

Abstract
Metabolic reprogramming exists in a variety of cancer cells, with the most relevance to glucose as a source of energy and carbon for survival and proliferation. Of note, Nrf1 was shown to be essential for regulating glycolysis pathway, but it is unknown whether it plays a role in cancer metabolic reprogramming, particularly in response to glucose starvation. Herein, we discover that Nrf1α−/− hepatoma cells are sensitive to rapid death induced by glucose deprivation, such cell death appears to be rescued by Nrf2 interference, but HepG2 (wild-type, WT) or Nrf2−/− cells are roughly unaffected by glucose starvation. Further evidence revealed that Nrf1α−/− cell death is resulted from severe oxidative stress arising from aberrant redox metabolism. Strikingly, altered gluconeogenesis pathway was aggravated by glucose starvation of Nrf1α−/− cells, as also accompanied by weakened pentose phosphate pathway, dysfunction of serine-to-glutathione synthesis, and accumulation of reactive oxygen species (ROS) and damages, such that the intracellular GSH and NADPH were exhausted. These demonstrate that glucose starvation leads to acute death of Nrf1α−/−, rather than Nrf2−/−, cells resulting from its fatal defects in the redox metabolism reprogramming. This is owing to distinct requirements of Nrf1 and Nrf2 for regulating the constructive and inducible expression of key genes involved in redox metabolic reprogramming by glucose deprivation. Altogether, this work substantiates the preventive and therapeutic strategies against Nrf1α-deficient cancer by limiting its glucose and energy demands.
1. Introduction
Metabolic reprogramming is involved in deregulating anabolism and catabolism of glucose, fatty acids, and amino acids, which is existing in a variety of cancer cells [1], to facilitate those uncontrolled cell growth and proliferation. Usually, cancer cells increase their uptake of nutrients, mainly including glucose and glutamine. Of note, the ensuing metabolism of glucose, as a major nutrient to fuel cell growth and proliferation, comprises glycolysis pathway, gluconeogenesis pathway, pentose phosphate pathway (PPP), and serine synthesis pathway (SSP), all of which occur in the cytoplasm, besides the tricarboxylic acid cycle (i.e., TCA cycle) occurring in the mitochondria [2]. Among these, glycolysis is a central pathway of glucose metabolism but also can be branched towards many anabolic pathways via its metabolic intermediates [3]. In cancer cells, decreases in both their oxidative phosphorylation and aerobic glycolysis are accompanied by increases in the another glycolytic flux, which is independent of oxygen concentration to support the enhanced anabolic demands (of e.g., nucleotides, amino acids, and lipids) by providing glycolytic intermediates as raw material [4, 5]. Thereby, such metabolic changes constitute one of the typical hallmarks of tumor cells [1, 6].
Clearly, cell life and death decisions are influenced by its cellular metabolism [7], particularly the metabolism of cancer cells, which is the most relevant to glucose as a source of energy and carbon. A recent study has uncovered the lower glycolytic rates leading to enhanced cell death by apoptosis [8]. By contrast, the another enforced glycolysis can also effectively inhibit apoptosis [9, 10]. As for the more nutrient uptake than that of normal cells, cancer cells frequently undergo certain metabolic stress due to the shortages in supply of oxygen, nutrients, and growth factors. As such, the rapidly proliferating cancer cells were also unable to stop their anabolic and energy requirements, which eventually leads to cell death [11]. Thereby, such a nutrient limitation has been proposed as an effective approach to inhibit the proliferation of cancer cells. For this end, glucose starvation is also considered as a major form of metabolic stress in cancer cells [12]. However, whether the determination of these cell life-or-death fates is influenced in response to metabolic stress induced by glucose starvation remains to be not well understood.
Glucose metabolism is also regulated by the proto-oncogene c-Myc, which was involved in glycolysis by regulating the glycolytic enzymes [13] and also promoted serine biosynthesis upon nutrient deprivation in cancer cells [14]. The another key oncogene HIF-1 was also identified to act as a central regulator of glucose metabolism [15, 16]. Besides, the tumor suppressor p53 can also play a key negative regulatory role in glycolysis by reducing the glucose uptake [17]. Herein, we determined whether two antioxidant transcription factors Nrf1 (also called Nfe2l1, as a tumor repressor) and Nrf2 (as a tumor promoter) are required for glycolysis and other glucose metabolic pathways and also involved in the redox metabolic reprogramming induced by glucose deprivation.
Among the cap'n'collar (CNC) basic-region leucine zipper (bZIP) family of transcription factor, Nrf1 and Nrf2 are two important members for maintaining redox homeostasis by binding = antioxidant response elements (AREs) of their downstream gene promoters [18]. However, ever-mounting evidence revealed that the water-soluble Nrf2 activation promotes cancer progression and metastasis [19–21]. Notably, Nrf2 also has a direct or another indirect role in all the hallmarks of cancer, such as mediating metabolic reprogramming [22] and altering redox homeostasis [23]. By contrast, the membrane-bound Nrf1 is subjected to alternative translation and proteolytic processing of this CNC-bZIP protein to yield multiple distinct isoforms of between 140 kDa and 25 kDa; they included TCF11/Nrf1α (120~140 kDa), Nrf1β (~65 kDa), Nrf1γ (~36 kDa), and Nrf1δ (~25 kDa). Among them, Nrf1α was identified to exist as a major isoform in HepG2 cells as described previously [24]. The specific knockout of Nrf1α (as a dominant tumor repressor) leads to obvious malignant proliferation and tumor metastasis of Nrf1α−/−-derived hepatoma in xenograft model mice [25]. Besides, Nrf1 was also considered to be involved in hepatic lipid metabolism by directly regulating Lipin1 and PGC-1 genes [26]. Moreover, Nrf1 was also found to contribute to the negative regulation of the cystine/glutamate transporter and lipid-metabolizing enzymes [27]. Interestingly, Nrf1 was also positively involved in glycolysis pathway by regulating the Slc2a2, Gck [28], and HK1 genes [29]. However, it is not clear about a role of Nrf1 in mediating the cancer cellular response to metabolic stress, especially stimulated by glucose deprivation.
In this study, we observed a surprising change in the growth of Nrf1α−/− cells starved in a nonglucose medium. It was found that Nrf1α−/− cells were more sensitive to glucose deprivation, leading to acute death within 12 h of glucose deprivation, while both cases of WT and Nrf2−/− were almost unaffected. As such, the glucose starvation-induced death of Nrf1α−/− cells was also rescued by Nrf2 interference. Further examinations revealed that a large amount of reactive oxygen species (ROS) was accumulated by glucose deprivation in Nrf1α−/− cells, leading to severe oxidative stress. Such a redox imbalance was also attributable to the fact that the intracellular reducing agents (i.e., GSH) were exhausted during glucose deprivation. This was due to fatal defects of Nrf1α−/− cells, resulting in aberrant expression of some key genes (e.g., CAT, GPX1, GSR, PCK1/2, G6PD, PHGDH, and ATF4) that are responsible for the redox metabolism reprogramming of Nrf1α−/−, but not Nrf2−/−, cells, albeit these genes were differentially regulated by Nrf1 and/or Nrf2. Collectively, these demonstrate that Nrf1 and Nrf2 play distinct and even opposite roles in mediating cancer cellular responses to the metabolic stress induced by glucose starvation. Notably, Nrf1 acts as a pivotal player to determine the steady-state level of distinct intracellular redox homeostasis.
2. Results
2.1. Nrf1α−/− Cells Are Susceptible to the Cellular Death from Glucose Deprivation
To explore whether both CNC-bZIP factors exert distinct or opposite roles in mediating the cancer cellular response to metabolic stress, Nrf1α−/−- and Nrf2−/−-deficient HepG2 cell lines (both had been established by Qiu et al. [30]), along with WT cells, were subjected to glucose-free starvation for distinct lengths of time. Subsequently, changes in these cell morphology after glucose deprivation were observed by microscopy. Within 6 h of glucose starvation, no obvious abnormalities of all three cell lines were shown (Figure S1A). In fact, they were growing well, with no changes in the small number of dead cells (Figure S1B). However, when the time of glucose starvation was extended to 12 h, Nrf1α−/− cells displayed apparent morphological characteristics of cell death (resembling the necrosis and/or necroptosis, Figure 1(a)), of which such dead cells were stained by trypan blue to 49.26% (Figure 1(b)). By sharp contrast, both WT and Nrf2−/− cell lines were largely unaffected by 12 h of glucose starvation (Figures 1(a) and 1(b)). These cell death or survival was further corroborated by flow cytometry analysis of dual staining cells with fluorescent Annexin V and propidium iodide (PI), showing that Nrf1α−/− cells were more susceptible to the cell death induced by glucose deprivation for 12 h, when compared with other two examined cell lines of WT and Nrf2−/− (Figure 1(c)).
Figure 1.
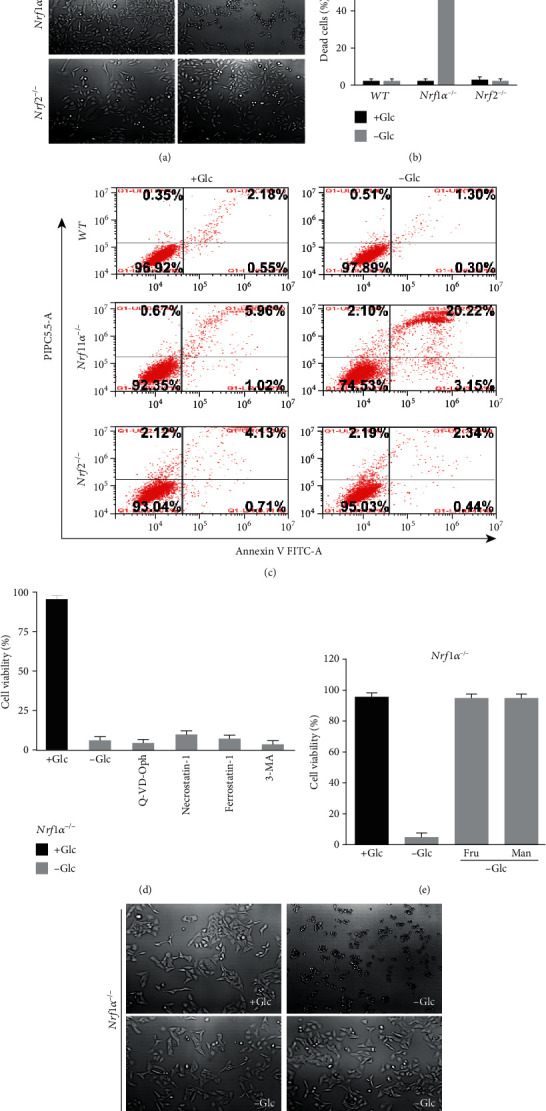
The response of Nrf1α−/− cells to glucose starvation. (a) Morphological changes of WT, Nrf1α−/−, and Nrf2−/− cells, which had been subjected to glucose deprivation for 12 h, were observed by microscopy (with an original magnification of 200x). (b) The percentage of their dead cells was calculated after being stained by trypan blue. (c) The apoptosis of glucose-starved cells was analyzed by flow cytometry, after being incubated with Annexin V-FITC and PI. (d) Nrf1α−/− cell viability was determined by incubation for 24 h with q-VD-OPH (10 μM), Necrostatin-1 (100 μM), Ferrostatin-1 (2 μM), or 3-methyladenine (2 mM) in the glucose-free media, each of which was resolved in DMSO as a vehicle. (e) Nrf1α−/− cell survival was recovered from glucose deprivation by being cultured for 24 h in alternative media containing 25 mM of fructose (Fru) or mannose (Man). (f) Morphology of Fru/Man-recovered Nrf1α−/− cells was visualized by microscopy (with an original magnification of 200x).
As the glucose starvation was further extended to 24 h, almost all Nrf1α−/− cells were subjected to the cellular death (Figure S1C). Such prolonged glucose starvation-induced death of Nrf1α−/− cells was incremented to 95.6% of their examined cells, but only a small number (20%) of WT cells were showed to its cellular death (Figure S1D). Meanwhile, Nrf2−/− cells appeared to be remaining robust resistant to the putative cellular death stimulated by glucose deprivation for 24 h, however (Figure S1, C & D).
Next, several inhibitors of distinct signaling pathways towards cell death were here employed so as to determine which types of Nrf1α−/− cell death are resulted from glucose deprivation. Unexpectedly, treatment of Nrf1α−/− cells with Q-VD-OPH (acting as a pan-caspase inhibitor to block the apoptosis pathway), Necrostatin-1 (as a necroptosis inhibitor), Ferrostatin-1 (as a ferroptosis inhibitor), and 3-methyladenine (3-MA, as an autophagy inhibitor) demonstrated that they all had not exerted any cytoprotective effects against the cellular death caused by glucose deprivation (Figure 1(d)). Thereby, it is inferable that Nrf1α−/− cell death from glucose withdrawal may pertain to a major nonapoptotic form of cellular necrosis, but the detailed mechanism of Nrf1α−/− cell death awaits further study.
Since glucose deprivation, but not the glycolytic inhibition, leads to death of Nrf1α−/− cells, we hence investigated whether the cellular death was rescued by other sugar sources, such as fructose or mannose, because both could also serve as a potent alternative to glucose. As shown in Figures 1(e) and 1(f), the results unraveled that fructose and mannose were metabolically utilized in Nrf1α−/− cells insomuch as to resist against the cell death induced by glucose deprivation. This demonstrates that the lack of sugar source is responsible for determining the death fate of Nrf1α−/− cells.
2.2. Nrf1α−/− Cell Death Is Driven by Glucose Deprivation Leading to Severe Endogenous Oxidative Stress
Clearly, cell life or death fate decisions are selectively determined by the intracellular energy metabolism and redox homeostasis [31]. Thereby, we herein examined whether endogenous oxidative stress of Nrf1α−/− cells is stimulated by glucose starvation contributing to the cellular responsiveness to death. As anticipated, Figures 2(a) and 2(b)) showed that Nrf1α−/− cell death arising from the removal of glucose enabled to be sufficiently rescued after addition of 2-deoxyglucose (2DG, as an analogue of glucose) to the sugar-free culture medium. This is due to the fact that 2DG has a potent ability to inhibit the glycolysis, and thus this treatment of Nrf1α−/− cells enabled the existing available glucose to enter the PPP route in order to generate certain amounts of NADPH (that acts as a major metabolically reducing agent required for the setting of intracellular basal redox state [32]).
Figure 2.
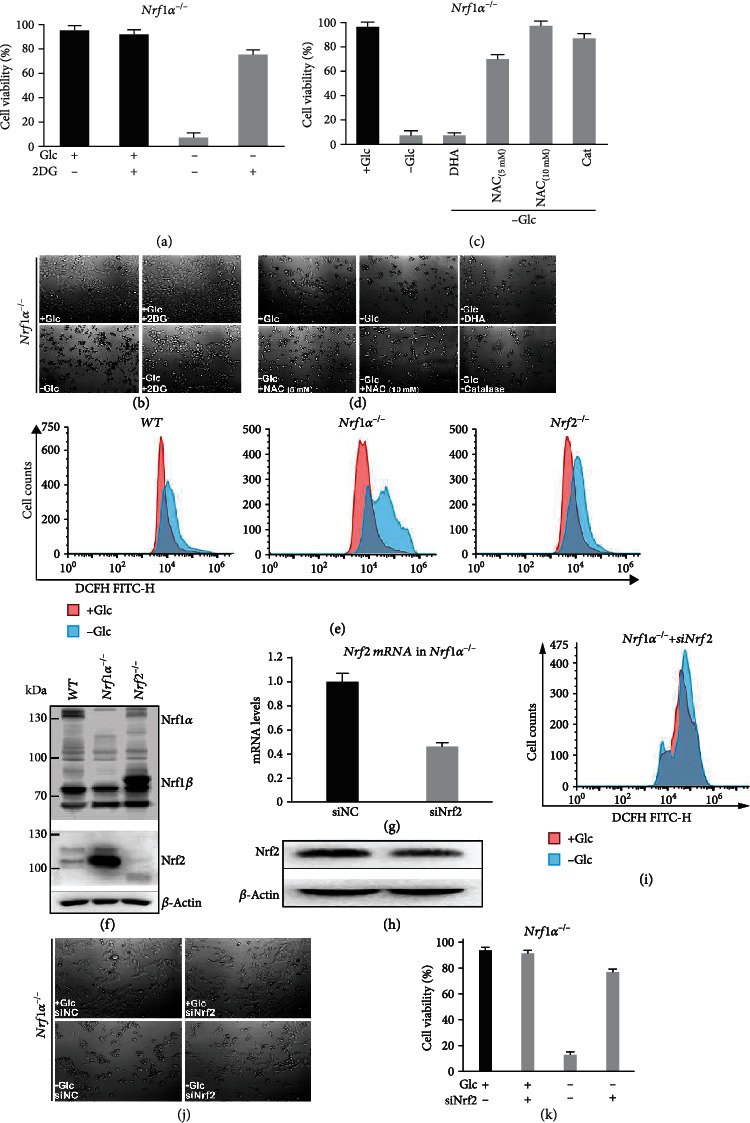
Distinct redox states of starved or rescued Nrf1α−/− cells with different morphological changes. (a) Nrf1α−/− cells had been rescued by incubation for 24 h with 2-deoxyglucose (2DG, 10 mM in the glucose-free media), before the cell viability was determined by trypan blue staining. (b) Morphology of 2DG-rescued Nrf1α−/− cells was visualized by microscopy (with an original magnification of 200x). (c) Nrf1α−/− cell viability was calculated after they had been cultured for 24 h in glucose-free media containing N-acetyl-cysteine (NAC at 5-10 mM), catalase (CAT at 50 units/ml), or dehydroascorbic acid (DHA at 100 μM). (d) Morphological changes of Nrf1α−/− cells, which had been treated with NAC, CAT or DHA, were observed by microscopy (with an original magnification of 200x). (e) Distinct ROS levels were determined by flow cytometry analysis of WT, Nrf1α−/−, and Nrf2−/− cells that had been starved, or not starved, for 12 h. (f) Abundances of Nrf1 and Nrf2 proteins were determined by Western blotting of WT, Nrf1α−/−, and Nrf2−/− cells. (g, h) The interference of siNrf2 in Nrf1α−/− cells was identified by RT-qPCR (g) and Western blotting (h). (i) Changes in ROS levels of Nrf1α−/−+siNrf2 cells were analyzed by flow cytometry after 12 h of glucose deprivation. (j) The effects of siNrf2 on glucose-starved cell morphology were observed by microscopy (with an original magnification of 200x). (k) Nrf1α−/−+siNrf2 cell viability was evaluated after they had been subjected to glucose starvation for 24 h.
Further examinations revealed that glucose starvation-induced death of Nrf1α−/− cells was significantly mitigated or completely rescued by treatment with N-acetyl-L-cysteine (NAC, an antioxidant agent to increase glutathione synthesis) or catalase (CAT, an enzyme that catalyzes the breakdown of hydrogen peroxide) (Figures 2(c) and 2(d)). By contrast, Nrf1α−/− cell death triggered by glucose starvation was almost unaffected by treatment with dehydroascorbic acid (DHA, as a stable oxidative product of L-ascorbic acid). These suggest that severe oxidative stress may contribute to the rapid death of Nrf1α−/− cells from glucose deprivation. This notion is further evidenced by flow cytometry analysis of distinct cellular oxidative states (Figure 2(e)). The results unraveled that glucose deprivation caused an obvious accumulation of ROS in Nrf1α−/− cells, with the oxidative fluorescent images becoming widened and right-shifted, when compared with the other two cases of WT and Nrf2−/− (only with modestly increased ROS levels) (Figure 2(e)). Collectively, these imply a fatal defect of Nrf1α−/−, rather than Nrf2−/−, cells in the antioxidant cytoprotective response against the cellular death attack from glucose withdrawal stress.
2.3. Rapid Death of Glucose-Starved Nrf1α−/− Cells Can Be Rescued by Interference with Nrf2
Based on a similar structure and function of Nrf1 and Nrf2 [33], it is postulated that the loss of Nrf1α is likely compensated by Nrf2. As anticipated, Western blotting revealed that aberrant high expression of Nrf2 was determined in Nrf1α−/− cells, when compared with that of WT cells (Figure 2(f)); this is consistent with our previous finding by Qiu et al. [30] (in which subcellular distribution of Nrf2 in Nrf1α−/− cells was shown). Conversely, considerable lower expression levels of Nrf1α-derived protein isoforms were maintained in Nrf2−/− cells, albeit with a compensatory higher expression of the short Nrf1β (Figure 2(f)).
Since such hyperexpression of Nrf2 in Nrf1α−/− cells serves as a complement to Nrf1α knockout, it is thus inferable that Nrf2 may also contribute to mediating a putative response of Nrf1α−/− cells to death attack by glucose starvation. Thereby, we here tried to interfere with the Nrf2 expression by siRNA transfection into glucose-starved Nrf1α−/− cells. As shown in Figures 2(g) and 2(h)), both mRNA and protein expression levels of Nrf2 were significantly knocked down by its specific siRNA (i.e., siNrf2) in Nrf1α−/− cells. More interestingly, glucose starvation-induced death of Nrf1α−/− cells was strikingly ameliorated by the interference of siNrf2 (Figures 2(j) and 2(k)). This is also substantiated by further evidence obtained from flow cytometry analysis of the cell death (Figure S1E). Such effectively siNrf2-alleviated death of Nrf1α−/− cells was accompanied by a significant reduction in the intracellular ROS accumulation by glucose deprivation (Figure 2(i)). Together, these demonstrate that hyperactive Nrf2 can also make a major contribution to the accumulation of ROS products in Nrf1α−/− cells, leading to the cellular death driven by glucose starvation.
2.4. The Failure of Redox Defense Systems in Nrf1α−/− Cells Contributes to Its Lethality of Glucose Starvation
The aforementioned evidence (as shown in Figures 2(c) and 2(e)) demonstrated that glucose starvation-induced death of Nrf1α−/− cells resulting from severe oxidative stress was effectively prevented by NAC and CAT. This suggests that the intracellular redox state is rebalanced by either NAC or CAT, because NAC facilitates the conversion of oxidized glutathione (GSSG) to reduced glutathione (GSH) by glutathione(-disulfide) reductase (GR or GSR), while CAT can catalyze hydrogen peroxide (H2O2) breakdown to water and oxygen, such that the cytotoxic effects of ROS on Nrf1α−/− cells are inhibited.
Herein, we further examined changes in basal and glucose starvation-inducible expression of CAT and GSR, as well as other redox cycling and relevant enzymes, including GPX1, PRX1, TRX1, TRX2, NOX4, SOD1, and SOD2 (Figure 3(a)), in Nrf1α−/− cells cultured in complete or glucose-free media. As shown in Figures 3(b) and 3(c), RT-qPCR analysis of Nrf1α−/− cells demonstrated significant increases in basal mRNA expression levels of CAT and GPX1 (glutathione peroxidase 1, which catalyzes the reduction of H2O2 and organic hydroperoxides by GSH, so as to protect cells against oxidative damages), when compared with those of WT cells. After glucose withdrawal from the culture of Nrf1α−/− cells, such basal expression of both CAT and GPX1 was abruptly inhibited close to the levels measured from WT cells (Besides GLUT1, other key metabolic enzymes). Similar marked changes in CAT and GPX1 expression were, however, not observed in Nrf2−/− cells.
Figure 3.
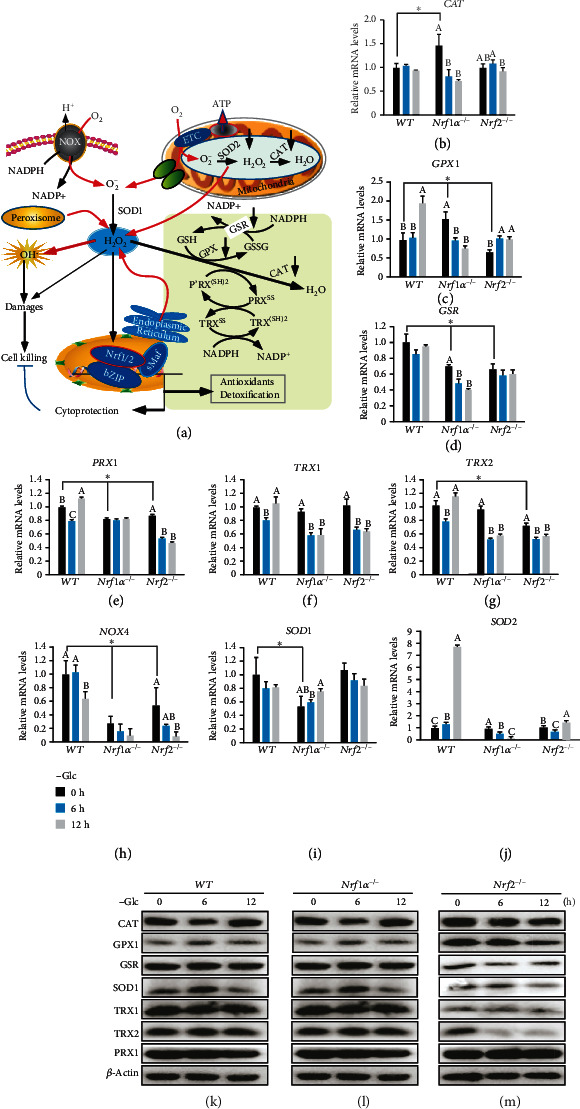
Dysfunction of redox signaling controls and defense systems in glucose-starvated Nrf1α−/− cells. (a) Schematic representation of intracellular ROS products, along with redox signaling controls and antioxidant defense systems. In this response, Nrf1 and Nrf2 can be induced to translocate the nucleus, in which its functional heterodimer with sMaf or other bZIP proteins is formed in order to transcriptionally regulate distinct subsets of ARE-driven genes, which are responsible for antioxidant, detoxification, and cytoprotection against a variety of cellular stress. (b–j) Alterations in mRNA expression levels of distinct genes, such as (b) GSR (glutathione disulfide reductase), (c) CAT (catalase), (d) GPX1 (glutathione peroxidase 1), (e) PRX1 (peroxidase 1), (f) TRX1 (thioredoxin 1), (g) TRX2 (thioredoxin 2), (h) NOX4 (NADPH oxidase 4), (i) SOD1 (superoxide dismutase 1), and (j) SOD2 (superoxide dismutase 2), in WT, Nrf1α−/−, and Nrf2−/− cells, which had been or had not been starved in glucose-free media for 0-12 h, were determined by RT-qPCR analysis. Then, the asterisk “∗” only represents a significant change in WT, Nrf1α−/−, and Nrf2−/− cell lines in the glucose-free culture for 0 h (P < 0.05), while the letters A, B, and C represent significant changes in the same cell line without glucose cultured for 0, 6, and 12 h (P < 0.05). (k–m) Changes in abundances of the following proteins CAT, GPX1, GSR, SOD1, TRX1, TRX2, and PRX1 were visualized by Western blotting of WT (k), Nrf1α−/− (l), and Nrf2−/− (m) cells that had been or had not been glucose-starved for 0-12 h.
By contrast, basal GSR expression was downregulated in Nrf1α−/− cells, and glucose deprivation also caused it to be further repressed to a considerable lower level, by comparison to the WT cells (Figure 3(d)). However, similar downregulation of GSR in Nrf2−/− cells was almost unaffected by glucose starvation. Conversely, expression of PRX1 (peroxiredoxin-1, also called thioredoxin peroxidase 1) was significantly diminished by glucose deprivation in Nrf2−/−, rather than Nrf1α−/−, cells, albeit its basal expression was similarly downregulated in these two deficient cell lines (Figure 3(e)). Further examinations of Nrf1α−/− and Nrf2−/− cells revealed that glucose starvation caused remarkable decreases in expression of TRX1 and TRX2 (i.e., both thioredoxin proteins involved in many reversible redox reactions) (Figures 3(f) and 3(g)). In addition, it is intriguing that deficiency of either Nrf1α or Nrf2 also led to significantly decreased expression of NOX4 (NADPH oxidase 4, which acts as an oxygen sensor and also catalyzes the reduction of molecular oxygen to various ROS) (Figure 3(h)), but the expression of SOD1 (superoxide dismutase 1) was only significantly decreased in Nrf1α−/− cells, while the expression of SOD2 is hardly affected by Nrf1α or Nrf2 deficiency (Figures 3(i) and 3(j)). Notably, only WT cells, but not Nrf1α−/− or Nrf2−/− cells, showed significant increases in glucose starvation-stimulated expression of GPX1 and SOD2 (Figures 3(c) and 3(j)), but not other examined gene transcripts.
Further Western blotting of Nrf1α−/− cells unraveled that the expression of CAT, GPX1, GSR, TRX1, and TRX2, but not PRX1 or SOD1, was substantially decreased by glucose deprivation for 12 h (Figures 3(l) and S2B). By contrast, abundances of these examined proteins except PRX1 were only marginally decreased to lesser extents by glucose starvation of Nrf2−/− cells (Figures 3(m) and S2C). Such minor effects cannot also be excluded to be attributable to a modest decrease of Nrf1α in Nrf2- deficient cells (Figure 2(f)). In addition, it should be noted that not any increases of the redox-relevant proteins were stimulated by glucose starvation for 6 h to 12 h, even in WT cells (Figures 3(k) and S2A). Together, it is postulated that Nrf1α−/−, but not Nrf2−/−, cells are likely to have certain fatal defects in setting the intracellular redox homeostasis along with antioxidant defense systems, such that the resulting redox imbalance contributes to severe endogenous oxidative stress and concomitant damages leading to Nrf1α−/− cell death, particularly after 12-h glucose starvation.
2.5. Deregulated Glucose Metabolism and Energy Demands of Nrf1α−/− Cells Are Deteriorated by Glucose Deprivation
Since aerobic glycolysis provides the main energy for cancer cells (as illustrated in Figure 4(a)), it is evitable that the intracellular production of ATP as a major energy source could thus be affected by glucose withdrawal from WT, Nrf1α−/−, and Nrf2−/− cell culture in sugar-free media. As shown in Figure 4(b), a substantial diminishment in the basal ATP levels of Nrf2−/− cells was determined, and even glucose deprivation-stimulated ATP products were also maintained to considerable lower levels, when compared to those corresponding values measured from WT cells. By sharp contrast, the basal ATP levels of Nrf1α−/− cells (with aberrant hyper-active Nrf2) were significantly elevated (Figure 4(b)), so as to meet the needs of its malignant growth and proliferation [25, 30]). Intriguingly, such higher ATP production by Nrf1α−/− cells was further promoted by glucose starvation for 6 h, and thereafter abruptly declined by 12-h prolonged starvation to basal levels of wild-type cells (Figure 4(b)). Accordingly, similar alternations in basal and glucose starvation-stimulated expression of GLUT1 (glucose transporter 1) were also determined in Nrf1α−/− cells (Figure 4(c)), so as to meet its highly metabolizable energy requirements. This is further supported by Western blotting of Nrf1α−/− cells, displaying a high expression pattern of GLUT1, particularly during glucose-free conditions (Figure 4(o)). Meanwhile, almost unaltered expression of GLUT1 in Nrf2−/− cells was observed, even upon glucose starvation (Figure 4(p)). But, this transporter abundances in WT cells were modestly decreased after glucose deprivation (Figure 4(n)).
Figure 4.
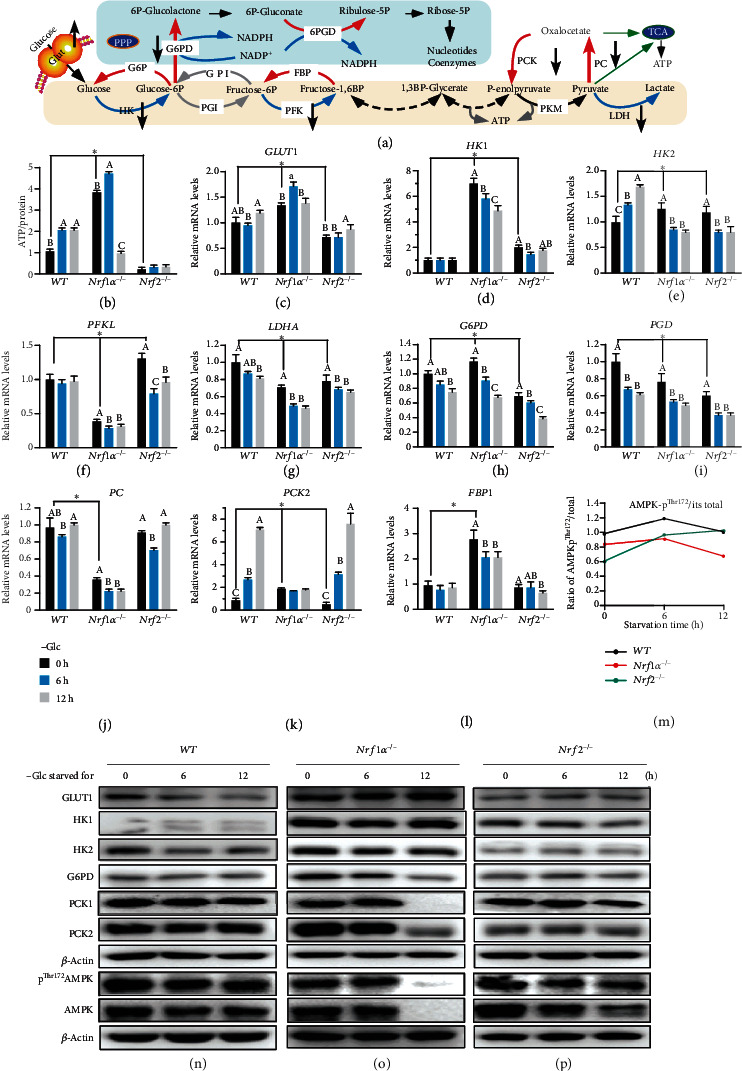
Deterioration of altered glucose metabolism and energy demands by glucose deprivation of Nrf1α−/− cells. (a) A schematic diagram to give a concise explanation of glycosis, gluconeogenesis, and pentose phosphate pathways (PPP). The key rate-limiting enzymes are indicated, apart from TCA (citric acid cycle). In some words, phospho- is represented by a single P letter. (b) Distinct ATP levels of WT, Nrf1α−/−, and Nrf2−/− cells were determined after glucose deprivation for 0-12 h. (c–l) Altered mRNA expression levels of key metabolic genes: (i) (c) GLUT1 (glucose transporter 1), (d) HK1 (hexokinase 1), (e) HK2, (f) PFKL (phosphofructokinase liver type), and (g) LDHA (lactate dehydrogenase A) involved in the glycolysis pathway; (ii) (h) G6PD (glucose-6-phosphate dehydrogenase) and (i) PGD (phosphogluconate dehydrogenase) as rate-limiting enzymes in the PPP; (iii) (j) PC (pyruvate carboxylase), (k) PCK2 (phosphoenolpyruvate carboxykinase 2), and (l) FBP1 (fructose bisphosphatase 1) responsible for the gluconeogenesis pathway, were analyzed by RT-qPCR analysis of WT, Nrf1α−/−, and Nrf2−/− cells that had been starved, or not starved, in the glucose-free media for 0-12 h. Then, the asterisk “∗” only represents a significant change in WT, Nrf1α−/−, and Nrf2−/− cell lines in the glucose-free culture for 0 h (P < 0.05), while the letters A, B, and C represent significant changes in the same cell line without glucose cultured for 0, 6, and 12 h (P < 0.05). (m) Phospho-AMPKThr172/AMPK ratios were calculated by the intensity of their immunoblots in Nrf1/2+/+, Nrf1α−/−, and Nrf2−/− cells. (n–p) Changes in protein abundances of GLUT1, HK1, HK2, G6PD, PCK1, PCK2, AMPK, and its phospho-AMPKThr172 were determined by Western blotting of Nrf1/2+/+ (n), Nrf1α−/− (o), and Nrf2−/− (p) cells, after having been glucose-starved, or not, for 0-12 h.
Besides GLUT1, other key metabolic enzymes (e.g., HK1/2, PFKL, and LDHA) required for the glycolysis pathway of cancer cells were also investigated herein. Among them, only hexokinase 2 (HK2) was transcriptionally activated by glucose starvation of WT cells, but not of Nrf1α−/− or Nrf2−/− cells. Although basal mRNA expression of HK2 was obviously upregulated, but rather its glucose starvation-stimulated expression was significantly suppressed, in Nrf1α−/− or Nrf2−/− cells (Figure 4(e)). Similarly, a substantial increase in basal HK1 expression occurred in Nrf1α−/− cells, whereas its transcriptional expression was strikingly reduced by glucose deprivation. By contrast, Nrf2−/− cells also showed a considerable high level of basal HK1 expression, albeit its mRNA transcription was unaffected by glucose starvation (Figure 4(d)). Further examinations by Western blotting revealed that both HK1 and HK2 protein expression levels were modestly decreased by glucose starvation of Nrf1α−/− cells, but appeared to be unaffected in glucose-starved WT and Nrf2−/− cells (Figures 4(n)–4(p) and S2, D-F). Moreover, apparent decreases in basal PFKL (phosphofructokinase, liver type) and LDHA (lactate dehydrogenase A) expression levels were observed in Nrf1α−/− cells (Figures 4(f) and 4(g)), of which the latter LDHA expression was further decreased by glucose starvation, while PFKL was only slightly reduced by this stimulation. Contrarily, Nrf2−/− cells showed a substantial increment in basal PFKL expression, but its mRNA transcription expression was markedly suppressed upon glucose deprivation (Figure 4(f)). By comparison, a modest decrease in basal expression of LDHA was also observed in Nrf2−/− cells, but its expression was roughly unaffected (Figure 4(g)). These collective data demonstrate distinct roles of Nrf1 and Nrf2 in controlling the expression of those key genes responsible for glycolysis.
Since the above observation (Figures 2(a) and 2(b)) uncovered that glucose-starved Nrf1α−/− cell death was rescued by 2DG, this glucose analogue could render the metabolic flow to enter the PPP and promote NADPH production. As anticipated, RT-qPCR analysis of the rate-limiting enzymes of the PPP showed that mRNA expression levels of G6PD (glucose-6-phosphate dehydrogenase) and PGD (phosphogluconate dehydrogenase) were significantly decreased after glucose deprivation of WT, Nrf1α−/−, and Nrf2−/− cells (Figures 4(h) and 4(i)), albeit bidirectionally positive and negative regulation of their basal expression by Nrf1α−/− or Nrf2−/− was determined. Furtherly, Western blotting also unraveled that G6PD protein abundances were significantly decreased in glucose-starved Nrf1α−/−, rather than WT and Nrf2−/−, cell lines (Figures 4(n)–4(p) and S2, D-F).
From the aforementioned evidence, it is postulated that the gluconeogenesis pathway should be enhanced after glucose deprivation, so that the resulting products were allowed to enter the PPP and other (redox) metabolic pathways. Thus, we investigated the expression of certain key enzymes involved in the gluconeogenesis. As shown in Figure 4(k), a significant increment in transcriptional expression of PCK2 (phosphoenolpyruvate carboxykinase 2) in WT and Nrf2−/− cells was stimulated by glucose starvation for 6 h to 12 h. However, no changes in mRNA expression of PCK2 were detected in glucose-starved Nrf1α−/− cells, although its basal expression was upregulated (Figure 4(k)). This is further supported by Western blotting of PCK1 and PCK2, revealing that both protein abundances were significantly decreased by glucose starvation, especially for 12 h, in Nrf1α−/− cells, but rather almost unaffected in glucose-starved WT and Nrf2−/− cells (Figures 4(n)–4(p) and S2, D-F). Further examinations of PC (pyruvate carboxylase) and FBP1 (fructose-bisphosphatase 1) unraveled that their mRNA levels were markedly reduced by glucose deprivation in Nrf1α−/− cells, but largely unaltered in glucose-starved WT or Nrf2−/− cell lines (Figures 4(j) and 4(l)). In addition, it should be noted that Nrf1α−/− cells manifested downregulation of basal PC expression, along with upregulation of basal FBP1 expression.
Next, we examined changes in active phosphorylation of AMPK (AMP-activated protein kinase), since it acts as a key regulator of energy metabolism [34]. As illustrated in Figure 4(m), the ratio of phosphorylated AMPKThr172 to total protein (as calculated by stoichiometry of their immunoblots as shown in Figures 4(n)–4(p)) was increased by glucose withdrawal from Nrf2−/− cells, but conversely decreased in glucose-starved Nrf1α−/− cells. The latter notion was also further supported by the fact that almost all of total AMPK and its phospho-AMPKThr172 proteins were evidently abolished by 12-h glucose starvation of Nrf1α−/− cells (Figure 4(o)), besides their significant reduction in glucose-starved Nrf2−/− cells (Figure 4(p)). Overall, the inactivation of cellular energy switch, along with blockage of its gluconeogenesis and ablation of both its PPP and glycolysis, is inferable as a crucial determinant of Nrf1α−/− cell death, resulting from glucose deprivation to deteriorate its altered energy metabolic demands.
2.6. Upregulation of Serine-to-Glutathione Synthesis by Glucose Deprivation Is Fatally Abolished in Nrf1α−/− Cells, Leading to Severe Endogenous Oxidative Stress
As illustrated in Figure 5(a), de novo serine synthesis from glycolytic metabolite 3-phosphoglycerate (3PG) by SSP contributes to major carbons for glutathione biosynthesis and also provides precursors for purine and pyrimidine biosynthetic pathway via the folate cycle. In the successive biochemical course, 3PG, as a key intermediate of glycolytic pathway, is allowed to flow into the SSP and thus limit ATP production, while oxidation of 3PG converts NAD+ to NADH so as to affect intracellular redox state. As a result, serine can also be converted to cysteine and glycine by key enzymes (Figure 4(a)), for maintaining intracellular glutathione homeostasis. Therefore, we herein investigated the putative effects of glucose deprivation on serine-to-glutathione synthesis pathways.
Figure 5.
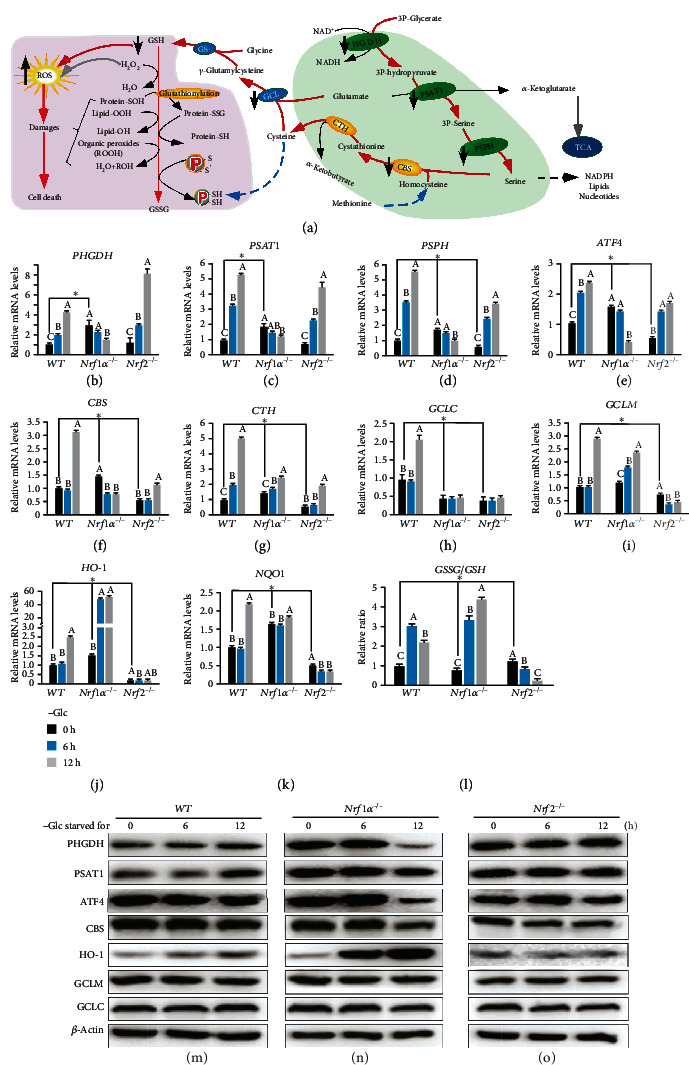
Fatal abolishment of de novo serine-to-glutathione biosynthesis by glucose deprivation of Nrf1α−/− cells. (a) A schematic to give a concise explanation of de novo serine synthesis pathway (SSP), along with ensuing transsulfuration to yield cysteine and glutathione (GSH). Both major buffers of GSH/GSSG and NADPH/NADP+ are tightly regulated by redox cycling switches and relevant defense systems against ROS and oxidative damages. (b–k) Altered mRNA expression levels of key biosynthetic genes as follows: (i) (b) PHGDH (phosphoglycerate dehydrogenase), (c) PSAT1 (phosphoserine aminotransferase 1), (d) PSPH (phosphoserine phosphatase) involved in the SSP, along with its regulator (e) ATF4 (activating transcription factor 4); (ii) (f) CBS (cystathionine beta-synthase) and (g) CTH (cystathionine gamma-lyase) essential for the transsulfuration to yield cysteine; (iii) (h) GCLC (glutamate-cysteine ligase catalytic subunit) and (i) GCLM (glutamate-cysteine ligase modifier subunit) to catalyze glutathione biosynthesis; (iv) as well as antioxidant genes, such as (j) HO-1 (heme oxygenase 1) and (k) NQO1 (NAD(P)H quinone dehydrogenase 1), were determined by RT-qPCR of WT, Nrf1α−/−, and Nrf2−/− cells that had been starved, or not starved, in the glucose-free media for 0-12 h. (l) Effects of glucose deprivation on the intracellular GSSG/GSH ratios in WT, Nrf1α−/−, and Nrf2−/− cells were assessed. The asterisk “∗” only represents a significant change in WT, Nrf1α−/−, and Nrf2−/− cell lines in the glucose-free culture for 0 h (P < 0.05), while the letters A, B, and C represent significant changes in the same cell line without glucose cultured for 0, 6, and 12 h (P < 0.05). (m–o) Changed abundances of PHGDH, PSAT1, ATF4, CBS, HO-1, GCLM, and GCLC proteins in WT (m), Nrf1α−/− (n), and Nrf2−/− (o) cells were visualized by Western blotting after glucose deprivation for 0-12 h.
As expected, significant increases in mRNA expression of those rate-limiting enzymes, such as PHGDH (phospho-glycerate dehydrogenase), PSAT1 (phosphoserine aminotransferase 1), and PSPH (phosphoserine phosphatase), required in the SSP, as well as their upstream regulatory factor ATF4 (activating transcription factor 4) [35], were triggered by glucose deprivation in WT and Nrf2−/− cells (Figures 5(b)–5(e)). By contrast, transcriptional induction of PHGDH, PSAT1, PSPH, and ATF4 by glucose deprivation was completely blocked or suppressed to lesser extents in starved Nrf1α−/− cells, albeit with an exception of evident increases in their basal mRNA expression upregulated by loss of Nrf1α (Figures 5(b)–5(e)). Accordingly, Western blotting showed that glucose starvation of WT cells for 12 h led to modest increases in abundances of PHGDH and PSAT1, rather than ATF4, to greater extents (Figures 5(m) and S2G). However, all three protein expression levels were strikingly decreased by glucose deprivation in Nrf1α−/− cells, but unaltered in glucose-starved Nrf2−/− cells (Figures 5(n) and 5(o) and S2, H & I).
Further insights into cysteine metabolism by transsulfuarion enzymes, such as CBS (cystathionine β-synthase) and CTH (cystathionine γ-lyase), revealed that both were transcriptionally upregulated by glucose starvation in WT cells (Figures 5(f) and 5(g)). Similarly, Nrf2−/− cells also manifested modest induction of CBS and CTH by glucose deprivation, notwithstanding the downregulation of their basal expression by loss of Nrf2 (Figures 5(f) and 5(g)). Conversely, the upregulation of basal CBS and CTH expression occurred in Nrf1α−/− cells (with hyper-active Nrf2). However, glucose starvation triggered opposite effects on transcriptional expression of CBS and CTH in Nrf1α−/− cells; the former CBS mRNA levels were evidently suppressed, while the latter CTH mRNA expression was marginally induced (Figures 5(f) and 5(g)). In addition, glucose deprivation also led to an obvious decrease in CBS protein levels in Nrf1α−/− or Nrf2−/− cells, when compared to its abundances measured from WT cells (Figures 5(m)–5(o) and S2, J-L).
Next, the effects of glucose deprivation on glutamate-cysteine ligase catalytic and modifier subunits (GCLC and GCLM, both comprising a key rate-limiting enzyme of glutathione biosynthesis) was investigated herein. As anticipated, both GCLC and GCLM mRNA expression was significantly induced by glucose deprivation of WT cells for 12 h (Figures 5(h) and 5(i)). Such induction of GCLC expression by glucose deprivation was completely abolished in Nrf1α−/− or Nrf2−/− cells, in which a considerable lower basal expression of GCLC was maintained by comparison with wild-type levels of HepG2 cells (Figure 5(h)). By contrast, basal and glucose starvation-stimulated GCLM expression levels were upregulated in Nrf1α−/− cells, but rather downregulated in Nrf2−/− cells (Figure 5(i)). However, GCLC and GCLM protein levels were almost unaffected after glucose starvation of the above examined three cell lines (Figures 5(m)–5(o) and S2, J-L).
Besides GCLC and GCLM, both HO-1 (heme oxygenase 1, also called HMOX1) and NQO1 (NAD(P)H quinone dehydrogenase 1) serve as downstream antioxidant genes of Nrf2 [36]. Here, an investigation by RT-qPCR revealed that glucose starvation for 12 h caused significant induction of HO-1 and NQO1 expression in WT cells (Figures 5(j) and 5(k)). By comparison, basal mRNA expression levels of HO-1 and NQO1 were upregulated in Nrf1α−/− cells, but rather downregulated in Nrf2−/− cells. Interestingly, remarkable induction of HO-1, rather than NQO1, by glucose deprivation was determined in Nrf1α−/− cells (Figures 5(j) and 5(k)). Conversely, transcriptional induction of HO-1 and NQO1 by glucose deprivation was roughly abolished or even slightly repressed in Nrf2−/− cells. Furthermore, Western blotting showed that HO-1 protein abundances were significantly induced by glucose starvation of WT and Nrf1α−/− cells for 6-12 h but appeared to be completely abolished in Nrf2−/− cells (Figures 5(m)–5(o) and S2, J-L). Moreover, further examination unraveled that glucose deprivation caused a significant increase in the GSSG/GSH ratio in WT or Nrf1α−/− cells, but this ratio was reversely decreased in Nrf2−/− cells (Figure 5(l)). Together, these findings demonstrate that the death of Nrf1α−/− cells is a consequence of severe endogenous oxidative stress and damages induced by glucose starvation. This is attributable to fatal defects of Nrf1α−/− cells in the redox metabolic reprogramming, such that the intracellular GSH/GSSG imbalance is further deteriorated by glucose deprivation, even though certain antioxidant response genes are aberrantly activated by the hyperexpression of Nrf2 in Nrf1α-deficient cells.
2.7. Distinct Requirements of Nrf1 and Nrf2 for the Redox Metabolic Reprogramming in Response to Glucose Deprivation
To determine distinct roles of Nrf1 and Nrf2 in mediating cellular redox metabolic responses to glucose deprivation, we examined the expression of Nrf1 and Nrf2 per se after glucose starvation of different genotypic cells for 6-12 h. The RT-qPCR showed that the transcriptional expression of Nrf1 and Nrf2 was significantly induced by glucose deprivation in WT cells (Figures 6(a) and 6(b)). However, such inducible mRNA expression levels of Nrf1 and Nrf2 by glucose starvation, as well as their basal expression, were almost completely abolished in Nrf1α−/− and Nrf2−/− cells, respectively (Figures 6(a) and 6(b)). Interestingly, glucose starvation of Nrf2−/− cells for 12 h also caused a modest increase in Nrf1 mRNA expression (Figure 6(a)), even though evident decreases in its basal mRNA levels (Figure 6(a)) and its Nrf1α-derived proteins (Figure 2(f)) were determined. Conversely, Nrf1α−/− cells only manifested a modest induction of Nrf2 mRNA expression by glucose deprivation, but with no changes in its basal mRNA levels (Figure 6(b)), albeit its proteins were strikingly accumulated by loss of Nrf1α (Figure 2(f)). Collectively, these demonstrate the bidirectional interregulatory roles of between Nrf1 and Nrf2 in distinct contributions to the cellular response triggered by glucose deprivation, in which Nrf1α is a dominant player.
Figure 6.
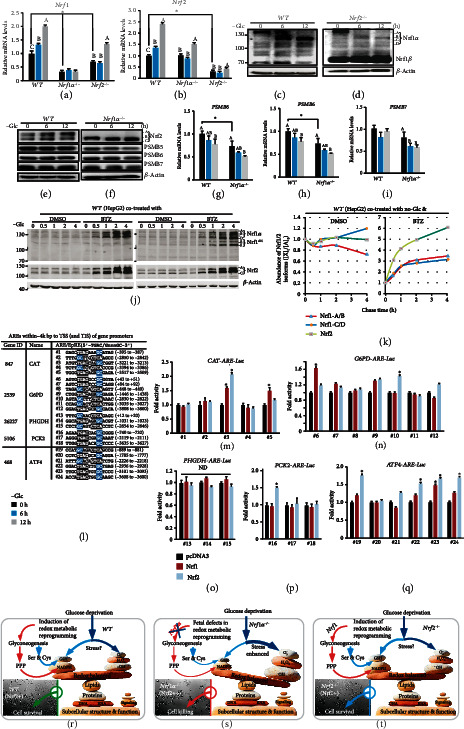
Distinct requirements of Nrf1 and Nrf2 for the cytoprotective response to glucose deprivation. (a, b) Distinct mRNA levels of Nrf1 (Nfe2l1) (a) and Nrf2 (Nfe2l2) (b) were determined by RT-qPCR analysis of WT, Nrf1α−/−, and Nrf2−/− cells, which had been starved, or not starved, for 0-12 h in the glucose-free media. Then, the asterisk “∗” only represents a significant change in WT, Nrf1α−/−, and Nrf2−/− cell lines in the glucose-free culture for 0 h (P < 0.05), while the letters A, B, and C represent significant changes in the same cell line without glucose cultured for 0, 6, and 12 h (P < 0.05). (c, d) Changes in Nrf1-derived protein isoforms in WT (c) and Nrf2−/− (d) cells were visualized by Western blotting after glucose deprivation for 0-12 h. (e, f) Western blotting of Nrf2, PSMB5, PSMB6, and PSMB7 proteins in WT (e) and Nrf1α−/− cells (f) was conducted after 0-12 h of glucose deprivation. (g–i) Alterations in mRNA levels of PSMB5 (g), PSMB6 (h), and PSMB7 (i) in WT and Nrf1α−/− cells were determined after glucose starvation for 0-12 h. the asterisk “∗” only represents a significant change in WT and Nrf1α−/− cell lines in the glucose-free culture for 0 h (P < 0.05), while the letters A, B, and C represent significant changes in the same cell line without glucose cultured for 0, 6, and 12 h (P < 0.05). (j) WT cells were or were co-treated for 0-4 h in the glucose-free media containing 1 μmol/L bortezomib (BTZ, a proteasomal inhibitor) or 0.1% DMSO vehicle, followed by Western blotting with antibodies against Nrf1 or Nrf2. (k) The intensity of the immunoblots representing distinct Nrf1-derived isoforms or Nrf2 proteins, respectively, in the above-treated WT cells (j) was quantified by the Quantity One 4.5.2 software, and then shown graphically. (l) 24 of the indicated ARE-adjoining sequences searched from the promoter regions of CAT, G6PD, PHGDH, PCK2, and ATF4 were cloned into the pGL3-Promoter vector, and the resulting contrasts served as ARE-driven luciferase (ARE-Luc) reporter genes. (m-q) WT cells were cotransfected with each of the above indicated ARE-Luc or non-ARE-Luc (as a background control) plasmids, together with an expression construct for Nrf1, Nrf2, or empty pcDNA3.1 vector, then allowed for 24-h recovery before the luciferase activity measured. The results were calculated as a fold change (mean ± S.D., n = 9) of three independent experiments. Then, the asterisk “∗” represents a significant change induced by expression Nrf1 or Nrf2, relative to that obtained from the empty pcDNA3.1 vector, in cotransfection with the same ARE-Luc (P < 0.05 and change folds >1.4). ND, nonsignificant difference. (r–t) Three distinct models are proposed to provide a better understanding of molecular basis for survival or death decisions made by glucose-starved WT (r), Nrf1α−/− (s), and Nrf2−/− (t) cells. In redox metabolic reprogramming caused by glucose deprivation, the glycosis was diminished or abolished, and thus replaced by increased glyconeogenesis. As a result, many of their intermediates are diverted to enter the PPP and serine-to-glutathione biosynthesis pathways, in order to yield certain amounts of GSH and NADPH. These two reducing agents enable cytoprotective adaptation to oxidative stress induced by glucose deprivation (r). However, rapid death of Nrf1α−/− cells results from its fatal defects in the redox metabolic reprogramming in cellular response to glucose starvation, as accompanied by severe oxidative stress and damage accumulation (s). Thereby, Nrf1 is reasonable as a dominant player in the key gene regulation of redox metabolic reprogramming caused by glucose deprivation. As a result, the existence of Nrf1 in Nrf2−/− cells can still endow their survival with its redox metabolic reprogramming in a rebalanced redox state (t).
Intriguingly, Nrf1 and Nrf2 exhibited two different but similar trends in their protein levels. Glucose starvation of WT or Nrf2−/− cells stimulated conversion of Nrf1 glycoprotein-A and then proteolytic processing to yield mature cleaved protein-C/D isoforms before transcriptionally regulating target genes. Thereby, Figures 6(c) and 6(d) showed that glycoprotein-A of Nrf1 was gradually disappeared from 6 h to 12 h in glucose-starved WT or Nrf2−/− cells and then was replaced by gradual enhancement of active cleaved Nrf1 protein-C/D. By contrast, Nrf2 proteins in WT or Nrf1α−/− cells were marginally increased by glucose starvation for 6-12 h (Figures 6(e) and 6(f)). Such distinct abundances in both Nrf1 and Nrf2 proteins, together with both discrepant mRNA expression levels (Figures 6(a) and 6(b)), are attributable to their distinct stability and transactivity during glucose deprivation.
Given that Nrf1, but not Nrf2, exerts an essential biological role in transcriptional expression of proteasomes (PSM), we hence examined potential effects of glucose deprivation on Nrf1-target PSM genes, to gain a better understanding of disparate contributions of Nrf1 and Nrf2 to death of Nrf1α−/−, but not Nrf2−/−, cells suffered from glucose starvation. As anticipated, both mRNA and protein levels of PSMB5, PSMB6, and PSMB7 (encoding the core enzymatic active β5, β1, and β2 subunits, respectively) were significantly abolished or suppressed in glucose-starved Nrf1α−/− cells, besides downregulation of their basal expression levels to varying extents (Figures 6(e)–6(i)). Such being the case, all three core subunits PSMB5, PSMB6, and PSMB7 in WT cells were also not induced by glucose deprivation. Reversely, mRNA expression of PSMB5 in WT cells was significantly suppressed to less than 20% of its basal level during glucose starvation from 6 h to 12 h, but with no obvious changes in expression of PSMB6 and PSMB7 (Figures 6(g)–6(i)). Altogether, these demonstrate that negative regulation of proteasomal expression by glucose deprivation is much likely to result in the accumulation of oxidatively damaged proteins (including Nrf2), particularly in glucose-starved Nrf1α−/− cells.
To clarify distinct contributions of Nrf1 and Nrf2 to mediating cellular responses induced by glucose starvation, we determined conversion of these two CNC-bZIP proteins-derived isoforms and their stability during glucose deprivation as shown in Figure 6(j), time-course analysis revealed that the full-length Nrf1α glycoprotein-A was gradually converted into deglycoprotein-B, and ensuing processed protein-C/D in glucose-starved WT cells (of note, similar processing of Nrf1 had been interpreted in details, as elsewhere [37]). Consequently, Nrf1α glycoprotein-A and transient deglycoprotein-B became gradually fainter within 4 h of glucose starvation, while its protein-C/D abundances were conversely enhanced (Figure 6(k), left panel). All these Nrf1α-derived isoforms were accumulated by cotreatment with the proteasomal inhibitor bortezomib (BTZ) (Figures 6(j) and 6(k)). Furthermore, the stability of Nrf1 precursor protein-A/B and its mature processed protein-C/D in glucose-starved WT cells was estimated by their distinct half-lives, which were determined to be 0.24 h (=14.4 min) and 2.53 h (=151.8 min), respectively, after treatment with cycloheximide (CHX) (Figure S3). Of note, even in the presence of BTZ, glucose deprivation stimulated a rapid processing mechanism of Nrf1α glycoprotein-A and deglycoprotein-B (with a collective half-life of 0.41 h = 24.6 min) to yield certain amounts of its processed protein-C/D (with a more than 4-h half-life, Figures 6(j) and S3). By contrast, Nrf2 protein levels were almost unaffected by glucose starvation of WT cells, but their abundances were further enhanced by BTZ (Figures 6(j) and 6(k)). Moreover, Nrf2 protein stability under glucose deprivation conditions was determined by its half-life, which was estimated to be 0.42 h (=25.2 min) after CHX treatment, but also extended by BTZ to 1.10 h (=66 min) (Figure S3). Thereby, it is inferable that discrepant stability of Nrf1 and Nrf2 is dedicated to distinct roles of both CNC-bZIP factors in mediating disparate cellular responses to glucose starvation.
Next, to gain insights into direct roles of Nrf1 and Nrf2 in mediating key genes transcriptional responses required for redox metabolic reprogramming, we established 24 of the indicated luciferase reporter genes driven by consensus ARE sequences from the CAT, G6PD, PHGDH, PCK2, and ATF4 promoter regions (Figure 6(l)). These ARE-driven Luciferase reporter assays revealed that transcriptional expression of CAT-ARE(#3)-Luc and ATF4-ARE(#23)-Luc was significantly activated by Nrf1 and Nrf2 (Figures 6(m)–6(q)). By contrast, Nrf2 alone also enabled transactivation of G6PD-ARE(#10)-Luc, PCK2-ARE(#16)-Luc and ATF4-ARE(#19, #22, and #24)-Luc (Figures 6(m)–6(q)), while only expression of CAT-ARE(#5)-Luc was upregulated by Nrf1 (Figure 6(m)). However, expression of all three PHGDH-ARE-Luc reporters appeared to be unaffected by either Nrf1 or Nrf2 (Figure 6(o)). Curiously, we should also notice that it is not hard to understand such seemingly-contradictory discrepancies between transactivation of these ARE-driven reporter genes mediated by Nrf1 (and Nrf2) (Figures 6(l)–6(q)) and relative high expression levels of the corresponding genes CAT (Figure 3(b)), G6PD (Figure 4(h)), PCK2 (Figure 4(k)), and PHGDH (Figure 5(b)) in Nrf1α−/− cells (albeit aberrant accumulation of Nrf2 being retained as shown in Figures 2(f)–2(h)), when compared with their controls.
3. Discussion
In the previous study, we reported that knockout of Nrf1α leads to malignant proliferation and tumor metastasis [25, 30]. Such malignant growth and proliferation of cancer cells are also dictated by nutrient availability [38], because they require large amounts of nutrients intake. On this basis, we herein discover that glucose starvation prevents the malignant proliferation and even causes a lot of cell death in Nrf1α-deficient hepatoma cells. This is fully consistent with the therapeutic strategy against cancer by its nutrients limiting [39].
It is, to our surprise, that glucose starvation leads to rapid cellular death of Nrf1α−/− cells within 12 h, albeit with aberrant accumulation of Nrf2 in this deficient cells, whereas Nrf2−/− cells manifest a strong resistance to the lethality of glucose deprivation, even though Nrf1 is downregulated. This finding demonstrates that both Nrf1 and Nrf2 may be disparately involved in setting the thresholds of distinct cellular patho-physiological (e.g., redox metabolic) responses. This notion is also supported by the evidence showing a small number of WT cell deaths after glucose starvation for 24 h. Further examinations of cell death induced by glucose deprivation reveal that it is different from classical caspase-activated apoptosis, necroptosis, ferroptosis, and autophagy. Notably, a similar phenomenon was also observed after glucose starvation of other cell lines [40]. Contrarily, it is inferable that abnormal survival of Nrf1α−/− cells are, in its malignant growth and proliferation state, maintained by a highly energy-consuming mechanism so as to require its ever-incrementing amounts of glucose and other nutrients. By contrast, the energy-consumption of Nrf2−/− cells could be reduced to a considerable lower level than that of WT cells. Thus, the putative demand for glucose is positively correlated with the subcutaneous tumorigenicity of distinct cancer xenografts in nude mice, as reported by Qiu et al. [30]. Indeed, this is also corroborated by the previous finding that glucose uptake is substantially increased by silencing of Nrf1 in pancreatic islet β-cells [29].
Further insights into acute death of glucose-starved Nrf1α−/− cells have unveiled that such a strong lethality should be ascribed to severe endogenous oxidative stress and damage accumulation. This notion is substantiated by several lines of experimental evidence as followed. Firstly, the accumulation of ROS in Nrf1α−/− cells, albeit with hyperactive Nrf2, is significantly augmented by glucose deprivation, as accompanied by depletion of GSH. These, together, result in a striking increment of the GSSG/GSH ratio during glucose starvation of Nrf1α−/− cells, in order to disrupt the intracellular redox balance and/or relevant signaling controls, as described by other groups [41, 42]. Secondly, the death of Nrf1α−/− cells induced by glucose starvation can be effectively prevented by both NAC and catalase, but not DHA. Similar results were obtained from the treatment of other cell lines with NAC and catalase to rescue its glucose starvation-induced death [43]. Thirdly, it is found that the GSSG/GSH ratio of Nrf2−/− cells is, conversely, diminished and even abolished during glucose starvation. Furtherly, silencing of Nrf2 in Nrf1α−/− cells can also enable them to be alleviated from the cytotoxic ROS accumulation, so that glucose starvation-induced death of Nrf1α−/− cells is sufficiently rescued by Nrf2 knockdown. Such surprising result implies that Nrf2 may also contribute to ROS production in Nrf1α−/− cells under basal and glucose deprivation conditions, albeit it has been accepted as a master regulator of antioxidant cytoprotective genes [44]. This is also supported by the finding that Nrf1α−/− cells are maintained at higher ROS levels in almost unstressed conditions, while Nrf2−/− cells are preserved at relatively lower ROS levels than those of WT cells (in this study). Similarly, a de facto contribution of Nrf2 to amplifying oxidative stress by upregulation of KLF9 was reported by Zucker et al. [45]. Herein, our transcriptome sequencing revealed that KLF9 expression level is too low to be detectable (Figure S4), but its family members KLF4, KLF6, KLF10, and KLF13 are significantly upregulated by constitutive active Nrf2 (caNrf2). By contrast, only KLF4, KLF5, and KLF16 were upregulated in Nrf1α−/− cells, but almost unaffected by Nrf2−/−, whereas two paralogs SP1 and SP3 were partially increased in Nrf2−/− cells (Figure S4B), which await further study. Fourthly, glucose starvation-induced expression of PSMB5, PSMB6, and PSMB7 (encoding the core subunits β5, β1, and β2) is substantially suppressed or even abolished in Nrf1α−/− cells (albeit retaining accumulation of Nrf2), besides their downregulated basal expression. Such proteasomal dysfunction is likely to contribute to the accumulation of oxidative damaged proteins (including Nrf2) to exacerbate endogenous oxidative stress in Nrf1α−/− cells. Taken together with our recent work [46], these lines of experimental evidence demonstrate that Nrf1 is more potent than Nrf2 at mediating intrinsic cytoprotective responses against cytotoxic effects of glucose deprivation, and other bona fide cellular stressors, such as tunicamycin alone or plus tert-butylhydroquinone.
In-depth insights into the endogenous molecular basis for oxidative stress, contributing to the lethality of glucose deprivation, unravel that dysfunctional redox defense systems, along with altered redox signaling, are deteriorated in glucose-starved Nrf1α−/− cells. In fact, we found that, though a large amount of ROS accumulation leads to acute death in Nrf1α−/− cells, but conversely, a relatively low concentration of ROS is also accumulated, facilitating the maintenance of the malignant Nrf1α−/− cell growth and proliferation. This finding is in full agreement with the double-edged effects of ROS, acting as two distinct and even opposite players in cell growth, differentiation, progression, and death [47]. It is known that distinct high concentrations of ROS are involved in a variety of pathological processes, including cancer, ischemia, and immune and endocrine system deficiencies [47, 48]. The excessive ROS can also induce cell death by promoting the intrinsic apoptotic pathway [49]. Nonetheless, the low concentration of ROS is indispensable for various physiological processes, such as signal transduction and immune responses [48]. Such physiological ROS levels should be maintained in a steady-state by the homeostatic redox controls, including antioxidant defense systems, which comprise SOD, GPX1, GSR, CAT, and other redox proteins. As a result, the intracellular superoxide anions, arising from aggressive mitochondrial metabolism [50] and other sources (as illustrated in Figure 3(a)), are converted by SOD to H2O2 and oxygen, and then H2O2 is decomposed by CAT into water and oxygen [51, 52]. Furtherly, oxidized glutathione (GSSG) can be reduced by GSR to yield the sulfhydryl GSH [53]. GPX1 catalyzes the reduction of H2O2 and organic hydroperoxides by glutathione, so as to protect cells against oxidative damage [54]. In this study, we demonstrate that basal expression of CAT and GPX1 is evidently upregulated in Nrf1α−/− cells (retaining hyper-active Nrf2 to activate the former consensus ARE-luc reporter), but substantially suppressed by glucose starvation. By contrast, basal expression of GSR and SOD1 is obviously downregulated in Nrf1α−/− cells, and also further diminished by glucose deprivation. In addition, differential decreases of TRX1, TRX2, and SOD2 occur only after glucose deprivation. Overall, dysfunctions of redox signaling controls and/or antioxidant defense systems are aggravated by glucose starvation of Nrf1α−/− cells, which contributes to the cellular lethality of this stress, albeit aberrant accumulation of hyperactive Nrf2. However, an exception to this is that NADPH oxidase 4 (NOX4), as a ROS-producing source, may also be coregulated by Nrf1 and Nrf2, based on the evidence that basal and starvation-stimulated expression of NOX4 is significantly downregulated in Nrf1α−/− or Nrf2−/− cells. Besides, CYBA (also called p22phox, which acts as a partner regulator of NADPH oxidases) was also decreased in caNrf2 cells or Nrf1α−/− cells (with accumulated Nrf2), but unaltered in Nrf2−/− cells (Figure S4A). Yet, the detailed mechanism remains to be further explored in the future works.
To ameliorate the severe endogenous oxidative stress induced by glucose deprivation and thus facilitate survival and proliferation of cancer cells, they tend to redistribute those intermediates from both glycolysis and gluconeogenesis to other metabolic pathways (e.g., PPP and SSP, in Figures 4(a) and 5(a)). As stated by [55–57], the aerobic glycolysis, as a distinctive metabolic pattern of cancer cells from normal cells, provides a lot of intermediates for pentose phosphate pathway (PPP), gluconeogenesis and serine-to-glutathione synthesis pathway. Thus, this can enable cancer cells to reduce products of ROS from mitochondria and other subcellular compartments, but also enhance the generation of NADPH and GSH from PPP and glutathione synthesis, respectively, such that both NADPH/NADP+ and GSH/GSSG ratio are restored to a newly redox-balanced level. However, we here found that such altered glucose metabolism pathways are dysregulated in Nrf1α−/− cells, and further deteriorated by glucose starvation, leading to the starved cell death. Among them mainly include increased glucose uptake, modestly reduced glycolysis, dysfunction of gluconeogenesis, PPP, and SSP. As a matter of fact, glucose deprivation results in an abject failure of glucose uptake and ensuing glycolysis. Thereby, this confers gluconeogenesis to gain the crucial importance, because gluconeogenesis is a potent alternative source of biosynthetic precursors under glucose deprivation, albeit its intermediates are shared from glycolytic pathways. Herein, we have proposed a conceptual model (as illustrated in Figure 6(r)–6(t)), based on the evidence that glucose starvation of Nrf1α−/− cells caused significant decreases in abundances of PCK2 and PCK1 (as key rate-limiting enzymes of gluconeogenesis), leading to an enhancement of the starved cell death, but similar results were not obtained from glucose-starved WT and Nrf2−/− cells. Conversely, de facto transcriptional expression of PCK2 in WT or Nrf2−/− cells was strikingly activated by glucose deprivation. Similar upregulation of PCK2 by low glucoses was also determined in A549 and H23 lung cancer cells, but its interference and inhibition also significantly enhanced their apoptosis induced by glucose deprivation [58]. This notion is further corroborated by another evidence showing that PCK2, but not PCK1, is highly expressed in different cancer cell lines [56, 59].
The resulting intermediates of gluconeogenesis in glucose-starved cancer cells are allowed for diversion to enter the PPP and serine-to-glutathione synthesis pathways, in order to restore the intracellular redox (e.g., NADPH/NADP+ and GSH/GSSG) balances. Herein, we found that basal expression of G6PD, but not PGD (as two key enzymes to catalyze generation of NADPH), was upregulated in Nrf1α−/− cells, but glucose deprivation caused significant decreases of both expression in the exacerbated cellular death process. Similar decreased PPP flux, as accompanied by reduced NADPH levels and instead increased oxidative stress, was approved as a major fatal cause of the lethality of glucose starvation, because cell death was also accelerated by inhibiting G6PD [32]. However, it is full of curiosity that basal and glucose deprivation-stimulated expression levels of G6PD and PGD were substantially downregulated by Nrf2−/− cells to be considerably lower than those of Nrf1α−/− cells, but rather Nrf2−/− cells displayed a strong resistance to the lethality of glucose starvation. Such paradoxical observations indicate that other mechanisms, beyond PPP, are also involved in the response to glucose deprivation and its lethal cellular process. Thereby, distinct intracellular energy demands of ATP to determine cell survival or death decisions are also investigated, revealing that its basal ATP products are substantially augmented in Nrf1α−/− cells, but abruptly repressed by glucose deprivation for 12 h to a much lower level than that of WT cells. By contrast, basal ATP levels of Nrf2−/− cells are markedly diminished to the lowest level, and also almost unaffected by glucose deprivation. Such disparate energy-consuming demands of between Nrf1α−/− and Nrf2−/− cell lines dictate their decision of survival or death, depending on gluconeogenesis and other nutrient sources in particular glucose deprivation conditions. In addition, we also discover that abundances of AMPK (as a key regulator of energy metabolism [34]) and phosphorylated AMPKThr172 (leading to its activation responsible for maintaining redox metabolic homeostasis [60]) are significantly suppressed by glucose starvation of Nrf1α−/− and Nrf2−/− cells, but with distinct (decreased or increased) ratios of phospho-AMPKThr172/AMPK. Altogether, the inactivation of AMPK to reduce ATP products is inferable as a main cause of blocking energy supply for glucose-starved Nrf1α−/− cells, which results in a large number of these cell deaths occurring after 12 h of glucose deprivation. In addition, it should be noted that this finding appears to be contradictory to two previous reports: one revealed that AMPK activation by glucose-starvation triggers autophagy of MEFs through phosphorylation of GAPDH at Ser122 and ensuing Sirt1 activation [61]; and the another showed that apoptotic death is promoted by AMPK activation in U2OS cells' response to glucose starvation [62]. Such discrepant results may be attributable to different experimental settings of different cell types with distinct responsive phenotypes to glucose deprivation.
Notably, serine biosynthesis is a vital turning point for glucose metabolism; its one hand provides an intermediate for anabolism, while its another hand directly affects cellular antioxidant capacity to generate cysteine and glutathione (as illustrated in Figure 5(a), [63, 64]). However, the effects of glucose limitation on serine-to-glutathione synthesis in cancer cells are, for the first time, determined here. From all this, we discovered that all key genes (i.e., PHGDH, PSAT1, PSPH, CBS, CTH, GCLC, and GCLM) for rate-limiting de novo serine-to-glutathione biosynthesis, along with ATF4 (as a putative upstream regulator of serine synthesis [35]) and also two antioxidant genes (HO-1 and NQO1), are substantially induced by glucose withdrawal from WT cells. Among them, basal expression levels of PHGDH, PSAT1, PSPH, and CBS were evidently upregulated in Nrf1α−/− cells, but unaffected or partially reduced by glucose deprivation. However, no significant changes in basal expression of these genes were observed in Nrf2−/− cells, but they were still induced by glucose starvation, as compared to those of WT cells. Such fatal defects of Nrf1α−/−, but not Nrf2−/−, cells in the serine biosynthesis and the ensuing transsulfuration to yield cysteine demonstrate that Nrf1α plays a dominant regulator in the successive processes. By contrast, Nrf2 is a master regulator of both GSH biosynthesis (GCLC and GCLM) and antioxidant responsive genes (HO-1 and NQO1) to glucose deprivation, albeit Nrf1 is also involved in controlling GCLC expression.
4. Conclusion
In the present study, we found that glucose deprivation induces conversion of Nrf1 glycoprotein and then proteolytic processing to give rise to its mature cleaved CNC-bZIP factor, in order to transcriptionally regulate distinct target genes expression. Of note, Nrf1-target proteasomal expression is required for posttranslational processing of key proteins (e.g., Nrf2), but it is herein found that the core proteasomal subunits are inhibited by glucose deprivation. Under these conditions, gluconeogenesis becomes a major alternative source of biosynthetic precursors, and its intermediates are shared from glycolytic pathway and then diverted into other synthetic pathways (e.g., PPP and SSP). Thereby, altered glucose metabolism and energy demands of Nrf1α−/− cells are significantly aggravated by glucose deprivation, even though hyperactive Nrf2 is accumulated by loss of Nrf1α. Importantly, induction of serine-to-glutathione synthesis by glucose starvation is fatally abolished in Nrf1α−/− cells, leading to severe endogenous oxidative stress and relevant damage; this is even accompanied by upregulation of antioxidant responsive genes by Nrf2. Such fatal defects of Nrf1α−/−, but not of Nrf2−/−, cells in the redox metabolism reprogramming caused by glucose deprivation, lead rapidly to a large number of the former cell deaths, because the intracellular ROS are elevated, along with the reduced ATP production (Figures 6(r)–6(t)). Overall, these findings demonstrate that Nrf1 acts as a dominant player in the redox metabolic reprogramming and thus fulfill its intrinsic cytoprotective response against the fatal cytotoxicity of glucose deprivation. Thereby, the existence of Nrf1 still enables Nrf2−/− cells to be endowed with a strong constructive resistance to glucose starvation. Conversely, it is reasoned that Nrf2 cannot fulfill a fully cytoprotective function against severe oxidative stress and damage, although it serves as a master regulator of antioxidant response genes (e.g., HO-1, NQO1), which are still upregulated in Nrf1α−/− cells. Collectively, these demonstrate distinct contributions of Nrf1 and Nrf2 to either constructive or stress-inducible expression of different subsets of critical genes for the redox metabolism. Moreover, there exist certain interregulatory crosstalks between Nrf1 and Nrf2 in the redox metabolism reprogramming by coregulating expression of key responsive genes (e.g., CAT, G6PD, PCK2, GCLC, ATF4) to glucose deprivation.
5. Materials and Methods
5.1. Cell Culture and Reagents
The human hepatocellular carcinoma HepG2 cells (WT) were obtained originally from the American Type Culture Collection (ATCC, Manassas, VA, USA). The fidelity was conformed to be true by its authentication profiling and STR (short tandem repeat) typing map (by Shanghai Biowing Applied Biotechnology Co., Ltd). On this base, Nrf1α−/− and Nrf2−/− (or Nrf2-/-ΔTA) were established by Qiu et al. [30]. Before experimentation, they were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 25 mmol/L high glucose, 10% (v/v) fetal bovine serum (FBS), 100 units/ml penicillin-streptomycin, and cultured in a 37°C incubator with 5% CO2. In addition, it is noted that all key reagents and resources used in this study were listed in Table S1.
5.2. Assays of Cell Death from Glucose Deprivation
Equal numbers (300, 000 or 3 × 105) of WT, Nrf1α−/− and Nrf2−/− cells were seeded in 6-well plates and allowed for growth in DMEM containing 25 mmol/L glucose and 10% FBS for 24 h. After reaching 80% of their confluence, they were then transferred to be cultured in fresh glucose-free DMEM for 6-24 h. Subsequently, the cell morphological changes in survival or death induced by glucose deprivation were observed by microscopy. These cells were then counted, after staining by trypan blue. In addition, Nrf1α−/−+siNrf2 cells were also prepared here, and then subjected to the cell death assay, after they were allowed for glucose deprivation.
To rescue the cell death, fructose and mannose (25 mmol/L) were added to the glucose-free media, respectively. The addition of 2DG (10 mmol/L) was to restore the PPP in the absence of glucose. In order to identify distinct types of cell death, q-VD-OPH (10 μmol/L, as a pan-caspase inhibitor), Necrostatin-1 (100 μmol/L), Ferrostatin-1 (2 μmol/L), and 3-methyladenine (2 mmol/L, an autophagy inhibitor) were added to the glucose-free media, respectively. Furthermore, NAC (5-10 mmol/L), CAT (50 units/ml), and DHA (100 μmol/L) were added in the glucose-free media to examine effects of antioxidants or prooxidants on cell death. After being incubated for 12-24 h, these cell morphological changes were visualized by microscopy, and the trypan-stained cells were also counted.
5.3. Analysis of Cell Apoptosis and ROS by Flow Cytometry
Equal numbers (300, 000 or 3 × 105) of experimental cells (WT, Nrf1α−/−, Nrf2−/−, and Nrf1α−/−+siNrf2) were allowed for 24 h of growth in DMEM containing 25 mmol/L glucose and 10% FBS for. After reaching 80% of their confluence, these cells were subjected to glucose deprivation by being cultured in a fresh glucose-free medium for 12 h. Subsequently, these cells were incubated with Annexin V-FITC and propidium iodide (PI) for 15 min, before the cell apoptosis was analyzed by flow cytometry. Furtherly, the intracellular ROS levels were also determined, according to the instruction of ROS assay kit (Beyotime, Shanghai, China). The resulting data were further analyzed by the FlowJo 7.6.1 software.
5.4. Assays of ATP Levels and GSSG/GSH Ratios
ATP levels are determined according to the instruction of enhanced ATP assay kit (Beyotime, Shanghai, China); GSSG/GSH ratios are determined according to the instruction of GSH and GSSG Assay Kit (Beyotime, Shanghai, China).
5.5. Knockdown of Nrf2 in Nrf1α−/− Cells by Its siRNAs
A pair of double-stranded small RNAs targeting for the interference with Nrf2 (i.e., siNrf2) were synthesized by TranSheep Bio Co.Ltd. (Shanghai, China). The oligonucleotide sequences are as follows: FW, 5′-GUAAGAAGCCAGAUGU UAAdTdT-3′; REV, 5′-UUAACAUCUGGCUUCUUACdTdT-3′. Subsequently, Nrf1α−/− cells were transfected with 80 nmol/L of the siNrf2 oligonucleotides in the mixture of Lipofectamine 3000 (Invitrogen, California, USA). Thereafter, the siNrf2-interfered cells were identified by RT-qPCR and Western blotting, before being experimented.
5.6. Real-Time Quantitative PCR Analysis of Gene Expression
After all experimental cells reached 80% of their confluence, they were subjected to glucose starvation by being transferred in fresh glucose-free media. Their total RNAs were extracted after glucose deprivation for 0-12 h, before being subjected to the reactions with a reverse transcriptase to synthesize the first strand of cDNAs. Subsequently, the mRNA levels of examined genes in different cell lines were determined by RT-qPCR with the indicated pairs of their forward and reverse primers (as listed in Table S1). All the RT-qPCRs were carried out in the GoTaq real-time PCR detection systems by a CFX96 instrument (Bio-rad, Hercules, CA, USA). The resulting data were analyzed by the Bio-Rad CFX Manager 3.1 software (Bio-rad).
5.7. Western Blotting Analysis of Key Functional Proteins
After all experimental cells reached 80% of their confluence, they were subjected to glucose starvation by being transferred in fresh glucose-free media. After 6-12 h of glucose deprivation, their total proteins were extracted by lysis buffer (0.5% SDS, 0.04 mol/L DTT, pH 7.5) containing protease inhibitor cOmplete Tablets EASYpack or phosphatase inhibitor PhosSTOP EASYpack (each 1 tablet per 15 mL, Roche, Basel, Switzerland), and diluted in 6×SDS-PAGE sample loading buffer (Beyotime, Shanghai, China). Subsequently, total lysates were denatured immediately at 100°C for 10 min, before equal amounts of proteins were separated by SDS-PAGE gels containing 8-12% polyacrylamide and visualized by Western blotting with distinct primary antibodies (as listed in Table S1). Among included those antibodies against Nrf1; Nrf2, ATF4, GLUT1, GPX1, TRX1, TRX2, PRX1, CBS, HO1, GCLM, GCLC, GSR, SOD1, PCK1, PCK2 and G6PD, CAT, AMPK and p-AMPKThr172, PHGDH, PSAT1, HK1, HK2, PSMB5, PSMB6, and PSMB7. In addition, β-Actin served as an internal control to verify the amounts of proteins that were loaded in each of the wells.
5.8. Assays of ARE-Driven Luciferase Reporter Gene Activity
The core ARE consensus sites within -4K-bp sequences to the transcription start sites (TSS) or extended to the translation initiation sites (TIS) of ATF4, CAT, G6PD, PHGDH, and PCK2 promoter regions were searched. Each of the core ARE and adjoining sequences was then inserted into the indicated site of the pGL3-promoter vector. The fidelity of all resultant constructs was confirmed by sequencing with indicated primer pairs (Table S1). Subsequently, equal numbers (150, 000 or 1.5 × 105) of HepG2 cells were seeded in 12-well plates and allowed for 24-h growth in DMEM containing 25 mmol/L glucose and 10% FBS. After reaching 80% confluence, the cells were transfected with an expression construct for human Nrf1 or Nrf2, along with each of the above ARE-Luc reporters plus pRL-TK (serves as an internal control). Approximately 24 hours after transfection, ARE-driven luciferase activity was measured by using the dual-luciferase reporter assay. The resulting data were calculated as fold changes (mean ± S.D., n = 9), relative to the basal activity (at a given value of 1.0) obtained from the transfection of cells with an empty pcDNA3.1 and each of ARE-driven reporter genes.
5.9. Statistical Analysis
Statistical significance was assessed by using the ANOVA with Holm-Sidak test. The data presented herein are shown as a fold change (mean ± S.D., n = 9), each of which represents at least three independent experiments, that were each performed in triplicate.
Acknowledgments
We are greatly thankful to Drs. Lu Qiu (at Zhengzhou University, China) and Yonggang Ren (North Sichuan Medical College, Sichuan, China) for having established those relevant cell lines used in this study. We also thank Mr. Shaofan Hu and other members (at Chongqing University, China) for giving invaluable help with this work. Of note, the study was supported by the National Natural Science Foundation of China (NSFC, with a key program 91429305 and another project 81872336) awarded to Yiguo Zhang (at Chongqing University, China). This work is, in part, funded by Sichuan Department of Science and Technology grant (2019YJ0482) to Dr. Yuancai Xiang (at Southwest Medical University, Sichuan, China).
Data Availability
All the data needed to evaluate the findings of this study are available in this publication along with the “Supplemental Information” that can be found online. Additional other data related to this paper may also be requested from the corresponding author (with a lead contact at the email: yiguozhang@cqu.edu.cn, or eaglezhang64@gmail.com).
Conflicts of Interest
The authors declare no conflict of interest. Besides, it should also be noted that the preprinted version of this paper had been initially posted at doi: 10.1101/2019.12.13.875369.
Authors' Contributions
Y.-P.Z. performed the most experiments with the help of Z.Z. and collected relative data, except that Y.X. did the experimental analysis of the half-lives of Nrf1 and Nrf2. Y.-P.Z. also made a draft of this manuscript with most figures and supplemental tables. Y.Z. designed and supervised this study, analyzed all the data, helped to prepare all figures with cartoons, and wrote and revised the paper.
Supplementary Materials
Figure S1: distinct cellular responses to glucose deprivation. Figure S2: the quantified results of western blot data in Figures 3, 4, and 5 by Quantity One 4.5.2 software. Figure S3: time-dependent effects of glucose deprivation on Nrf1 and Nrf2 with distinct half-lives. Figure S4: the mRNA expression of CYBA, NOXO1, and RAC1 in WT, Nrf1a-/-, Nrf1a-/- + siNrf2, Nrf2-/-, and caNrf2 from transcriptomic sequencing. Table S1: the key resources used in this work.
References
- 1.Hanahan D., Weinberg R. A. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Li Z., Zhang H. Reprogramming of glucose, fatty acid and amino acid metabolism for cancer progression. Cellular and Molecular Life Sciences. 2016;73(2):377–392. doi: 10.1007/s00018-015-2070-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lunt S. Y., Vander Heiden M. G. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annual Review of Cell and Developmental Biology. 2011;27(1):441–464. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- 4.Hay N. Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nature Reviews. Cancer. 2016;16(10):635–649. doi: 10.1038/nrc.2016.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pavlova N. N., Thompson C. B. The emerging hallmarks of cancer metabolism. Cell Metabolism. 2016;23(1):27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Warburg O. On the origin of cancer cells. Science. 1956;123(3191):3309–3314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 7.Munoz-Pinedo C., El Mjiyad N., Ricci J. E. Cancer metabolism: current perspectives and future directions. Cell Death & Disease. 2012;3(1, article e248) doi: 10.1038/cddis.2011.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu D., Jin J., Yu H., et al. Chrysin inhibited tumor glycolysis and induced apoptosis in hepatocellular carcinoma by targeting hexokinase-2. Journal of Experimental & Clinical Cancer Research. 2017;36(1):p. 44. doi: 10.1186/s13046-017-0514-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vander Heiden M. G., Plas D. R., Rathmell J. C., Fox C. J., Harris M. H., Thompson C. B. Growth factors can influence cell growth and survival through effects on glucose metabolism. Molecular and Cellular Biology. 2001;21(17):5899–5912. doi: 10.1128/MCB.21.17.5899-5912.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee H.-C., Lin S.-C., Wu M.-H., Tsai S.-J. Induction of pyruvate dehydrogenase kinase 1 by hypoxia alters cellular metabolism and inhibits apoptosis in endometriotic stromal cells. Reproductive Sciences. 2018;26(6):734–744. doi: 10.1177/1933719118789513. [DOI] [PubMed] [Google Scholar]
- 11.Choo A. Y., Kim S. G., Vander Heiden M. G., et al. Glucose addiction of TSC null cells is caused by failed mTORC1-dependent balancing of metabolic demand with supply. Molecular Cell. 2010;38(4):487–499. doi: 10.1016/j.molcel.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.El Mjiyad N., Caro-Maldonado A., Ramirez-Peinado S., Munoz-Pinedo C. Sugar-free approaches to cancer cell killing. Oncogene. 2011;30(3):253–264. doi: 10.1038/onc.2010.466. [DOI] [PubMed] [Google Scholar]
- 13.Mikawa T., LLeonart M. E., Takaori-Kondo A., Inagaki N., Yokode M., Kondoh H. Dysregulated glycolysis as an oncogenic event. Cellular and Molecular Life Sciences. 2015;72(10):1881–1892. doi: 10.1007/s00018-015-1840-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun L., Song L., Wan Q., et al. cMyc-mediated activation of serine biosynthesis pathway is critical for cancer progression under nutrient deprivation conditions. Cell Research. 2015;25(4):429–444. doi: 10.1038/cr.2015.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang D., Li C., Zhang H. Hypoxia and cancer cell metabolism. Acta Biochimica et Biophysica Sinica. 2014;46(3):214–219. doi: 10.1093/abbs/gmt148. [DOI] [PubMed] [Google Scholar]
- 16.Kim J. W., Tchernyshyov I., Semenza G. L., Dang C. V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metabolism. 2006;3(3):177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 17.Schwartzenberg-Bar-Yoseph F., Armoni M., Karnieli E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Research. 2004;64(7):2627–2633. doi: 10.1158/0008-5472.CAN-03-0846. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y., Xiang Y. Molecular and cellular basis for the unique functioning of Nrf1, an indispensable transcription factor for maintaining cell homoeostasis and organ integrity. The Biochemical Journal. 2016;473(8):961–1000. doi: 10.1042/BJ20151182. [DOI] [PubMed] [Google Scholar]
- 19.Satoh H., Moriguchi T., Takai J., Ebina M., Yamamoto M. Nrf2 prevents initiation but accelerates progression through the Kras signaling pathway during lung carcinogenesis. Cancer Research. 2013;73(13):4158–4168. doi: 10.1158/0008-5472.CAN-12-4499. [DOI] [PubMed] [Google Scholar]
- 20.Tao S., Rojo de la Vega M., Chapman E., Ooi A., Zhang D. D. The effects of NRF2 modulation on the initiation and progression of chemically and genetically induced lung cancer. Molecular Carcinogenesis. 2018;57(2):182–192. doi: 10.1002/mc.22745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang H., Liu X., Long M., et al. NRF2 activation by antioxidant antidiabetic agents accelerates tumor metastasis. Science Translational Medicine. 2016;8(334, article 334ra51) doi: 10.1126/scitranslmed.aad6095. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y. Y., Chen J., Liu X. M., Zhao R., Zhe H. Nrf2-mediated metabolic reprogramming in cancer. Oxidative Medicine and Cellular Longevity. 2018;2018:7. doi: 10.1155/2018/9304091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hayes J. D., Dinkova-Kostova A. T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends in Biochemical Sciences. 2014;39(4):199–218. doi: 10.1016/j.tibs.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y., Li S., Xiang Y., Qiu L., Zhao H., Hayes J. D. The selective post-translational processing of transcription factor Nrf1 yields distinct isoforms that dictate its ability to differentially regulate gene expression. Scientific Reports. 2015;5(1) doi: 10.1038/srep12983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ren Y., Qiu L., Lu F., et al. TALENs-directed knockout of the full-length transcription factor Nrf1α that represses malignant behaviour of human hepatocellular carcinoma (HepG2) cells. Scientific Reports. 2016;6(1) doi: 10.1038/srep23775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hirotsu Y., Hataya N., Katsuoka F., Yamamoto M. NF-E2-related factor 1 (Nrf1) serves as a novel regulator of hepatic lipid metabolism through regulation of the Lipin1 and PGC-1beta genes. Molecular and Cellular Biology. 2012;32(14):2760–2770. doi: 10.1128/MCB.06706-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsujita T., Peirce V., Baird L., et al. Transcription factor Nrf1 negatively regulates the cystine/glutamate transporter and lipid-metabolizing enzymes. Molecular and Cellular Biology. 2014;34(20):3800–3816. doi: 10.1128/MCB.00110-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirotsu Y., Higashi C., Fukutomi T., et al. Transcription factor NF-E2-related factor 1 impairs glucose metabolism in mice. Genes to Cells. 2014;19(8):650–665. doi: 10.1111/gtc.12165. [DOI] [PubMed] [Google Scholar]
- 29.Zheng H., Fu J., Xue P., et al. CNC-bZIP protein Nrf1-dependent regulation of glucose-stimulated insulin secretion. Antioxidants & Redox Signaling. 2015;22(10):819–831. doi: 10.1089/ars.2014.6017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qiu L., Wang M., Hu S., et al. Oncogenic activation of Nrf2, though as a master antioxidant transcription factor, liberated by specific knockout of the full-length Nrf1α that acts as a dominant tumor repressor. Cancers. 2018;10(12):p. 520. doi: 10.3390/cancers10120520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perales-Clemente E., Folmes C. D. L., Terzic A. Metabolic regulation of redox status in stem cells. Antioxidants & Redox Signaling. 2014;21(11):1648–1659. doi: 10.1089/ars.2014.6000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jeon S. M., Chandel N. S., Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012;485(7400):661–665. doi: 10.1038/nature11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y., Crouch D. H., Yamamoto M., Hayes J. D. Negative regulation of the Nrf1 transcription factor by its N-terminal domain is independent of Keap1: Nrf1, but not Nrf2, is targeted to the endoplasmic reticulum. The Biochemical Journal. 2006;399(3):373–385. doi: 10.1042/BJ20060725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hardie D. G., Schaffer B. E., Brunet A. AMPK: an energy-sensing pathway with multiple inputs and outputs. Trends in Cell Biology. 2016;26(3):190–201. doi: 10.1016/j.tcb.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao E., Ding J., Xia Y., et al. KDM4C and ATF4 cooperate in transcriptional control of amino acid metabolism. Cell Reports. 2016;14(3):506–519. doi: 10.1016/j.celrep.2015.12.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nam L. B., Keum Y. S. Binding partners of NRF2: functions and regulatory mechanisms. Archives of Biochemistry and Biophysics. 2019;678, article 108184 doi: 10.1016/j.abb.2019.108184. [DOI] [PubMed] [Google Scholar]
- 37.Xiang Y., Wang M., Hu S., et al. Mechanisms controlling the multistage post-translational processing of endogenous Nrf1α/TCF11 proteins to yield distinct isoforms within the coupled positive and negative feedback circuits. Toxicology and Applied Pharmacology. 2018;360:212–235. doi: 10.1016/j.taap.2018.09.036. [DOI] [PubMed] [Google Scholar]
- 38.Vander Heiden M. G., DeBerardinis R. J. Understanding the intersections between metabolism and cancer biology. Cell. 2017;168(4):657–669. doi: 10.1016/j.cell.2016.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sullivan M. R., Vander Heiden M. G. Determinants of nutrient limitation in cancer. Critical Reviews in Biochemistry and Molecular Biology. 2019;54(3):193–207. doi: 10.1080/10409238.2019.1611733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leon-Annicchiarico C. L., Ramirez-Peinado S., Dominguez-Villanueva D., Gonsberg A., Lampidis T. J., Munoz-Pinedo C. ATF4 mediates necrosis induced by glucose deprivation and apoptosis induced by 2-deoxyglucose in the same cells. The FEBS Journal. 2015;282(18):3647–3658. doi: 10.1111/febs.13369. [DOI] [PubMed] [Google Scholar]
- 41.Nogueira V., Hay N. Molecular pathways: reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clinical Cancer Research. 2013;19(16):4309–4314. doi: 10.1158/1078-0432.CCR-12-1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Y., Song X. D., Liu W., Zhang T. Y., Zuo J. Glucose deprivation induces mitochondrial dysfunction and oxidative stress in PC12 cell line. Journal of Cellular and Molecular Medicine. 2003;7(1):49–56. doi: 10.1111/j.1582-4934.2003.tb00202.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gonzalez-Menendez P., Hevia D., Alonso-Arias R., et al. GLUT1 protects prostate cancer cells from glucose deprivation-induced oxidative stress. Redox Biology. 2018;17:112–127. doi: 10.1016/j.redox.2018.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zimta A.-A., Cenariu D., Irimie A., et al. The role of Nrf2 activity in cancer development and progression. Cancers. 2019;11(11):p. 1755. doi: 10.3390/cancers11111755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zucker S. N., Fink E. E., Bagati A., et al. Nrf2 amplifies oxidative stress via induction of Klf9. Molecular Cell. 2014;53(6):916–928. doi: 10.1016/j.molcel.2014.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu Y. P., Zheng Z., Hu S., et al. Unification of opposites between two antioxidant transcription factors Nrf1 and Nrf2 in mediating distinct cellular responses to the endoplasmic reticulum stressor tunicamycin. Antioxidants. 2020;9(1):p. 4. doi: 10.3390/antiox9010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.MatÉs J. É. M., Pérez-Gómez C., De Castro I. N. Antioxidant enzymes and human diseases. Clinical Biochemistry. 1999;32(8):595–603. doi: 10.1016/S0009-9120(99)00075-2. [DOI] [PubMed] [Google Scholar]
- 48.Monnier V., Girardot F., Audin W., Tricoire H. Control of oxidative stress resistance by IP3 kinase in Drosophila melanogaster. Free Radical Biology & Medicine. 2002;33(9):1250–1259. doi: 10.1016/S0891-5849(02)01019-5. [DOI] [PubMed] [Google Scholar]
- 49.Marchi S., Giorgi C., Suski J. M., et al. Mitochondria-ros crosstalk in the control of cell death and aging. Journal of Signal Transduction. 2012;2012:17. doi: 10.1155/2012/329635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chang S. H., Garcia J., Melendez J. A., Kilberg M. S., Agarwal A. Haem oxygenase 1 gene induction by glucose deprivation is mediated by reactive oxygen species via the mitochondrial electron-transport chain. The Biochemical Journal. 2003;371(3):877–885. doi: 10.1042/bj20021731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Michiels C., Raes M., Toussaint O., Remacle J. Importance of se-glutathione peroxidase, catalase, and Cu/Zn-SOD for cell survival against oxidative stress. Free Radical Biology & Medicine. 1994;17(3):235–248. doi: 10.1016/0891-5849(94)90079-5. [DOI] [PubMed] [Google Scholar]
- 52.Aebi H. [13] Catalase in vitro. Methods in Enzymology. 1984;105:121–126. doi: 10.1016/S0076-6879(84)05016-3. [DOI] [PubMed] [Google Scholar]
- 53.Pyne P., Alam M., Ghosh W. A novel soxO gene, encoding a glutathione disulfide reductase, is essential for tetrathionate oxidation in Advenella kashmirensis. Microbiological Research. 2017;205:1–7. doi: 10.1016/j.micres.2017.08.002. [DOI] [PubMed] [Google Scholar]
- 54.Lubos E., Loscalzo J., Handy D. E. Glutathione peroxidase-1 in health and disease: from molecular mechanisms to therapeutic opportunities. Antioxidants & Redox Signaling. 2011;15(7):1957–1997. doi: 10.1089/ars.2010.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Patra K. C., Hay N. The pentose phosphate pathway and cancer. Trends in Biochemical Sciences. 2014;39(8):347–354. doi: 10.1016/j.tibs.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vincent E. . E., Sergushichev A., Griss T., et al. Mitochondrial phosphoenolpyruvate carboxykinase regulates metabolic adaptation and enables glucose-independent tumor growth. Molecular Cell. 2015;60(2):195–207. doi: 10.1016/j.molcel.2015.08.013. [DOI] [PubMed] [Google Scholar]
- 57.Locasale J. W. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nature Reviews. Cancer. 2013;13(8):572–583. doi: 10.1038/nrc3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Leithner K., Hrzenjak A., Trotzmuller M., et al. PCK2 activation mediates an adaptive response to glucose depletion in lung cancer. Oncogene. 2015;34(8):1044–1050. doi: 10.1038/onc.2014.47. [DOI] [PubMed] [Google Scholar]
- 59.Mendez-Lucas A., Hyrossova P., Novellasdemunt L., Vinals F., Perales J. C. Mitochondrial phosphoenolpyruvate carboxykinase (PEPCK-M) is a pro-survival, endoplasmic reticulum (ER) stress response gene involved in tumor cell adaptation to nutrient availability. The Journal of Biological Chemistry. 2014;289(32):22090–22102. doi: 10.1074/jbc.M114.566927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ren Y., Shen H. M. Critical role of AMPK in redox regulation under glucose starvation. Redox Biology. 2019;25, article 101154 doi: 10.1016/j.redox.2019.101154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chang C., Su H., Zhang D., et al. AMPK-dependent phosphorylation of GAPDH triggers Sirt1 activation and is necessary for autophagy upon glucose starvation. Molecular Cell. 2015;60(6):930–940. doi: 10.1016/j.molcel.2015.10.037. [DOI] [PubMed] [Google Scholar]
- 62.Okoshi R., Ozaki T., Yamamoto H., et al. Activation of AMP-activated protein kinase induces p53-dependent apoptotic cell death in response to energetic stress. The Journal of Biological Chemistry. 2008;283(7):3979–3987. doi: 10.1074/jbc.M705232200. [DOI] [PubMed] [Google Scholar]
- 63.Kalhan S. C., Hanson R. W. Resurgence of serine: an often neglected but indispensable amino acid. The Journal of Biological Chemistry. 2012;287(24):19786–19791. doi: 10.1074/jbc.R112.357194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Amelio I., Cutruzzola F., Antonov A., Agostini M., Melino G. Serine and glycine metabolism in cancer. Trends in Biochemical Sciences. 2014;39(4):191–198. doi: 10.1016/j.tibs.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: distinct cellular responses to glucose deprivation. Figure S2: the quantified results of western blot data in Figures 3, 4, and 5 by Quantity One 4.5.2 software. Figure S3: time-dependent effects of glucose deprivation on Nrf1 and Nrf2 with distinct half-lives. Figure S4: the mRNA expression of CYBA, NOXO1, and RAC1 in WT, Nrf1a-/-, Nrf1a-/- + siNrf2, Nrf2-/-, and caNrf2 from transcriptomic sequencing. Table S1: the key resources used in this work.
Data Availability Statement
All the data needed to evaluate the findings of this study are available in this publication along with the “Supplemental Information” that can be found online. Additional other data related to this paper may also be requested from the corresponding author (with a lead contact at the email: yiguozhang@cqu.edu.cn, or eaglezhang64@gmail.com).