

Abstract
This commentary article is co-authored by a patient with Usher syndrome type 2A. The patient kindly shares her experience of living a life marked by the severe dual sensory loss associated with Usher syndrome. The experiences of the patient are put into perspective by two ophthalmologists associated with the Ophthalmological Department at Rigshospitalet, Denmark.
Keywords: Blindness, Hearing impairment, Psychological disease burden, Usher syndrome
Plain Language Summary
In this patient–physician perspective a patient with Usher syndrome shares her experience. Usher syndrome affects both hearing and vision. The patient describes how being deprived of these two important sensory functions have formed her life choices, from travelling the world while she still had vision to choosing not to have children due to fear of passing on the disease. Most importantly, the psychological impacts of the disease are shared and put into perspective by her ophthalmologists.
Key Summary Points
| Blind patients are often also invisible to the world. |
| Understanding the patient’s perspectives can improve patient care and management. |
| Patients who manage to live a full life despite severe handicap serve as role models for other patients. |
| The psychological impact of a blinding and deafening disease is enormous. |
Patient Perspective
The course of my illness was gradual, changing and progressive. What I could do one year, I could not do the next, loss followed upon loss. It was a great mental burden and worst of all was becoming totally blind as you become completely dependent on other people when you live alone (my husband died in 2013). You have no private life when you need help to do everything. Prejudice against the blind is another mental burden as you are seen as a second-class citizen and people do not talk directly to you but to your helper.
You organize your life to accommodate your loss of function, but you are constantly thinking of how you will cope in the future. You keep living, you perform your work and you make decisions out of necessity. For example, I chose not to have children since fetal diagnostics were not yet available. I also changed my subspeciality as a doctor from Pediatrics to Psychiatry.
My special challenge has been to complete a medical specialization to Senior Specialist from 1973 to 1991. At this time, it was normal to live close to your place of work so you could get there quickly when you were in the front line of duty for acute patients. This meant that with my progressive loss of vision, I needed to move several times to obtain my basic authorization (Surgery–Psychiatry–Obstetrics and Gynecology–Medicine) until I could obtain my introductory training in Paediatrics. Once the diagnosis had been made, I started my specialist training in Pediatric Psychiatry (Introductory position plus Postgraduate position in Århus—Postgraduate position in Hillerød—first Postgraduate position in Vejle—back to first Postgraduate position at Århus University Hospital). Then followed a Senior Specialist position in Herning and then the last Senior Specialist position at Odense University Hospital. Finally, I started a private practice in Aarhus. Altogether nine moves while I was in the hospital service. It was very difficult to move from apartment to apartment and get to know a new town and a new hospital with poor vision.
The last 10 years my hearing has worsened significantly. I have new hearing aids fitted every 4 years and manage relatively well in one-on-one situations, but large crowds and background noise, such as music in shops, are really a problem and I don’t talk to anybody when I walk the streets. In my work as a therapist, I often have to ask clients to repeat what they said. I know that cochlear implants are available, but a deaf–blind friend told me it would take 2 years to learn to use them.
With regard to retinitis pigmentosa being treatable with gene therapy, I believe that regardless of whether you have always been blind or lost your sight later in life, you will always wish to regain your sight, as it is a terrible loss. Gene therapy is an exciting development, and I hope that all the genes causing this condition will eventually be discovered. I hope the condition can be eradicated, both with gene therapy and with fetal diagnostics. But spontaneous mutations will probably continue to occur.
Physician Perspective
We are grateful that Mrs. Lønborg-Møller is willing to share her experience of living with Usher type 2A syndrome with us. As an eye doctor working with severely, visually disabled patients it is important to have beacons of light showing the way forward for other patients. Mrs Lønborg-Møller is indeed such a beacon of light. She has accomplished more in her life than most people manage with normal vision, as can be seen from the resume in Table 1. It is evident from her story, that not only is she a very intelligent woman who has been able to accommodate her life to living with severe dual sensory handicaps, but the disease has also forced her to make life-altering decisions. The decisions have often been made in advance to the visual deterioration based on advice from esteemed eye doctors, such as Professor Ernst Goldschmidt and Professor Thomas Rosenberg.
Table 1.
A patient’s odyssee
| Timeline | Patient’s story | Doctor’s note |
|---|---|---|
| 1960’s | Scooter accident as streetlights were turned off at midnight. Missed ferry because of having to drive slow at night | Diagnosed with myopia |
| Mid-1970’s | Working as a doctor in Africa, notices severe night blindness | |
| Late 1970’s |
Medical training Several accidents at home; falls over bicycles and vacuum cleaner; cannot find objects (e.g. tools) Changes medical subspeciality from Paediatrics to Psychiatry because of retinal problems and fear of losing vision |
First visit to eye hospital. Chloroquin retinopathy suspected. Normal visual fields (Goldman); bi-phasic dark adaptometry. Retina without pigmentary changes, but also a few thin arterioles and macular changes (spoke-wheel pattern) |
| 1980’s | Quits job as a psychiatrist to travel the world while there is still vision. Works for a year as a doctor on a ship crossing the Atlantic Ocean and travels the Caribbean | Diagnosed with retinitis pigmentosa. Dark adaptometry showed cone response but no rod response. ERGa described with a- and b-wave present. Retina with discrete pigmentary changes, no bone spicules, thin arterioles, normal optic nerves |
| 1990’s |
Serves on the board of the Danish Association of the Blind and the Danish Retinitis Pigmentosa Association Changes career from hospital-based consultant to private practice because of difficulties reading the medical files of patients and problems navigating the corridors of the hospital Works as a consultant in paediatric cases for the Danish High Court Stops driving Several accidents, including a fall from 1.5 m into an unmarked hole resulting in a broken ankle. Ankle has been broken a total of three times Poor hearing and vision make independent travelling dangerous |
Visual acuity 20/32; Goldman visual fields 5 degrees with small peripheral islands. Dark adaptometry and ERG shows flat curves. Atrophic optic nerves; no vessels present in peripheral retina; few bone spicules; cystoid macular edema Referred for a hearing tests; results confirm Usher-type hearing loss |
| 2000– |
Mobility training Participated in Retina International World Congress in Japan and Brazil |
Visual acuity 6/12 and 6/32;, Goldman visual fields < 5 degrees. Peripheral retina with classical bone spicules. Genetic testing reveals two USH2A mutations reported by Dreyer [8] |
| 2016 |
Becomes totally blind Forced to sell vacation house in Turkey because international travel was economically unrealistic as travel companion was no longer reimbursed |
No light perception |
aThe medical records did not contain information on the first electroretinography (ECG) regarding whether it was a scotopic or photopic ERG
Unfortunately, Mrs Lønborg-Møllers story also reminds us that although visually disabled persons accommodate to their handicap, society does not as easily accommodate the visually disabled. Feeling a second-class citizen is something no one should experience, and as eye doctors we have a responsibility to tell patients and society that inferior eyesight does not mean that the person is inferior. For many patients, the symptoms of the disease, such as traffic accidents in the dark, feeling not up to the skills of teenage peers when walking home from parties in the dark, misplacing items, falling over things, among others, make a significant psychological impact and contribute to a feeling of loneliness and inferiority. Making room for patients to share their experiences can aid in guiding patients on how to avoid or manage the difficulties. Letting patients know that their experiences are shared by most other patients with severe retinal disease will help the patient understand that their experiences are caused by the disease—not because the patient is a second-class citizen.
Ms. Lønborg-Møller, and other patients with Usher syndrome have a dual sensory loss. Whereas she has lost the ability to see light, her hearing is stable using hearing aids, albeit some tones have been lost and she is not able to follow conversations from multiple persons at the same time. Whereas we do not at present have therapies to restore visual acuity, hearing aids and cochlear implants are available to assist hearing. Promising results have been reported after cochlear implantation in patients with USH2A mutations, even in an adult group ranging from 52 to 82 years of age [1].
Usher syndrome is a rare disease with a prevalence ranging from 3 to 6 in 100,000 individuals [2]. It was first described as a clinical variant of retinitis pigmentosa by the German ophthalmologist Albrecht von Graefe in 1858 [3]. The disease is named after the Scottish ophthalmologist Charles Usher, who in 1914 published his studies on the inheritance of a disease characterized by both blindness and deafness [4]. The inheritance was suggestive of a genetic disease, and today Usher syndrome is known to be caused by at least 11 genes [2, 5]. These genes code for a range of proteins, such as motor proteins, transmembrane adhesion proteins and receptors, and a common denominator is that the proteins are highly expressed in the cells of the retina and the inner ear [2, 5]. Mutations in one of these genes lead to dysfunction of the protein which in turn affects the function of the eye and the ear. Visual symptoms begin with night blindness and a loss of peripheral vision, but these later progress to loss of central vision. Impaired vestibular function of the inner ear may affect motor development. From a clinical perspective, Usher syndrome is categorized into three clinical subtypes that differ in clinical presentation and prognosis/timeline of visual impairment (Table 2; see references [2, 5, 6]). Briefly, Usher syndrome type 1 is associated with the most severe early-life affection of the eye and the ear, whereas types 2 and 3 present with a less profound affection at early age. However, with time, most patients develop blindness regardless of subtype due to retinal degradation (Fig. 1), although genetically based therapies may be available in the future [7]. Making a genetic diagnosis is therefore extremely important to ensure that the patient will have the possibility to participate in future clinical trials or have access to genetically based therapies.
Table 2.
Clinical subtypes of Usher syndrome
| Characteristic features | Clinical subtype | ||
|---|---|---|---|
| Type 1 | Type 2 | Type 3 | |
| Prevalence | 1.5 per 100.000 | 2.2 per 100.000 | 0.1 per 100.000 |
| Visual impairment onset | |||
| Night vision | First decade | Second decade | Variable, but within three decades |
| Central vision | Midlife | Midlife | Midlife |
| Auditory function | Congenitally profound deafness | Congenitally moderate-to-severe deafness | Progressive deafness in first or second decade |
| Vestibular function | Absent | Normal | Variable |
| Motor development | Delayed | Normal | Normal |
Fig. 1.
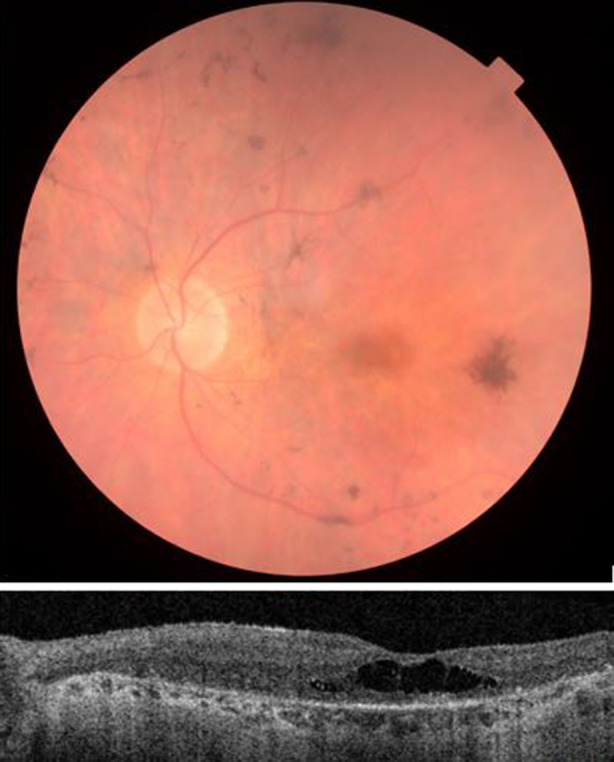
Fundus photograph (upper image) shows retinal degeneration with attenuated arterioles, pigmentary changes and central macular degeneration. Optical coherence tomography (lower image) shows retinal dystrophy with absence of distinct retinal layers, atrophic outer retina and choroid and cystoid foveal degeneration
Acknowledgements
We thank the patient for participating and allowing us to give our perspective on the disease.
Funding
No funding was obtained for this work or for the publication of this paper.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
Helene Lønborg-Møller, Yousif Subhi and Line Kessel have nothing to disclose.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Footnotes
Digital Features
To view digital features for this article go to 10.6084/m9.figshare.12206309.
References
- 1.Hartel BP, van Nierop JWI, Huinck WJ, et al. Cochlear implantation in patients with usher syndrome type IIa increases performance and quality of life. Otol Neurotol. 2017;38(6):e120–e127127. [DOI] [PubMed]
- 2.Toms M, Bitner-Glindzicz M, Webster A. Usher syndrome: a review of the clinical phenotype, genes and therapeutic strategies. Exp Rev Ophthalmol. 2015;10(3):241–256. doi: 10.1586/17469899.2015.1033403. [DOI] [Google Scholar]
- 3.von Graefe A. Exceptionelles Verhalten des Gesichtsfeldes bei Pigmententartung der Netzhaut. Archiv für Ophthalmologie. 1858;4:250–253. [Google Scholar]
- 4.Usher C. On the inheritance of Retinitis pigmentosa with notes of cases. R Lond Ophthalmol Hosp Rep. 1914;19:130–236. [Google Scholar]
- 5.Yan D, Liu XZ. Genetics and pathological mechanisms of Usher syndrome. J Hum Genet. 2010;55(6):327–335. doi: 10.1038/jhg.2010.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenberg T, Haim M, Hauch A-M, Parvinget A. The prevalence of Usher syndrome and other retinal dystrophy-hearing impairment associations. Clin Genet. 1997;51(5):314–21. [DOI] [PubMed]
- 7.Moore NA, Morral N, Ciulla T, Bracha P. Gene therapy for inherited retinal and optic nerve degenerations. Expert Opin Biol Ther. 2017;18(1):37–49. doi: 10.1080/14712598.2018.1389886. [DOI] [PubMed] [Google Scholar]
- 8.Dreyer B, Brox V, Tranebjaerg L, et al. Spectrum of USH2A mutations in Scandinavian patients with Usher syndrome type II. Hum Mutat. 2008;29(3):451. doi: 10.1002/humu.9524. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.