

Abstract
Unconjugated bilirubin, the end product of heme catabolism and antioxidant, induced brain damage in human neonates is a well-recognized clinical syndrome. However, the cellular and molecular mechanisms underlying bilirubin neurotoxicity remain unclear. To characterize the sequence of events leading to bilirubin-induced neurotoxicity, we investigated whether bilirubin-induced glial activation was involved in bilirubin neurotoxicity by exposing co-cultured rat glial cells and cerebellar granule neurons (CGN) to bilirubin. We found that bilirubin could markedly induce the expression of TNF-α and iNOS in glial cells, and even at low concentrations, the co-culture of glial cells with neurons significantly enhances neurotoxicity of bilirubin. Pretreatment of the co-cultured cells with minocycline protected CGN from glia-mediated bilirubin neurotoxicity and inhibited overexpression of TNF-α and iNOS in glia. Furthermore, we found that high doses of bilirubin were able to induce glial injury, and minocycline attenuated bilirubin-induced glial cell death. Our data suggest that glial cells play an important role in brain damage caused by bilirubin, and minocycline blocks bilirubin-induced encephalopathy possibly by directly and indirectly inhibiting neuronal death pathways.
Keywords: Minocycline, Bilirubin, Glia, Neurotoxicity, Neuroprotection
1. Introduction
Minocycline is a semisynthetic second-generation tetracycline with neuroprotective, antioxidant, and anti-inflammatory effects that are completely distinct from its antimicrobial actions (Shultz and Zhong, 2017). This compound could directly inhibit free radical production as well as exhibit free-radical scavenging activity (Kraus et al., 2005). Interestingly, in addition to showing minocycline protects brains against many brain injury insults through the anti-oxidant mechanism (Shultz and Zhong, 2017), we also clearly show that minocycline can markedly attenuate brain damage induced by excess bilirubin, a physiological antioxidant in Gunn rats (Lin et al., 2005).
Bilirubin is a catabolic end product of heme metabolism and is excreted from the liver via conjugating with glucuronide by hepatic glucuronyl transferase. Conjugation solubilizes free bilirubin in water and reduces the serum levels of unconjugated bilirubin. Normally, bilirubin is a physiologic antioxidant (Baranano et al., 2002; Dore and Snyder, 1999; Stocker et al., 1987) and mildly increased blood bilirubin is able to lower the risk for ischemic stroke and coronary artery disease (Djousse et al., 2001; Perlstein et al., 2008; Schwertner et al., 1994). Most recently, it has been demonstrated that bilirubin’s redox activity could directly prevent excitotoxicity and neuronal death by scavenging O2· – (Vasavda et al., 2019). However, in the neonate, failure of the bilirubin excretion pathway or its conjugation in the liver can result in an accumulation of unconjugated bilirubin leading to hyperbilirubinemia (Gourley, 1997). Neonatal jaundice is usually the consequence of a transient deficiency of bilirubin conjugation caused by breast-feeding, prematurity, glucose-6-phosphate dehydrogenase (G6PD) deficiency, or a variety of hemolytic diseases (Kaplan and Hammerman, 1998). Toxic levels of unconjugated bilirubin are also encountered in adults with recessively inherited Crigler-Najjar type I disease (Ebrahimi and Rahim, 2018; Erlinger et al., 2014; Green and Gollan, 1997; Rossi et al., 2005). Severe hyperbilirubinemia can cause bilirubin-induced neurologic dysfunction, potentially leading to permanent brain damage (Yueh et al., 2017). It is often associated with characteristic neuropathology and neurodegeneration of the cerebellum (particularly loss of Purkinje cells and granule neurons), the basal ganglia, hippocampus, and cochlear nucleus (Bortolussi et al., 2015; Dennery et al., 2001).
The exact cellular and molecular mechanisms underlying bilirubin-induced brain damage are currently poorly understood. It has been shown in neurons, high levels of bilirubin interfere with DNA and protein synthesis, interact directly with cell membrane phospholipids and alter intracellular pH (Chuniaud et al., 1996; Dennery et al., 2001; Rodrigues et al., 2002; Rosenstein et al., 1983). Additionally, bilirubin inhibits several mitochondrial enzymes (Dennery et al., 2001) and Ca2+-mediated calmodulin kinase activity (Machaca, 2003; Rodrigues et al., 2002), as well as activates the function of the N-methyl-d-aspartate (NMDA) receptor (Chen et al., 2016; Hoffman et al., 1996; McDonald et al., 1998) and p38 MAP kinase phosphorylation (Lin et al., 2005; NaveenKumar et al., 2015). Furthermore, many reports demonstrated that bilirubin also directly induced glial death (Brites et al., 2009; Feng et al., 2018; Kumral et al., 2005; Vodret et al., 2017). It appears that neurons are more vulnerable to bilirubin toxicity than astrocytes (Silva et al., 2002), although astrocytes are more competent in releasing glutamate and inflammatory factors than neurons when exposed to bilirubin (Falcao et al., 2006).
Studies suggest that minocycline blocks neuronal death by inhibiting the release of cytochrome c and p38 MAP Kinase. Additionally, minocycline can also inhibit inflammation from glia (Lin et al., 2005). However, it has never been shown whether minocycline also inhibits antioxidants, such as bilirubin-induced neuronal death and inflammation. Given the potential therapeutic efficacy of minocycline on hyperbilirubinemia, understanding its neuroprotective mechanism(s) underlying this disease is of great importance.
Inflammatory factors such as the inducible nitric oxide (iNOS) and cytokines including TNF-α play key roles in brain injury (Lee et al., 2004) and, if the secretion is imbalanced, are detrimental to neurons (Xie et al., 2004). Currently, there is no direct evidence to support that glial cells are directly involved in bilirubin-induced neuronal death.
In the present study, we demonstrate that bilirubin as a physiological antioxidant can induce the expressions of TNFα and iNOS in glia and such inductions can be blocked by minocycline. Additionally, bilirubin is able to induce neuronal death in the mixed culture of CGN and glial cells even at subtoxic levels. Pretreatments of CGN with minocycline significantly attenuated bilirubin-induced neuronal death in the presence of glia. Furthermore, bilirubin at high doses was able to induce glial death and minocycline could block bilirubin-induced glial toxicity. Our data demonstrate that bilirubin induced CGN neuronal death could be potentiated by glia, especially at lower doses and minocycline is able to block bilirubin-induced glial activation and death, as well as glia-mediated neuronal death.
2. Materials and methods
2.1. Primary culture of CGN and glial cells
CGN and glial cells used in this study were prepared from 8-day-old Sprague-Dawley rat pups (Harlan Laboratories, IN) as previously described (Du et al., 2001). Briefly, freshly dissected cerebella were dissociated in the presence of trypsin and DNase I and planted on poly-l-lysine coated dishes. Cells were seeded at a density of 1.5 × 106 cells/ml in basal medium Eagle supplemented with 10 % FBS, 25mM KCl, and gentamicin (0.1 mg/ml). For glial cell culture, 3 days later, the cells were passaged once to get rid of neurons. The mixed glial cultures were grown in this culture medium for two weeks and the glial cells were then passaged twice for experiments. For co-culture experiments, CGN at 1.5 × 106 cells/ml were cultured onto removable 10-mm Nunc tissue inserts with treatments with cytosine arabinoside (10 μM) 24 h after initial plating. When the glial cells reach confluence, the inserts containing 10-day cultured CGN were placed onto wells containing glial cells and used for the experiments immediately. Viable neurons will be quantified by counting fluorescein (green) positive cells which result from the de-esterification of fluorescein diacetate (FDA) by living neurons in inserts (Du et al., 2001). The viability of glial cells was quantified by using MTT (Du et al., 2003). Values are expressed as a % of control cultures for each experiment and the data is represented as the mean ± standard error of replicate experiments.
2.2. Western blot analysis
Western blot was performed on whole-cell extracts (10 μg) that were prepared by lysing cells in RIPA buffer containing 1 % Nonidet P-40, 0.1 % SDS, 50mM Tris (pH 8.0), 50mM NaC1, 0.05 % deoxycholate, and protease inhibitor (Roche, Indianapolis). Proteins were size-fractionated (SDS-PAGE) on a 4–12 % polyacrylamide gradient gel and transferred onto nitrocellulose (Hybond N, Amersham, CA, USA). The blots were then probed with polyclonal antibodies specific for TNFα, iNOS, and β-actin (Santa Cruz, CA) followed by horseradish peroxidase-linked antibodies (Santa Cruz, CA). Bound antibody was visualized using enhanced chemiluminescence (Amersham, Arlington Heights, IL).
3. Results
3.1. Minocycline treatments blocked bilirubin-induced expression of iNOS and TNFα in primary rat glial cells
In order to examine whether bilirubin affected expressions of iNOS and TNFα in glial cells, we incubated primary rat glial cells with 1μM bilirubin (equivalent to 0.625 μg/ml) for 24 h. iNOS and TNFα expression in glial cells were measured by western blot. After incubation of bilirubin, the iNOS levels were increased by 292.2 ± 16.6 % as compared to control cells (p < 0.001). TNFα levels were also significantly induced (455.3 ± 46.9 %) as compared to controls (p < 0.001) (Fig. 1). Pretreatments of cells with minocycline (10 μM) significantly inhibited bilirubin-induced expressions of iNOS (from 292.2 ± 16.6 % to 158.2 ± 16.39 %, p < 0.01) and TNFα (from 455.3 %±46.9% to 190.4 ± 52.2 %, p < 0.01) (Fig. 1).
Fig. 1. Minocycline treatments blocked bilirubin-induced expressions of TNFα and iNOS in rat cerebellar glia.
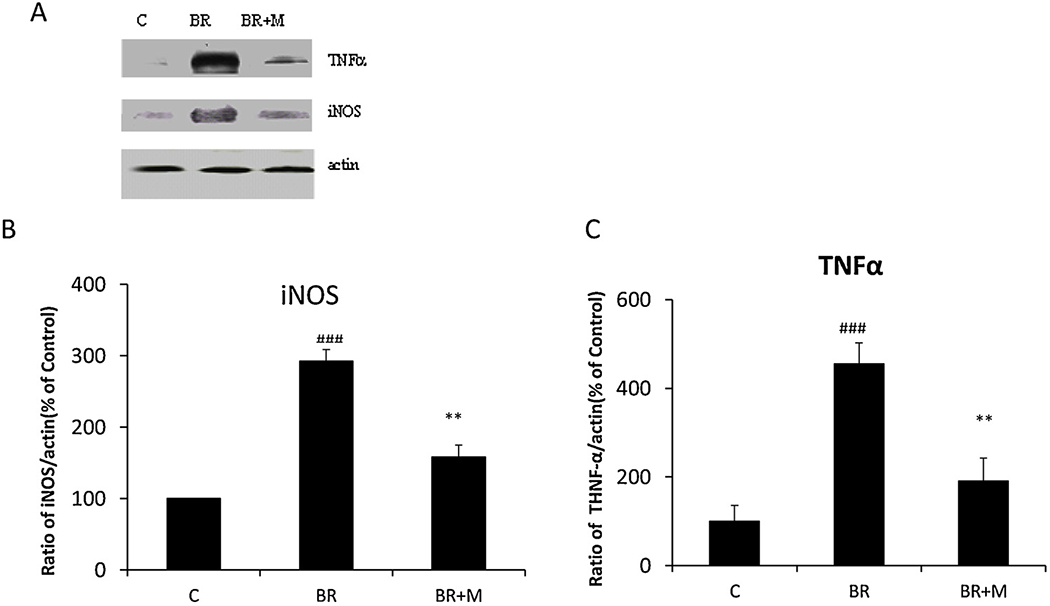
Mixed glial cells were pretreated with or without minocycline (10 μM, 2h) followed by bilirubin (1 μM) treatments for additional 24 h (BR + M vs. BR). The lysates were immunoblotted with anti-iNOS and TNFα antibodies. The anti-β-actin antibody was used to confirm an equal amount of protein loading in each gel lane. All cultures were treated in triplicate at the indicated times and data are represented as the mean ± SEM (### p < 0.001, treated with bilirubin vs. control; ** p < 0.01, treated with minocycline and bilirubin vs. bilirubin only by Student’s t-test). This experiment was repeated three times.
3.2. Glia enhances bilirubin-induced CGN neuronal death, and minocycline exerts neuroprotection
To further examine whether minocycline protects CGN against glia-mediated bilirubin neurotoxicity, we treated co-cultures of glia and CGN with bilirubin and minocycline. As shown in Fig. 2A, we have found that glial cells significantly enhance neuronal death from 81.4 ± 8.4% to 62.5 ± 7.6 % (p < 0.05) and 71.2 ± 8.8% to 45.5 ± 10.6 % (p < 0.05) followed by exposure of co-cultures to 1 μM and 2 μM bilirubin for 24 h. Pretreatment of cells with minocycline significantly attenuated 1 μM bilirubin-induced neuronal death (p < 0.05) (Fig. 2B).
Fig. 2.
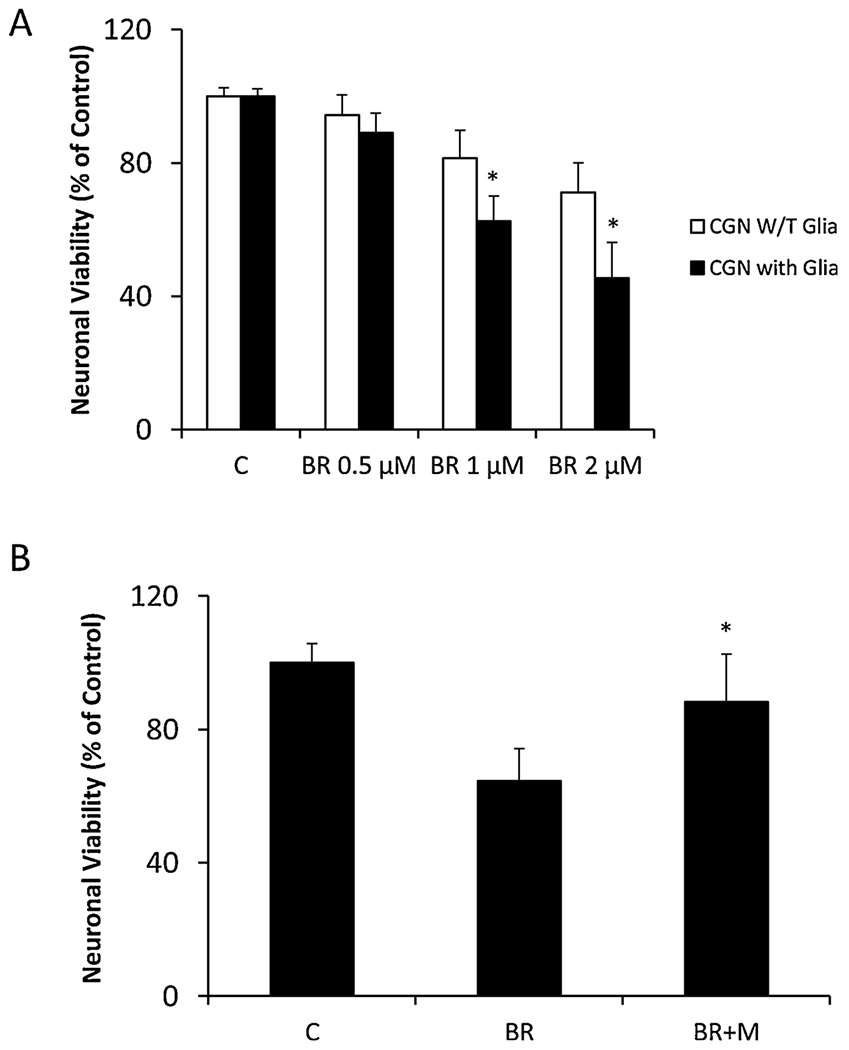
Minocycline is able to block inflammation-mediated bilirubin-induced neurotoxicity. A. Glia potentiates subtoxic bilirubin-induced CGN neuronal death. Rat CGN in the insert were co-cultured on the confluent layer of glia for 10 days and then treated with bilirubin (0.5–2 μM) for additional 24 h. Neuronal viability in the inserts was assessed by FDA, as compared to pure CGN cultures. All cultures were treated in triplicate at the indicated times and data are represented as the mean ± SEM (*p < 0.05, treated vs. control by Student’s t-test). This experiment was repeated three times with similar results. B. Minocycline protects CGN against glia-mediated bilirubin neurotoxicity. Rat CGN in the insert co-cultured with glia were pretreated with minocycline (10 μM) followed by the bilirubin (1 μM) treatments. Neuronal viability in the inserts was assessed by FDA. All cultures were treated in triplicate at the indicated times and data are represented as the mean ± SEM (*p < 0.05, treated with minocycline vs. bilirubin only by Student’s t-test). This experiment was repeated three times with similar results.
3.3. Minocycline attenuates bilirubin-induced glial cell death
Interestingly, exposure of primary glial cells to bilirubin at 1 μM and 2 μM did not s induce cell death. In contrast, 5 μM and 10 μM bilirubin markedly induced primary glial cell death as compared with the control group (Fig. 3A). 2h pretreatment of minocycline (10 μM) significantly blocks bilirubin (5 μM) -induced glial death (from 48.5 ± 1.3% to 93.5 ± 2.5 %, Fig. 3B).
Fig. 3. Minocycline attenuates bilirubin-induced glial cell death.
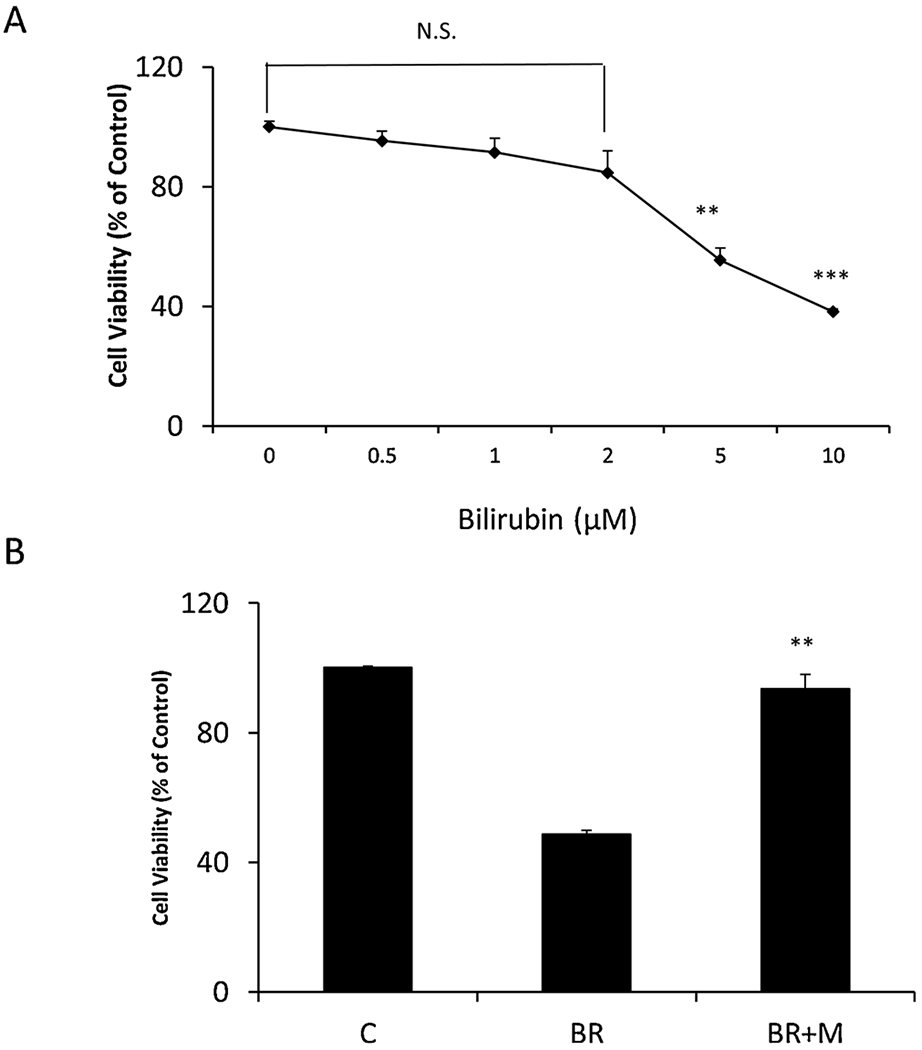
A. Rat cerebellar glia were treated with bilirubin (0.5–10 μM) for 24 h. B. Cerebellar glia were pretreated with minocycline (10 μM) for 2h followed by 24 h bilirubin treatments (5μM). Cell viability was assessed by MTT. All cultures were treated in triplicate at the indicated times and data are represented as the mean ± SEM (N,S, not significant, **p < 0.01, ***p < 0.001 treated vs. control by Student’s t-test). This experiment was repeated three times with similar results.
4. Discussion
Although bilirubin is a well-recognized neurotoxin for neonates, the cellular mechanisms underlying its toxicity are poorly understood. Additionally, how minocycline protects cells against the neurotoxicity of bilirubin, a physiology antioxidant, remains to be elucidated. Glial activation is observed in many neurodegenerative disorders (Chen et al., 2010; Lull and Block, 2010) and usually mediated by free radicals (McGeer and McGeer, 1998; von Bernhardi et al., 2015). Here, by using a co-culture system, we demonstrated that glial cells could be activated by anti-oxidant bilirubin and activated glial inflammation greatly enhanced neuronal death induced by bilirubin at sub toxic levels (1 and 2 μM), as compared to without glial involvement. Minocycline blocks bilirubin-induced neuronal death in the absence or presence of glia. Interestingly, bilirubin at 1 μM induced expression of inflammatory factors, iNOS, and TNFα, but did not significantly induce glial cell death. This finding indicates that bilirubin at lower doses induces neurotoxicity possibly via the activation of glia. At high doses, bilirubin directly induces both neuronal and glial cell death. Minocycline not only blocks bilirubin-induced CGN neuronal death as we previously reported but also blocks bilirubin-induced glial cell death. To the best of our knowledge, our findings are the first to demonstrate that antioxidant bilirubin could directly activate glia and glial activation potentiates bilirubin neurotoxicity. Minocycline significantly protects both neurons and glial cells from bilirubin toxicity. The fact that glial activation enhances CGN neurotoxicity when treated with bilirubin, suggests that neuroprotective therapeutic development has to consider effects of bilirubin-induced glial activation/inflammation on neuronal death as both are the major cause for kernicterus in mild hyperbilirubinemia, although in severe bilirubinemia, bilirubin can directly induce glial and neuronal death. In summary, data obtained from this study explain, at least partially, why minocycline is so protective in the treatment of hyperbilirubinemia in vivo and suggest that minocycline not only inhibits free radical, but also antioxidant-induced neurotoxicity. Our results may lead to the development of new therapies for the treatment of both mild and severe hyperbilirubinemia.
Acknowledgements
Project supported by Foundation of Heilongjiang Educational Committee, PR China (Grant No.12521305).
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Baranano DE, Rao M, Ferris CD, Snyder SH, 2002. Biliverdin reductase: a major physiologic cytoprotectant. Proc. Natl. Acad. Sci. U.S.A 99, 16093–16098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortolussi G, Codarin E, Antoniali G, Vascotto C, Vodret S, Arena S, Cesaratto L, Scaloni A, Tell G, Muro AF, 2015. Impairment of enzymatic antioxidant defenses is associated with bilirubin-induced neuronal cell death in the cerebellum of Ugt1 KO mice. Cell Death Dis. 6, e1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brites D, Fernandes A, Falcao AS, Gordo AC, Silva RF, Brito MA, 2009. Biological risks for neurological abnormalities associated with hyperbilirubinemia. J. Perinatol 29 (Suppl. 1), S8–13. [DOI] [PubMed] [Google Scholar]
- Chen CJ, Ou YC, Lin SY, Raung SL, Liao SL, Lai CY, Chen SY, Chen JH, 2010. Glial activation involvement in neuronal death by Japanese encephalitis virus infection. J. Gen. Virol 91, 1028–1037. [DOI] [PubMed] [Google Scholar]
- Chen XJ, Zhou HQ, Ye HB, Li CY, Zhang WT, 2016. The effect of bilirubin on the excitability of mitral cells in the olfactory bulb of the rat. Sci. Rep 6, 32872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuniaud L, Dessante M, Chantoux F, Blondeau JP, Francon J, Trivin F, 1996. Cytotoxicity of bilirubin for human fibroblasts and rat astrocytes in culture. Effect of the ratio of bilirubin to serum albumin. Clin. Chim. Acta 256, 103–114. [DOI] [PubMed] [Google Scholar]
- Dennery PA, Seidman DS, Stevenson DK, 2001. Neonatal hyperbilirubinemia. N. Engl. J. Med 344, 581–590. [DOI] [PubMed] [Google Scholar]
- Djousse L, Levy D, Cupples LA, Evans JC, D’Agostino RB, Ellison RC, 2001. Total serum bilirubin and risk of cardiovascular disease in the Framingham offspring study. Am. J. Cardiol 87, 1196–1200 A1194,, 1197. [DOI] [PubMed] [Google Scholar]
- Dore S, Snyder SH, 1999. Neuroprotective action of bilirubin against oxidative stress in primary hippocampal cultures. Ann. N. Y. Acad. Sci 890, 167–172. [DOI] [PubMed] [Google Scholar]
- Du Y, Ma Z, Lin S, Dodel RC, Gao F, Bales KR, Triarhou LC, Chernet E, Perry KW, Nelson DL, Luecke S, Phebus LA, Bymaster FP, Paul SM, 2001. Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A 98, 14669–14674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Wei X, Dodel R, Sommer N, Hampel H, Gao F, Ma Z, Zhao L, Oertel WH, Farlow M, 2003. Human anti-beta-amyloid antibodies block beta-amyloid fibril formation and prevent beta-amyloid-induced neurotoxicity. Brain 126, 1935–1939. [DOI] [PubMed] [Google Scholar]
- Ebrahimi A, Rahim F, 2018. Crigler-Najjar Syndrome: current perspectives and the application of clinical genetics. Endocr. Metab. Immune Disord. Drug Targets 18, 201–211. [DOI] [PubMed] [Google Scholar]
- Erlinger S, Arias IM, Dhumeaux D, 2014. Inherited disorders of bilirubin transport and conjugation: new insights into molecular mechanisms and consequences. Gastroenterology 146, 1625–1638. [DOI] [PubMed] [Google Scholar]
- Falcao AS, Fernandes A, Brito MA, Silva RF, Brites D, 2006. Bilirubin-induced immunostimulant effects and toxicity vary with neural cell type and maturation state. Acta Neuropathol. 112, 95–105. [DOI] [PubMed] [Google Scholar]
- Feng J, Li M, Wei Q, Li S, Song S, Hua Z, 2018. Unconjugated bilirubin induces pyroptosis in cultured rat cortical astrocytes. J. Neuroinflammation 15, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourley GR, 1997. Bilirubin metabolism and kernicterus. Adv. Pediatr 44, 173–229. [PubMed] [Google Scholar]
- Green RM, Gollan JL, 1997. Crigler-Najjar disease type I: therapeutic approaches to genetic liver diseases into the next century. Gastroenterology 112, 649–651 094451. [DOI] [PubMed] [Google Scholar]
- Hoffman DJ, Zanelli SA, Kubin J, Mishra OP, Delivoria-Papadopoulos M, 1996. The in vivo effect of bilirubin on the N-methyl-D-aspartate receptor/ion channel complex in the brains of newborn piglets. Pediatr. Res 40, 804–808. [DOI] [PubMed] [Google Scholar]
- Kaplan M, Hammerman C, 1998. Severe neonatal hyperbilirubinemia. A potential complication of glucose-6-phosphate dehydrogenase deficiency. Clin. Perinatal 5, 575–590 viii.0/8. [PubMed] [Google Scholar]
- Kraus RL, Pasieczny R, Lariosa-Willingham K, Turner MS, Jiang A, Trauger JW, 2005. Antioxidant properties of minocycline: neuroprotection in an oxidative stress assay and direct radical-scavenging activity. J. Neurochem 94, 819–827. [DOI] [PubMed] [Google Scholar]
- Kumral A, Gene S, Gene K, Duman N, Tatli M, Sakizli M, Ozkan H, 2005. Hyperbilirubinemic serum is cytotoxic and induces apoptosis in murine astrocytes. Biol. Neonate 87, 99–104. [DOI] [PubMed] [Google Scholar]
- Lee SM, Yune TY, Kim SJ, Kim YC, Oh YJ, Markelonis GJ, Oh TH, 2004. Minocycline inhibits apoptotic cell death via attenuation of TNF-alpha expression following iNOS/NO induction by lipopolysaccharide in neuron/glia co-cultures. J. Neurochem 91, 568–578. [DOI] [PubMed] [Google Scholar]
- Lin S, Wei X, Bales KR, Paul AB, Ma Z, Yan G, Paul SM, Du Y, 2005. Minocycline blocks bilirubin neurotoxicity and prevents hyperbilirubinemia-induced cerebellar hypoplasia in the Gunn rat. Eur. J. Neurosci 22, 21–27. [DOI] [PubMed] [Google Scholar]
- Lull ME, Block ML, 2010. Microglial activation and chronic neurodegeneration. Neurotherapeutics 7, 354–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machaca K, 2003. Ca2+-calmodulin-dependent protein kinase II potentiates store-operated Ca2+ current. J. Biol. Chem 278, 33730–33737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JW, Shapiro SM, Silverstein FS, Johnston MV, 1998. Role of glutamate receptor-mediated excitotoxicity in bilirubin-induced brain injury in the Gunn rat model. Exp. Neurol. 150, 21–29. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG, 1998. Glial cell reactions in neurodegenerative diseases: pathophysiology and therapeutic interventions. Alzheimer Dis. Assoc. Disord 12 (Suppl. 2), S1–6. [PubMed] [Google Scholar]
- NaveenKumar SK, Thushara RM, Sundaram MS, Hemshekhar M, Paul M, Thirunavukkarasu C, Basappa, Nagaraju G, Raghavan SC, Girish KS, Kemparaju K, Rangappa KS, 2015. Unconjugated bilirubin exerts pro-apoptotic effect on platelets via p38-MAPK activation. Sci. Rep 5, 15045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlstein TS, Pande RL, Creager MA, Weuve J, Beckman JA, 2008. Serum total bilirubin level, prevalent stroke, and stroke outcomes: NHANES 1999-2004. Am. J. Med 121, 781–788 e781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues CM, Sola S, Brites D, 2002. Bilirubin induces apoptosis via the mitochondrial pathway in developing rat brain neurons. Hepatology 35, 1186–1195. [DOI] [PubMed] [Google Scholar]
- Rosenstein BS, Ducore JM, Cummings SW, 1983. The mechanism of bilirubin-photosensitized DNA strand breakage in human cells exposed to phototherapy light. Mutat. Res 112, 397–406. [DOI] [PubMed] [Google Scholar]
- Rossi F, Francese M, Iodice RM, Falcone E, Vetrella S, Punzo F, De Vita S, Perrotta S, 2005. [Inherited disorders of bilirubin metabolism]. Minerva Pediatr. 57, 53–63. [PubMed] [Google Scholar]
- Schwertner HA, Jackson WG, Tolan G, 1994. Association of low serum concentration of bilirubin with increased risk of coronary artery disease. Clin. Chem 40, 18–23. [PubMed] [Google Scholar]
- Shultz RB, Zhong Y, 2017. Minocycline targets multiple secondary injury mechanisms in traumatic spinal cord injury. Neural Regen. Res 12, 702–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva RF, Rodrigues CM, Brites D, 2002. Rat cultured neuronal and glial cells respond differently to toxicity of unconjugated bilirubin. Pediatr. Res 51, 535–541. [DOI] [PubMed] [Google Scholar]
- Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN, 1987. Bilirubin is an antioxidant of possible physiological importance. Science 235, 1043–1046. [DOI] [PubMed] [Google Scholar]
- Vasavda C, Kothari R, Malla AP, Tokhunts R, Lin A, Ji M, Ricco C, Xu R, Saavedra HG, Sbodio JI, Snowman AM, Albacarys L, Hester L, Sedlak TW, Paul BD, Snyder SH, 2019. Bilirubin links heme metabolism to neuroprotection by scavenging superoxide. Cell Chem. Biol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vodret S, Bortolussi G, Jasprova J, Vitek L, Muro AF, 2017. Inflammatory sig-nature of cerebellar neurodegeneration during neonatal hyperbilirubinemia in Ugt1 (−/−) mouse model. J. Neuroinflammation 14, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bernhardi R, Eugenin-von Bernhardi L, Eugenin J, 2015. Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci 7, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Smith CJ, Van Eldik LJ, 2004. Activated glia induce neuron death via MAP kinase signaling pathways involving JNK and p38. Glia 45, 170–179. [DOI] [PubMed] [Google Scholar]
- Yueh MF, Chen S, Nguyen N, Tukey RH, 2017. Developmental, genetic, dietary, and xenobiotic influences on neonatal hyperbilirubinemia. Mol. Pharmacol 91, 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]