and through disruption of the myelin sheaths by the mixed lineage kinase domain-like protein, MLKL (Ying et al., 2018). Inhibition of autophagy in SCs, genetically or pharmacologically, slows the clearance of myelin proteins and increases the number of intact myelin sheaths that remain after injury (Gomez-Sanchez et al., 2015; Jang et al., 2016).
Generally, inhibition of mTOR will activate autophagy. However, after injury, mTOR activity is upregulated in repair SCs (Norrmén et al., 2018), suggesting that activation of myelinophagy occurs via an mTOR-independent pathway. Notably, calcineurin has been recently identified as a novel regulator of autophagy (Medina et al., 2015). In agreement, we find that ablation of CnB in mice impairs myelin clearance after injury and primary SCs lacking CnB have reduced autophagic function. These data suggest a defect in myelinophagy that may be due in part to loss of activation of autophagy by calcineurin.
In summary, we present data that suggest calcineurin is not a significant promyelinating factor as previously believed, but rather a novel regulator of autophagy after injury.
Materials and Methods
Mice
Mice carrying floxed alleles of Ppp3r1 (CnB1) (B6;129S-Ppp3r1tm2Grc/J) and Thy1-YFP (Tg(Thy1-YFP)HJrs) (Feng et al., 2000; Porrero et al., 2010) were obtained from The Jackson Laboratory. Mice carrying mP0TOTA (Cre) transgene (previously described by Feltri et al., 1999a) were maintained in house and bred to CnB1 floxed mice to generate CnBf/f (WT) and P0Cre//CnBf/f (CnBscko) mice. The reporter line, Thy1-YFP, was used to generate YFP//P0Cre//CnBf/f (CnBscko-YFP) and YFP//CnBf/f (WT-YFP) mice. All lines were maintained in C57B/L6 background (Charles River Laboratories). Animal housing and experimentation strictly adhered to Department of Laboratory and Animal Resource core facility at Roswell Park Cancer Institute and University at Buffalo Institutional Animal Care and Use Committee protocols (Institutional Animal Care and Use Committee approval, UB1188 and 1197). Both male and female littermates were used for experiments. Genomic DNA for genotyping was extracted in 75 µl 25 mm NaOH/0.2 mm EDTA at 95°C for 45 min and neutralized with 75 µl of 40 mm Tris HCl, pH 5.5. PCR primers and protocols were as follows: P0-Cre (Feltri et al., 2002): 5′-ccaccacctctccattgcac-3', 5′-gctggcccaaatgttgctgg-3′ PCR program: 94°C 4 min, 30 cycles (94°C 30 s, 56°C 1 min, 72°C 1 min)72°C 1 min. CnB1 flox (Neilson et al., 2004): forward primer 5′-TTCGAGGACAGCTATACAGAGAAA-3′, reverse primer 5′-GACCTCCAGCCTCCACATAC-3′ PCR program: 94°C 2 min, 7 cycles (94°C 20 s, 65°C 15 s, 68°C 10 s), 28 cycles (94°C 15 s, 50°C 15 s, 72°C 10 s), 72°C 2 min. Thy1 YFP- oIMR7303 5′-TCTGAGTGGCAAAGGACCTTAGG-3′, oIMR8744 5′- CAAATGTTGCTTGTCTGGTG-3′, oIMR8745 5′- GTCAGTCGAGTGCACAGTTT-3′, oIMR9296 5′- CGCTGAACTTGTGGCCGTTTACG-3′ PCR program: 94° 3 min, 35 cycles (94°C 30 s, 6°C 1 min, 72°C 1 min), 72°C 2 min, 4°C.
Morphology
P1, P10, P28, 6 month, and crushed WT and mutant mice were killed, and sciatic nerves were collected and fixed in 2% glutaraldehyde (Polysciences, 00216a-10). Semithin section and EM section preparation was performed as previously described (Quattrini et al., 1996). In nerve crush samples, sections were cut 2 mm from the crush site into the distal portion. Images for semithin were acquired with a 100× objective on a DM 6000 microscope (Leica Microsystems), whereas for EM a FEI Tecnai G2 Spirit bio twin electron microscope was used. The g-ratio was measured and analyzed as previously described (Sidoli et al., 2016).
Nerve crush
Mice 42-50 d old were used for crush experiments. Mice were maintained under isoflurane anesthesia during surgery. The general surgery area was shaved and cleaned. The skin over the area of the sciatic nerve was cut, and a small opening in the fascia and muscle was created to expose a portion of the sciatic nerve. The sciatic nerve was crushed once for 15 s with clamp forceps dipped in liquid nitrogen for 2 s. The muscle was replaced over the nerve, and the injury site was closed using Vetbond surgical glue. Carprofen (5 mg/kg) was administered subcutaneously daily for 3 d after surgery.
Western blotting
Tissue or cells were lysed in RIPA buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% Triton X-100, 0.5% Nadeoxycholate, 1% SDS) supplemented with phosphatase and protease inhibitors: protease inhibitor cocktail (Roche Diagnostic), NaF, PMSF, Na2Vo3, C2 Cocktail (Sigma Millipore), and C3 Cocktail (Sigma Millipore). Lysates were disrupted on a cell homogenizer for 10 min at 4°C, left on ice for 20 min, centrifuged at 13,000 rpm for 10 min at 4°C, and the supernatant was removed for protein concentration analysis with the BCA protein assay kit (Thermo Fisher Scientific). Samples were prepared with 6× Laemmli buffer (360 mm Tris-HCl, pH 6.8, 30% glycerol, 10% SDS, 0.012% bromophenol blue, 0.6 M DTT) and lysis buffer, and boiled for 5 min; 5-20 µg of protein was resolved using SDS-PAGE under denaturing conditions with a mini-Protean II gel electrophoresis apparatus. Separated proteins were transferred to a PVDF blotting membrane, in a mini-gel transfer tank. Membranes were blocked with 5% milk powder in TBS Tween 20 (0.05%), incubated overnight in primary antibodies in 3% BSA in TBST, washed in TBST and incubated for 1 h at room temperature with secondary antibody. Secondary antibodies were either HRP-conjugated secondary antibody diluted in 5% milk TBST or LI-COR, secondary fluorescent antibody diluted in LI-COR, Odyssey buffer. Blots incubated with HRP-conjugated secondary antibody were developed with ECL chemiluminescent reagent on the Chemi-doc, while LI-COR, antibodies were developed using the Odyssey.
Mouse SC culture
Plates were prepared the day before and were coated with 0.01 mg/ml poly-L-lysine (Sigma Millipore, P-5899) for 1 h at room temperature followed overnight with Laminin 211 (Biolamina, LN211-03) at 13.3 µg/ml before drying. Sciatic nerves from 10-d-old pups were dissected under aseptic conditions in penicillin/streptomycin-supplemented Leibovitz medium. Epineurium and extra tissue were removed before digesting overnight at 37°C with 0.25% Dispase II and 0.05% Collagenase Type I in high-glucose DMEM. The following day, digested tissue was homogenized manually and filtered through a 70 µm strainer before being plated on coverslips in SC media: DMEM high glucose, 10% FBS, 2 mm L-glutamine, penicillin/streptomycin, 2 μm forskolin (EMD Millipore, 344270), 2 ng/ml Neuregulin (R&D Systems, 396-HB-05).
In vitro autophagic flux
The procedure was previously reported by Gomez-Sanchez et al. (2015). Primary mouse SCs were grown in culture for 4 d before being treated with 15 mm ammonium chloride for 3 h. Cells were collected for Western blot (WB) and lysed in RIPA lysis buffer; 5 µg of protein was separated by SDS-PAGE and probed for rabbit anti-LC3B and rabbit anti-GAPDH in 5% BSA/TBST. Autophagic flux was calculated by normalizing LC3II density to GAPDH density, then subtracting the untreated group from the treated group.
Ex vivo experiments
Sciatic nerves from 42- to 54-d-old WT and mutant mice were dissected under aseptic conditions in penicillin/streptomycin-supplemented Leibovitz medium and transferred to 5% FBS in DMEM supplemented with penicillin/streptomycin at 37°C and 5% CO2.
Immunofluorescence
Sciatic nerves were fixed in 4% ice-cold PFA for 30 min and cryopreserved in sucrose 20% overnight before embedding in OCT for cryostat sectioning. For SOX10, KROX20, and OCT6, nerves were frozen in OCT unfixed and sections were postfixed with 4% PFA. Sections were permeabilized in methanol for 3 min; blocking was achieved in 5% BSA, 2% NGS, and 0.5% Triton X-100 in PBS. Slides were incubated overnight at 4°C with primary antibodies, washed with PBS, and incubated for 1 h at room temperature with secondary antibodies. Nuclei were highlighted with DAPI.
RNA extraction and qPCR
Total RNA was isolated from the distal portion of crushed sciatic nerves using TRIzol reagent (Invitrogen) following the manufacturer's instructions. cDNA was prepared using Invitrogen Kit (Superscript III) following the manufacturer's instructions. The SYBR Green System was used for qPCR. Reaction mixture was as follows: 1 µl cDNA (5 ng), 1 µl forward primer, 1 µl reverse primer, 12.5 µl SYBR Master mix, 9.5 µl H20. RT-PCR program was as follows: 50°C for 3 min, 95°C for 10 min, 95°C for 15 min, 60°C for 1 min for 40 cycles. All the samples were analyzed in triplicate, and the relative expression of the target RNAs was expressed normalized to UBC expression. UBC was chosen as the housekeeping gene because of the stability of its mRNA in crushed nerves (Gambarotta et al., 2014; Norrmén et al., 2018). Transcription factor EB (TFEB): forward 5'-gcgagagctaacagatgctga-3', reverse 5'-ccggtcattgatgttgaacc-3′ (Medina et al., 2015). UBC forward 5′-AGCCCAGTGTTACCACCAAG-3′, UBC reverse 5′-ACCCAAGAACAAGCACAAGG-3′ (Gambarotta et al., 2014).
Antibodies
Western blotting
Antibodies used for western blotting were AKT 1:1000 (Cell Signaling Technology, 9272), beta tubulin 1:10,000 (Novus Biologicals, NB600-936), Pan-CnA 1:1000 (Cell Signaling Technology, 2614), CnB 1:1000 (Millipore 07-069), c-JUN 1:1000 (Cell Signaling Technology, 9165), ERK 1:1000 (Cell Signaling Technology, 9102s), GAPDH 1:5000 (Sigma Millipore, G9545), KROX20 (gift from Deis Meijer), LC3B 1:1000 (Novus Biologicals, NB100-2220), MAG 1:1000 (Invitrogen 34-6200), MBP 1:1000 (BioLegend, 836504), OCT6 (gift from Deis Meijer), pAKT (ser473) 1:1000 (Cell Signaling Technology, 9271), pS6 1:1000 (Cell Signaling Technology, 4858), pERK1/2 1:1000 (Cell Signaling Technology, 9101S), PMP22 1:1000 (Sigma Millipore, Sab4502217), P0 1:1000 (Aves Labs, PZO), S6 1:1000 (Cell Signaling Technology, 2217S), donkey anti-chicken HRP 1:10,000 (Abcam, Ab16349), donkey anti-chicken 800W 1:10,000 (LI-COR, 926-32218), donkey anti-rabbit 1:10,000 (Novus Biologicals, NB7185), goat anti-rabbit IgG 800CW 1:10,000 (LI-COR, 92632211), goat anti-rabbit IgG 670CW 1:10,000 (LI-COR, 926-68071), and goat anti-mouse IgG 670CW 1:10,000 (LI-COR, 926-68070).
Immunofluorescence
Antibodies used for immunofluorescence stainings were F480 1:300 (Bio-Rad, MCA497GA), Ki67 1:300 (Thermo Fisher Scientific, 14-5698-80), KROX20 1:8000 (gift from Deis Meijer), MBP 1:1000 (BioLegend, 808403), Neurofilament 1:1000 (BioLegend, 822701), OCT6 1:1000 (gift from Deis Meijer), P0 1:800 (Aves Labs, PZO), SOX10 1:500 (Cell Signaling Technology, 89356s), TFEB 1:500 (Bethyl Laboratories, A303-673A-M), donkey anti-rabbit IgG 488, donkey anti-rabbit IgG 594, donkey anti-mouse IgG 488, donkey anti-rat IgG Cy3, and donkey anti-chicken IgY Cy3 1:800 (Jackson ImmunoResearch Laboratories, 711-546-152, 711-585-152, 715-546-150, 712-165-153, 703-165-155).
Experimental design and statistical analysis
For all experiments, at least 3 animals per genotype were used. Data collection and analyses were performed blinded to the conditions of the experiments. Researchers blinded to conditions or genotype performed morphologic and immunofluorescence analyses. No statistical methods were used to predetermine sample size, but our sample sizes are similar to those generally used in the field. All the experiments were analyzed by Student's t test or two-way ANOVA. A value of p < 0.05 was considered significant. Error bars indicate SEM.
Results
SC-specific ablation of CnB causes a mild radial sorting delay but no hypomyelination
To study the role of calcineurin in SCs, we crossed a mouse carrying a floxed allele of the Ppp3r1 gene (CnB) (Neilson et al., 2004) with a mouse expressing recombinase under the Mpz promoter (P0-Cre), which resulted in a conditional KO of CnB in SCs after EXIII.5 (Feltri et al., 1999a, 2002), hereafter called CnBscko. The loss of CnB protein in SCs was confirmed by isolating SCs from the sciatic nerves of P5-P6 pups and measuring protein expression by WB. As expected, CnB was reduced in the SCs from the CnBscko mice (Fig. 1a). Interestingly, CnA, the enzymatic subunit of the calcineurin, is also reduced (Fig. 1a), suggesting a role for CnB in the stability of the CnA subunit.
Figure 1.
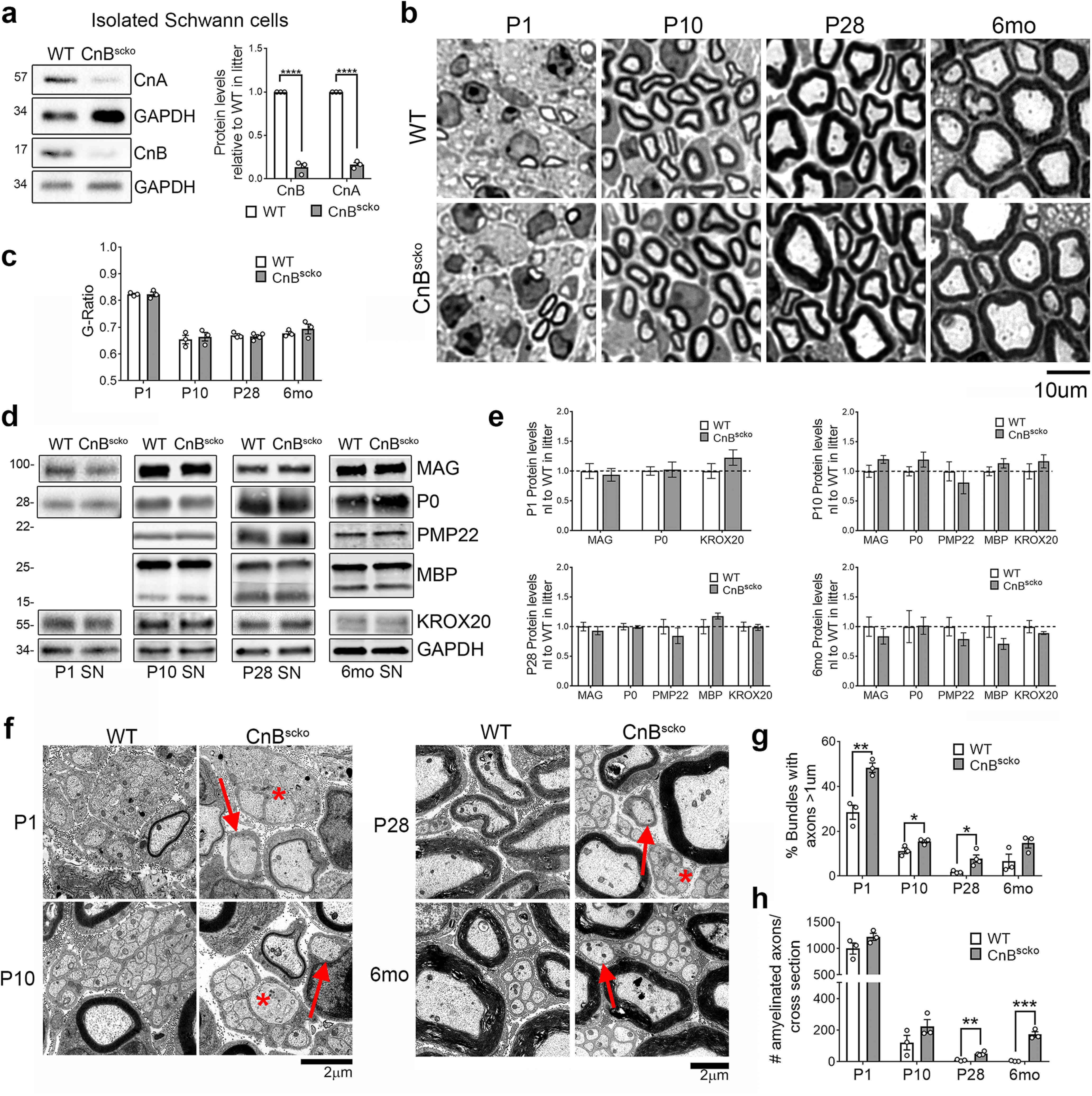
a, Representative immunoblot and quantification of CnA and CnB expression in SCs isolated from sciatic nerves of WT and CnBscko pups, normalized to GAPDH, and reported relative to the WT expression level. All pups of the same genotype from one litter (2-4 mice) were pooled for a single experiment, which represents one WB band. This was repeated 3 times to generate n = 3. Error bar indicates SEM. ****p < 0.0001 (Student's t test). b, Semithin images of sciatic nerves from P1, P10, P28, and 6-month-old WT and CnBscko mice. Scale bar, 10 µm. c, Quantification of myelin thickness measured by g-ratio (axon diameter/fiber diameter) from EM images. N = 3 mice/genotype. Error bar indicates SEM. d, Representative WB images of MAG, P0, PMP22, MBP, KROX20, and GAPDH in sciatic nerve from WT and CnBscko mice aged P1, P10, P28, and 6 months. PMP22 and MBP could not be detected in the WBs from P1 animals. e, Quantification of WBs in d for P1, P10, P28, and 6 months WT and CnBscko nerves. Protein levels were normalized to GAPDH and reported compared with WT. N = 4 pooled groups (P1) (3 or 4 mice per group), n = 10 mice (P10), n = 9 mice (P28), n = 4 mice (6 months) f, EM images of bundles with unsorted axons (*) and amyelinated fibers (arrows) from P1, P10, P28, and 6 months old CnBscko and WT mice. Scale bar, 2 μm. g, Quantification of the percentage of total bundles counted from 20 to 30 EM images with unsorted axons in CnBscko and WT mice at P1, 10, P28, and 6 months. Error bar indicates SEM. **p < 0.01, *p < 0.05. h, Quantification of the number of amyelinated axons per whole sciatic nerve cross section in P1, P10, P28, and 6 months CnBscko and WT mice. Error bar indicates SEM. **p < 0.01, ***p < 0.001.
We next analyzed the morphology of sciatic nerves from WT and CnBscko mice at the onset of myelination (P1), during active myelination (P10), after myelination (P28), and in adulthood (6 months) (Fig. 1b). At all these time points, there is no difference in myelin thickness as measured by g ratio (Fig. 1c). This was also reflected in myelin protein expression, which was equal in WT and CnBscko sciatic nerves from mice of all ages (Fig. 1d,e). However, in the CnBscko mice, there is a mild delay in the axonal radial sorting process (Fig. 1f). At P1, P10, and P28, there is a higher percentage of bundles with unsorted axons in CnBscko sciatic nerves compared with WT (Fig. 1g). We also observe an increase in the number of amyelinated axons at P28 and 6 months compared with WT (Fig. 1h). Interestingly, the number of amyelinated fibers in 6-month-old CnBscko mice is greater than the number in P28 CnBscko mice, suggesting that some of these fibers have been demyelinated rather than never myelinated (Fig. 1h). These data suggest that calcineurin loss causes a mild radial sorting delay, but overall calcineurin plays a limited role in developmental myelination and in achievement of the correct myelin thickness.
Proliferation and differentiation of SCs is normal in CnBscko sciatic nerves
Previous literature reported that the loss of CnB reduced KROX20 expression in the neonatal sciatic nerves, suggesting failure of neural crest cells to mature into myelinating SCs (Kao et al., 2009). Therefore, we assessed KROX20 expression in P10 CnBscko mice by immunofluorescence. Interestingly, we observed no reduction in nuclear expression compared with WT (Fig. 2a,b). This was further confirmed by no reduction in KROX20 protein expression in CnBscko nerves by WB during development (Fig. 2d,e). Furthermore, analysis of the promyelinating transcription factor, OCT6, is unchanged in CnBscko sciatic nerves compared with WT sciatic nerves (Fig. 2a,c). These data indicate that the deletion of CnB in SC precursors does not prevent their maturation into promyelinating SCs nor the upregulation of the master myelin transcription factor, KROX20.
Figure 2.
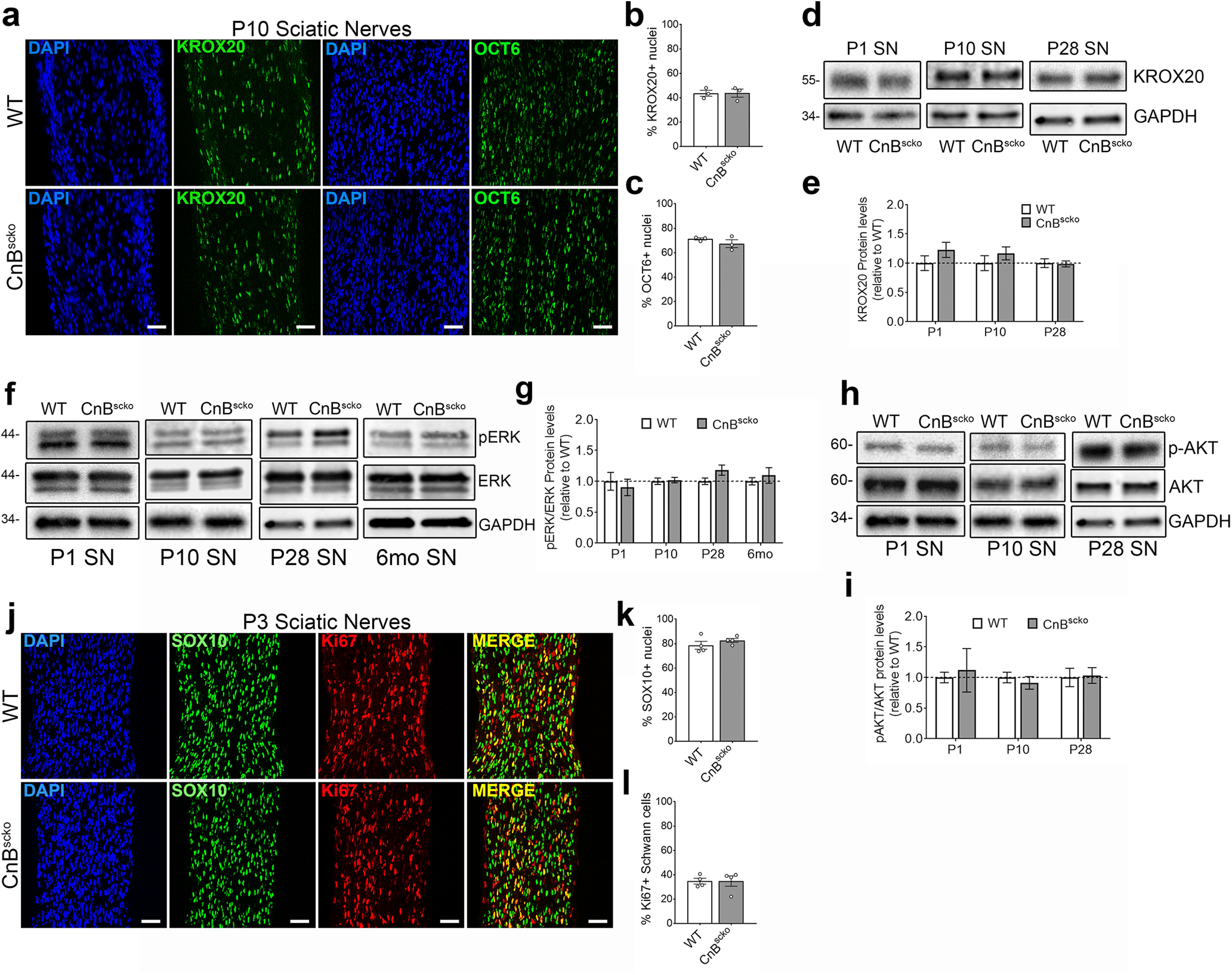
a, Representative images from longitudinal sections of P10 WT and CnBscko sciatic nerves. Sections were stained with KROX20 or OCT6 and for DAPI. Scale bar, 50 µm. b, Quantification of percentage of nuclei that are KROX20+ from a. N = 3 mice/genotype. Error bar indicates SEM. c, Quantification of percentage of nuclei that are OCT6+ from a. N = 3 mice/genotype. Error bar indicates SEM. d, Representative WB images of KROX20 and GAPDH in sciatic nerve from mice aged P1, P10, and P28. e, Quantification of WB in d. Protein levels of KROX20 were normalized to GAPDH and then reported compared with WT. N = 4 pooled groups (P1) (3 or 4 mice per group), n = 10 mice (P10), n = 9 mice (P28). Error bar indicates SEM. f, Representative WB images of P-ERK, ERK, and GAPDH in WT or CnBscko sciatic nerve from mice aged P1, P10, P28, and 6 months. g, Quantification of WB in f. Protein levels of P-ERK and ERK were normalized to GAPDH and then reported as P-ERK/ERK levels compared with WT. N = 4 pooled groups (P1) (3 or 4 mice per group), n = 10 mice (P10), n = 9 mice (P28), n = 4 mice (6 months). Error bar indicates SEM. h, Representative WB images of P-AKT, AKT, and GAPDH in sciatic nerve from mice aged P1, P10, and P28. i, Quantification of WB in h. Protein levels of P-AKT and AKT were normalized to GAPDH and then reported as P-AKT/AKT levels compared with WT. N = 4 pooled groups (P1) (3 or 4 mice per group), n = 10 mice (P10), n = 9 mice (P28). Error bar indicates SEM. j, Representative images from longitudinal sections of P3 WT and CnBscko sciatic nerves. Sections were stained with SC marker, SOX10, and the proliferation marker, Ki67. Scale bar, 50 µm. k, Quantification of percentage of total nuclei that are SOX10+ in P3 WT and CnBscko sciatic nerves. N = 4 mice/genotype. Error bar indicates SEM. l, Quantification of percentage of SOX10+ cells that are Ki67+ in P3 WT and CnBscko sciatic nerves. N = 4 mice/genotype. Error bar indicates SEM.
Since calcineurin was proposed to work downstream of NRG1, we analyzed the other known pathways downstream of NRG1 in SCs to determine whether there was compensation for the loss of calcineurin. We analyzed phosphorylation of ERK in P1, P10, P28, and 6 month sciatic nerves (Fig. 2f) and of AKT in P1, P10, and P28 sciatic nerves by WB (Fig. 2h). We found no difference between P-ERK expression between WT and CnBscko sciatic nerves at P1, P10, P28, and 6 months (Fig. 2f,g) nor in P-AKT expression at P1, P10, and P28 (Fig. 2h,i). Our data suggest that there is no compensation for the loss of calcineurin downstream of NRG1 via the ERK or AKT pathways.
We also analyzed whether the loss of calcineurin activity influenced the proliferation of SCs by quantifying the percentage of Ki67-positive SCs (SOX10) in P3 WT and CnBscko sciatic nerves but found no differences (Fig. 2j-l).
In conclusion, CnBscko mice exhibit no hypomyelination and only a mild delay in radial sorting in the sciatic nerve compared with WT mice. Furthermore, KROX20 expression remains unchanged in these animals. Based on our data, calcineurin either does not play an essential role in normal myelination during development or is redundant if the other signaling pathways downstream of NRG1 are functional.
The loss of CnB in SCs delays degeneration after injury
We next determined whether calcineurin played a role after injury and remyelination by analyzing crushed WT and CnBscko sciatic nerves. Following injury, the portion of the nerve distal to the site of injury undergoes Wallerian degeneration. This includes a series of coordinated events with initial degeneration of the distal stump and clearance of debris before allowing axonal regrowth and remyelination. At 7 d post crush (dpc), we observed more myelin sheaths surrounding both intact and degenerated axons in CnBscko sciatic nerves compared with WT nerves (Fig. 3a,b). Interestingly, when we visualized axonal degeneration using mice expressing yellow fluorescent protein (YFP) in neurons driven by the Thy1 promoter (Feng et al., 2000; Ey et al., 2007), we observed less degeneration of axons in CnBscko mice compared with WT mice at 3 dpc (Fig. 3c). When quantified, axon fragments in CnBscko mice were longer than fragments in WT mice at 3 dpc (Fig. 3d). These data demonstrate a potential relationship between myelin clearance and axonal degeneration.
Figure 3.

a, Representative images of WT and CnBscko sciatic nerves 7 d after nerve crush. Red boxes represent intact myelin sheaths. Scale bars, 20 µm. b, Quantification of the number of intact myelin sheaths (a, red boxes) per whole nerve cross section of WT and CnBscko mice 7 d after sciatic nerve crush. N = 3 mice/genotype. Error bars indicate SEM. *p < 0.05 (Student's t test). c, Representative images of YFP-labeled axons distal to the crush site of WT and CnBscko mice at 3 dpc. Scale bar, 100 µm. d, Quantification of YFP-labeled axons in c. The axon fragments in each image were measured and averaged, three 40× images per animal were obtained, and the final number reported for each animal was an average of the three images. N = 5 or 6 mice. Error bar indicates SEM. **p < 0.01. e, MCP-1 mRNA expression in the distal portion of WT and CnBscko nerves at 3 dpc. Expression reported relative to average WT expression. N = 3 mice/genotype. Error bar indicates SEM. f, Representative images from longitudinal sections of the distal stump of crushed WT and CnBscko sciatic nerves at 3 and 7 dpc. Macrophages are stained in red by F480. Scale bar, 50 µm. g, Quantification of the macrophages from the longitudinal sections in f. N = 5 mice/genotype for 3 dpc and N = 4 mice/genotype for 7 dpc. Error bar indicates SEM.
Since the process of myelin clearance relies on both macrophages and SCs, we wanted to determine whether macrophage activity contributed to this phenotype. Measurement of the chemoattractant MCP-1 by qPCR showed no difference in expression in WT and CnBscko sciatic nerves at 3 dpc, suggesting that loss of calcineurin does not affect the ability of SCs and fibroblasts to recruit macrophages after injury (Fig. 3e). Additionally, the number of F480+ cells in the distal portion of WT and CnBscko crushed sciatic nerves was not different in the distal portions of WT and CnBscko nerves at both 3 and 7 dpc (Fig. 3f,g). Next, we used an ex vivo model of sciatic nerve degeneration to evaluate the SC autonomous effect of loss of CnB after injury (Fig. 4a). When sciatic nerves are left in culture (ex vivo), macrophages cannot enter the sciatic nerve, eliminating the contributing activity of infiltrating macrophages on degeneration and myelin clearance. CnBscko sciatic nerves had significantly more intact myelin sheaths and axons by 12 d ex vivo compared with WT nerves (Fig. 4b). Likewise, primary SCs isolated from the sciatic nerves of WT and CnBscko pups were left in culture to digest their own myelin and analyzed at 1, 4, and 7 DIV (Fig. 4c). The primary SCs isolated from CnBscko pups had a greater percentage of SCs still containing P0 myelin protein compared with primary SCs from WT pups after 7 d in culture (Fig. 4d). Together, these data suggest that the impairment in myelin clearance seen in CnBscko mice at 7 dpc is not because of differences in immune cell infiltration, but is rather because of a SC autonomous defect.
Figure 4.

a, Reconstructed cross sections and high-powered representative images from sciatic nerve ex vivo stained for myelin basic protein (MBP) and neurofilament (NF). Scale bars: cross section, 200 µm; high-powered image, 20 µm. b, Quantification of the number of intact axons and MBP rings remaining in a cross section of WT and CnBscko sciatic nerves after 12 d ex vivo (12 DEV). N = 4 animals/genotype. Error bar indicates SEM. **p < 0.01; ***p < 0.001; Student's t test. c, Representative images of primary mouse SCs isolated from the sciatic nerves of P10 WT and CnBscko pups, stained for S100 (SC) and P0. Scale bar, 2 µm. d, Quantification of the percentage of SCs (marked by S100 staining) still containing myelin protein P0 after 7 d in culture. N = 4 coverslips/genotype per time point. Each coverslip contained SCs from 1 individual animal. Error bar indicates SEM. Two-way ANOVA: ****p < 0.0001 (WT 1 DIV vs 7 DIV); ***p < 0.001 (CnB 1 DIV vs 7 DIV); ###p < 0.001 (WT 7 DIV vs CnB 7 DIV).
Calcineurin does not regulate SC trans-differentiation and proliferation after injury
Within a few hours of injury, SCs proliferate and begin a trans-differentiation process into repair SCs. Inability to dedifferentiate halts the SC's ability to degrade myelin, support axonal regrowth, and remyelinate new axons, thereby preventing recovery after injury (Arthur-Farraj et al., 2012). The master regulator of this trans-differentiation program, c-JUN, is expressed at the same level in CnBscko distal stumps compared with WT distal stumps at 3 and 7 dpc by WB (Fig. 5a-d) and is expressed by the same percentage of cells by immunofluorescence staining of c-JUN (Fig. 5e,g) compared with WT. Recent work has shown that mTORC1 activation is upstream of c-JUN in SCs after injury (Norrmén et al., 2018). Analysis of the phosphorylation levels of the ribosomal protein S6, a substrate downstream of mTORC1 activity, shows there is no significant difference in mTOR activity between CnBscko and WT distal nerves at 3 and 7 dpc (Fig. 5a-d). Furthermore, OCT6 expression, which is a marker of immature SCs and increased following injury, and P-ERK expression, which activates SC recruitment of immune cells and promotes regeneration, are both activated in the distal stumps of nerves from CnBscko mice relative to expression in the distal stumps of WT mice (Fig. 5a-d). Finally, proliferation of SCs, an important early step to promote repair after injury is also equal in WT and CnBscko nerves at 3 dpc (Fig. 5f,h). Together, these data suggest that SCs trans-differentiate properly in CnBscko nerves after injury and the phenotype observed in CnBscko SCs is because of a problem in myelin clearance rather than in SC transition into a repair phenotype.
Figure 5.

a, b, Representative WB images from WT and CnBscko nerves at 3 and 7 dpc. c, d, Quantification of WBs shown in a, b. Protein levels were normalized to GAPDH and then reported normalized to average WT levels. Error bar indicates SEM. n = 9 mice/3 dpc, and n = 12 mice/7 dpc. e, Representative images of longitudinal sections from the distal portion of WT and CnBscko sciatic nerves, stained for c-JUN at 3 and 7 dpc. Scale bar, 50 µm. f, Representative images of longitudinal sections from the distal portion of WT and CnBscko sciatic nerves, stained for SOX10 and Ki67 at 3 and 7 dpc. Scale bar, 50 µm. g, Quantification of cJUN+ cells in WT and CnBscko sciatic nerves at 3 and 7 dpc. Error bars indicate SEM. n = 5 mice/3 dpc, and n = 4 mice/7 dpc. h, Quantification of the percentage of SOX10+ cells that were also Ki67+ in WT and CnBscko sciatic nerves at 3 and 7 dpc. Error bars indicate SEM. n = 5 mice/3 dpc, and n = 4 mice/7 dpc.
Deletion of CnB blunts TFEB activation and impairs autophagy
Recent studies have demonstrated that SCs use autophagy to clear myelin debris following injury. Conditional deletion of Atg7 (Autophagy related gene 7) in SCs prevents autophagy and results in a phenotype similar to that observed in CnBscko nerves after crush (Gomez-Sanchez et al., 2015; Jang et al., 2016). This similarity extends to the fact that SC trans-differentiation and proliferation are normal in ATG7scko nerves. Therefore, we hypothesized that calcineurin, a novel regulator of autophagy, may be involved in activating myelinophagy in SCs after injury. To test this, we measured autophagic flux in isolated SCs from WT and CnBscko pups, as previously performed by Gomez-Sanchez et al. (2015). This technique allowed us to eliminate measuring the autophagic activity of macrophages and other cells within the nerve. We treated the isolated SCs with ammonium chloride (NH4Cl) and measured LC3II expression. Ammonium chloride inhibits the fusion of autophagosomes and lysosomes, thereby preventing degradation and causing an accumulation of autophagosomes (Seglen and Reith, 1976; Gurney et al., 2015). This accumulation can then be quantified via measurement of LC3II protein, a component of autophagosomes. In WT SCs, treatment with NH4Cl for 3 h causes a robust increase in LC3II expression, as expected. Interestingly, this was not observed in CnBscko SCs. On the contrary, untreated CnBscko SCs already expressed high levels of LC3II (Fig. 6a). This is consistent with the fact that primary CnBscko SCs have more P0 protein remaining when left in culture (Fig. 4) and with the finding that P0 is sequestered in the autophagosome after injury (Gomez-Sanchez et al., 2015). The observation that, when treated, LC3II expression does not increase in CnBscko cells indicates that CnBscko SCs are not generating more autophagosomes over this period. From this blot, autophagic flux was calculated, and SCs lacking CnB demonstrated a reduced autophagic flux compared with WT SCs (Fig. 6b). Together, we concluded that loss of CnB hinders autophagic activity in SCs after injury and impairs the clearance of myelin.
Figure 6.

a, Representative WB measuring LC3II expression in WT and CnBscko SCs. SCs cultured at 4 DIV were treated with NH4Cl for 3 h before lysis for WB. Each band represents the SCs isolated from the sciatic nerves of 1 P10 pup. N = 3 pups/treatment group. b, Autophagic flux was calculated by subtracting the average LC3II/GAPDH expression in untreated cells, from the LC3II/GAPDH expressions in treated cells, respectively. Error bar indicates SEM. *p < 0.05 (Student's t test). c, Representative images of the proximal and distal portions of a WT sciatic nerve at 7 dpc. Nerves were stained for F480 and TFEB. Scale bar, 50 µm. d, Quantification of the TFEB-positive nuclei. N = 4 mice. Error bar indicates SEM. ***p < 0.001 (Student's t test). e, Representative images of the distal portion of WT and CnBscko nerves at 3 and 7 dpc. Nerves were stained for F480 and TFEB. White arrows indicate TFEB-positive SCs. Scale bar, 50 µm. f, Quantification of TFEB-positive cells in the distal portion of WT and CnBscko sciatic nerves at 3 dpc. N = 5 mice/genotype. Error bar indicates SEM. *p < 0.05 (Student's t test). g, Quantification of TFEB mRNA expression in the distal portion of WT and CnBscko sciatic nerves at 3 dpc. Expression reported relative to average WT 3 dpc levels. N = 3 mice/genotype. Error bar indicates SEM. *p < 0.05. h, Quantification of TFEB-positive cells in the distal portion of WT and CnBscko sciatic nerves at 7 dpc. N = 5 mice/genotype. Error bar indicates SEM.
In 2015, a report identified calcineurin as a regulator of lysosomal biogenesis and autophagy through regulation of the TFEB (Medina et al., 2015). Knockdown of either the CnA or CnB subunit, or pharmacological inhibition of calcineurin with cyclosporine in vitro, reduced TFEB translocation to the nucleus and autophagic flux (Medina et al., 2015; Martina et al., 2016). Therefore, we asked whether the loss of calcineurin in CnBscko mice alters TFEB translocation after nerve crush. However, little is known about the role of TFEB in nerve crush. Therefore, to determine whether TFEB is activated after nerve injury, we first analyzed TFEB expression in the proximal and distal portions of crushed WT nerves (Fig. 6c). At 7 dpc, there is a robust increase in nuclear TFEB in the distal portion compared with the proximal portion of crushed WT nerves in both macrophages and SCs (Fig. 6d). We next repeated this experiment in the distal nerves of WT and CnBscko mice at 3 and 7 dpc (Fig. 6e). We quantified the number of TFEB-positive macrophages and SCs and observed a reduction in the number of TFEB-positive SC nuclei, but not in the number of TFEB-positive macrophage nuclei in CnBscko nerves, compared with WT at 3 dpc (Fig. 6f). This was supported by a reduction in TFEB mRNA expression in CnBscko nerves at 3 dpc, which is consistent with the fact that TFEB positively regulates its own transcription (Fig. 6g) (Sardiello et al., 2009; Settembre et al., 2013; Sardiello, 2016). In contrast, there was no longer a difference between groups when the experiment was repeated from nerves at 7 dpc (Fig. 6h). Medina et al. (2015) showed that knockdown of calcineurin did not fully abrogate TFEB nuclear translocation and activation, indicating that there are other phosphatases acting on TFEB. Therefore, we hypothesize that the reestablishment of TFEB at 7 dpc is caused by compensation from other phosphatases activating TFEB.
In summary, we propose that the defect in autophagy in our model could be a result of the initial inhibition of TFEB activation in CnBscko nerves. Here we provide a possible mechanism for the impaired myelinophagy phenotype we observe in CnBscko sciatic nerves.
Calcineurin is not necessary in SCs for remyelination after injury
To determine whether the delay that we observed in CnBscko nerves in TFEB and autophagy activation affects axonal regeneration and remyelination, we analyzed WT and CnBscko nerves at 21 dpc (Fig. 7a). Despite the observed delay in myelin clearance, CnBscko sciatic nerves were comparable to WT nerves at 21 dpc. There was no difference in myelin thickness (Fig. 7b), or in the number of myelinated fibers, amyelinated fibers (blue arrows), or cells with myelin debris (red arrows) that remain in CnBscko nerves compared with WT nerves at 21 dpc (Fig. 7c). These data suggest a role for calcineurin in the degeneration and clearance of myelin following injury, but not a role for calcineurin in axonal regeneration and remyelination. This phenotype is remarkably similar to what was observed in ATG7scko mice after crush (Jang et al., 2016), as these mice have impaired initiation of autophagy and show significant reductions in myelin clearance at 5 dpc, but show no difference in axonal regeneration and remyelination by 28 dpc. Together, it is plausible to hypothesize there are other unknown SC autonomous mechanisms that play a role in myelin digestion after injury and can compensate for reduced autophagy in these models, allowing for proper remyelination.
Figure 7.

a, Representative images of WT and CnBscko sciatic nerves at 21 dpc. Scale bars, 10 µm. b, Quantification of myelin thickness by g-ratio in WT and CnBscko sciatic nerves 21 dpc; 100-200 axons/animal were measured from EM images. N = 4 mice/genotype. Error bars indicate SEM. c, Quantification of the number of myelinated fibers, amyelinated fibers (blue arrows), and cells with myelin ovoids (red arrows) per total nerve cross section in WT and CnBscko sciatic nerves 21 dpc. N = 4 mice/genotype.
Discussion
Calcineurin in SCs is not required for myelination
In Kao et al. (2009), a mouse carrying a floxed allele of the Ppp3r1 gene (CnB) was crossed to a mouse expressing cre recombinase under the Wnt1 promoter. Use of the Wnt1cre mouse caused recombination of CnB in all neural crest cell derivatives at EVIII.5, including SC precursors, sensory neurons, and endoneurial fibroblasts (Danielian et al., 1998; Joseph et al., 2004; Foster et al., 2008). Kao et al. (2009) reported that the loss of CnB impaired radial sorting and caused hypomyelination at the day of birth. However, these mice die shortly after birth (Jessen and Mirsky, 2010), so it unknown whether the hypomyelination observed was permanent. It is possible that the hypomyelination observed was transient; and, if the Wnt1cre/Cnb mice had survived postnatally, myelination would have proceeded normally. Alternatively, the additive effects of CnB deletion in SC precursors and other neural crest derivatives resulted in the more dramatic myelination phenotype observed by Kao et al. (2009). Here, we generated a mouse that conditionally ablates Ppp3r1 (CnB) in immature SCs, using P0-Cre mice that recombines almost exclusively in the SC lineage starting at EXIII.5 (Feltri et al., 1999a,b), allowing us to evaluate the proposed role of calcineurin in myelinating SCs without additional confounding factors. Proliferation and maturation of SCs as measured by KROX20 and OCT6 levels were normal in CnBscko sciatic nerves compared with WT sciatic nerves. Our data show that loss of calcineurin in SCs causes only a mild radial sorting delay but does not cause hypomyelination. This phenotype is reflected in normal myelin protein levels at all points in development. Furthermore, expression of KROX20 in CnBscko sciatic nerves remains at WT levels with or without calcineurin signaling. These data are in line with the in vitro data from Kipanyula et al. (2013), which demonstrate that NFAT activation alone is insufficient to upregulate Krox20 transcription.
Calcineurin regulates myelin clearance in SCs after injury
In peripheral nerve injury, the process of breaking down and clearing myelin begins with SCs in a process coined “myelinophagy.” In the CnBscko model, myelin clearance is impaired both in vivo after crush and in vitro. Our paper demonstrates a connection between calcineurin and “myelinophagy” in SCs. We offer a potential mechanism via a reduction in TFEB activation at 3 dpc. To our knowledge, this is one of the first eukaryotic in vivo models to show a defect in autophagic activity when calcineurin activity is inhibited. The mechanism by which calcineurin is activated after crush still needs to be illuminated. Interestingly, Smith and Hall (1988) demonstrated spontaneous vesicular demyelination of peripheral nerves following injection with the calcium ionophore, ionomycin. It was hypothesized that this observation was caused by an increase in intracellular Ca2+ within the SC, which may activate phospholipase A2 or some other enzyme. These initial observations are aligned with our data and suggest that the phenotype observed could be because of calcineurin activation via ionomycin treatment, causing autophagic destruction of myelin.
Deletion of CnB decreases TFEB activation after crush
This is the first model to describe activation of TFEB after peripheral nerve injury and one of the first models to show in vivo regulation of TFEB by calcineurin. SC clearance of myelin is accomplished by autophagy (Gomez-Sanchez et al., 2015), but independent of mTOR activity. In most systems, inhibition of mTOR will activate autophagy, but in injured sciatic nerves high levels of autophagy are seen in parallel with mTOR upregulation (Gomez-Sanchez et al., 2015; Jang et al., 2016; Norrmén et al., 2018). The transcription factor, TFEB, has emerged as a master regulator of lysosomal and autophagic regulation (Settembre and Ballabio, 2011; Settembre et al., 2012; Medina et al., 2015). Under normal conditions, TFEB, a substrate of mTORC1, remains phosphorylated and sequestered in the cytoplasm. In times of stress, such as starvation or exercise, inhibition of mTORC1 kinase activity allows for the translocation of TFEB into the nucleus (Martina et al., 2012; Roczniak-Ferguson et al., 2012; Settembre et al., 2012). The robust activation of TFEB we see in the distal portion of the sciatic nerve after injury, despite high mTORC1 activity, further points to an mTORC1-independent activator of autophagy. Our model suggests that calcineurin may be a possible activator of TFEB and autophagy after injury. When calcineurin is present, dephosphorylation of TFEB overwhelms inhibition via mTORC1, allowing for translocation to the nucleus and activation of autophagy. However, in our mice where CnB is knocked out, TFEB is unable to overcome phosphorylation by mTORC1 delaying the activation of autophagy and slowing myelin clearance (Fig. 8). The first in vitro models that identified calcineurin as a phosphatase for TFEB described that knockdown or pharmacological inhibition of calcineurin produced a partial reduction of TFEB activity (Medina et al., 2015), suggesting there are other phosphatases that can activate TFEB that have not been identified. In our model, these other phosphatases may compensate for loss of calcineurin because nuclear TFEB expression in CnBscko nerves catches up to nuclear TFEB expression in WT nerves by 7 dpc.
Figure 8.

Schematic representation of the model of activation of TFEB in WT and CnBscko after injury. Calcineurin acts a phosphatase for TFEB, whereas mTORC1 is a known inhibitor of TFEB. After injury, when calcineurin is present, the activation of TFEB by calcineurin dephosphorylation overwhelms the inhibitory effect of mTORC1, causing an increase in nuclear TFEB. However, when calcineurin is deleted, such as in CnBscko animals, high mTORC1 activity causes TFEB phosphorylation and inhibition of activity. We hypothesize that decreased TFEB activation in CnBscko nerves impairs autophagy activation and delays myelin clearance after injury.
Calcineurin does not activate SC trans-differentiation
The transcription factor c-JUN is the major activator of SC reprogramming into repair SCs (Parkinson et al., 2008; Arthur-Farraj et al., 2012). Recent work identified that translational upregulation of c-JUN is dependent on activation of mTORC1 (Norrmén et al., 2018). Signals activating mTORC1 after injury have not been identified, but it has been suggested that NRG1 signaling is involved. Following nerve injury, SCs switch from relying on axonal NRG1 Type III signals to auto and paracrine NRG1 Type I signaling from SCs (Fricker et al., 2011; Stassart et al., 2013). Norrmén et al. (2018) pondered whether this switch from NRG1 Type III signaling to Type I signaling could be responsible for the activation of mTORC1, restricting this pathway to upregulation only after axonal contact is lost. NRG1 Type I reactivates PI3K-AKT and MEK-ERK1/2 pathways after injury (Fricker et al., 2011; Norrmén et al., 2018); and comparable to its effect during myelination, the loss of ERK signaling has severe consequences in injury and repair. Inhibition of MEKK1 impaired SC proliferation, trans-differentiation, and recruitment of immune cells after injury (Napoli et al., 2012). Surprisingly, loss of calcineurin showed none of these characteristics, suggesting that NRG1 Type I signaling does not signal through calcineurin after injury and that calcineurin is not needed for trans-differentiation.
Peripheral nerve regeneration occurs despite reduced autophagy
CnBscko mice are able to recover comparably to WT mice by 21 dpc. Although this is surprising, it is also similar to what was observed in ATG7scko mice after crush (Jang et al., 2016). Like our results, ATG7scko mice have no deficiency in SC proliferation or trans-differentiation, but show significant reductions in myelin clearance. Together, it appears that autophagy is downstream of trans-differentiation in SCs. In c-JUN or mTORC1-deficient mice (Arthur-Farraj et al., 2012; Napoli et al., 2012; Norrmén et al., 2018), where trans-differentiation is inhibited, regeneration and remyelination do not occur, signifying that trans-differentiation is the first trigger for all subsequent processes after injury.
As reported previously, the clearance of myelin immediately after injury is SC mediated, while later, the recruitment of macrophages aids in myelin phagocytosis and removal (Hirata and Kawabuchi, 2002). As ATG7scko and CnBscko mice do not show delayed regeneration and remyelination despite the fact that myelin debris inhibits neurite regeneration (Mukhopadhyay et al., 1994; DeBellard et al., 1996), it is plausible to hypothesize there are other SC-autonomous mechanisms that compensate for decreased autophagic activity, such as the phagocytic pathway (Lutz et al., 2017) or the recently reported myelin destruction pathway involving activation of MLKL (Ying et al., 2018). Data have shown that all three pathways are concurrently upregulated within the first week after injury (Gomez-Sanchez et al., 2015; Jang et al., 2016; Lutz et al., 2017; Ying et al., 2018), but how they interact is unknown. However, interruption of any single pathway is enough to cause a delay in myelin clearance despite the complexity and apparent overlap of activity. Future experiments should look at the interplay between these pathways and the potential compensatory mechanisms they may play to ensure successful nerve recovery. Moreover, compensation by infiltrating nonmacrophage phagocytic cells or even endoneurial fibroblasts that can digest myelin (Schubert and Friede, 1981) could be responsible for the destruction and clearance of myelin in CnB or ATG7 SC KO mice.
Impact in human disease
Targeting myelin clearance pathways could be an effective pharmacological approach to promote regeneration and functional recovery after nerve injury. Indeed, enhanced activation of autophagy after crush using the mTOR inhibitor, rapamycin, accelerated early myelin degeneration and remyelination (Huang et al., 2016). Rapamycin-treated rats had thicker myelin 6 weeks after nerve crush compared with vehicle-treated rats, suggesting that pharmaceutical targeting of this pathway can enhance recovery. As calcineurin is highly expressed in multiple tissue types, manipulation of the autophagic pathways via calcineurin activation may prove to have too many off-target effects. However, more targeted approaches, such as direct TFEB activators (Song et al., 2016; Wang et al., 2017), could be an alternative.
Interestingly, inhibition of calcineurin in nerve injury has already been performed in humans. The calcineurin inhibitors, tacrolimus and cyclosporine, have been used in peripheral nerve regrafting for their immunosuppressive activity (Midha et al., 1993) and positive effect on neurite growth (Gold et al., 1994, 1995; Udina et al., 2003). It would be interesting to evaluate whether these treatments have any effect on SCs.
Footnotes
The authors declare no competing financial interests.
This work was supported by National Institutes of Health Grants R01-NS NS55256 and R56NS096104 to L.W., Grant F30NS105405-02 to C.B.R., and Grant R01NS100464 to M.L.F. We thank Ed Hurley for the processing of sciatic nerves for semithin and electron microscopy; Dies Meijer (University of Edinburgh) for KROX20 and OCT6 antibodies; and Nadav Weinstock for useful discussion and time spent imaging nerve sections on the confocal microscope.
References
- Arthur-Farraj PJ, Latouche M, Wilton DK, Quintes S, Chabrol E, Banerjee A, Woodhoo A, Jenkins B, Rahman M, Turmaine M, Wicher G, Mitter R, Greensmith L, Behrens A, Raivich G, Mirsky R, Jessen KR (2012) c-Jun reprograms Schwann cells of injured nerves to generate a repair cell essential for regeneration. Neuron 75:633–647. 10.1016/j.neuron.2012.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielian PS, Muccino D, Rowitch DH, Michael SK, McMahon AP (1998) Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr Biol 8:1323–1326. 10.1016/S0960-9822(07)00562-3 [DOI] [PubMed] [Google Scholar]
- DeBellard ME, Tang S, Mukhopadhyay G, Shen YJ, Filbin MT (1996) Myelin-associated glycoprotein inhibits axonal regeneration from a variety of neurons via interaction with a sialoglycoprotein. Mol Cell Neurosci 7:89–101. 10.1006/mcne.1996.0007 [DOI] [PubMed] [Google Scholar]
- Domenech-Estevez E, Baloui H, Meng X, Zhang Y, Deinhardt K, Dupree JL, Einheber S, Chrast R, Salzer JL (2016) Akt regulates axon wrapping and myelin sheath thickness in the PNS. J Neurosci 36:4506–4521. 10.1523/JNEUROSCI.3521-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ey B, Kobsar I, Blazyca H, Kroner A, Martini R (2007) Visualization of degenerating axons in a dysmyelinating mouse mutant with axonal loss. Mol Cell Neurosci 35:153–160. 10.1016/j.mcn.2007.02.014 [DOI] [PubMed] [Google Scholar]
- Feltri ML, D'Antonio M, Previtali S, Fasolini M, Messing A, Wrabetz L (1999a) P0-Cre transgenic mice for inactivation of adhesion molecules in Schwann cells. Ann NY Acad Sci 883:116–123. 10.1111/j.1749-6632.1999.tb08574.x [DOI] [PubMed] [Google Scholar]
- Feltri ML, D'Antonio M, Quattrini A, Numerato R, Arona M, Previtali S, Chiu SY, Messing A, Wrabetz L (1999b) A novel P0 glycoprotein transgene activates expression of lacZ in myelin-forming Schwann cells. Eur J Neurosci 11:1577–1586. 10.1046/j.1460-9568.1999.00568.x [DOI] [PubMed] [Google Scholar]
- Feltri ML, Graus Porta D, Previtali SC, Nodari A, Migliavacca B, Cassetti A, Littlewood-Evans A, Reichardt LF, Messing A, Quattrini A, Mueller U, Wrabetz L (2002) Conditional disruption of β1 integrin in Schwann cells impedes interactions with axons. J Cell Biol 156:199–210. 10.1083/jcb.200109021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR (2000) Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron 28:41–51. 10.1016/s0896-6273(00)00084-2 [DOI] [PubMed] [Google Scholar]
- Foster K, Sheridan J, Veiga-Fernandes H, Roderick K, Pachnis V, Adams R, Blackburn C, Kioussis D, Coles M (2008) Contribution of neural crest-derived cells in the embryonic and adult thymus. J Immunol 180:3183–3189. 10.4049/jimmunol.180.5.3183 [DOI] [PubMed] [Google Scholar]
- Fricker FR, Lago N, Balarajah S, Tsantoulas C, Tanna S, Zhu N, Fageiry SK, Jenkins M, Garratt AN, Birchmeier C, Bennett DL (2011) Axonally derived neuregulin-1 is required for remyelination and regeneration after nerve injury in adulthood. J Neurosci 31:3225–3233. 10.1523/JNEUROSCI.2568-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambarotta G, Ronchi G, Friard O, Galletta P, Perroteau I, Geuna S (2014) Identification and validation of suitable housekeeping genes for normalizing quantitative real-time PCR assays in injured peripheral nerves. PLoS One 9:e105601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebbels S, Oltrogge JH, Kemper R, Heilmann I, Bormuth I, Wolfer S, Wichert SP, Möbius W, Liu X, Lappe-Siefke C, Rossner MJ (2010) Elevated phosphatidylinositol 3, 4, 5-trisphosphate in glia triggers cell-autonomous membrane wrapping and myelination. J Neurosci 30:8953–8964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold BG, Katoh K, Storm-Dickerson T (1995) The immunosuppressant FK506 increases the rate of axonal regeneration in rat sciatic nerve. J Neurosci 15:7509–7516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold BG, Storm-Dickerson T, Austin DR (1994) The immunosuppressant FK506 increases functional recovery and nerve regeneration following peripheral nerve injury. Restor Neurol Neurosci 6:287–296. 10.3233/RNN-1994-6404 [DOI] [PubMed] [Google Scholar]
- Gomez-Sanchez JA, Carty L, Iruarrizaga-Lejarreta M, Palomo-Irigoyen M, Varela-Rey M, Griffith M, Hantke J, Macias-Camara N, Azkargorta M, Aurrekoetxea I, De Juan VG, Jefferies HBJ, Aspichueta P, Elortza F, Aransay AM, Martínez-Chantar ML, Baas F, Mato JM, Mirsky R, Woodhoo A, et al. (2015) Schwann cell autophagy, myelinophagy, initiates myelin clearance from injured nerves. J Cell Biol 210:153–168. 10.1083/jcb.201503019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney MA, Huang C, Ramil JM, Ravindran N, Andres AM, Sin J, Linton PJ, Gottlieb RA (2015) Measuring cardiac autophagic flux in vitro and in vivo. Apoptosis and Cancer, pp 187–197. New York, NY: Humana Press. [DOI] [PubMed] [Google Scholar]
- Hirata K, Kawabuchi M (2002) Myelin phagocytosis by macrophages and nonmacrophages during Wallerian degeneration. Microsc Res Tech 57:541–547. 10.1002/jemt.10108 [DOI] [PubMed] [Google Scholar]
- Huang HC, Chen L, Zhang HX, Li SF, Liu P, Zhao TY, Li CX (2016) Autophagy promotes peripheral nerve regeneration and motor recovery following sciatic nerve crush injury in rats. J Mol Neurosci 58:416–423. 10.1007/s12031-015-0672-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang SY, Shin YK, Park SY, Park JY, Lee HJ, Yoo YH, Kim JK, Park HT (2016) Autophagic myelin destruction by Schwann cells during Wallerian degeneration and segmental demyelination. Glia 64:730–742. 10.1002/glia.22957 [DOI] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R (2010) Control of Schwann cell myelination. F1000 Biol Rep 2:19 10.3410/B2-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen KR, Brennan A, Morgan L, Mirsky R, Kent A, Hashimoto Y, Gavrilovi J (1994) The Schwann cell precursors and its fate: a study of cell death and differentiation during gliogenesis in rat embryonic nerves. Neuron 12:509–527. 10.1016/0896-6273(94)90209-7 [DOI] [PubMed] [Google Scholar]
- Joseph NM, Mukouyama YS, Mosher JT, Jaegle M, Crone SA, Dormand EL, Lee KF, Meijer B, Anderson DJ, Morrison SJ (2004) Neural crest stem cells undergo multilineage differentiation in developing peripheral nerves to generate endoneurial fibroblasts in addition to Schwann cells. Development 131:5599–5612. 10.1242/dev.01429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J, Cai W, Jang SY, Shin YK, Suh DJ, Kim JK, Park HT (2011) Transient lysosomal activation is essential for p75 nerve growth factor receptor expression in myelinated Schwann cells during Wallerian degeneration. Anat Cell Biol 44:41–49. 10.5115/acb.2011.44.1.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao SC, Wu H, Xie J, Chang CP, Ranish JA, Graef IA, Crabtree GR (2009) Calcineurin/NFAT signaling is required for neuregulin-regulated Schwann cell differentiation. Science 323:651–654. 10.1126/science.1166562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipanyula MJ, Woodhoo A, Rahman M, Payne D, Jessen KR, Mirsky R (2013) Calcineurin–nuclear factor of activated T cells regulation of Krox‐20 expression in Schwann cells requires elevation of intracellular cyclic AMP. J Neurosci Res 91:105–115. 10.1002/jnr.23131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz AB, Chung WS, Sloan SA, Carson GA, Zhou L, Lovelett E, Posada S, Zuchero JB, Barres BA (2017) Schwann cells use TAM receptor-mediated phagocytosis in addition to autophagy to clear myelin in a mouse model of nerve injury. Proc Natl Acad Sci USA 114:E8072–E8080. 10.1073/pnas.1710566114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina JA, Chen Y, Gucek M, Puertollano R (2012) MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 8:903–914. 10.4161/auto.19653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina JA, Diab HI, Brady OA, Puertollano R (2016) TFEB and TFE3 are novel components of the integrated stress response. EMBO J 35:479–495. 10.15252/embj.201593428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, Montefusco S, Scotto-Rosato A, Prezioso C, Forrester A, Settembre C, Wang W, Gao Q, Xu H, Sandri M, Rizzuto R, De Matteis MA, Ballabio A (2015) Lysosomal calcium signaling regulates autophagy through calcineurin and TFEB. Nat Cell Biol 17:288–299. 10.1038/ncb3114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midha R, Mackinnon SE, Evans PJ, Best TJ, Hare GM, Hunter DA, Falk-Wade JA (1993) Comparison of regeneration across nerve allografts with temporary or continuous cyclosporin A immunosuppression. J Neurosurg 78:90–100. 10.3171/jns.1993.78.1.0090 [DOI] [PubMed] [Google Scholar]
- Morris JK, Lin W, Hauser C, Marchuk Y, Getman D, Lee KF (1999) Rescue of the cardiac defect in ErbB2 mutant mice reveals essential roles of ErbB2 in peripheral nervous system development. Neuron 23:273–283. 10.1016/s0896-6273(00)80779-5 [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay G, Doherty P, Walsh FS, Crocker PR, Filbin MT (1994) A novel role for myelin-associated glycoprotein as an inhibitor of axonal regeneration. Neuron 13:757–767. 10.1016/0896-6273(94)90042-6 [DOI] [PubMed] [Google Scholar]
- Napoli I, Noon LA, Ribeiro S, Kerai AP, Parrinello S, Rosenberg LH, Collins MJ, Harrisingh C, White IJ, Woodhoo A, Lloyd AC (2012) A central role for the ERK-signaling pathway in controlling Schwann cell plasticity and peripheral nerve regeneration in vivo. Neuron 73:729–742. 10.1016/j.neuron.2011.11.031 [DOI] [PubMed] [Google Scholar]
- Neilson JR, Winslow MM, Hur EM, Crabtree GR (2004) Calcineurin B1 is essential for positive but not negative selection during thymocyte development. Immunity 20:255–266. 10.1016/S1074-7613(04)00052-4 [DOI] [PubMed] [Google Scholar]
- Newbern JM, Li X, Shoemaker SE, Zhou J, Zhong J, Wu Y, Bonder D, Hollenback S, Coppola G, Geschwind DH, Landreth GE, Snider WD (2011) Specific functions for ERK/MAPK signaling during PNS development. Neuron 69:91–105. 10.1016/j.neuron.2010.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norrmén C, Figlia G, Pfistner P, Pereira JA, Bachofner S, Suter U (2018) mTORC1 is transiently reactivated in injured nerves to promote c-Jun elevation and Schwann cell dedifferentiation. J Neurosci 38:4811–4828. 10.1523/JNEUROSCI.3619-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata T, Iijima S, Hoshikawa S, Miura T, Yamamoto S, Oda H, Nakamura K, Tanaka S (2004) Opposing extracellular signal-regulated kinase and Akt pathways control Schwann cell myelination. J Neurosci 24:6724–6732. 10.1523/JNEUROSCI.5520-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HT, Kim JK, Tricaud N (2019) The conceptual introduction of the “demyelinating Schwann cell” in peripheral demyelinating neuropathies. Glia 67:571–581. 10.1002/glia.23509 [DOI] [PubMed] [Google Scholar]
- Parkinson DB, Bhaskaran A, Arthur-Farraj P, Noon LA, Woodhoo A, Lloyd AC, Feltri ML, Wrabetz L, Behrens A, Mirsky R, Jessen KR (2008) c-Jun is a negative regulator of myelination. J Cell Biol 181:625–637. 10.1083/jcb.200803013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porrero C, Rubio-Garrido P, Avendaño C, Clascá F (2010) Mapping of fluorescent protein-expressing neurons and axon pathways in adult and developing Thy1-eYFP-H transgenic mice. Brain Research 1345:59–72. [DOI] [PubMed] [Google Scholar]
- Quattrini A, Previtali S, Feltri ML, Canal N, Nemni R, Wrabetz L (1996) β4 integrin and other Schwann cell markers in axonal neuropathy. Glia 17:294–306. [DOI] [PubMed] [Google Scholar]
- Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, Walther TC, Ferguson SM (2012) The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal 5:ra42 10.1126/scisignal.2002790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardiello M. (2016) Transcription factor EB: from master coordinator of lysosomal pathways to candidate therapeutic target in degenerative storage diseases. Ann NY Acad Sci 1371:3–14. 10.1111/nyas.13131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, Bandi S, Parenti G, Cattaneo E, Ballabio A (2009) A gene network regulating lysosomal biogenesis and function. Science 325:473–477. 10.1126/science.1174447 [DOI] [PubMed] [Google Scholar]
- Schubert T, Friede R (1981) The role of endoneurial fibroblasts in myelin degradation. J Neuropathol Exp Neurol 40:134–154. 10.1097/00005072-198103000-00006 [DOI] [PubMed] [Google Scholar]
- Seglen PO, Reith A (1976) Ammonia inhibition of protein degradation in isolated rat hepatocytes: quantitative ultrastructural alterations in the lysosomal system. Exp Cell Res 100:276–280. 10.1016/0014-4827(76)90148-8 [DOI] [PubMed] [Google Scholar]
- Settembre C, Ballabio A (2011) TFEB regulates autophagy: an integrated coordination of cellular degradation and recycling processes. Autophagy 7:1379–1381. 10.4161/auto.7.11.17166 [DOI] [PubMed] [Google Scholar]
- Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, Facchinetti V, Sabatini DM, Ballabio A (2012) A lysosome‐to‐nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 31:1095–1108. 10.1038/emboj.2012.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O, Huynh T, Carissimo A, Palmer D, Klisch TJ, Wollenberg AC, Di Bernardo D, Chan L, Irazoqui JE, Ballabio A (2013) TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol 15:647–658. 10.1038/ncb2718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheean ME, McShane E, Cheret C, Walcher J, Müller T, Wulf-Goldenberg A, Hoelper S, Garratt AN, Krüger M, Rajewsky K, Meijer D, Birchmeier W, Lewin GR, Selbach M, Birchmeier C (2014) Activation of MAPK overrides the termination of myelin growth and replaces Nrg1/ErbB3 signals during Schwann cell development and myelination. Genes Dev 28:290–303. 10.1101/gad.230045.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidoli M, Musner N, Silvestri N, Ungaro D, D'Antonio M, Cavener DR, Feltri ML, Wrabetz L (2016) Ablation of PERK in Schwann cells improves myelination in the S63del Charcot-Marie-Tooth 1B mouse. J Neurosci 36:11350–11361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith K, Hall S (1988) Peripheral demyelination and remyelination initiated by the calcium-selective ionophore ionomycin: in vivo observations. J Neurol Sci 83:37–53. 10.1016/0022-510x(88)90018-4 [DOI] [PubMed] [Google Scholar]
- Song JX, Sun YR, Peluso I, Zeng Y, Yu X, Lu JH, Xu Z, Wang MZ, Liu LF, Huang YY, Chen LL, Durairajan SS, Zhang HJ, Zhou B, Zhang HQ, Lu A, Ballabio A, Medina DL, Guo Z, Li M (2016) A novel curcumin analog binds to and activates TFEB in vitro and in vivo independent of MTOR inhibition. Autophagy 12:1372–1389. 10.1080/15548627.2016.1179404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stassart RM, Fledrich R, Velanac V, Brinkmann BG, Schwab MH, Meijer D, Sereda MW, Nave KA (2013) A role for Schwann cell–derived neuregulin-1 in remyelination. Nat Neurosci 16:48–54. 10.1038/nn.3281 [DOI] [PubMed] [Google Scholar]
- Taveggia C, Zanazzi G, Petrylak A, Yano H, Rosenbluth J, Einheber S, Xu X, Esper RM, Loeb JA, Shrager P, Chao MV, Falls DL, Role L, Salzer JL (2005) Neuregulin-1 Type III determines the ensheathment fate of axons. Neuron 47:681–694. 10.1016/j.neuron.2005.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udina E, Ceballos D, Gold BG, Navarro X (2003) FK506 enhances reinnervation by regeneration and by collateral sprouting of peripheral nerve fibers. Exp Neurol 183:220–231. 10.1016/s0014-4886(03)00173-0 [DOI] [PubMed] [Google Scholar]
- Wang X, Li SH, Zhu L, Nian QG, Yuan S, Gao Q, Hu Z, Ye Q, Li XF, Xie DY, Shaw N, Wang J, Walter TS, Huiskonen JT, Fry EE, Qin CF, Stuart DI, Rao Z (2017) Small-molecule TFEB pathway agonists that ameliorate metabolic syndrome in mice and extend C. elegans lifespan. Nat Commun 8:14 10.1038/s41467-017-02332-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Z, Pan C, Shao T, Liu L, Li L, Guo D, Zhang S, Yuan T, Cao R, Jiang Z, Chen S, Wang F, Wang X (2018) Mixed lineage kinase domain-like protein MLKL breaks down myelin following nerve injury. Mol Cell 72:457–468. 10.1016/j.molcel.2018.09.011 [DOI] [PubMed] [Google Scholar]