

Daxx loss causes coordinate dysregulation of ERVs and protein-coding genes after physiologic stress and impairs tissue recovery.
Abstract
Tumor sequencing studies have emphasized the role of epigenetics and altered chromatin homeostasis in cancer. Mutations in DAXX, which encodes a chaperone for the histone 3.3 variant, occur in 25% of pancreatic neuroendocrine tumors (PanNETs). To advance our understanding of physiological functions of Daxx, we developed a conditional Daxx allele in mice. We demonstrate that Daxx loss is well tolerated in the pancreas but creates a permissive transcriptional state that cooperates with environmental stress (inflammation) and other genetic lesions (Men1 loss) to alter gene expression and cell state, impairing pancreas recovery from inflammatory stress in vivo. The transcriptional changes are associated with dysregulation of endogenous retroviral elements (ERVs), and dysregulation of endogenous genes near ERVs is also observed in human PanNETs with DAXX mutations. Our results reveal a physiologic function of DAXX, provide a mechanism associated with impaired tissue regeneration and tumorigenesis, and expand our understanding of ERV regulation in somatic cells.
INTRODUCTION
One of the major findings from cancer genome sequencing projects was the large proportion of genetic mutations in epigenetic regulators found across broad cancer types. This includes enzymes involved in DNA methylation and histone modifications as well as chromatin remodelers (1, 2). One of the most unexpected discoveries was recurrent mutations in genes that encode histones. Missense mutations in the histone 3 variant H3.3 (encoded by the H3F3A and H3F3B genes) were first identified in the invariably fatal pediatric malignancy diffuse intrinsic pontine glioma and have subsequently been identified in several other cancers (3, 4). Moreover, the protein chaperones that are responsible for loading H3.3 into chromatin, DAXX and ATRX, are also mutated in cancers. Mutually exclusive loss-of-function mutations, predominantly indels, in DAXX or ATRX occur in 43% of pancreatic neuroendocrine tumors (PanNETs) (5). This high frequency of mutations suggests an important tumor suppressor role for this histone chaperone complex and, more generally, for this epigenetic regulatory axis.
Biochemical and cell-based studies demonstrate that DAXX and ATRX mediate H3.3 deposition into heterochromatic regions of the DNA, including telomeres and pericentromeric DNA (6). Notably, all PanNETs with DAXX or ATRX mutations activate the alternative lengthening of telomeres pathway, implicating telomere dysfunction in tumorigenesis (7). Additional studies have demonstrated that Daxx, Atrx, and H3.3 cooperate with histone methylation to mediate the epigenetic silencing of transposable elements in the genome of mouse embryonic stem cells (8). Derepression of endogenous retroviral elements (ERVs) can lead to active transposition and affect the expression of endogenous genes through their strong long terminal repeat (LTR) promoters; however, our understanding of the mechanisms that maintain silencing in somatic tissues as well as how dysregulation affects normal tissue function and contributes to disease remains limited. Several other and often opposing functions have also been suggested for Daxx, including roles in promoting apoptosis, inhibiting apoptosis and affecting the DNA damage response (9), further emphasizing the need to evaluate Daxx function(s) in physiologically relevant models.
Mouse models have been pivotal to understanding the physiologically relevant functions of genes and the molecular mechanisms downstream of their dysregulation that contribute to disease. Daxx deletion is lethal at an early embryonic stage in mice and embryos exhibit apoptosis, yet the reasons for this lethality remain poorly understood (10). We have therefore generated and characterized a conditional Daxx allele in mice, allowing for both spatial and temporal control of Daxx expression and the ability to investigate the essential biology downstream of Daxx loss. Using this model, we demonstrated that Daxx loss is well tolerated in the developing pancreas, with no robust phenotypic changes under homeostatic conditions and no impact on mouse survival. However, whole-transcriptome analysis indicated dysregulation of heterochromatin with Daxx loss. Because both genetics and environment interact to maintain tissue homeostasis, we combined Daxx deficiency with Men1 loss (a lesion that frequently co-occurs with DAXX mutation in human cancers), inflammatory stress, and the combination of the two. Comprehensive transcriptome and chromatin accessibility profiling revealed that Daxx loss leads to dysregulation of endogenous retrovirus silencing, with coordinate gene expression changes that are associated with altered cell state and impaired pancreas recovery and regeneration in vivo. An analysis of human PanNET RNA sequencing (RNA-seq) data provided supporting evidence of coordinate dysregulation of ERVs and protein-coding genes downstream of DAXX mutation.
RESULTS
Daxx loss is well tolerated in the developing pancreas
The early lethality of Daxx-null mice (10) requires a conditional Daxx allele to study the effects of Daxx loss in both a tissue- and temporal-specific manner. We previously identified a single-nucleotide deletion in the intron 2 loxP site in the available Daxxtm2Led allele, thereby rendering recombination inefficient in vivo (11). However, we were able to obtain a null allele (Daxx∆3) from this mouse and confirmed the previously reported embryonic lethality, demonstrating that deletion of exon 3 does result in a null allele (11). To generate a functional conditional allele, we designed a CRISPR-Cas9 strategy to insert the missing nucleotide (fig. S1, A to C). We evaluated recombination of this new conditional allele (Daxxfl) compared with Daxxtm2Led, using the constitutive CMV-CreTg driver. Robust induction of recombination was observed in vivo by polymerase chain reaction (PCR) genotyping of tail biopsies in Daxxfl/+CMV-CreTg mice compared with Daxxtm2Led/+CMV-CreTg mice (fig. S1D).
Because DAXX mutations are almost exclusively present in PanNETs, we chose to evaluate the physiologic consequences of Daxx loss in the pancreas. Moreover, the pancreas is a robustly regenerative organ with inherent cell plasticity (12), making it an ideal system to study the effects of epigenetic dysregulation. The pancreas is specified between E8.5 and E9.5 in mouse development, largely driven by the expression of the homeobox transcription factor Pdx1 (13). We therefore used the Pdx1-CreTg (C) line to drive deletion of Daxx throughout the pancreas (14). Daxx loss was well tolerated because Daxxfl/flPdx1-CreTg (DC) mice were born at the expected Mendelian ratio. We euthanized a small number of mice at 6 to 8 weeks of age to comprehensively evaluate pancreas development. We first confirmed successful recombination of the Daxxfl allele through quantitative PCR genotyping analysis (fig. S1E). Western blot analysis of total pancreas lysates (Fig. 1A) demonstrated loss of protein expression in DC pancreases. Pancreas weight, as a percentage of total mouse weight, was the same in DC mice as in wild-type (WT) controls (Fig. 1B). In addition, the pancreas was histologically normal (Fig. 1C). To evaluate the long-term consequences of Daxx loss in the pancreas, we aged a cohort of mice and found no reduction in survival (Fig. 1D) or significant alterations of β cell function as measured by nonfasting blood glucose in 12-month-old mice (Fig. 1E). We obtained similar results using a β cell–specific rat insulin II promoter–driven Cre recombinase (RIP-CreTg) to induce Daxx deletion. Combined, these data suggest that Daxx is not required for pancreas specification or normal function in vivo.
Fig. 1. Daxx loss is well tolerated in the developing pancreas.
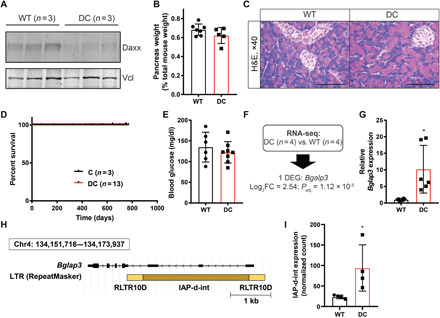
(A) Western blot analysis of Daxx protein expression in the pancreas of wild-type (WT) or Daxxfl/flPdx1-CreTg (DC) mice with vinculin (Vcl) as a control. Each lane is one independent pancreas. (B) Pancreas weight presented as a percentage of total mouse weight (mean ± SD). (C) Representative hematoxylin and eosin (H&E)–stained sections of WT and DC mouse pancreases. Image magnification is ×40; scale bars, 100 μm. (D) Kaplan-Meier survival analysis, compared with Pdx1-CreTg (C) controls. (E) Nonfasting blood glucose measurements of 12-month-old mice (mean ± SD). (F) Schematic representation of basal RNA-seq analysis. DEG, differentially expressed gene. (G) Quantitative reverse transcription PCR validation of Bglap3 expression, shown as mean ± SD. *P < 0.05, Student’s t test. (H) Schematic representation of the murine Bglap3 locus, with intragenic ERV, IAP-d-int, flanked by RLTR10D LTRs. (I) Normalized read counts for IAP-d-int from RNA-seq data (mean ± SD). *P = 0.03, Wilcoxon test.
To evaluate the pancreas at a molecular level, we next conducted basal transcriptome analysis through RNA-seq in adult (6- to 8-week-old) DC mice compared with WT controls. This analysis revealed significant differential expression of a single gene, Bglap3, a largely unstudied osteocalcin-related gene (15) in the Daxx-deficient pancreas compared with controls (log2FC = 2.54, Padj = 1.12 × 10−5; Fig. 1F). We confirmed this gene expression difference in independent mice; Bglap3 expression was an average of 10-fold higher in the DC mouse pancreas compared with controls (Fig. 1G). Bglap3 also emerged as a transcriptional readout of endogenous retrovirus dysregulation in multiple cell types because it contained an intragenic ERV, IAP-d-int (Fig. 1H). Deletion of components of the Setdb1 histone methyltransferase complex in mouse embryonic stem cells and fibroblasts leads to loss of H3K9me3 and robust overexpression of Bglap3 (16, 17). We further interrogated our RNA-seq data for expression of the IAP-d-int (18) and found a significant increase in expression of the ERV as well (P = 0.03, Wilcoxon test; Fig. 1I). Combined, these data suggest that Daxx loss may alter heterochromatin, which we speculate may create a permissive environment and cooperate with additional environmental stresses or genetic lesions to affect biology in vivo.
Daxx and Atrx interact to form the H3.3 chaperone complex. Correspondingly, Atrx loss is also well tolerated in the developing pancreas (fig. S1F) and is associated with variable but increased expression of Bglap3 (fig. S1G), suggesting that deregulation of the Bglap3 locus is associated with the histone chaperone function of Daxx and Atrx.
The combined loss of Daxx and Men1 leads to cystic degeneration of the exocrine pancreas
Genetic lesions cooperate to affect pancreas diseases. For example, Kras mutation alone is insufficient to drive robust cancer phenotypes in mice, despite occurring in most pancreatic ductal adenocarcinomas (19). However, when combined with loss of the p53 tumor suppressor, Kras mutation promotes rapid and aggressive disease (20). We therefore evaluated the effects of Daxx loss in the context of Men1 deficiency. Our choice to use Men1 loss as a cooperating genetic lesion was based on the high frequency of concurrent mutations in DAXX and MEN1 in human PanNETs (5, 21). In the endocrine pancreas, Men1 functions as part of a multiprotein mixed lineage leukemia (MLL) histone methyltransferase complex, mediating H3K4me3 and promoting the expression of important cell cycle inhibitors, Cdkn1b and Cdkn2c (22, 23).
To evaluate the long-term consequences of combined Daxx and Men1 loss throughout the pancreas, we aged a cohort of mice with the following genotypes: Pdx1-CreTg (C), Daxxfl/flPdx1-CreTg (DC), Men1fl/flPdx1-CreTg (MC), and Daxxfl/flMen1fl/flPdx1-CreTg (DMC; fig. S2A). The C and DC mice from this cohort were the same mice presented in Fig. 1D and showed no differences in survival. Consistent with previous reports, homozygous inactivation of Men1 led to islet cell hyperplasia and PanNET development in the endocrine pancreas, but no major phenotypic changes to the exocrine pancreas in mice aged up to 2 years (Fig. 2A, a and b) (24). However, the combined loss of Daxx and Men1 caused many changes to the exocrine pancreas, detected by histological analysis. Representative sections are provided in Fig. 2A (c to f). Specifically, there was evidence of acinar atrophy, including fatty replacement of acinar cells (Fig. 2A, c) and acinar-to-ductal metaplasia (Fig. 2A, e), suggesting cellular plasticity and an altered stress response. The metaplastic areas were composed of ductal proliferation, often accompanied by cyst formation (Fig. 2A, c and d), chronic inflammation, and surrounding fibrosis, the latter characteristic of activated stellate cells. In some cases, epithelial changes were seen, including partial or complete lining of cells exhibiting enlarged, hyperchromatic, pencilate nuclei. In one case, the cells lining the cysts (partially in some cysts and completely in others) had morphology reminiscent of signet ring cells (i.e., globoid cells with abundant eosinophilic cytoplasm pushing the nucleus to the periphery; Fig. 2A, f). We quantified the frequency of different lesions in the pancreas. The cystically dilated ducts found in DMC pancreases were absent in MC pancreases (P < 0.0001, chi-square test compared with MC; Fig. 2B). Metaplasias (Fig. 2A, b and e) were identified in most mice from both genotypes (Fig. 2C); however, the pancreas area occupied by these lesions was increased in DMC mice (Fig. 2D). Moreover, the proportion of mice with a metaplastic area >1% of the total pancreas area was significantly higher in DMC mice than in MC mice (P < 0.0001, chi-square test compared with MC; Fig. 2D). These data demonstrate that Daxx loss, in combination with Men1 deficiency, results in an impaired homeostatic response in the pancreas. We speculated that the large endocrine tumor burden caused by Men1 deficiency (see below) induced tissue stress/injury, with the potential to obstruct vessels and ducts and induce damage and inflammation. Correspondingly, immune cell infiltration was significantly higher in DMC pancreases than in MC pancreases, as assayed by immunohistochemistry for the CD45 pan-immune marker (Fig. 2, E and F), indicative of an inflammatory response. These data uncovered an important role for Daxx in maintaining pancreas homeostasis in vivo.
Fig. 2. The combined loss of Daxx and Men1 leads to cystic degeneration of the pancreas.
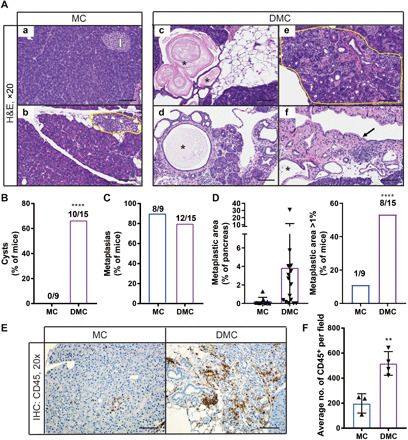
(A) Representative hematoxylin and eosin–stained images of pancreas sections from Men1fl/flPdx1-CreTg (MC) and Daxxfl/flMen1fl/flPdx1-CreTg (DMC) mice. Image magnification is ×20; scale bars, 100 μm. I indicates pancreatic islet; yellow outline indicates areas of acinar to ductal metaplasia; * denotes cysts; arrow indicates cells with signet ring morphology. (B) Quantification of the percentage of mice with cystic and (C) metaplastic lesions. ****P < 0.0001, chi-square test compared with MC. (D) Quantification of metaplastic area (mean ± SD) and proportion of mice with metaplastic lesions covering more than 1% of the total pancreas area. ****P < 0.0001, chi-square test compared with MC. (E) Immunohistochemical (IHC) staining for the pan-immune marker CD45. Image magnification is ×20; scale bars, 100 μm. (F) Quantification of average CD45+ cells per ×20 field (mean ± SD). **P < 0.01, Student’s t test.
This aged cohort also provided us with the opportunity to evaluate whether Daxx loss cooperates with Men1 deficiency to promote PanNETs in mice. Men1 loss, driven by several different pancreas Cre-recombinase lines, is sufficient on its own to promote multifocal hyperplasia and PanNET development, with a long disease latency (24–28). We observed no difference in overall survival between DMC and MC mice (fig. S2B), with mice from both genotypes developing well-differentiated PanNETs (fig. S2, C and D). Quantification of the endocrine compartment (islets) further revealed no significant changes in tumorigenesis (fig. S2E) and indicated that Daxx is not a strong endocrine tumor suppressor in mice.
Daxx loss cooperates with inflammation to affect the transcriptional state of the pancreas
Genetic alterations also cooperate with environmental stress to affect cellular and organismal responses. Experimental mouse models of inflammation and pancreatitis are well established (29, 30) and exhibit acinar-to-ductal metaplasia and immune cell infiltration, similar to the phenotypes that we observed in our aged cohort. Repeated intraperitoneal injection of the cholecystokinin analog caerulein is extensively used to study both acute and chronic pancreatitis. Caerulein induces acinar cells to locally secrete digestive enzymes, resulting in edema, infiltration of immune cells, and loss of identifiable acinar cells. The acinar cells undergo either cell death or a transient transdifferentiation to a metaplastic intermediate that expresses ductal markers. To induce chronic inflammation, we treated adult mice (6 to 10 weeks of age) with caerulein over 3 weeks, administered as four daily doses (hourly), 3 days per week (Monday, Wednesday, and Friday), as per established protocols (Fig. 3A) (31). We included four different genotypes in these experiments: WT, DC, MC, and DMC mice. A small number of WT mice were euthanized 24 hours following the final caerulein dose to confirm pancreatitis (fig. S3, A and B). To evaluate the molecular response, we then euthanized mice 5 days following the final caerulein dose. In all genotypes, the pancreas weight (as a percentage of total mouse weight) was reduced in treated mice compared with untreated mice, but only the DMC pancreas was significantly reduced in size compared with WT-treated controls (fig. S3C), again suggesting cooperation between Daxx and Men1 loss. Histologically, both MC and DMC pancreas sections showed more severe atrophy than either WT or DC pancreas sections (fig. S3D). These data are consistent with our recent report demonstrating that Men1 loss potentiates the inflammatory response following acute caerulein-induced pancreatitis (32).
Fig. 3. Daxx loss cooperates with inflammation and Men1 deficiency to affect the transcriptional state of the pancreas.
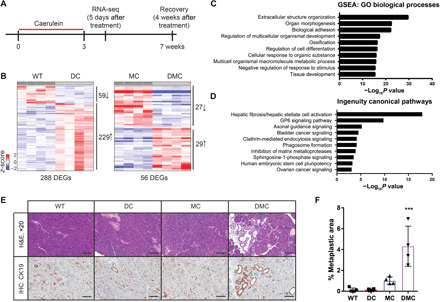
(A) Schematic representation of experimental outline. (B) Heatmaps representing Daxx-dependent gene expression changes in DC compared with WT mouse pancreases and in DMC compared with MC mouse pancreases following treatment with caerulein. (C) Gene Ontology (GO) biological processes pathway analysis of DEGs in DC mouse pancreases compared with WT controls. GSEA, Gene Set Enrichment Analysis. (D) Ingenuity Pathway Analysis of DEGs in DC mouse pancreases compared with WT controls. (E) Representative histologic images and immunohistochemistry for cytokeratin 19 (CK19) of pancreas sections 4 weeks following the final dose of caerulein. Image magnification is ×20; scale bars, 100 μm. (F) Quantification of metaplastic areas as a percentage of total pancreas area (mean ± SD); ***P < 0.001, analysis of variance with Dunnett’s posttest comparing all groups with WT controls.
We next conducted global transcriptome analysis through RNA-seq of the total pancreas. We identified Daxx-dependent gene expression changes through comparisons of DC with WT and DMC with MC pancreases (Fig. 3B and table S1). What was immediately apparent was the much higher number of differentially expressed genes (DEGs) in caerulein-treated DC mice compared with WT mice, considering that the basal expression analysis identified only a single DEG. In addition, 80% of the DEGs were up-regulated in DC mice compared with WT mice, supporting the establishment of a more permissive chromatin state. We further focused on the comparison between DC and WT mice because it yielded a greater number of DEGs and is not complicated by Men1 loss. In addition, the DMC versus MC comparison identified too few genes for comprehensive pathway analysis. We conducted pathway analysis using the Molecular Signatures Database of Gene Set Enrichment Analysis and evaluated enrichments in the Gene Ontology biological processes. We identified pathways associated with tissue development, differentiation, and responses to stimuli (Fig. 3C). We then focused on the genes driving these pathways and found that several, including transcription factors, are associated with specific cell populations in the pancreas according to single-cell transcriptome analysis (table S2) (33). Specific markers of pancreatic ductal cells, Mmp7 and Onecut2, were among the DEGs. In response to environmental or genetic stresses, acinar cells in the pancreas are known to undergo acinar-to-ductal metaplasia, and these data suggest that Daxx loss may contribute to establishing this transdifferentiated state. We also noted several genes associated with the mesenchymal cell types, which are not targeted by Pdx1-CreTg. The resident mesenchymal cell type in the pancreas is the stellate cell, which does not undergo recombination (34). Typically, these cells are quiescent, but they can be stimulated by cytokines to an activated state. Several established activators of stellate cells (Fgf1, Fgf2, and Tgfb3) are up-regulated in the DC pancreas, supporting paracrine activation of stellate cells. Once activated, stellate cells secrete factors that can promote growth and survival, as well as produce the fibrotic reaction that is characteristic of pancreatitis and pancreatic ductal adenocarcinoma. Single-cell sequencing defined a signature of activated stellate cells (35), and 6 of 11 of these genes were significantly up-regulated in our dataset (table S2). Ingenuity Pathway Analysis (36) of our DEGs identified hepatic fibrosis/hepatic stellate cell activation as the most significant pathway (Fig. 3D), and there are many similarities between pancreatic and hepatic stellate cells (34). Combined, these data demonstrated that, in the context of caerulein-induced pancreatitis, Daxx loss induced gene expression changes associated with pancreas cell state and suggested a role in restricting cellular plasticity.
Given the DEGs, and the potential paracrine activation of stellate cells in the pancreas, we hypothesized that the transcriptional changes and the potential cellular plasticity induced by Daxx loss could impair tissue repair and regeneration. We therefore repeated the chronic pancreatitis experiments, but this time, we allowed mice to recover for 4 weeks following the final dose. Pancreases from WT and DC mice completely recovered at the 4-week time point (Fig. 3, E and F). Pancreases from MC mice also recovered, despite the increased tissue damage in these mice. DMC mice, however, had a significant proportion of persistent metaplastic lesions in their pancreases. Metaplasia was confirmed by immunohistochemical staining for cytokeratin 19 (CK19), a marker of ductal differentiation (Fig. 3, E and F). Combined, these data suggest that Daxx loss cooperated with inflammation and Men1 deficiency to affect the transcriptional state of the pancreas and impair tissue recovery following pancreatitis, consistent with the phenotypic response in our aged cohort.
ATAC sequencing identifies chromatin accessibility changes associated with ERVs
To date, the most important finding indicating the functional role for Daxx in vivo is the mutual exclusivity of DAXX and ATRX mutations in PanNETs, indicating that their shared function as a histone chaperone complex for H3.3 is important for tumor suppression. To understand the impact of Daxx loss on chromatin structure, we conducted ATAC sequencing (ATAC-seq) experiments because chromatin immunoprecipitation (ChIP) experiments for endogenous H3.3 have not provided sufficient signal for genome-wide ChIP sequencing analysis. A similar approach was recently taken to evaluate Atrx loss in glioma stem cells (37). Using established protocols for processing frozen tissues (38), we conducted ATAC-seq of pancreas samples from four MC mice and four DMC mice (two males and two females each) isolated 5 days following the final caerulein injection. We focused on these two genotypes because of the tissue repair and cell state defects (Fig. 3, E and F). The distribution of peaks across genomic regions was very similar between the two genotypes (Fig. 4A). We next identified 265 differentially accessible regions in the DMC pancreas compared with the MC pancreas, with 136 regions (51%) open and 129 (49%) closed (table S3). The relative proportions of open and closed regions were similar to what was reported for Atrx loss in glioma stem cells (37), although the number of regions that we identified was considerably smaller. As a positive control in our data, an 840–base pair (bp) region containing exon 3 of Daxx (region 140; table S3), which is deleted from the DMC pancreas and not from the MC pancreas, was identified as closed (false discovery rate = 0.01661). Overall, differentially accessible regions were enriched within 15 Mb of chromosome ends (P = 4.73 × 10−12, Fisher’s exact test), consistent with the role of Daxx loading H3.3 into telomeres.
Fig. 4. ATAC-seq identifies chromatin accessibility changes associated with ERVs.
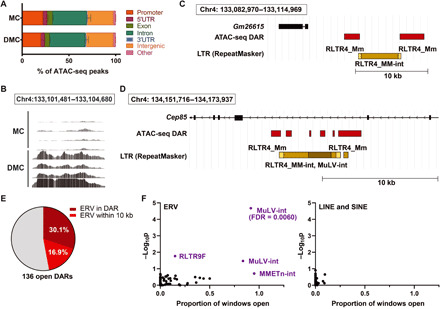
(A) Distribution of peaks across different types of genomic regions in MC and DMC pancreases. (B) Example of the most significant open region in DMC pancreases compared with MC pancreases (control). (C and D) Schematic representation of the genomic loci of the most significantly open regions (in red) in DMC pancreases compared with MC pancreases, with LTR elements from RepeatMasker depicted. DAR, differentially accessible region. (E) Proportion of open regions identified by ATAC-seq that contain ERVs and differentially accessible regions within 10 kb of an ERV. (F) Global analysis of transposable elements in DMC pancreases compared with MC pancreases (controls) depicted as a proportion of the windows within the called peak or region that were open. FDR, false discovery rate.
Because of the strong association between Daxx and H3.3 and heterochromatin, we next focused on differentially open regions in the DMC pancreas compared with the MC pancreas (table S3). The first and fourth most significant regions, 3.2 kb (false discovery rate = 1.08 × 10−8, Fig. 4B) and 2.0 kb (false discovery rate = 1.90 × 10−7), respectively, flank an approximately 5-kb region of chromosome 4qD2.2 containing the ERV RLTR4_MM-int with RLTR4_Mm LTRs (Fig. 4C). Daxx-mediated H3.3 deposition was previously linked to ERV silencing in mouse embryonic stem cells in vitro (8), and our data indicate that this function is conserved in somatic cells in vivo. In addition, the second most significant open region was just downstream of another RLTR4_Mm-int and an adjacent MuLV-int element, this time contained in an intron of the Cep85 gene (Fig. 4D and table S3). Five additional significantly open regions were also near these ERV sequences (Fig. 4D). We next manually evaluated all open regions for proximity to ERVs, and we found that 41 of 136 (30.1%) of the open regions contained an ERV sequence, and an additional 23 of 136 (16.9%) were within 10 kb of ERVs (Fig. 4E and table S3).
Because ERV sequences occur in many, sometimes thousands, copies in the genome, they present a computational challenge as short sequencing reads of these repetitive regions cannot uniquely map and are generally excluded from the analysis. This likely explains why our analysis captured the regions flanking the ERV, but not the ERV itself. To address this limitation and directly evaluate chromatin accessibility of transposable elements throughout the genome, we assembled a mouse transposable element reference genome with sequences representing all 219 elements annotated in Dfam, an open collection of DNA transposable element sequence alignments (39). These 219 elements collectively encompass ERV, LINE (long interspersed nuclear element), SINE (short interspersed nuclear element), and a small number of satellite repeats. We then repeated the ATAC-seq analysis pipeline comparing caerulein-treated DMC to MC pancreases, but this time, we mapped the reads specifically to these transposable element sequences, allowing for a genome-wide but not locus-specific analysis. In total, 213 peaks or regions at least 200 bp in length were called in this analysis. Four regions in ERVs were found to be differentially open in DMC pancreases (unadjusted P < 0.05; Fig. 4F), including one region in MuLV-int that was highly significant (false discovery rate = 0.0060; table S3 and Fig. 4F). Collectively, these ATAC-seq analyses demonstrate that Daxx loss alters chromatin accessibility near ERVs in vivo.
Daxx-dependent gene expression changes are associated with ERVs
We next wanted to specifically investigate our transcriptome data for associations with ERV dysregulation. To identify genes that associate the strongest with Daxx loss in the caerulein-treated pancreas, we compared the DEGs identified through the DC versus WT and DMC versus MC analyses and identified nine common genes, significantly more than would be expected by chance (hypergeometric P = 9.88 × 10−7; Fig. 5A and fig. S4). The differential expression was significantly correlated for these nine genes (Pearson correlation R2 = 0.9768, P < 0.0001; Fig. 5B). The most significantly differentially expressed of the overlapping genes was Bglap3, which showed the most marked up-regulation with the combined loss of Daxx and Men1 (Fig. 5C). IAP-d-int elements revealed an expression pattern that paralleled Bglap3 (Fig. 5D), indicating coordinate regulation, similar to what we observed in the basal context (Fig. 1, F to I) and what has previously been reported following loss of H3K9me3 due to either Zfp932 or Setdb1 depletion (17, 40). Of the other eight genes, five showed a consistent pattern of up-regulation with loss of Daxx: Ajuba, Neto1, Pcdh11x, Sult5a1, and Cd209a. All five of these genes have an ERV, with retained internal proviral sequences, within 30 kb of the gene, and, in four of the five, the ERV is within only 10 kb (table S4).
Fig. 5. Daxx-dependent gene expression changes are associated with ERV dysregulation.
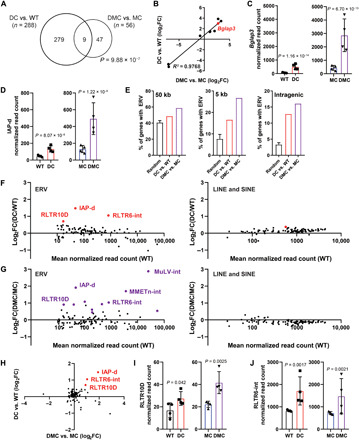
(A) Venn diagram depicting overlap of significant DEGs between mouse genotypes. Hypergeometric P = 9.88E-07. (B) Correlated expression (R2 = 0.9768) of the nine common Daxx-dependent gene expression changes in (A), with Bglap3 denoted in red. (C) Bglap3 normalized read counts from RNA-seq data (mean ± SD). Unadjusted P values are indicated. (D) Normalized IAP-d counts from RNA-seq analysis (mean ± SD). Unadjusted P values are indicated. (E) Percentage of DEGs with associated ERVs, compared with three randomly generated gene lists (mean ± SD). (F and G) Differential expression of transposable element families from RNA-seq data. Data points in red or purple reflect increased expression with an unadjusted P < 0.05. (H) Correlated ERV expression between DC versus WT and DMC versus MC pancreases. (I and J) Normalized RLTR10D and RLTR6-int counts from RNA-seq analysis. Unadjusted P values are indicated.
To evaluate whether our DEG sets were globally enriched for genes with nearby ERV elements, we compared our data with three randomly generated gene sets (Random Gene Set Generator, www.molbiotools.com) containing 100 genes. We manually annotated ERVs with internal sequences up to 50 kb from the gene (table S4). The proportion of genes with an annotated ERV was higher in our DEGs compared to the randomly generated gene sets (Fig. 5E). This was especially notable at very close distances to the gene (<5 kb). Intragenic ERVs were present in 12.8% of the genes differentially expressed when comparing DC mice with WT mice and 16.1% of the genes differentially expressed between DMC and MC mice, compared with only 3.3% of the genes in the randomly generated gene sets. Our combined ATAC-seq and RNA-seq analyses strongly indicate that dysregulation of ERVs occurs downstream of Daxx loss, which subsequently results in robust gene expression changes following inflammatory stress. Combined, these data demonstrate that a major function of Daxx is to regulate the epigenome and that dysregulation of ERVs leads to the coordinate dysregulation of protein-coding genes in vivo.
Last, we globally interrogated our transcriptome data for differential expression of transposable elements. As in our analysis of the ATAC-seq data, we used the assembled list of transposable elements from Dfam to map RNA-seq reads, thereby giving a global (not locus-specific) quantification of transposable element expression. We evaluated transposable element expression in DC versus WT and DMC versus MC mice, focusing on elements with a mean normalized read count in WT mice greater than 10 (Fig. 5, F and G, and table S5). The expression of a subset of ERVs was increased by Daxx loss, whereas LINE and SINE expression remained unchanged, indicating that this dysregulation is specific to ERVs. In the DC pancreas compared with WT, there were three ERVs with increased expression following Daxx loss (IAP-d, RLTR10D, and RLTR6-int, unadjusted P < 0.05; Fig. 5, D, F, and H to J). Consistently, Obp2b was the most significant DEG in DC mice treated with caerulein (log2FC = 3.87, Padj = 2.07 × 10−16), and it had an RLRT6-int element less than 10-kb upstream of the gene. The number of ERVs up-regulated with Daxx loss was even higher in the DMC versus MC comparison. The same three ERVs from the DC versus WT comparison were up-regulated, as well as eight additional elements (unadjusted P < 0.05; Fig. 5, G to J, and table S5). MuLV-int also demonstrated significantly increased accessibility from our ATAC-seq data, suggesting that MuLV-int elements are strongly dysregulated in this context. Combined, these data further indicate that ERV dysregulation occurs downstream of Daxx loss in vivo.
Transcriptome data from human PanNETs supports coordinate dysregulation of ERVs and protein-coding genes
Transposable elements, including ERVs, evolved independently and are not conserved between mouse and human germlines, with different elements integrating in different regions of the genome. We therefore wanted to determine whether the mechanisms downstream of Daxx loss and the coordinate dysregulation of ERVs and coding genes might be conserved. We turned to a published study of human PanNETs, which included RNA-seq data for 33 samples (21). Because we cannot yet fully appreciate the contributions of Atrx to our phenotypes and mechanisms in the mouse, we excluded the three samples with ATRX mutations (SA514934, SA506710, and SA506700) and one of two samples (SA528774) from a single donor. We then conducted a differential gene expression analysis between the five tumors with DAXX mutations and the 24 tumors that were WT for DAXX (Fig. 6A). Despite the relatively low sample size in this analysis, we identified four genes with significant gene expression changes in DAXX mutant PanNETs compared with controls (Fig. 6B and table S6). As a positive control, DAXX expression was significantly reduced (log2FC = −1.729, Padj = 0.00114) in DAXX mutant tumors. This is consistent with published data showing that DAXX mutant tumors are negative for DAXX protein according to immunohistochemistry (5). Two genes were found to be significantly up-regulated in DAXX mutant tumors: FABP3 (log2FC = 6.784, Padj = 1.03 × 10−7) and SERINC2 (log2FC = 3.97, Padj = 0.00131). These genes are located adjacent to each other on chromosome 1p35.2, and there is a large ERV locus just 18.8 kb downstream of SERINC2 (Fig. 6C). The ERV locus is nearly 20 kb in length and contains LTRs (LTR10A and LTR10E) and internal proviral sequences (HERVIP10F-int; Fig. 6C). ENCODE (Encyclopedia of DNA Elements) data showed that this region is marked by the repressive H3K9me3 heterochromatin mark in several cancer cell lines (41). A recent study in lung adenocarcinoma identified HERVIP10F-int and SERINC2 as a differentially expressed transposable element-gene pair, in which both the ERV and the adjacent gene were up-regulated (42). These data indicate the presence of coordinate regulation and the potential for HERVIP10F derepression to affect the expression of nearby genes, much like the example of IAP-d-int and Bgalp3 from our mouse data. These data suggest that the mechanisms that we have identified downstream of Daxx loss in our mouse model may be conserved in human PanNETs and that transposable element derepression may contribute to tumorigenesis.
Fig. 6. Human PanNET RNA-seq data support dysregulation of ERVs and protein-coding genes.
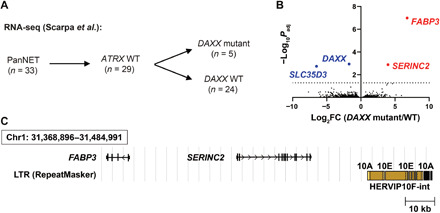
(A) Schematic representation of human PanNET RNA-seq analysis. (B) Volcano plot highlighting the top 1000 DEGs in DAXX mutant human PanNETs, with four genes reaching statistical significance. Red indicates up-regulated genes and blue indicates down-regulated genes. (C) Schematic representation of the genomic locus containing the significantly up-regulated genes, SERINC2 and FABP3, and adjacent ERV, HERVIP10F-int (indicated in brown) with LTRs 10A and 10E (indicated in yellow).
DISCUSSION
Genetic mutations frequently interact with the environment and other genetic alterations to promote disease. This is especially true for mutations that affect the chromatin landscape, and 50% of human cancers have genetic mutations in epigenetic regulators (1, 2). To understand the physiological importance of Daxx, a chaperone for the H3.3 histone variant, we have generated a conditional mouse model and used it to extensively interrogate the transcriptome and biology downstream of Daxx loss. Combined, our findings demonstrate that Daxx deletion creates a permissive chromatin state that cooperates with environmental stress (inflammation) and other genetic lesions (Men1 loss) to affect the transcriptional profile and cell state, which ultimately impairs pancreas recovery in vivo. The transcriptional changes are associated with dysregulation of ERVs, thereby expanding our understanding of the somatic regulation of ERVs beyond DNA and histone methylation in vivo, and implicating ERV dysregulation in pancreatic neuroendocrine tumorigenesis.
Chromatin is maintained by a complex interplay between DNA methylation, histone posttranslational modification, and histone variants, which cooperate to shape and restrict the transcriptional landscape of specific cell types. Alterations in the epigenetic landscape can provide a more permissive (or less restricted) transcriptional state. The concept of permissive chromatin was recently supported by a study evaluating the role of Setdb1 in silencing ERVs in mouse embryo fibroblasts (40). Specifically, the data suggest that dysregulation of histone methylation alone is insufficient to drive robust transcriptional changes; however, the altered chromatin landscape can cooperate with context-dependent transcriptional factors to significantly affect the expression of ERVs. The basal overexpression of Bglap3 in Daxx-depleted pancreases suggests epigenetic dysregulation in our model; however, we observed robust phenotypic and transcriptomic changes only in the context of tissue stress, suggesting a primed state that is waiting for a signal. These findings further suggest that proteins that are normally induced in response to injury may affect ERV expression or promoter/enhancer activity. Our RNA-seq data showed up-regulation of five pancreas cell type–specific transcription factors (table S2). This is an intriguing model to explain the specificity of DAXX mutations in human PanNETs, implying that epigenetic dysregulation may cooperate with islet-specific transcriptional regulation to promote tumorigenesis.
In addition to creating a more permissive transcriptional landscape, mutations that alter chromatin maintenance mechanisms can have profound biological consequences, including giving cells the capacity to alter their state. A classic example of this is polycomb protein mutations in Drosophila, which cause transdifferentiation across cell lineages (43). Deposition of the histone variant H3.3 has previously been associated with cell fate transitions (44). Loss of H3.3 early during reprogramming of fibroblasts to induced pluripotent stem cells enhanced the efficiency of the process, whereas H3.3 loss late in the process of differentiation results in an impaired ability to establish a new cell state. Our work suggests that Daxx restricts cellular plasticity in vivo. In the pancreas, cellular plasticity is exemplified by acinar-to-ductal metaplasia. Following inflammatory stress, loss of Daxx causes an increase in the expression of ductal genes, compensatory ductal proliferation, and a failure to restore the normal acinar compartment in the background of Men1 deficiency. These results parallel the H3.3 studies in induced pluripotent stem cells.
Additional work to understand the contributions of Men1 loss will be important because DAXX and MEN1 mutations frequently co-occur in PanNETs. Although Men1 loss may merely increase tissue damage in response to treatment with caerulein, as we have reported (32), there could also be a mechanistic link between the two mutations. Like Daxx, Men1 has several reported functions. Men1 regulates the epigenome as part of the MLL histone methyl transferase complex, which promotes the activating H3K4me3 mark (23). The loss of Men1 could thereby further perturb the chromatin landscape. In addition, Men1 can negatively regulate the activity of transcription factors, including Jund (45). This provides a simple model of cooperation in which an aberrantly active transcription factor could be coupled to an altered or permissive chromatin landscape.
In addition to understanding genetic cooperation between Daxx and Men1, it will also be of interest to explore how Daxx loss affects other mechanisms that maintain heterochromatin and the silencing of ERVs. Our results are similar to those studies evaluating loss of H3K9me3 (40, 46, 47). Depending on the cell type, Setdb1 depletion causes specific families of ERVs to be derepressed, as we also observed from our RNA-seq analysis of caerulein-treated pancreases. These data further suggest that additional cell-intrinsic factors contribute to the transcriptional outputs and downstream biology. In addition to histone methylation, DNA methylation also plays an important role in establishing and maintaining heterochromatin and ERV silencing. Comprehensive epigenomic profiling and integration will be essential to explore and understand the chromatin-based alterations downstream of Daxx loss. In addition, as longer read sequencing technologies advance, we will be in a stronger position to interrogate locus-specific transposable elements and their epigenetic regulation.
Last, this work has provided insights into the molecular mechanisms downstream of Daxx loss in vivo. Although our data strongly implicate the dysregulation of ERV silencing as an important biological consequence downstream of Daxx loss, transposable elements have evolved independently in the mouse and human genomes. Hence, the specific elements and their localization in the genome are not conserved between species. Therefore, the mouse may not be a faithful model for PanNETs with DAXX mutations. Moving forward, understanding how Daxx loss affects chromatin organization, transposable element silencing, and subsequent gene expression in humans will be essential to advance our understanding of relevant disease pathogenesis. We analyzed publicly available human PanNET RNA-seq data and identified significant up-regulation of two genes, SERINC2 and FABP3. These are adjacent genes nearby a large ERV locus. Moreover, the coordinate dysregulation of HERVIP10F-int and SERINC2 has been previously reported in lung cancer (42), and derepression of HERVIP10F-int was also found in HCC1944 breast cancer cells compared with human mammary epithelial cell controls (48). Even the small number of tumors analyzed in our study implicates transposable element derepression driving altered expression of endogenous genes in human PanNETs.
MATERIALS AND METHODS
Mice
All mouse experiments were performed in compliance with the National Institute of Health guidelines for animal research and approved by The University of Texas MD Anderson Cancer Center Institutional Animal Care and Use Committee. Men1 conditional knockout mice (The Jackson Laboratory, stock no. 005109), Atrx conditional knockout mice, and Pdx1-CreTg (The Jackson Laboratory, stock no. 014647) mice were previously described (14, 25, 49). Mice were maintained on a mixed background (C57BL/6, FVB, 129). Unless otherwise stated, Cre-negative littermates were used as functionally WT controls throughout these studies.
To generate the conditional Daxxfl allele, we used sequence surrounding the mutant loxP site in the Daxxtm2Led mice (Jackson Laboratory, stock no.008669) to score possible single-guide RNA (sgRNA) sequences using the CRISPR Design Platform (crispr.mit.edu). Our approach used a protospacer adjacent motif (PAM) sequence 15 bp upstream of the loxP mutation, within the retained flippase recognition target (FRT) site, and a single-stranded donor oligonucleotide that introduces a BamH1 sequence in place of the six nucleotides immediately upstream of the PAM sequence (fig. S1A). This allowed us to use restriction digest to screen pups for efficient homology-directed repair, and it destroyed the n20 sequence to prevent retargeting of the allele. The injection solution contained Cas9 mRNA (5 ng/μl) (PrecisionX hspCas9 SmartNuclease mRNA, System Biosciences), sgRNA (2.5 ng/μl), and single-stranded donor oligonucleotides (5 ng/μl), prepared in tris-EDTA buffer (5 mM tris, 0.1 mM EDTA). This was injected into the pronucleus of 200 to 250 Daxxtm2Led/+ zygotes, which were subsequently implanted into pseudo-pregnant recipient WT females (20 to 25 per mouse). All injections and implantations were performed by the MD Anderson Genetically Engineered Mouse Facility. In all, 10 mice were obtained and screened. We observed nonhomologous end-joining in two mice (20%), and homology-directed repair in four mice (40%). Notably, three of four of these mice incorporated the BamH1 sequence but not the repaired loxP sequence, demonstrating that homology-direct repair can occur with as little as 25 bp of homology. We obtained one mouse with homology-directed repair that incorporated both the BamH1 sequence and the fixed loxP sequence (fig. S1, B and C). We confirmed germline transmission and subsequently screened the seven highest scoring potential off target sites as determined by the CRISPR Design Platform using PCR and Sanger sequencing and found no evidence of off-target cutting at these loci (data not shown). We then backcrossed for two generations to WT C57BL/6 mice.
Caerulein-induced chronic pancreatitis
Chronic pancreatitis was induced using established protocols (31). Caerulein (MedChemExpress) was reconstituted in sterile phosphate-buffered saline. Adult mice (6 to 10 weeks old) were injected with caerulein (individual dose, 50 μg/kg), given as four hourly intraperitoneal injections, three times per week for three consecutive weeks.
Histology and immunohistochemistry
Tissues harvested from mice were fixed in 10% neutral-buffered formalin and embedded in paraffin. Four-micrometer sections were stained with hematoxylin and eosin and examined using light microscopy. Tissue processing, paraffin embedding, sectioning, and hematoxylin and eosin staining were performed by the MD Anderson Department of Veterinary Medicine and Surgery Histology Laboratory. Immunohistochemistry was performed using standard methods, and tissue sections were stained with antibodies against CK19 (ab133496, Abcam) and CD45 (550539, BD Pharmingen). Visualization was performed using avidin-biotin complex and DAB (3,3’-diaminobenzidine) kits (Vector Laboratories) and slides were counterstained with hematoxylin.
Image quantification
Acinar-to-ductal metaplasias and islets were quantified from whole pancreas sections scanned on an Aperio ScanScope XT (Leica Biosystems Inc.) and manually annotated using Aperio ImageScope software (Leica Biosystems Inc.). The percentage of total pancreas areas was calculated for each sample.
Western blot analysis
Tissue samples were flash-frozen, pulverized, and lysed in SDS lysis buffer [1% SDS, 6.5 mM tris-HCl (pH 6.8), 25% glycerol, 10% β-mercaptoethanol]. Protein extracts were separated by SDS–polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes, and probed with antibodies against Daxx (M-112, Santa Cruz Biotechnology) and vinculin (V1931, Sigma-Aldrich). Proteins were visualized using Li-COR secondary antibodies and scanned on a Li-COR Odyssey Imager.
RNA isolation
Flash-frozen tissue was pulverized and homogenized in TRIzol RNA isolation reagent (Invitrogen). RNA was then purified as per manufacturer’s protocols or using the RNeasy Mini Kit (Qiagen).
RNA-seq and analysis
Barcoded, Illumina-compatible, strand-specific total RNA libraries were prepared using the TruSeq Stranded Total RNA Sample Preparation Kit (Illumina). Briefly 250 ng of deoxyribonuclease I–treated total RNA was depleted of cytoplasmic and mitochondrial ribosomal RNA using Ribo-Zero Gold (Illumina). After purification, the RNA was fragmented using divalent cations and double-stranded complementary DNA (cDNA) was synthesized using random primers. The ends of the resulting double-stranded cDNA fragments were repaired, 5′-phosphorylated, and 3′-A–tailed, and then Illumina-specific indexed adapters were ligated. The products were purified and then enriched with 12 cycles of PCR to create the final cDNA library.
The libraries were quantified using the Qubit double-stranded DNA HS Assay (Thermo Fisher Scientific), and then 16 libraries were multiplexed per pool. The library pool was quantified by quantitative PCR using the KAPA Library Quantification Kit (KAPABiosystems) and assessed for size distribution using the Fragment Analyzer (Advanced Analytical) and then sequenced with the Illumina HiSeq 4000 sequencer using the 76-nucleotide paired-end format. For the basal analysis, four mice of each genotype (eight in total) were sequenced in half of a sequencing lane. For the caerulein-induced gene expression analyses, 16 samples (four mice for each of four genotypes) per lane were sequenced on two lanes to increase sequence depth.
STAR (version 2.6.0b) was used to align reads to the GRCm38.p6 reference genome (50). Samtools (version 1.8) was used to sort, convert between formats, and calculate mapping statistics (51). FastQC (0.11.5) was used to check for FASTQ read qualities. Gene annotation was carried out using GENCODE M19 (Ensembl 94) annotation, which was downloaded from the GENCODE project (52). Aligned reads were summarized at the gene level using high-throughput sequencing count (53). Differential gene expression was determined using R (3.5.1) and Bioconductor package DESeq2 (54). Read count was first prefiltered to remove extremely low expressed genes. DEDeq2 was then used to carry out read count filtering, normalization, dispersion estimation, and identification of differential expression. DESeq2 modeled the counts using a negative binomial distribution, followed by the Walk test. The final adjusted P value was obtained using the Benjamini and Hochberg method.
ATAC-seq and analysis
Flash-frozen pancreatic tissue was pulverized and used for ATAC-seq following the OMNI-ATAC protocol (38). Briefly, tissue was added to a prechilled Dounce Homogenizer with cold homogenization buffer. Nuclei were pelleted and purified using an OptiPrep gradient and then collected. A total of 50,000 nuclei were used in the transposition reaction as per standard protocol using TDE1 (tagmentation enzyme, Illumina). DNA was purified using a Zymo DNA Clean and Concentrator kit. Barcodes were added through PCR amplification. Following library quantification, samples were pooled with all eight samples sequenced on a single lane of an Illumina HiSeq 4000, with 76-nucleotide paired-end sequencing.
Quality assessment of sequencing data was performed using FastQC (0.11.5) to check FASTQ read quality. Adapter sequences at the 3′ end of reads were removed using trim_galore (0.4.5) and cutadapt (1.18). Reads were aligned to the GRCm38.96 reference genome using Bowtie2 (2.3.4.2) (55). Samtools (1.8) was used to sort, convert between formats, and filter alignment reads (51). Chromosome M (mitochondria) was removed from the alignments, and duplicated reads were also removed using PICARD (2.9.0). Reads that were mapped to multiple locations were removed, and only reads that mapped to unique chromosomal locations and paired properly were retained. Peaks were called using MACS2 (2.1.2) (56), and annotation was carried out using HOMER (4.10) (57). To count reads, we used paired-end mode with a maximum fragment length of 800 bp, a window size of 20 nucleotides, and a spacing of 20. When merging adjacent windows into the same genomic region, a cutoff of 10 bp was used. Windows that were less than 10 bp apart were considered adjacent and were grouped into the same cluster. To identify differentially accessible chromatin regions, we used the R package csaw (1.16.1), which carried out a window-based approach to compare between groups of samples (58).
Transposable element analysis of ATAC-seq and RNA-seq
The sequences for each of the transposable element families were extracted from Dfam, using the chromosome location of the nonredundant element with the smallest e-value as the reference sequence. A reference genome for transposable elements was then assembled specifically for this project, and sequencing reads were aligned to this genome using Bowtie (2.3.4.2). Analysis followed the same RNA-seq and ATAC-seq pipelines as described above.
Statistical analysis
Data are presented as means ± SD. All statistical analyses were performed using GraphPad Prism 6 software, and P < 0.05 was considered statistically significant. Comparisons between two groups were made using the Student’s t test, and comparisons among multiple groups were made using analysis of variance with the Dunnett’s multiple comparisons test to compare experimental samples to WT controls.
Supplementary Material
Acknowledgments
The Atrx conditional knockout mouse was provided by R. Gibbons. We thank S. Arur, N. Copeland, N. Jenkins, and A. Maitra, as well as members of the Lozano laboratory for helpful discussions. We also acknowledge Scientific Publications, Research Medical Library at MD Anderson for editing this manuscript. Funding: This research was supported by grants from the Neuroendocrine Tumor Research Foundation and NIH (NIH/NCI R21 CA208463) to G.L. and a fellowship from the Canadian Institutes of Health Research to A.R.W. Sequencing experiments were performed with The MD Anderson Advanced Technology Genomics Core and gene targeting to generate the Daxxfl allele was conducted by The MD Anderson Genetically Engineered Mouse Facility. Both core facilities were supported, in part, by a Cancer Center Support Grant from the National Cancer Institute (CA016672). Author contributions: A.R.W. and G.L. conceived the initial concept, developed the project, interpreted data, and wrote the manuscript. Experiments were conducted by A.R.W., C.S., S.M.M., G.P.C., and N.K.A. All computational analyses were done by Y.Q., with supervision from X.S. F.M., M.P.K., and M.C.B. contributed to experimental design and interpretation. J.S.E. performed pathological evaluation of mouse tissue samples. All authors reviewed and provided suggestions on the manuscript. Competing interests: G.L. is a current member of the Board of Scientific Advisors for the Neuroendocrine Tumor Research Foundation. N.K.A.’s contributions were made while affiliated with The University of Texas MD Anderson Cancer Center. As per the requirement of the journal, he is currently an employee of AstraZeneca and may or may not own stock options. The other authors declare no competing interests. Data and materials availability: RNA-seq and ATAC-seq data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database with accession nos. GSE145175, GSE150017, and GSE150011.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/32/eaba8415/DC1
REFERENCES AND NOTES
- 1.You J. S., Jones P. A., Cancer genetics and epigenetics: Two sides of the same coin? Cancer Cell 22, 9–20 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shen H., Laird P. W., Interplay between the cancer genome and epigenome. Cell 153, 38–55 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwartzentruber J., Korshunov A., Liu X.-Y., Jones D. T. W., Pfaff E., Jacob K., Sturm D., Fontebasso A. M., Quang D.-A. K., Tönjes M., Hovestadt V., Albrecht S., Kool M., Nantel A., Konermann C., Lindroth A., Jäger N., Rausch T., Ryzhova M., Korbel J. O., Hielscher T., Hauser P., Garami M., Klekner A., Bognar L., Ebinger M., Schuhmann M. U., Scheurlen W., Pekrun A., Frühwald M. C., Roggendorf W., Kramm C., Dürken M., Atkinson J., Lepage P., Montpetit A., Zakrzewska M., Zakrzewski K., Liberski P. P., Dong Z., Siegel P., Kulozik A. E., Zapatka M., Guha A., Malkin D., Felsberg J., Reifenberger G., von Deimling A., Ichimura K., Collins V. P., Witt H., Milde T., Witt O., Zhang C., Castelo-Branco P., Lichter P., Faury D., Tabori U., Plass C., Majewski J., Pfister S. M., Jabado N., Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Behjati S., Tarpey P. S., Presneau N., Scheipl S., Pillay N., Van Loo P., Wedge D. C., Cooke S. L., Gundem G., Davies H., Nik-Zainal S., Martin S., Laren S. M., Goody V., Robinson B., Butler A., Teague J. W., Halai D., Khatri B., Myklebost O., Baumhoer D., Jundt G., Hamoudi R., Tirabosco R., Amary M. F., Futreal P. A., Stratton M. R., Campbell P. J., Flanagan A. M., Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat. Genet. 45, 1479–1482 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiao Y., Shi C., Edil B. H., de Wilde R. F., Klimstra D. S., Maitra A., Schulick R. D., Tang L. H., Wolfgang C. L., Choti M. A., Velculescu V. E., Diaz L. A. Jr., Vogelstein B., Kinzler K. W., Hruban R. H., Papadopoulos N., DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 331, 1199–1203 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldberg A. D., Goldberg A. D., Banaszynski L. A., Noh K.-M., Lewis P. W., Elsaesser S. J., Stadler S., Dewell S., Law M., Guo X., Li X., Wen D., Chapgier A., De Kelver R. C., Miller J. C., Lee Y.-L., Boydston E. A., Holmes M. C., Gregory P. D., Greally J. M., Rafii S., Yang C., Scambler P. J., Garrick D., Gibbons R. J., Higgs D. R., Cristea I. M., Urnov F. D., Zheng D., Allis C. D., Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 140, 678–691 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heaphy C. M., de Wilde R. F., Jiao Y., Klein A. P., Edil B. H., Shi C., Bettegowda C., Rodriguez F. J., Eberhart C. G., Hebbar S., Offerhaus G. J., Lendon R. M., Rasheed B. A., He Y., Yan H., Bigner D. D., Oba-Shinjo S. M., Marie S. K. N., Riggins G. J., Kinzler K. W., Vogelstein B., Hruban R. H., Maitra A., Papadopoulos N., Meeker A. K., Altered telomeres in tumors with ATRX and DAXX mutations. Science 333, 425 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elsässer S. J., Noh K.-M., Diaz N., Allis C. D., Banaszynski L. A., Histone H3.3 is required for endogenous retroviral element silencing in embryonic stem cells. Nature 522, 240–244 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mahmud I., Liao D., DAXX in cancer: Phenomena, processes, mechanisms and regulation. Nucleic Acids Res. 47, 7734–7752 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Michaelson J. S., Bader D., Kuo F., Kozak C., Leder P., Loss of Daxx, a promiscuously interacting protein, results in extensive apoptosis in early mouse development. Genes Dev. 13, 1918–1923 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wasylishen A. R., Estrella J. S., Pant V., Chau G. P., Lozano G., Daxx functions are p53-independent in vivo. Mol. Cancer Res. 16, 1523–1529 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Puri S., Folias A. E., Hebrok M., Plasticity and dedifferentiation within the pancreas: Development, homeostasis, and disease. Cell Stem Cell 16, 18–31 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bastidas-Ponce A., Scheibner K., Lickert H., Bakhti M., Cellular and molecular mechanisms coordinating pancreas development. Development 144, 2873–2888 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Hingorani S. R., Petricoin E. F. III, Maitra A., Rajapakse V., King C., Jacobetz M. A., Ross S., Conrads T. P., Veenstra T. D., Hitt B. A., Kawaguchi Y., Johann D., Liotta L. A., Crawford H. C., Putt M. E., Jacks T., Wright C. V. E., Hruban R. H., Lowy A. M., Tuveson D. A., Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4, 437–450 (2003). [DOI] [PubMed] [Google Scholar]
- 15.Desbois C., Hogue D. A., Karsenty G., The mouse osteocalcin gene cluster contains three genes with two separate spatial and temporal patterns of expression. J. Biol. Chem. 269, 1183–1190 (1994). [PubMed] [Google Scholar]
- 16.Groh S., Schotta G., Silencing of endogenous retroviruses by heterochromatin. Cell. Mol. Life Sci. 74, 2055–2065 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ecco G., Cassano M., Kauzlaric A., Duc J., Coluccio A., Offner S., Imbeault M., Rowe H. M., Turelli P., Trono D., Transposable elements and their KRAB-ZFP controllers regulate gene expression in adult tissues. Dev. Cell 36, 611–623 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johanning G. L., Malouf G. G., Zheng X., Esteva F. J., Weinstein J. N., Wang-Johanning F., Su X., Expression of human endogenous retrovirus-K is strongly associated with the basal-like breast cancer phenotype. Sci. Rep. 7, 41960 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cancer Genome Atlas Research Network , Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 32, 185–203.e13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hingorani S. R., Wang L., Multani A. S., Combs C., Deramaudt T. B., Hruban R. H., Rustgi A. K., Chang S., Tuveson D. A., Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7, 469–483 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Scarpa A., Chang D. K., Nones K., Corbo V., Patch A.-M., Bailey P., Lawlor R. T., Johns A. L., Miller D. K., Mafficini A., Rusev B., Scardoni M., Antonello D., Barbi S., Sikora K. O., Cingarlini S., Vicentini C., Kay S. M., Quinn M. C. J., Bruxner T. J. C., Christ A. N., Harliwong I., Idrisoglu S., Lean S. M., Nourse C., Nourbakhsh E., Wilson P. J., Anderson M. J., Fink J. L., Newell F., Waddell N., Holmes O., Kazakoff S. H., Leonard C., Wood S., Xu Q., Nagaraj S. H., Amato E., Dalai I., Bersani S., Cataldo I., Dei Tos A. P., Capelli P., Davì M. V., Landoni L., Malpaga A., Miotto M., Whitehall V. L. J., Leggett B. A., Harris J. L., Harris J., Jones M. D., Humphris J., Chantrill L. A., Chin V., Nagrial A. M., Pajic M., Scarlett C. J., Pinho A., Rooman I., Toon C., Wu J., Pinese M., Cowley M., Barbour A., Mawson A., Humphrey E. S., Colvin E. K., Chou A., Lovell J. A., Jamieson N. B., Duthie F., Gingras M.-C., Fisher W. E., Dagg R. A., Lau L. M. S., Lee M., Pickett H. A., Reddel R. R., Samra J. S., Kench J. G., Merrett N. D., Epari K., Nguyen N. Q., Zeps N., Falconi M., Simbolo M., Butturini G., Van Buren G., Partelli S., Fassan M.; Australian Pancreatic Cancer Genome Initiative, Khanna K. K., Gill A. J., Wheeler D. A., Gibbs R. A., Musgrove E. A., Bassi C., Tortora G., Pederzoli P., Pearson J. V., Waddell N., Biankin A. V., Grimmond S. M., Whole-genome landscape of pancreatic neuroendocrine tumours. Nature 543, 65–71 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Milne T. A., Hughes C. M., Lloyd R., Yang Z., Rozenblatt-Rosen O., Dou Y., Schnepp R. W., Krankel C., Li Volsi V. A., Gibbs D., Hua X., Roeder R. G., Meyerson M., Hess J. L., Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc. Natl. Acad. Sci. U.S.A. 102, 749–754 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hughes C. M., Rozenblatt-Rosen O., Milne T. A., Copeland T. D., Levine S. S., Lee J. C., Hayes D. N., Shanmugam K. S., Bhattacharjee A., Biondi C. A., Kay G. F., Hayward N. K., Hess J. L., Meyerson M., Menin associates with a trithorax family histone methyltransferase complex and with the Hoxc8 locus. Mol. Cell 13, 587–597 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Shen H.-C. J., He M., Powell A., Adem A., Lorang D., Heller C., Grover A. C., Ylaya K., Hewitt S. M., Marx S. J., Spiegel A. M., Libutti S. K., Recapitulation of pancreatic neuroendocrine tumors in human multiple endocrine neoplasia type I syndrome via Pdx1-directed inactivation of Men1. Cancer Res. 69, 1858–1866 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crabtree J. S., Scacheri P. C., Ward J. M., Mc Nally S. R., Swain G. P., Montagna C., Hager J. H., Hanahan D., Edlund H., Magnuson M. A., Garrett-Beal L., Burns A. L., Ried T., Chandrasekharappa S. C., Marx S. J., Spiegel A. M., Collins F. S., Of mice and MEN1: Insulinomas in a conditional mouse knockout. Mol. Cell. Biol. 23, 6075–6085 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crabtree J. S., Scacheri P. C., Ward J. M., Garrett-Beal L., Emmert-Buck M. R., Edgemon K. A., Lorang D., Libutti S. K., Chandrasekharappa S. C., Marx S. J., Spiegel A. M., Collins F. S., A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors. Proc. Natl. Acad. Sci. U.S.A. 98, 1118–1123 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shen H.-C. J., Ylaya K., Pechhold K., Wilson A., Adem A., Hewitt S. M., Libutti S. K., Multiple endocrine neoplasia type 1 deletion in pancreatic α-cells leads to development of insulinomas in mice. Endocrinology 151, 4024–4030 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu J., Herrera P. L., Carreira C., Bonnavion R., Seigne C., Calender A., Bertolino P., Zhang C. X., α cell-specific Men1 ablation triggers the transdifferentiation of glucagon-expressing cells and insulinoma development. Gastroenterology 138, 1954–1965 (2010). [DOI] [PubMed] [Google Scholar]
- 29.Hyun J. J., Lee H. S., Experimental models of pancreatitis. Clin. Endosc. 47, 212–216 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guerra C., Schuhmacher A. J., Cañamero M., Grippo P. J., Verdaguer L., Pérez-Gallego L., Dubus P., Sandgren E. P., Barbacid M., Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 11, 291–302 (2007). [DOI] [PubMed] [Google Scholar]
- 31.McAllister F., Bailey J. M., Alsina J., Nirschl C. J., Sharma R., Fan H., Rattigan Y., Roeser J. C., Lankapalli R. H., Zhang H., Jaffee E. M., Drake C. G., Housseau F., Maitra A., Kolls J. K., Sears C. L., Pardoll D. M., Leach S. D., Oncogenic Kras activates a hematopoietic-to-epithelial IL-17 signaling axis in preinvasive pancreatic neoplasia. Cancer Cell 25, 621–637 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wasylishen A. R., Sun C., Chau G. P., Qi Y., Su X., Kim M. P., Estrella J. S., Lozano G., Men1 maintains exocrine pancreas homeostasis in response to inflammation and oncogenic stress. Proc. Natl. Acad. Sci. U.S.A. 117, 6622–6629 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muraro M. J., Dharmadhikari G., Grün D., Groen N., Dielen T., Jansen E., van Gurp L., Engelse M. A., Carlotti F., de Koning E. J. P., van Oudenaarden A., A single-cell transcriptome atlas of the human pancreas. Cell Syst. 3, 385–394.e3 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haqq J., Howells L. M., Garcea G., Metcalfe M. S., Steward W. P., Dennison A. R., Pancreatic stellate cells and pancreas cancer: Current perspectives and future strategies. Eur. J. Cancer 50, 2570–2582 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Baron M., Veres A., Wolock S. L., Faust A. L., Gaujoux R., Vetere A., Ryu J. H., Wagner B. K., Shen-Orr S. S., Klein A. M., Melton D. A., Yanai I., A single-cell transcriptomic map of the human and mouse pancreas reveals inter- and intra-cell population structure. Cell Syst. 3, 346–360.e4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krämer A., Green J., Pollard J. Jr., Tugendreich S., Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30, 523–530 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Danussi C., Bose P., Parthasarathy P. T., Silberman P. C., Van Arnam J. S., Vitucci M., Tang O. Y., Heguy A., Wang Y., Chan T. A., Riggins G. J., Sulman E. P., Lang F., Creighton C. J., Deneen B., Miller C. R., Picketts D. J., Kannan K., Huse J. T., Atrx inactivation drives disease-defining phenotypes in glioma cells of origin through global epigenomic remodeling. Nat. Commun. 9, 1057 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Corces M. R., Trevino A. E., Hamilton E. G., Greenside P. G., Sinnott-Armstrong N. A., Vesuna S., Satpathy A. T., Rubin A. J., Montine K. S., Wu B., Kathiria A., Cho S. W., Mumbach M. R., Carter A. C., Kasowski M., Orloff L. A., Risca V. I., Kundaje A., Khavari P. A., Montine T. J., Greenleaf W. J., Chang H. Y., An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hubley R., Finn R. D., Clements J., Eddy S. R., Jones T. A., Bao W., Smit A. F. A., Wheeler T. J., The Dfam database of repetitive DNA families. Nucleic Acids Res. 44, D81–D89 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kato M., Takemoto K., Shinkai Y., A somatic role for the histone methyltransferase Setdb1 in endogenous retrovirus silencing. Nat. Commun. 9, 1683 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.The ENCODE Project Consortium , An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.M. Arroyo, R. Larrosa, M. G. Claros, R. Bautista, Expression change correlations between transposons and their adjacent genes in lung cancers reveal a genomic location dependence and highlights cancer-significant genes, in Proceedings of the International Work-Conference on Bioinformatics and Biomedical Engineering (Springer International Publishing, 2019), pp. 84–92. [Google Scholar]
- 43.Lee N., Maurange C., Ringrose L., Paro R., Suppression of Polycomb group proteins by JNK signalling induces transdetermination in Drosophila imaginal discs. Nature 438, 234–237 (2005). [DOI] [PubMed] [Google Scholar]
- 44.Fang H. T., EL Farran C. A., Xing Q. R., Zhang L.-F., Li H., Lim B., Loh Y.-H., Global H3.3 dynamic deposition defines its bimodal role in cell fate transition. Nat. Commun. 9, 1537 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Agarwal S. K., Guru S. C., Heppner C., Erdos M. R., Collins R. M., Park S. Y., Saggar S., Chandrasekharappa S. C., Collins F. S., Spiegel A. M., Marx S. J., Burns A. L., Menin interacts with the AP1 transcription factor JunD and represses JunD-activated transcription. Cell 96, 143–152 (1999). [DOI] [PubMed] [Google Scholar]
- 46.Karimi M. M., Goyal P., Maksakova I. A., Bilenky M., Leung D., Tang J. X., Shinkai Y., Mager D. L., Jones S., Hirst M., Lorincz M. C., DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell 8, 676–687 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tan S.-L., Nishi M., Ohtsuka T., Matsui T., Takemoto K., Kamio-Miura A., Aburatani H., Shinkai Y., Kageyama R., Essential roles of the histone methyltransferase ESET in the epigenetic control of neural progenitor cells during development. Development 139, 3806–3816 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Hon G. C., Hawkins R. D., Caballero O. L., Lo C., Lister R., Pelizzola M., Valsesia A., Ye Z., Kuan S., Edsall L. E., Camargo A. A., Stevenson B. J., Ecker J. R., Bafna V., Strausberg R. L., Simpson A. J., Ren B., Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res. 22, 246–258 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Garrick D., Sharpe J. A., Arkell R., Dobbie L., Smith A. J. H., Wood W. G., Higgs D. R., Gibbons R. J., Loss of Atrx affects trophoblast development and the pattern of X-inactivation in extraembryonic tissues. PLOS Genet. 2, e58 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R., STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li H., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.; 1000 Genome Project Data Processing Subgroup , The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Harrow J., Frankish A., Gonzalez J. M., Tapanari E., Diekhans M., Kokocinski F., Aken B. L., Barrell D., Zadissa A., Searle S., Barnes I., Bignell A., Boychenko V., Hunt T., Kay M., Mukherjee G., Rajan J., Despacio-Reyes G., Saunders G., Steward C., Harte R., Lin M., Howald C., Tanzer A., Derrien T., Chrast J., Walters N., Balasubramanian S., Pei B., Tress M., Rodriguez J. M., Ezkurdia I., van Baren J., Brent M., Haussler D., Kellis M., Valencia A., Reymond A., Gerstein M., Guigó R., Hubbard T. J., GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Anders S., Pyl P. T., Huber W., HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Langmead B., Salzberg S. L., Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nusbaum C., Myers R. M., Brown M., Li W., Liu X. S., Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Heinz S., Benner C., Spann N., Bertolino E., Lin Y. C., Laslo P., Cheng J. X., Murre C., Singh H., Glass C. K., Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lun A. T. L., Smyth G. K., csaw: A Bioconductor package for differential binding analysis of ChIP-seq data using sliding windows. Nucleic Acids Res. 44, e45 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/32/eaba8415/DC1