

Abstract
Ketamine, a dissociative anesthetic, is experiencing a clinical resurgence as a fast-acting antidepressant. In the central nervous system, ketamine acts primarily by blocking NMDA receptor currents. Although it is generally safe in a clinical setting, it can be addictive, and several of its derivatives are being investigated as preferable alternatives. 2R,6R-Hydroxynorketamine (HNK), a ketamine metabolite, reproduces some of the therapeutic effects of ketamine and appears to lack abuse liability. Here, we report a systematic investigation of the effects of HNK on macroscopic responses elicited from recombinant NMDA receptors expressed in human embryonic kidney 293 cells. We found that, like ketamine, HNK reduced NMDA receptor currents in a dose-, pH-, and voltage-dependent manner. Relative to ketamine, it had 100-fold-lower potency (46 µM at pH 7.2), 10-fold-slower inhibition onset, slower apparent dissociation rate, weaker voltage dependence, and complete competition by magnesium. Notably, HNK inhibition was fully effective when applied to resting receptors. These results revealed unexpected properties of hydroxynorketamine that warrant its further investigation as a possible therapeutic in pathologies associated with NMDA receptor dysfunction.
SIGNIFICANCE STATEMENT
NMDA receptors are excitatory ion channels with fundamental roles in synaptic transmission and plasticity, and their dysfunction associates with severe neuropsychiatric disorders. 2R,6R-Hydroxynorketamine, a metabolite of ketamine, mimics some of the neuroactive properties of ketamine and may lack its abuse liability. Results show that 2R,6R-hydroxynorketamine blocks NMDA receptor currents with low affinity and weak voltage dependence and is effective when applied to resting receptors. These properties highlight its effectiveness to a subset of NMDA receptor responses and recommend it for further investigation.
Introduction
Ketamine (KET) is a synthetic anesthetic valued for its rapid onset and short half-life. It has been in clinical use for more than 50 years, and it is generally considered safe and effective (Domino et al., 1965; Featherstone et al., 2012; Lodge and Mercier, 2015). A drawback is that its dissociative and euphoric properties make KET a popular recreational drug with potential for abuse. Further, chronic use, especially at high doses, can produce neurodegeneration and urinary tract pathologies (Morgan et al., 2004; Muetzelfeldt et al., 2008; Chan et al., 2013). Despite these concerns, its unique pharmacologic actions make it the anesthetic of choice for several patient populations, especially the critically ill (Kurdi et al., 2014; Mazzeffi et al., 2015). Therefore, KET remains one of the medications on the World Health Organization’s List of Essential Medicines, and new uses continue to be developed. Recently, KET has been investigated aggressively for its rapid antidepressant effects, especially in patients with treatment-resistant depression (Berman et al., 2000). The mechanisms by which KET exerts its numerous biologic effects remain incompletely understood and are likely complex.
Like many other drugs, KET affects several molecular targets (Sleigh et al., 2014). The anesthetic, amnesic, dissociative, and hallucinogenic effects of KET are largely mediated by fast, reversible inhibition of N-methyl-d-aspartate (NMDA) receptor currents (Conseiller et al., 1972; Anis et al., 1983; Berry et al., 1984; MacDonald et al., 1991). NMDA receptors mediate excitatory transmission in the central nervous system and have critical roles in higher brain function, such as cognition, learning, and memory. They are glutamate- and glycine-gated ion channels whose calcium-rich currents mediate synaptic transmission and plasticity in brain and spinal cord (Lang et al., 2018). Previous research has established that KET acts as a classic open-channel blocker of NMDA receptor currents (MacDonald et al., 1987, 1991; Huettner and Bean, 1988; Dilmore and Johnson, 1998; Johnson et al., 2015).
Chemically, KET is an amphipathic phencyclidine derivative with both aqueous and lipophilic actions (Emnett et al., 2016; Zanos et al., 2018). The molecule contains an asymmetric carbon atom with S(+) and R(−) configurations, and even though the two enantiomers differ somewhat in their pharmacologic profile, KET is widely used as the racemic mixture (Zeilhofer et al., 1992). Moreover, enantiomers undergo rapid acid/base equilibria in the physiologic pH range, and the charged and uncharged molecular species may have distinct biologic effects (MacDonald et al., 1991; Dravid et al., 2007). Lastly, upon administration, KET is rapidly and extensively metabolized in the liver, and several of the resulting metabolites retain biologic activity (Leung and Baillie, 1986). The extent to which these various species mediate the effects of KET on neuronal excitability and its many biologic actions is unknown.
Recent evidence suggests that the KET metabolite 2R,6R-hydroxynorketamine (HNK) may mediate some of the behavioral effects and antidepressant properties of KET (Zarate et al., 2012; Zanos et al., 2016; Suzuki et al., 2017; Pham et al., 2018). The half-life of KET metabolites (2S,6S-HNK and 2R,6R-HNK) in human plasma post–KET infusion is 16.5 and 7.7 hours, respectively (Hasan et al., 2017), and given that HNK may lack the addictive effects of KET (Zanos et al., 2016), it may be a more desirable drug. Here, we undertake a systematic examination of the effects of HNK on NMDA receptor currents and compare these with those observed for KET.
Materials and Methods
Chemicals.
All electrophysiology reagents used for this work were of >99.0% purity and were purchased from Sigma-Aldrich. Racemic R,S-ketamine (HCl injection, United States Pharmacopeia (USP), 100 mg/ml) was purchased from Vedco (St. Joseph, MO; CAS: 6740-881). 2R,6R-Hydroxynorketamine and 1,2-bis(o-aminophenoxy) ethane-N,N,N′,N′-tetraacetic acid (BAPTA) were purchased from Tocris (Bioscience, Bristol, UK; CAS: 81395-70-2 and CAS: 126150-97-8, respectively). Polyethylenimine linear, mol. wt. 25,000, was purchased from (Polysciences, Inc.). All tissue culture reagents were purchased from Gibco. HEK-293 cell cultures were not routinely tested for mycoplasma contamination.
Cells and Protein Expression.
All methods were previously described (Kussius and Popescu, 2009; Amico-Ruvio and Popescu, 2010). Briefly, rat GluN1-1a (U08261) and GluN2A (M91561.1) or GluN2B (NM012574) were expressed together with GFP from pcDNA3.1(+) in HEK-293 cells (Research Resource Identifier (RRID): CVCL_0045; CRL-1573; American Type Culture Collection). HEK-293 cells were grown to approximately 50% confluence in Dulbecco's Modified Eagle Medium (DMEM) (12100-046; Gibco) with 10% FBS (10437-028; Gibco) and 1% penicillin-streptomycin and were transiently transfected with polyethylenimine linear, mol. wt. 25,000 (Polysciences, Inc.), using a DNA ratio GluN1:GluN2:GFP (1:1:1) (Longo et al., 2013).
Electrophysiology.
Whole-cell currents were recorded from HEK-293 cells 18–48 hours after transfection. Borosilicate pipettes were pulled and polished to a resistance of 4–6 MΩ and filled with intracellular solution containing (in millimolars) 125 CsCl, 10 HEPES, and 10 1,2-bis(o-aminophenoxy) ethane-N,N,N′,N′-tetraacetic acid (BAPTA) at pH 7.2 ± 0.02 (CsOH) with an osmolarity of (275 ± 10 mOsm). Bath solution consisted of PBS supplemented with 2 mM Ca2+ and Mg2+ (Gibco: 21300-025). Extracellular solutions contained (in millimolars) 150 NaCl, 2.5 KCl, 1.0 CaCl2, 10 HEPES, 0.01 EDTA, and 0.1 glycine at pH 7.2 ± 0.1 (NaOH) with an osmolarity of (290 ± 10 mOsm). Currents were elicited by adding glutamate (1 mM) in the perfusate. HEK-293 cells were clamped at −70 mV, and solution exchange was achieved with a perfusion pencil (Automate Scientific) attached to an 8-valve pressurized system (BPS-9; ALA Scientific). Solution exchange, estimated from open tip recordings, occurred with 10%–90% rise times shorter than 250 milliseconds. Currents were amplified and filtered at 2 kHz (Axopatch 200B, 4-pole Bessel; Molecular Devices, Sunnyvale, CA), sampled at 5 kHz (Digidata, 1322A; Molecular Devices), and written into digital files with pClamp 10.5 (Molecular Devices). For all recordings, seal quality was determined by monitoring series resistance.
Unless otherwise indicated, drug effects were evaluated by applying the indicated drug concentrations to the steady-state phase of the current elicited with glutamate (1 mM), as described previously (Kotermanski and Johnson, 2009). Kinetics of inhibition onset and offset were evaluated as previously described (Parsons and Gilling, 2007; Gilling et al., 2009) by recording the time for 10%–90% change in response upon glutamate application or washout, respectively.
The degree of voltage dependence was evaluated with two protocols. A voltage-ramp approach recorded the current amplitude during a gradual change in holding voltage from −70 to +80 mV over 2 seconds in the continuous presence of drug. A sequential approach recorded the steady-state current amplitude in the presence of drug for responses elicited consecutively at membrane voltages with 20-mV increments. State dependence was evaluated with protocols described previously (Paganelli and Popescu, 2015). For each cell, currents were elicited by applying glutamate (1 mM) in the presence of or together with the drug and compared with matched no-drug controls.
Data Analyses.
ChemAxon software was used to estimate the pKa value for KET and HNK. ChemAxon provides a pKa calculator plug-in that estimates the pKa of a molecule based on partial charge distribution (Marvin 18.1.0, 2018) (ChemAxon: http://www.chemaxon.com).
Digital files encoding macroscopic current traces were selected for analysis if they included at least three sweeps. Traces were analyzed using Clampfit (Molecular Devices, Union City, CA), Origin (Microal Software, Northhampton, MA), and/or GraphPad Prism 8 software. Dose-response curves were generated by measuring current amplitudes with (Idrug) and without drug (Ictr), where each value represents the average current observed during a 2.5-second period. Values were plotted as percentage of inhibited current response, and IC50 values were calculated by fitting the equation below to these data:
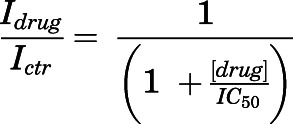 |
IC50 and nH were left as free parameters during fits and reported as means and 95% confidence intervals (CIs). Percent recovery and recovery τ from HNK block were calculated from hook tail currents observed upon simultaneous withdrawal of agonists and HNK by fitting single exponential functions to the rising phase of the current.
Response kinetics were measured as described previously (Parsons and Gilling, 2007). Briefly, onset and offset times (τ) were estimated by fitting exponential functions to the onset and offset phases of current inhibition and recovery, respectively. The inverse of time (1/τ) was plotted against concentration, and apparent rate constants (kon and koff) were calculated from fitting a linear equation to these data. The apparent dissociation constant, KD, was estimated as the ratio of koff/kon.
The degree of voltage dependence was determined in Clampfit using the Z-delta Boltzmann function (Woodhull, 1973; Hille, 1992) shown below:
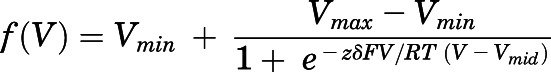 |
The maximum and minimum voltage values are represented by Vmax and Vmin, respectively; the voltage at which F(V) is half-maximal is Vmid; and the magnitude of the charge valence associated with the electric field (V) is zδ. R is the gas constant at the absolute temperature T, and F is the Faraday constant. We compared values for the slope of block by HNK and KET using an extra sum-of-squares F-test.
Statistical Analyses.
To analyze and compare electrophysiology data, we used GraphPad Prism 8.2.0 software. This work was an exploratory study, and the number of cells collected for each experiment was not predetermined before the work was done, and as such, P values reported are to be interpreted as descriptive only. To compare single-concentration HNK inhibition at three pH values, we used ANOVA Tukey’s multiple comparison test. To compare HNK IC50 and KET IC50 values at varying pH values for either N1/2A or N1/2B preparations, we reported the 95% confidence intervals. To evaluate state-dependent block by KET and HNK, we used paired t tests because both CTR and HNK dosing were done on the same cell using a linked protocol.
Results
Inhibitory Effect of HNK on NMDA Receptor Currents.
To examine the acute effects of HNK on NMDA receptor responses and relate these to the effects of the parent compound KET, we recorded whole-cell currents from HEK-293 cells expressing the adult isoform of NMDA receptors GluN1-1a/GluN2A (N1/N2A) in the absence and presence of these two drugs. Figure 1A illustrates chemical structures for HNK and KET and the predicted distribution of protonated species according to pH. KET and HNK are similar in molecular weight and total surface area (255.8 Å vs. 246.7 Å); however, there is a substantial difference in their topological polar surface area (29.1 Å vs. 63.3 Å), as illustrated in Fig. 1A. Given that, like KET, HNK displays a pKa value within the physiologic pH range (Balestrino and Somjen, 1988; Chesler and Kaila, 1992), we anticipated that, as for KET (MacDonald et al., 1991), the inhibitory effects of HNK will vary with extracellular pH and recoded whole-cell currents at several proton concentrations. We elicited inward (largely sodium) currents by applying glutamate (1 mM) in the continuous presence of glycine (0.1 µM) and perfused HNK (500 µM) after the current reached a steady-state level (Fig. 1B). In these conditions, we observed that inhibition levels by HNK were slightly higher at pH 6.8 and pH 7.2 (84% and 87%, respectively) relative to pH 7.6 (52%, n = 3, P = 0.003 and P = 0.001). These results show that a single concentration of HNK is more potent at acidic extracellular pH values, consistent with the premise that the protonated form of HNK mediates the observed acute inhibitory effect on NMDA receptor currents.
Fig. 1.
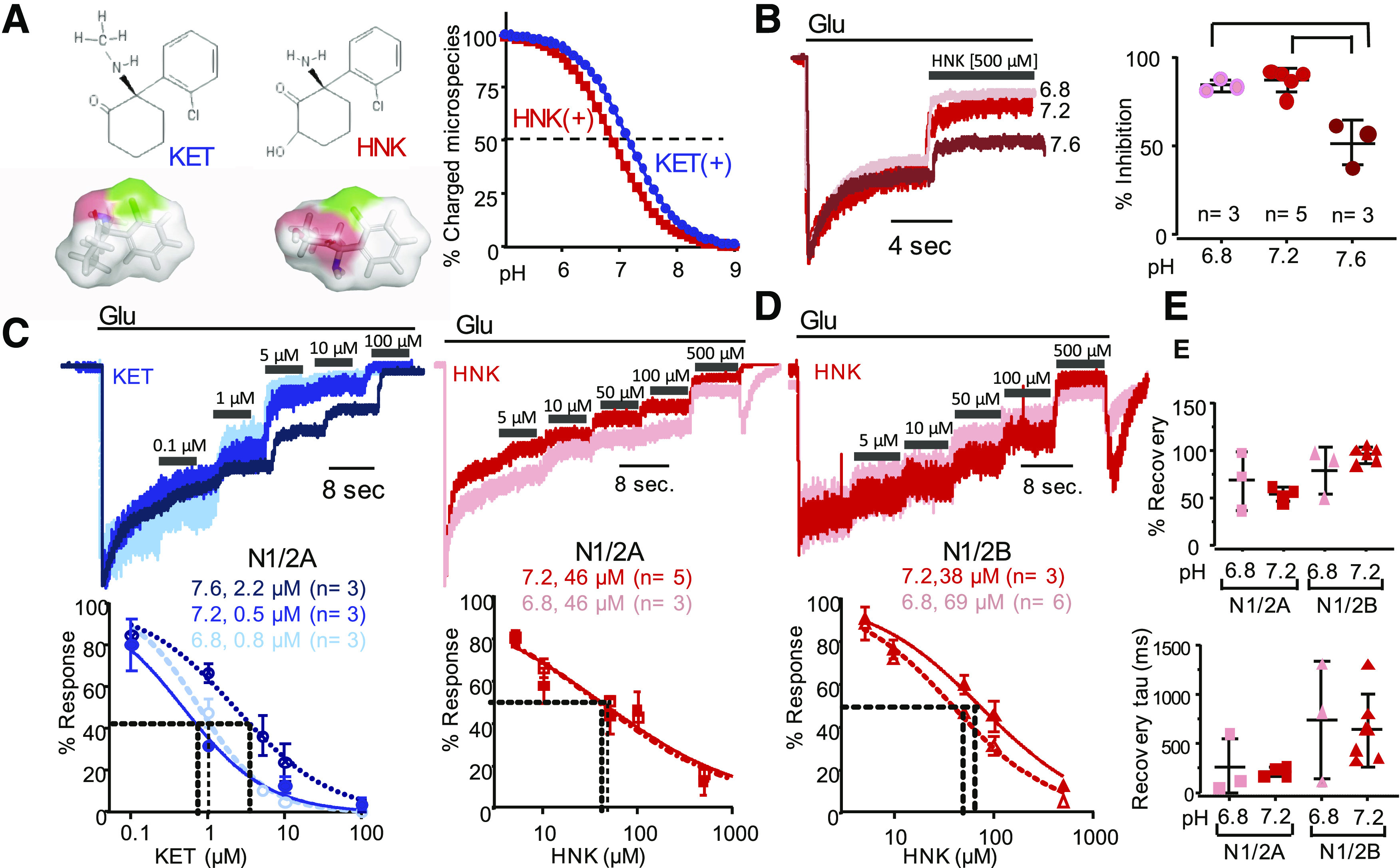
pH dependence of NMDA receptor current inhibition by HNK and KET. (A) Chemical structures for unprotonated KET and HNK and three-dimensional renderings with surface polarity (red, oxygen; blue, nitrogen; gray, hydrogen and carbon; and green, chloride) (left). Protonation equilibria predict pKa of 7.5 and 6.9 for KET and HNK, respectively (calculated in ChemAxon, described in Materials and Methods) (right). (B) Representative current traces recorded from N1/N2A receptors expressed in HEK-293 cells and associated extracellular pH normalized to peak current (Ipeak) responses. (C) Concentration-dependence for KET (left, blue) and HNK (right, red) inhibition of N1/N2A currents. (D) Concentration dependence for HNK inhibition of N1/N2B currents. (E) Extent (%) and kinetics (τ) of current recovery from HNK inhibition.
To determine the concentration of HNK responsible for half-maximal inhibition and compare it with that of KET, we measured the reduction in steady-state NMDA receptor current produced by applying either HNK or KET at increasing concentrations. Consistent with literature reports, the potency of KET increased substantially in more acidic conditions, with the following IC50 values: 2.2 µM (95% CI 1.6–2.9) at pH 7.6, 0.5 µM (95% CI 0.3–0.7) at pH 7.2, and 0.8 µM (95% CI 0.6–0.9) at pH 6.8 (Fig. 1C, left; Table 1).
TABLE 1.
KET and HNK potency on NMDA receptor isoforms
Data are means and 95% confidence intervals.
| N1/N2A | N1/N2B | ||||
|---|---|---|---|---|---|
| IC50 (µM) | Number of cells | IC50 (µM) | Number of cells | ||
| HNK | pH 6.8 | 46 (95% CI 31–68) | 3 | 39 (95% CI 32–47) | 3 |
| pH 7.2 | 46 (95% CI 38–56) | 5 | 69 (95% CI 26–184) | 6 | |
| KET | pH 6.8 | 0.8 (95% CI 0.6–0.9) | 3 | ||
| pH 7.2 | 0.5 (95% CI 0.3–0.7) | 3 | |||
| pH 7.6 | 2.2 (95% CI 1.6–2.9) | 3 | |||
Relative to KET, HNK was ∼100-fold less potent in physiologic proton concentrations (pH 7.2), with IC50 = 46 µM (95% CI 38–56) (Fig. 1C, right; Table 1). HNK had similarly low and pH-sensitive potencies when tested on currents elicited from the juvenile form of NMDA receptors GluN1-1a/GluN2B (N1/N2B), with the following IC50 values: 69 µM (95% CI 26–184) at pH 7.2 and 39 µM (95% CI 32–47) at pH 6.8 (Fig. 1D). This result is consistent with our observation that HNK is more potent in an acidic environment and with previous reports of differences on KET potencies across NMDA receptor isoforms (Yamakura et al., 1993; Hollmann et al., 2001; Dravid et al., 2007).
These results show that, like KET, the potency of HNK is pH-dependent and that in all conditions tested, HNK was substantially less effective than KET at reducing steady-state currents of NMDA receptors.
Kinetics of KET and HNK Inhibition of NMDA Receptor Currents.
To examine the kinetics of NMDA receptor inhibition by HNK and compare with KET, we recorded whole-cell currents from N1/N2A receptors at pH 7.2 and observed the time for inhibition onset and recovery upon application and removal, respectively, of three subsaturating drug concentrations (Fig. 2). We used KET at 0.5, 2, and 10 µM and HNK at 50, 100, and 500 µM. From these data, we estimated time constants by fitting exponential functions to the changing portions of the current trace, and we used these values to extrapolate apparent association (kon) and dissociation (koff) rate constants (Fig. 2). Results show that for KET, kon was 6 ± 1 × 105 M−1s−1, and koff was 0.7 second−1 (95% CI, 0.2–1.3), with a calculated apparent dissociation constant of 0.6 µM. These values are within the range of values reported in the literature (MacDonald et al., 1991). For HNK, we observed much slower apparent association rate constant (kon was 6 ± 3 × 103 M−1 s−1) and also a slower dissociation rate constant [koff was 1.3 s−1 (95% CI 1.28–1.3) ], predicting KD = 203 µM.
Fig. 2.

Kinetics of NMDA receptor current inhibition by KET and HNK. N1/2A currents with three concentrations of (A) KET (top, blue) or (B) HNK (bottom, red) (n = 5 for KET, n = 4 for HNK) (left). Rate constants for inhibition onset (solid) and offset (broken line) as determined by fitting exponential functions (solid line) to the rising or decaying phase of the current responses at each concentration, respectively (exemplary current trace with overlapping exponential fitting shown below in dotted boxes) (right).
These results are consistent with the observed ∼100-fold-lower potency of HNK relative to KET and suggest that this originates largely from slower apparent binding rates. In contrast, the slower apparent dissociation rate constant for HNK relative to KET would increase the drug’s potency. This may reflect a true slower dissociation of HNK from receptors or a slowing of receptor reopening rate. The recovery time from block has been proposed as an important factor in the therapeutic value of NMDA receptor open-channel blockers (Chen and Lipton, 2006) and thus merits further investigation.
Voltage Dependence of NMDA Receptor Current Inhibition by KET, HNK, and Mg2+.
We first examined the voltage dependence of HNK by recording whole-cell currents from N1/N2A receptors at pH 7.2 using voltage-ramp and sequential recording protocols. In the voltage-ramp protocol, we applied each drug to the steady-state phase of the current, and after the inhibition stabilized, we ramped the holding potential within 2 seconds from −80 to +60 mV. Figure 3A illustrates current traces in the absence of added drug (CTR) or after application of HNK (500 µM), KET (10 µM), or Mg2+ (1 mM). In control conditions, we observed a linear current-voltage (I/V) relationship across the entire voltage range, with reversal of ionic flux at 0 mV, as expected for this nonselective cation-permeable channel. When KET or Mg2+ was present, the relationship adopted a J-shape, indicative of reduced current influx at hyperpolarized membrane potentials (Nowak et al., 1984). We observed a similar voltage dependence of HNK block of NMDA receptor currents, strongly suggesting that HNK, like KET and Mg2+, binds in the membrane field and prevents current flow by physically obstructing the passage of ions.
Fig. 3.
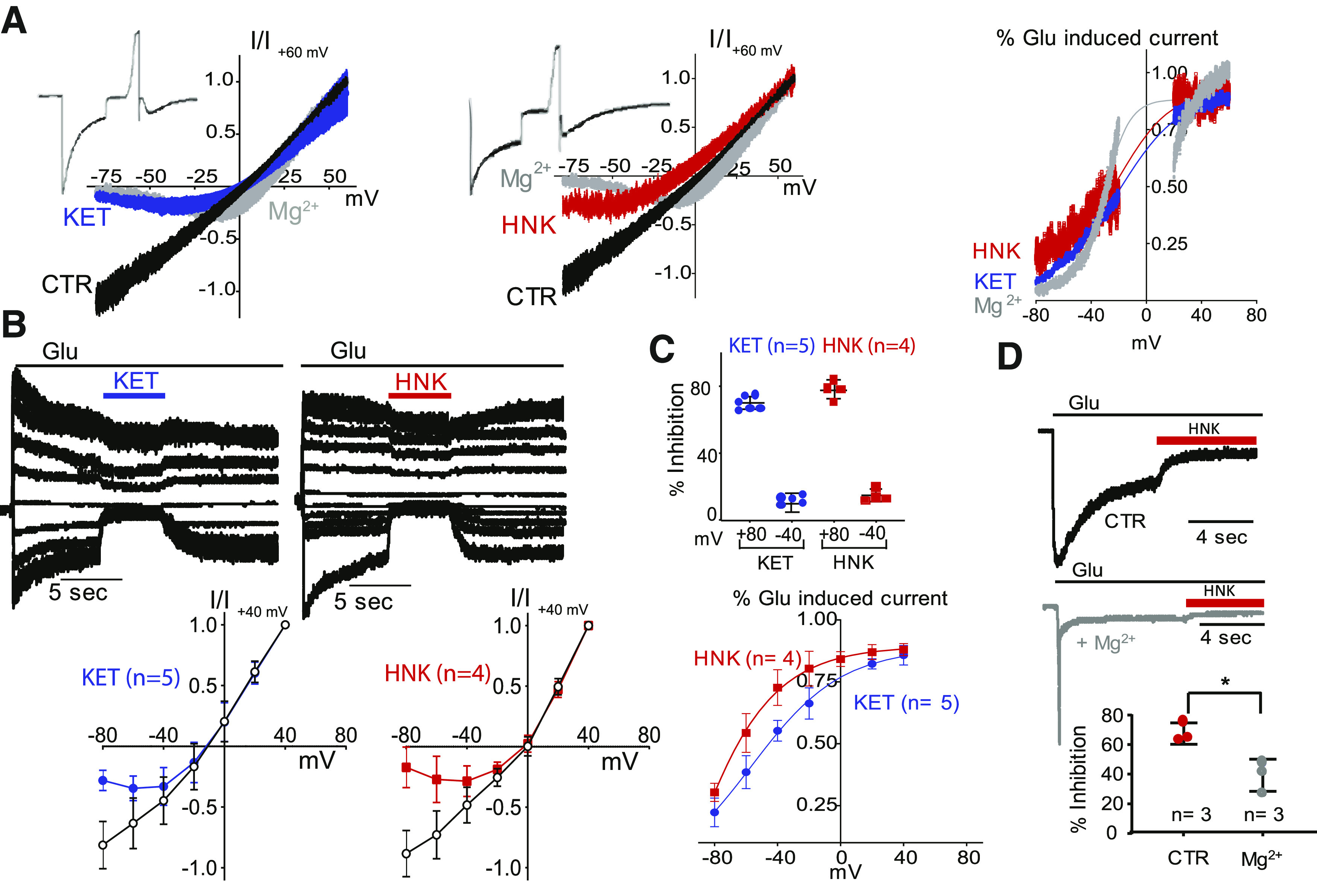
Voltage dependence of NMDA receptor current inhibition by KET, HNK, and Mg. N1/N2A currents were elicited with glutamate (pH 7.2), without (CTR, black), and with modulators applied onto the steady-state portion of the response: KET (10 µM, blue), HNK (500 µM, red), or Mg (1 mM, gray). (A) Exemplary steady-state currents recorded while ramping the holding potential (left, middle) and calculated fractional inhibition relative to membrane voltage (right). (B) Exemplary currents recorded sequentially at several holding potentials (top-left and middle panels) and calculated current-voltage (I/V) plots (below). (C) Calculated percent inhibition of N1/N2A currents at two holding potentials (top-right panel) and calculated fractional inhibition (bottom-right panel). (D) Reduced HNK inhibition of N1/N2A currents by Mg; * P = 0.02 (unpaired Student’s t test).
We sought to validate these results with a second approach. Instead of ramping the membrane potential after receptors were activated and exposed to drug, which recorded a continuous current trace across the range of potentials tested, we now recorded currents from drug-exposed receptors sequentially at several potentials and plotted the measured current inhibition at each voltage (Fig. 3B). Results from this approach are qualitatively similar with those from the first. They show that, as observed for KET, HNK inhibition was voltage-dependent.
A comparison of current block at holding potentials of both −80 and +40 mV (Fig. 3C, top panel) revealed that voltage dependence of block at these potentials is similar for KET and HNK. We used Ann Woodhull’s model to estimate the fractional current inhibition by each drug as a function of voltage and calculated a valence for the blocking charge (zδ) (Woodhull, 1973; Hille, 1992). The values calculated from our measurements, 1.4 for KET and 1.0 for HNK, indicated a weaker charge valence for HNK relative to KET or Mg2+ (Fig. 3C, bottom panel). We also measured and compared values for the slope of block by HNK and KET using the data illustrated in Fig. 3C (bottom panel). We found no statistically significant difference between the two groups: KET slope = 17 (n = 5, 95% CI 13–25) and HNK slope = 24 (n = 4, 95% CI 19–32, P = 0.15).
Whether KET and HNK interact with the same pore residues that bind Mg2+ is unclear. However, Mg2+ (1 mM) reduces the potency of KET inhibition by 10-fold (increases IC50 from 0.3 to 3 µM) (Kotermanski and Johnson, 2009). Given our evidence that, like KET, HNK is a voltage-dependent blocker, we set out to determine the potency of HNK in the presence of physiologic Mg2+ concentrations. For this, we repeated the experiments illustrated in Fig. 1A with solutions that included Mg2+ (1 mM). We found that 1 mM Mg2+ reduced the inhibitory effect of HNK (500 µM) from 68% to 39% (Fig. 3D), indicative of some level of competition. However, the effect was much smaller than that observed for KET. Therefore, in physiologic conditions, the relative effects of KET and HNK on NMDA receptor currents will depend not only on the relative concentrations of the two and membrane voltage but also on the local concentrations of protons and magnesium ions, which can fluctuate with neuronal activity and physiologic states (Tang et al., 1990; Traynelis and Cull-Candy, 1990, 1991). Further, the two drugs may access their respective binding sites preferentially through aqueous and lipophilic paths.
State Dependence of HNK Block.
Both KET and HNK are amphipathic molecules that can distribute into cellular membranes and access their binding sites through membrane diffusion in addition to aqueous routes. This model has been proposed for KET because of its ability to affect currents from receptors isolated within a patch pipette even when only applied in the bath (Orser et al., 1997). To determine whether HNK can access its blocking site when receptors are closed, we used a classic approach that compared the effects of the drug when applied to closed channels (prior to agonist application, preapplication) with those observed when the drug is applied at the same time with the agonist (coapplication) (Table 2).
TABLE 2.
Kinetics of HNK inhibition
Data are means and 95% confidence intervals.
| KET | HNK | |
|---|---|---|
| kon | 0.6 (95% CI −0.6 to 1.5) × 106 M−1 s−1 | 0.006 (95% CI 0.002–0.01) × 106 M−1 s−1* |
| koff | 0.7 (95% CI 0.2–1.3) s−1 | 1.3 (95% CI 1.28–1.3) s−1 |
| KD | 0.6 µM | 203 µM* |
P = 0.02 relative to KET.
With this approach, we determined that coapplication of HNK resulted in a substantial decrease in peak responses relative to agonist-only controls (Table 3), and this decrease was much more pronounced than that observed with KET coapplication. Additionally, there was a reduction in the rate of current desensitization, indicating some allosteric effects of the two drugs in addition to direct block of ionic flux (Fig. 4 (A-D); Table 3). A combined reduction in peak current responses and an acceleration of current desensitization are consistent with a dual pore block and allosteric gating effects for both KET and HNK. Together, these results suggest that both KET and HNK can access their respective blocking site by agonist-dependent and agonist-independent pathways. However, HNK appears effective when applied prior to channel opening, strongly indicating an affinity for resting receptors.
TABLE 3.
State-dependent actions of KET and HNK on NMDA receptor responses
Data are means  S.D.
S.D.
| Ipeak (pA) | Tau D (s) | Iss/Ipeak | |
|---|---|---|---|
| Glutamate | −1731  358 358 |
1.7  0.4 0.4 |
0.53  0.02 0.02 |
| Coapply KET (n = 4) | −1749  736 736 |
0.8  0.2 0.2 |
0.26  0.07 (P = 0.02)* 0.07 (P = 0.02)*
|
| Glutamate | −1347  701 701 |
1.6  0.3 0.3 |
0.54  0.07 0.07 |
| Coapply HNK (n = 4) | −1057  622 (P = 0.01)* 622 (P = 0.01)*
|
0.6  0.1 (P = 0.01)* 0.1 (P = 0.01)*
|
0.20  0.03 (P = 0.001)* 0.03 (P = 0.001)*
|
| Glutamate | −805  438 438 |
1.5  0.2 0.2 |
0.54  0.05 0.05 |
| Preapply KET (n = 3) | −441  242 (P = 0.02)* 242 (P = 0.02)*
|
1.1  1.5 1.5 |
0.18  0.03 (P = 0.0002)* 0.03 (P = 0.0002)*
|
| Glutamate | −2061  1760 1760 |
1.4  0.4 0.4 |
0.44  0.10 0.10 |
| Preapply HNK (n = 5) | −1471  1560 (P = 0.01)* 1560 (P = 0.01)*
|
1.3 0.4 0.4 |
0.36  0.14 0.14 |
Steady-state current (Iss), Peak current (Ipeak). Data are means ± S.D., *P values determined by paired t test.
Fig. 4.

State dependence of KET and HNK inhibition. Exemplary whole-cell current traces obtained from N1/N2A receptors (pH 7.2) from the same cell when KET (10 µM; top, blue) or HNK (500 µM; bottom, red) were coapplied (A,C) or preapplied (B,D) relative to the agonist glutamate (n = 1 cell for all conditions shown).
Discussion
KET is a widely used anesthetic whose effects on central neurotransmission are largely mediated by NMDA receptors. HNK derivatives are currently investigated for their ability to reproduce some of the biologic effects of KET. HNK is a minor (15%) metabolite of KET with higher water solubility (13 g/l vs. 3 g/l). To compare the mechanism by which the two drugs affect NMDA receptor current responses, we investigated HNK for its ability to antagonize macroscopic NMDA receptor currents elicited with maximally effective concentrations of glutamate and glycine and compared these with literature reports for KET and with our own results.
We found that acidic conditions increased the inhibitory potency of both KET and HNK, suggesting that in both cases, the protonated species is the more potent molecular form and that the biologic effect will vary with local pH across physiologic conditions and brain regions. Our results confirm that, relative to KET, HNK is a less potent inhibitor of NMDA receptor currents; however, the IC50 value we measured for HNK was lower than previously reported (Lumsden et al., 2019), which may be due to differences in the pH of solutions used for electrophysiology experiments, contaminating Mg2+ in slice preparations, or differences in cells used for electrophysiology. Discrepancies between IC50 values obtained for recombinant receptors expressed in Xenopus oocytes (Dravid et al., 2007) and for receptors in native to neuronal cultures (Emnett et al., 2016) have been reported previously for norketamine (50 µM vs. 2 µM). Moreover, because potency varies with pH, voltage, and application protocol for KET and HNK, the differences across experimental conditions are not surprising.
At pH 7.2, the half-maximal inhibitory concentration at N1/N2A receptors expressed in HEK cells was 0.5 µM for KET and 46 µM for HNK. This ∼100-fold-weaker potency for HNK originated primarily from a much slower apparent association constant for the metabolite (measured at −70 mV). However, the apparent dissociation constant was slower for HNK than for KET. This was an interesting result given that the therapeutic effect of NMDA receptor blockers may relate to their time on target. Our results also show that, like KET, HNK inhibition is voltage-dependent and competitive with Mg2+. KET antagonizes NMDA receptor current as a classic open-channel, use-dependent blocker. It enters the channel pore and physically obstructs cationic flow through the channel (MacDonald et al., 1991). Several other NMDA receptor physiologic and synthetic modulators act with a similar mechanism, including Mg2+, MK-801, and memantine. Moreover, open-channel blockers compete with each other for recognition sites within the pore (Liu et al., 2001); Kotermanski and Johnson, 2009). For this reason, their effects depend on contemporaneous concentrations of competitors, especially Mg2+ ions, whose concentrations fluctuate during physiologic conditions in the 1 to 2 mM range (Morris, 1992). Whether HNK also reduces NMDA receptor currents with an open-channel, use-dependent mechanism and whether its potency depends on Mg2+ ions have not yet been reported. We observed in this study that HNK inhibition displayed voltage dependence and was also less sensitive to displacement by Mg2+ relative to KET. These results are consistent with the previous report that KET is more effective than HNK at displacing MK-801 binding in brain slices (Zanos et al., 2016). It is important to note that these complex dependencies will influence the overall effect of these two drugs on NMDA receptor currents in situ.
An unexpected result was that administering HNK in advance of activation of the NMDA receptor caused immediate maximal inhibition, whereas KET administered in the same condition required some time to fully develop (Fig. 4). This may be due to the distinct binding and dissociation kinetics between the two drugs or to a larger proportion of HNK accessing its binding site through the membrane, where it is immediately effective in blocking current when the channel opens. This result is consistent with the previous observation that both KET and HNK block spontaneous synaptic currents mediated by NMDA receptors in hippocampal neurons (Suzuki et al., 2017), yet when the drugs are coapplied with subsaturating levels of the synthetic partial agonist NMDA, only KET (10 µM) reduced whole-cell currents from hippocampal interneurons, whereas HNK (10 µM) was not effective.
Taken together, our results show that HNK reduced NMDA receptor currents with a complex mechanism and that the magnitude of the observed effect will depend not only on the local concentration of HNK but also on the extracellular pH, the mode and duration of NMDA receptor activation, and the Mg2+ concentrations present. Aside from its potential to mediate some of the reported behavioral effects of KET, HNK may represent a derivative with unique pharmacologic profile. If found to be well tolerated in humans, it may have beneficial effects in any number of psychiatric and neurologic pathologies associated with overactivation of NMDA receptors.
Acknowledgments
The authors would like to thank Gary Iacobucci for his critical review of this manuscript.
Abbreviations
- CI
confidence interval
- CTR
control
- HEK
human embryonic kidney
- HNK
2R,6R-hydroxynorketamine
- KET
ketamine
- NMDA
N-methyl-d-aspartate
Authorship Contributions
Conducted experiments: Abbott.
Performed data analysis: Abbott.
Wrote or contributed to the writing of the manuscript: Abbott, Popescu.
Footnotes
Funding was from National Institutes of Health National Institute of Neurological Disorders and Stroke [Grants R21-NS098385 and R01-NS097016] (to G.K.P.) and National Institute of General Medical Sciences [Institutional Fellowship T32-GM099607] (to J.A.).
References
- Amico-Ruvio SA, Popescu GK. (2010) Stationary gating of GluN1/GluN2B receptors in intact membrane patches. Biophys J 98:1160–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anis NA, Berry SC, Burton NR, Lodge D. (1983) The dissociative anaesthetics, ketamine and phencyclidine, selectively reduce excitation of central mammalian neurones by N-methyl-aspartate. Br J Pharmacol 79:565–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balestrino M, Somjen GG. (1988) Concentration of carbon dioxide, interstitial pH and synaptic transmission in hippocampal formation of the rat. J Physiol 396:247–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, Krystal JH. (2000) Antidepressant effects of ketamine in depressed patients. Biol Psychiatry 47:351–354. [DOI] [PubMed] [Google Scholar]
- Berry SC, Anis NA, Lodge D. (1984) The effect of the dioxolanes on amino acid induced excitation in the mammalian spinal cord. Brain Res 307:85–90. [DOI] [PubMed] [Google Scholar]
- Chan KW, Lee TM, Siu AM, Wong DP, Kam CM, Tsang SK, Chan CC. (2013) Effects of chronic ketamine use on frontal and medial temporal cognition. Addict Behav 38:2128–2132. [DOI] [PubMed] [Google Scholar]
- Chen H-SV, Lipton SA. (2006) The chemical biology of clinically tolerated NMDA receptor antagonists. J Neurochem 97:1611–1626. [DOI] [PubMed] [Google Scholar]
- Chesler M, Kaila K. (1992) Modulation of pH by neuronal activity. Trends Neurosci 15:396–402. [DOI] [PubMed] [Google Scholar]
- Conseiller C, Benoist JM, Hamann KF, Maillard MC, Besson JM. (1972) Effects of ketamine (CI 581) on cell responses to cutaneous stimulations in laminae IV and V in the cat’s dorsal horn. Eur J Pharmacol 18:346–352. [DOI] [PubMed] [Google Scholar]
- Dilmore JG, Johnson JW. (1998) Open channel block and alteration of N-methyl-D-aspartic acid receptor gating by an analog of phencyclidine. Biophys J 75:1801–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domino EF, Chodoff P, Corssen G. (1965) Pharmacologic effects of Ci-581, a new dissociative anesthetic, in man. Clin Pharmacol Ther 6:279–291. [DOI] [PubMed] [Google Scholar]
- Dravid SM, Erreger K, Yuan H, Nicholson K, Le P, Lyuboslavsky P, Almonte A, Murray E, Mosely C, Barber J, et al. (2007) Subunit-specific mechanisms and proton sensitivity of NMDA receptor channel block. J Physiol 581:107–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emnett C, Li H, Jiang X, Benz A, Boggiano J, Conyers S, Wozniak DF, Zorumski CF, Reichert DE, Mennerick S. (2016) A clickable analogue of ketamine retains NMDA receptor activity, psychoactivity, and accumulates in neurons. Sci Rep 6:38808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Featherstone RE, Liang Y, Saunders J, Tatard-Leitman V, Ehrlichman RS, Siegel SJ. (2012) Subchronic ketamine treatment leads to permanent changes in EEG, cognition and the astrocytic glutamate transporter EAAT2 in mice. Neurobiol Dis 47:338–346. [DOI] [PubMed] [Google Scholar]
- Gilling KE, Jatzke C, Hechenberger M, Parsons CG. (2009) Potency, voltage-dependency, agonist concentration-dependency, blocking kinetics and partial untrapping of the uncompetitive N-methyl-D-aspartate (NMDA) channel blocker memantine at human NMDA (GluN1/GluN2A) receptors. Neuropharmacology 56:866–875. [DOI] [PubMed] [Google Scholar]
- Hasan M, Hofstetter R, Fassauer GM, Link A, Siegmund W, Oswald S. (2017) Quantitative chiral and achiral determination of ketamine and its metabolites by LC-MS/MS in human serum, urine and fecal samples. J Pharm Biomed Anal 139:87–97. [DOI] [PubMed] [Google Scholar]
- Hille B. (1992) Ion Channels of Excitable Membranes, 2nd ed, Sinauer Associates, Sunderland, MA. [Google Scholar]
- Hollmann MW, Liu HT, Hoenemann CW, Liu WH, Durieux ME. (2001) Modulation of NMDA receptor function by ketamine and magnesium. Part II: interactions with volatile anesthetics. Anesth Analg 92:1182–1191. [DOI] [PubMed] [Google Scholar]
- Huettner JE, Bean BP. (1988) Block of N-methyl-D-aspartate-activated current by the anticonvulsant MK-801: selective binding to open channels. Proc Natl Acad Sci USA 85:1307–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JW, Glasgow NG, Povysheva NV. (2015) Recent insights into the mode of action of memantine and ketamine. Curr Opin Pharmacol 20:54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotermanski SE, Johnson JW. (2009) Mg2+ imparts NMDA receptor subtype selectivity to the Alzheimer’s drug memantine. J Neurosci 29:2774–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurdi MS, Theerth KA, Deva RS. (2014) Ketamine: current applications in anesthesia, pain, and critical care. Anesth Essays Res 8:283–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kussius CL, Popescu GK. (2009) Kinetic basis of partial agonism at NMDA receptors. Nat Neurosci 12:1114–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang E, Mallien AS, Vasilescu AN, Hefter D, Luoni A, Riva MA, Borgwardt S, Sprengel R, Lang UE, Gass P, et al. (2018) Molecular and cellular dissection of NMDA receptor subtypes as antidepressant targets. Neurosci Biobehav Rev 84:352–358. [DOI] [PubMed] [Google Scholar]
- Leung LY, Baillie TA. (1986) Comparative pharmacology in the rat of ketamine and its two principal metabolites, norketamine and (Z)-6-hydroxynorketamine. J Med Chem 29:2396–2399. [DOI] [PubMed] [Google Scholar]
- Liu H T, Hollmann M W, Liu W H, Hoenemann C W, Durieux M E. (2001) Modulation of NMDA receptor function by ketamine and magnesium: Part I. Anesth Analg 92 (5):1173–1181, doi: 10.1097/00000539-200105000-00019 . [DOI] [PubMed] [Google Scholar]
- Lodge D, Mercier MS. (2015) Ketamine and phencyclidine: the good, the bad and the unexpected. Br J Pharmacol 172:4254–4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo PA, Kavran JM, Kim MS, Leahy DJ. (2013) Transient mammalian cell transfection with polyethylenimine (PEI). Methods Enzymol 529:227–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumsden EW, Troppoli TA, Myers SJ, Zanos P, Aracava Y, Kehr J, Lovett J, Kim S, Wang FH, Schmidt S, et al. (2019) Antidepressant-relevant concentrations of the ketamine metabolite (2R,6R)-hydroxynorketamine do not block NMDA receptor function. Proc Natl Acad Sci USA 116:5160–5169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald JF, Bartlett MC, Mody I, Pahapill P, Reynolds JN, Salter MW, Schneiderman JH, Pennefather PS. (1991) Actions of ketamine, phencyclidine and MK-801 on NMDA receptor currents in cultured mouse hippocampal neurones. J Physiol 432:483–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald JF, Miljkovic Z, Pennefather P. (1987) Use-dependent block of excitatory amino acid currents in cultured neurons by ketamine. J Neurophysiol 58:251–266. [DOI] [PubMed] [Google Scholar]
- Mazzeffi M, Johnson K, Paciullo C. (2015) Ketamine in adult cardiac surgery and the cardiac surgery Intensive Care Unit: an evidence-based clinical review. Ann Card Anaesth 18:202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan CJ, Riccelli M, Maitland CH, Curran HV. (2004) Long-term effects of ketamine: evidence for a persisting impairment of source memory in recreational users. Drug Alcohol Depend 75:301–308. [DOI] [PubMed] [Google Scholar]
- Morris M E. (1992) Brain and CSF magnesium concentrations during magnesium deficit in animals and humans: neurological symptoms. Magnes Res 5 (4):303–313 . [PubMed] [Google Scholar]
- Muetzelfeldt L, Kamboj SK, Rees H, Taylor J, Morgan CJ, Curran HV. (2008) Journey through the K-hole: phenomenological aspects of ketamine use. Drug Alcohol Depend 95:219–229. [DOI] [PubMed] [Google Scholar]
- Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. (1984) Magnesium gates glutamate-activated channels in mouse central neurones. Nature 307:462–465. [DOI] [PubMed] [Google Scholar]
- Orser BA, Pennefather PS, MacDonald JF. (1997) Multiple mechanisms of ketamine blockade of N-methyl-D-aspartate receptors. Anesthesiology 86:903–917. [DOI] [PubMed] [Google Scholar]
- Paganelli MA, Popescu GK. (2015) Actions of bupivacaine, a widely used local anesthetic, on NMDA receptor responses. J Neurosci 35:831–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons CG, Gilling K. (2007) Memantine as an example of a fast, voltage-dependent, open channel N-methyl-D-aspartate receptor blocker. Methods Mol Biol 403:15–36. [DOI] [PubMed] [Google Scholar]
- Pham TH, Defaix C, Xu X, Deng SX, Fabresse N, Alvarez JC, Landry DW, Brachman RA, Denny CA, Gardier AM. (2018) Common neurotransmission recruited in (R,S)-ketamine and (2R,6R)-hydroxynorketamine-induced sustained antidepressant-like effects. Biol Psychiatry 84:e3–e6. [DOI] [PubMed] [Google Scholar]
- Sleigh J, Harvey M, Voss L, Denny B. (2014) Ketamine – more mechanisms of action than just NMDA blockade. Trends Anaesthesia Crit Care 4:76–81. [Google Scholar]
- Suzuki K, Nosyreva E, Hunt KW, Kavalali ET, Monteggia LM. (2017) Effects of a ketamine metabolite on synaptic NMDAR function. Nature 546:E1–E3. [DOI] [PubMed] [Google Scholar]
- Tang CM, Dichter M, Morad M. (1990) Modulation of the N-methyl-D-aspartate channel by extracellular H+. Proc Natl Acad Sci USA 87:6445–6449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Cull-Candy SG. (1990) Proton inhibition of N-methyl-D-aspartate receptors in cerebellar neurons. Nature 345:347–350. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Cull-Candy SG. (1991) Pharmacological properties and H+ sensitivity of excitatory amino acid receptor channels in rat cerebellar granule neurones. J Physiol 433:727–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhull AM. (1973) Ionic blockage of sodium channels in nerve. J Gen Physiol 61:687–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakura T, Mori H, Masaki H, Shimoji K, Mishina M. (1993) Different sensitivities of NMDA receptor channel subtypes to non-competitive antagonists. Neuroreport 4:687–690. [DOI] [PubMed] [Google Scholar]
- Zanos P, Moaddel R, Morris PJ, Georgiou P, Fischell J, Elmer GI, Alkondon M, Yuan P, Pribut HJ, Singh NS, et al. (2016) NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature 533:481–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanos P, Moaddel R, Morris PJ, Riggs LM, Highland JN, Georgiou P, Pereira EFR, Albuquerque EX, Thomas CJ, Zarate CA, Jr., et al. (2018) Ketamine and ketamine metabolite pharmacology: insights into therapeutic mechanisms. Pharmacol Rev 70:621–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate CA, Jr., Brutsche N, Laje G, Luckenbaugh DA, Venkata SL, Ramamoorthy A, Moaddel R, Wainer IW. (2012) Relationship of ketamine’s plasma metabolites with response, diagnosis, and side effects in major depression. Biol Psychiatry 72:331–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeilhofer HU, Swandulla D, Geisslinger G, Brune K. (1992) Differential effects of ketamine enantiomers on NMDA receptor currents in cultured neurons. Eur J Pharmacol 213:155–158. [DOI] [PubMed] [Google Scholar]