

Abstract
With aging, the kidney undergoes inexorable and progressive changes in structural and functional performance. These aging-related alterations are more obvious and serious in diabetes mellitus (DM). Renal accelerated aging under DM conditions is associated with multiple stresses such as accumulation of advanced glycation end products (AGEs), hypertension, oxidative stress, and inflammation. The main hallmarks of cellular senescence in diabetic kidneys include cyclin-dependent kinase inhibitors, telomere shortening, and diabetic nephropathy-associated secretory phenotype. Lysosome-dependent autophagy and antiaging proteins Klotho and Sirt1 play a fundamental role in the accelerated aging of kidneys in DM, among which the autophagy-lysosome system is the convergent mechanism of the multiple antiaging pathways involved in renal aging under DM conditions. Metformin and the inhibitor of sodium–glucose cotransporter 2 are recommended due to their antiaging effects independent of antihyperglycemia, besides angiotensin-converting enzyme inhibitors/angiotensin receptor blockers. Additionally, diet intervention including low protein and low AGEs with antioxidants are suggested for patients with diabetic nephropathy (DN). However, their long-term benefits still need further study. Exploring the interactive relationships among antiaging protein Klotho, Sirt1, and autophagy-lysosome system may provide insight into better satisfying the urgent medical needs of elderly patients with aging-related DN.
1. Introduction
The increasing global morbidity of type 2 diabetes mellitus (T2DM) and chronic kidney disease (CKD) has provoked research efforts to overcome the growing prevalence of diabetic nephropathy (DN), which has been a global catastrophe due to limited efficacy with existing therapies and serious financial burden [1–3]. It is urgent to explore the unknown mechanisms underlying DN and discover curative efficient therapies.
As is well known, both T2DM and CKD are aging-related diseases. The morbidity of DM in people over 65 years old is more than twice that of people over 20 years old [1], and aging is a key factor attributing to nephron loss and resulting in CKD [4].
DM has been considered to be an inducer of accelerated cellular senescence and has been associated with aging-related cardiovascular diseases and kidney diseases due to high glucose levels [5]. However, the aging in a tissue-specific manner remains rarely explored. Globally, DM has been the leading cause of end-stage renal disease (ESRD), especially in elders [3, 6, 7]. Feasible histopathological patterns of individuals often imply the presence of other pathogenic factors, such as aging-related nephropathy, resulting in the complicated and difficult diagnosis and treatment of type 2 DN [8]. Thus, under the double risk factors of high glucose and aging, it is hypothesized that renal aging plays a vital role in the development of DN. Herein we will discuss current knowledge on renal aging-related mechanisms and potential therapeutic targets of DN.
2. The Role of Accelerated Kidney Aging in DN
Kidney aging is a complex process that interacts with many diseases, especially those that are more prevalent in the elderly population. Kidney aging is manifested in the decline of glomerular filtration rate (GFR), which is also the physiological characteristic of CKD [9, 10]. The GFR decreases by about 5%–10% per decade after 35 years of age, and the elderly, 70–75 years old, had 48% fewer intact nephrons than the younger patients aged 18–29 years old [11, 12]. It is often difficult to distinguish between chronological change and pathological changes, but some studies have focused on accelerated aging as a potential target to retard the process of renal diseases, including DN [13, 14].
In kidneys with premature aging due to the morbid state such as IgA nephropathy [13], the above characteristics may not be necessarily related to chronological change. For DN, the incidence of kidney disease in diabetic individuals partly depends on the aging-related nephron loss [15]. Except for the functional change of decreasing GFR, the structural changes are also observed as pathologic reduction in kidney size and renal histomorphology changes, including glomerulosclerosis, interstitial fibrosis, and tubular atrophy macroscopically and compensatory hypertrophy of renal cells, glomerular basement membrane (GBM) thickening, podocyte loss, and tubular epithelial cell (TEC) shrinking microscopically [16].
It has been demonstrated that the kidney appears aging phenotype which represents a proximate mechanism by which the kidney is damaged in DN [17, 18], leading to a complicated and difficult diagnosis and treatment of type 2 DN. Thus, accelerated kidney aging may be an important part of the pathogenesis of DN (Figure 1). However, the cellular and molecular mechanisms of kidney aging in diabetic individuals are complicated and poorly understood.
Figure 1.
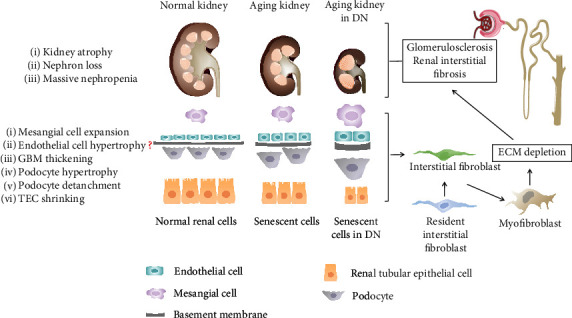
Normal kidney, kidney aging in nature, and kidney aging under DM conditions. Each normal kidney possesses thousands of nephrons. With aging and the onset of DN and the interaction of the two, nephrons are gradually lost and become massive, particularly when occurring in aging kidneys with underlying DM. Macroscopically, pathologic reduction is observed in kidney size and renal histomorphology changes, including glomerulosclerosis, interstitial fibrosis, and tubular atrophy. Microscopically, compensatory hypertrophy of renal cells, glomerular basement membrane (GBM) thickening, podocyte loss, and tubular epithelial cell (TEC) shrinking are seen, which contribute to driving an associated dysfunction like the pathologic changes in kidneys as mentioned above.
3. Factors Associated with Accelerated Kidney Aging in DN
DN is the renal manifestation of the hyperglycemia-driven process in vulnerable sites along with retinopathy. Multifactors are posed with DN beyond high glucose, such as oxidative stress and activation of the renin-angiotensin-aldosterone system (RAAS) [15]. Accumulation of advanced glycation end products (AGEs) [19] and inflammation also contribute to the process of DN [20]. Similarly, kidney aging is associated with clinical factors such as DM and hypertension, as well as tissue factors including angiotensin II, AGEs, oxidative stress, and so on [21]. Hence, herein we focus on AGEs, hypertension, inflammation, and oxidative stress in diabetic kidneys.
AGEs, which are accumulated in multiple tissues during aging, provide information of a nonenzymatic reaction of proteins and carbohydrates that can be detected with an increased level in each related organ of patients with DM [22]. AGEs, as a result of pathologically increased glycation due to chronic hyperglycemia, have a variable “pathological expression” in DM, kidney failure, and tissue aging [23]. Interestingly, reducing AGEs extends the lifespan of yeast [24]. Hence, AGEs can be a biomarker of aging and may lead to renal lesions in DM related to kidney aging [25]. In hyperglycemic circumstances, the accumulated AGEs induce the accelerated aging of kidney dysfunction by inducing podocyte damage and apoptosis of mesangial cells and the expression of transforming growth factor-β (TGF-β), the latter of which plays a pivotal role in fibrogenesis [26, 27]. AGEs stimulate the activation of the receptor for AGEs (RAGE), which induces oxidative stress and cellular dysfunction. In the kidney, the RAGE-AGE activation also contributes to the induction of oxidative stress, endoplasmic reticulum (ER) stress inflammatory, and fibrotic responses by activating different intracellular signaling pathways, such as phosphatidylinositol 3 kinase/protein kinase B (PI3K/Akt), mitogen-activated protein kinase/extracellular regulated protein kinases (MAPK/ERK), and nuclear factor kappa-B (NF-κB), all of which lead to functional and structural damages of kidneys, as well as premature aging [19, 28].
Hypertension is one of the clinical symptoms of DN due to the dysfunction of the RAAS, and RAAS inhibitors have been considered to be the most effective therapy for DN [29]. In the aging kidney, the RAAS is related to glomerular and tubular damage via oxidative stress and/or downregulating antiaging proteins, such as Sirtuins and Klotho [30, 31]. AGEs stimulate angiotensinogen production in renal proximal tubular cells, which results in inappropriate activation of RAAS and exacerbates the development of diabetic-related kidney lesions [32]. Thus, these findings suggest that the abnormal activation of RAAS aggravates kidney damage and may lead to accelerated senescence in DN.
Oxidative stress (OS) is considered a major factor in the pathogenesis of DN due to its contribution to hyperglycemia and hypertension [33]. OS contributes to aging as a result of causing increased damage to important cellular targets, increasing mutation rates, and inducing growth inhibition [34]. In diabetic kidneys, the main sources of OS include nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activation [35], mitochondrial dysfunction [36, 37], xanthine oxidase pathway abnormality, cyclooxygenase pathway dysregulation, and endothelial nitric oxide synthase uncoupling [38]. These sources of OS contribute to a range of harmful intracellular events, including DNA damage within the nucleus and mitochondria, and ultimately result in the death of renal intrinsic cells via apoptosis that ER stress and cellular senescence contribute to [39, 40]. Additionally, high glucose- (HG-) induced reactive oxygen species (ROS) production results in increased TGF-β1 expression, which is the core link of renal fibrosis in diabetic kidneys and results in the epithelial-mesenchymal transition [41]. Recent studies have reported that P66 plays a key role in the pathogenesis of DN because of its relation to OS. ROS metabolism was substantially increased in HG-induced mesangial cells in association with more cell death via apoptosis, as well as the acquisition of a senescent phenotype and a homozygous mutation at the P66 locus that confers delayed aging phenotypes in the diabetic kidneys [18]. The abnormal expression of p66 may have a correlation with the protein kinase C (PKC) β activation and then regulate NADPH oxidase, which could further promote the activation of OS [42].
Inflammation is the both cause and consequence of accelerated aging leading to renal damage [43, 44]. Inflammation plays an important role in the genesis of DM as well as in the development of diabetic complications, including DN. The metabolic alterations with the accumulation of toxic products, such as AGEs and hemodynamic factors and the activation of RAAS, enhance inflammation of the kidney [45, 46]. Excessive ROS production in kidney tissues activates inflammation-related signaling pathways, such as PKC, MAPK, and NF-κB, and leads to the production of a large number of cytokines and growth factors that trigger the onset of DN. This process then causes the deposition of the extracellular matrix (ECM) in glomeruli, the differentiation of tubular epithelial cells (TECs), and the interstitium and an increase in the synthesis of glomerular fibronectin (FN) proteins in the mesangial region [47]. Proteinuria, a marker of renal lesions, aggravates the local microinflammatory response and enhances interstitial cellular infiltration, leading to the overexpression of mesangial matrix production, glomerular basement membrane (GBM) thickening, and glomerulosclerosis [48]. Persistent microinflammation is increased in the aging kidney and provides potential mechanistic links between the epigenetic landscape of aging and renal dysfunction [49, 50].
Taken together, in diabetic kidneys, multiple stresses, such as AGEs accumulation, hypertension, oxidative stress, and inflammation, induce a negative environment that accelerates senescence that is manifested in renal functional decline and aberrant structural changes (Figure 2).
Figure 2.
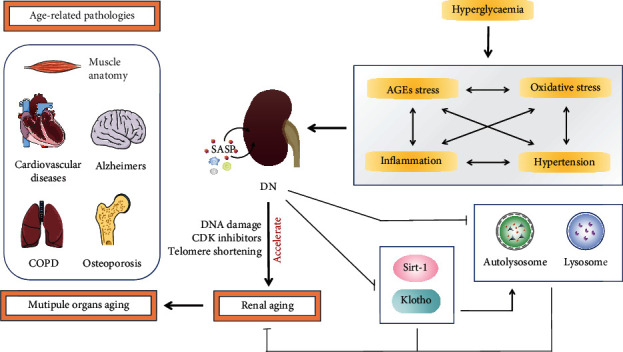
Schematic of accelerated kidney aging in DM and the relationship between kidney aging and systemic aging. In diabetic kidneys, AGE accumulation, oxidative stress, inflammation, and hypertension caused by hyperglycemia-induced metabolic impairment are central to the development and progression of DN and, hence, the acceleration of defining renal aging. In addition, the senescence of DN plays a key role in aging-related pathologies, such as cardiovascular diseases, Alzheimer's, chronic obstructive pulmonary emphysema (COPD), muscle atrophy, and osteoporosis.
4. Cellular Senescence and Kidney Aging in DN
It has been reported that cellular senescence plays a vital role in the aging and diseased kidney [51]. Cellular senescence can be widely observed in the aging kidney as an important cellular process that contributes to age-related kidney changes and CKD progression [52]. Besides, it is also involved in kidney aging. During aging, cellular senescence can be independently induced by stresses such as oxidative stress, which is called stress-induced premature senescence through p16INK4/retinoblastoma (Rb) or p53/p21Cip1 pathway. In kidneys, senescent cells are manifested in the arrest cell cycle, imbalance of apoptosis and proliferation, and senescence-associated secretory phenotype (SASP), leading to kidney aging by increasing sensitivity to injury and reducing repair after injury [53, 54]. In addition, clearance or depletion of senescent cells can relieve age-related damage and dysfunction in kidneys [51]. The conversion of a senescent phenotype is a significant step that underlies the pathogenesis of renal lesions as an early response to DM, which might be a target to retard DN progression [55].
Senescent cells provoke permanent cell cycle arrest by triggering cyclin-dependent kinase (CDK) inhibitors which mediate renal injury in DN. Studies on both progeroid and naturally aged mice showed that selective elimination of p16INK4-expressing senescent cells increased health and lifespan [56, 57], and in humans, p16INK4was shown as one of the top genes exhibiting elevated expression with age in multiple tissues, including kidneys [58]. In diabetic kidneys, accelerated senescent phenotypes were mainly observed in tubular cells and podocytes, and the positive correlation between glomerular p16INK4 with proteinuria indicated that glomerular cellular senescence takes partly responsibility for altered permeability [17]. AGE-induced p16INK4 expression and premature senescence were successfully relieved by an ER stress inhibitor and cyclic AMP-dependent transcription factor (ATF) 4 gene silencing in TECs of DN [14]. The expression of p21Cip1 was increased with an upregulation in senescence-associated β-galactosidase (SA-β-gal) staining in TECs [55], and the changes could be suppressed by insulin therapy. However, senescence induced by high glucose was inhibited in p21Cip1 knockdown mice, indicating that aging in renal TECs is mediated by a p21Cip1-dependent pathway [59]. TGF-𝛽1 induces p21Cip1 dependent hypertrophy of mesangial cells and plays an important role in the pathogenesis of chronic kidney diseases, including DN. TGF-𝛽1 increases p21Cip1 gene expression in renal mesangial cells and elevates the recruitment of the H3K4 methyltransferase SET7/9 to the p21Cip1 gene promoter [60]. It has been reported that there exists a complex crosslink between ER stress and p21Cip1 signaling in aging-related diseases [61]. In diabetic TECs, the ER stress marker is expressed at a higher level compared to that of controls and is positively correlated with enhanced SA-β-gal-positive cells and colocalization with RAGE. ER stress-mediated premature senescence is dependent on p21Cip1 activation, because the ER marker and p21Cip1 were colocalized in the same diabetic TECs in vivo and in vitro. Moreover, inducers of ER stress directly cause premature senescence of TECs by p21Cip1 activation. Thus, p21Cip1 signaling plays a deterministic role, which is promoted by RAGE, in the premature senescence of TECs that is mediated by the activation of ER stress [62]. P27Kip1 is another member of CDK2 inhibitors. Chronic hyperglycemia induces hypertrophy and damage to podocytes and mesangial cells related to p27Kip1 [63, 64]. In Type I DM, p27Kip1 knockout mice exhibited milder renal lesions compared to that of p27Kip1 +/+mice due to the regulation of TGF-β; additionally, angiotensin receptor blocker treatment alleviated renal hypertrophy by inhibiting p27Kip1 expression [64, 65]. Senescent phenotypes might have individualized expression in different renal cells, but controversial results have still been reported. For example, some studies showed that p16INK4 expression increased in diabetic TECs, whereas other studies have reported that overexpression of p21Cip1 but not other CDK inhibitors, such as p16INK4 and p27Kip1, is increased due to hyperglycemia [59]. These discrepancies might be related to the different stages of DN in different studies. However, further studies still need to further account for these discrepancies in the future.
Another feature of senescent cells is senescence-associated secretory phenotype (SASP). SASP is a significantly distinctive feature of senescent cells that includes diverse cytokines, chemokines, growth factors, proteases, and lipids, which may promote inflammation in aging-related diseases [66]. NF-κB is activated by DDR and p38 AMP-activated protein kinase (AMPK) in the production and secretion of SASP, and the main components of SASP during the mature period are soluble cytokines, such as C-X-C-motif chemokine ligand-1/2 (CXCL-1/2), interleukin- (IL-) 8, IL-1, matrix metalloproteinases (MMPs), and ECM proteins, that may contribute to the accumulation of ECM and renal interstitial fibrosis in CKD [66]. Premature senescence of intrarenal and extrarenal cells appearing with the overexpression of SASP leads to aggregative kidney aging and disease progression. The similarities of SASP and the CKD-associated secretory phenotype (CASP) have been compared with one another and may present a link between CKD and renal cellular senescence [67]. Considering the microinflammation in the mechanisms of DN, we summarize the relative specific secretory phenotypes in DN compared with those in CASP (Table 1).
Table 1.
Comparison between SASP and DN-associated secretory phenotype.
| SASP factors | Secretory profile for senescent cells [67, 227] | DN-associated secretory phenotype | |
|---|---|---|---|
| Cytokines | |||
| IL-1α,-1β,-2,-4,-6,-10,-13-17,-18,-20 | ↑ | ↑ | [228–239] |
| TNF-α | ↑ | ↑ | [229, 232, 233, 237–239] |
| TWEAK | ↑ | ↑ | [240] |
| ICAM-1 | ↑ | ↑ | [230, 233, 241–244] |
| VCAM-1 | ↑ | ↑ | [232] |
| Chemokines | |||
| CXCL-1,-5,-7 | ↑ | ↑ | [233, 243] |
| CCL-2,20 | ↑ | ↑ | [233] |
| CCL-4,-5 | ↑ | ↑ | [230, 243, 245] |
| IL-8 | ↑ | ↑ | [236] |
| MCP-1 | ↑ | ↑ | [235, 236, 238, 243–246] |
| MIP-1,-2 | ↑ | ↑ | [243, 244] |
| Eotaxin | × | ↑ | [241] |
| Other inflammatory factors | |||
| GM-CSF | ↑ | ↑ | [233] |
| G-CSF | × | ↑ | [234, 237] |
| IFN-γ | × | ↑ | [229] |
| Growth factors and regulators | |||
| IGFBP-3, -7 | ↑ | ↑ | [247, 248] |
| TGF-β | ↑ | ↑ | [96, 117, 234, 242, 246] |
| VEGF | ↑ | ↑ | [230, 233] |
| PDGF | ↑ | ↑ | [233] |
| FGF-2, 23 | ↑ | ↑ | [249, 250] |
| Proteases and regulators | |||
| MMP-2, -9, | ↑ | ↑ | [232, 251, 252] |
| TIMP-1 | ↓or × | ↓ | [251] |
| TIMP-2 | ↑ | ↑ | [242] |
| PAI-1 | ↑ | ↑ | [230, 232, 253] |
| Cathepsin B | ↑ | ↑ | [106, 117] |
| Insoluble factors (ECM) | |||
| Fibronectin | ↑ | ↑ | [239, 246, 253] |
| Collagens | Altered | ↑ | [231, 234, 239, 242, 246, 253] |
| Other factors | |||
| iNOS | ↑ | ↑ | [235] |
| ROS | Altered | ↑ | [96, 239] |
| COX-2 | ↑ | ↑ | [235] |
| NOX-4 | ↑ | ↑ | [254] |
| SOD | ↓ | ↓ | [96, 235, 238, 239, 254] |
| MDA | ↑ | ↑ | [96, 235, 238, 239, 254] |
Abbreviations: SASP: senescence-associated secretory phenotype; IL: interleukin; TNF-α: tumor necrosis factor α; TWEAK: apoptosis of tumor necrosis factor-like weak inducer; ICAM-1: intercellular adhesion molecule 1; VCAM-1: vascular cell adhesion molecule 1; CXCL: C-X-C-motif chemokine ligand; CCL: C-C-motif chemokine ligand; MCP: monocyte chemoattractant protein; MIP: macrophage inflammatory protein; GM-CSF: granulocyte-macrophage colony-stimulating factor; G-CSF: granulocyte colony-stimulating factor; IFN-γ: interferon-γ; IGFBP: insulin-like growth factor binding protein; TGF-β: transforming growth factor -β; VEGF: vascular endothelial growth factor; PDGF: platelet-derived growth factor; FGF: fibroblast growth factor; MMP: matrix metalloproteinase; TIMP: tissue inhibitors of metalloproteinases-1; PAI-1: plasminogen activator inhibitor -1; iNOS: inducible nitric oxide synthase; ROS: reactive oxygen species; COX-2: cyclooxygenase-2; NOX-4: NADPH oxidase-4; SOD: superoxide dismutase; MDA: administration; ∗Upward arrows, crosses, and downward arrows show secretory increase, no change and decrease in senescence, respectively.
Telomere shortening is another cause of triggering senescence. It has been shown that telomeric DNA is lost in the aging kidney of humans [68], and the shortening of telomere length may be associated with CKD occurrence and/or decline of kidney function [69]. In cultured TECs, high glucose accelerated telomere shortening may be mediated by oxidative stress because of hyperglycemia [17]. For mesangial cells, senescence was associated with telomere attrition induced by high glucose via the p53-p21-Rb signaling pathway. [70]. These results may contribute to a new strategy for the treatment of DN.
5. Main Cellular and Molecular Mechanisms of Accelerated Kidney Aging in DN
Molecules and signaling pathways related to the mechanisms of accelerated kidney aging in DM remain multiple and complicated. Herein, we review the mechanisms in four aspects (including Sirt1, Klotho, autophagy, and lysosome) because of their core and inevitable role related to the kidney aging in DM.
5.1. Sirt1 and Kidney Aging
Sirt1 protein expression can be detected in the normal kidney, and its expression level is decreased in the diabetic kidney [71, 72]. The decline of Sirt1 expression in diabetic-kidney tissue leads to mitochondrial damage and OS and plays a vital role in renal premature senescence by impairing antistress capacity and accumulating renal lesions [73]. The podocyte-specific loss of Sirt1 not only aggravates diabetic kidney injury but also leads to aggravated aging-induced glomerulosclerosis and albuminuria. This phenomenon is associated with reduced activation of transcription factors, such as peroxisome proliferator-activated receptor- (PPAR-) α coactivador-1 (PGC-1α)/PPARγ, Foxo3, Foxo4, and p65 NF-κB, via Sirt1-mediated deacetylation [71](Figure 3(b)).
Figure 3.
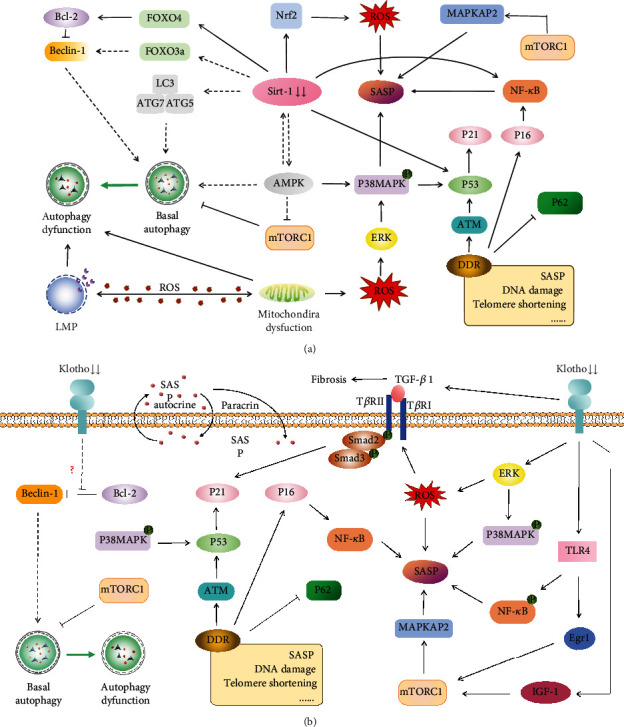
Klotho, Sirt1, and lysosome-dependent autophagy in DN. Under physiological conditions, the antiaging protein klotho and Sirt1 can enhance basal autophagy, protecting the kidney from DN and aging. Under DM conditions, the expression of Klotho and Sirt1 in the kidney is downregulated. Besides, the autophagy is not able to maintain cellular homeostasis and resist renal cell senescence. This overwhelmed suppression in DN accelerates kidney aging. The solid lines with black arrowheads indicate promoting function. The solid lines with black truncated ends stand for inhibiting function. The dotted lines in black show the disturbed and altered regulatory function in the diabetic kidney with aging. The solid line in green represents autophagy from normal to dysfunction. Abbreviations: DDR: DNA damage response; SASP: senescence-associated secretory phenotype; ROS: reactive oxygen species; ATM: ataxia telangiectasia mutated; MAPK: mitogen-activated protein kinase; ERK: extracellular signal-regulated kinase; NF-κB: nuclear factor-kappa B; Nrf2: nuclear factor E2-related factor 2; SIRT1: sirtuin 1; AMPK: adenosine monophosphate–activated protein kinase; mTOR: mammalian target of rapamycin; LC3: microtubule-associated protein 1A/1B–light chain 3; Atg5: autophagy-related 5; Atg7: autophagy-related 7; BCL2: B-cell lymphoma 2; P62: SQSTM1/sequestosome 1; FOXO3a: forkhead box O3a; FOXO4: forkhead box O4; TGF-β: transforming growth factor-β; LMP: lysosomal membrane permeabilization.
Caloric restriction was first proposed to be associated with antiaging in 1934, and Sirt1 has been reported as an antiaging molecule related to caloric restriction of aging-related diseases [74, 75]. In fatty diabetic Wistar rats, dietary restriction ameliorated renal abnormalities and decreased expression of Sirt1, increased expression of acetylated-NF-κB, and impaired autophagy. These results demonstrated that dietary restriction-mediated Sirt1 restoration exerted anti-inflammatory effects and improved autophagy dysregulation, which resulted in the amelioration of renal injuries in DM [76]. AGEs reduced the Sirt1 level but enhanced the expressions of FN and TGF-β1 in mesangial cells. The overexpression of Sirt1 further increased the nuclear accumulation of nuclear factor E2-related factor 2 (Nrf2) and promoted heme oxygenase 1(HO-1) and superoxide dismutase (SOD) 1 levels, whereas it decreased ROS, FN, and TGF-β1 levels induced by AGEs (Figure 3(b)). Thus, Sirt1 showed resistance against oxidative stress-mediated diabetic renal fibrosis [77]. The same results could also be demonstrated in human renal TEC line HK2 cells [78]. Sirt1 regulates mitochondrial biogenesis and turnover in relation to the deacetylation of PGC-1α [79]. HG accelerated mitochondrial dysfunction and downregulated Sirt1 expression. Activation of the Nrf2- antioxidant response element (ARE) antioxidative pathway ameliorates hyperglycemia-mediated mitochondrial dysfunction partly through Sirt1 [80–82] (Figure 3(b)).
These findings highlight that Sirt1 plays an important role in antisenescence in DN, by targeting members of SASP (e.g., NF-κB and TGF-β), as well as by relieving renal inflammation, fibrosis, and oxidative stress to alleviate mitochondrial damage of renal cells.
5.2. Klotho and Kidney Aging
Klotho is an aging suppressor gene that encodes a single-pass transmembrane protein with an extracellular portion, exhibits multiple pleiotropic effects, and is found in two forms: an intermembrane form and a secreted form [83]. The membrane klotho forms a complex with fibroblast growth factor 23 (FGF23) receptor, and this complex mediates phosphate homeostasis and vitamin D metabolism. Secreted Klotho acts as a humoral factor targeting distant organs with pleiotropic activities consisting oxidative stress regulation, growth factor signaling, and ion homeostasis and can be detected in blood, urine, and cerebrospinal fluid [84, 85].
In mice, the overexpression of Klotho extends lifespan, whereas mutations to the Klotho gene shorten lifespan. In humans, serum Klotho levels are lower in individuals that are 40 years old or older [86]. Klotho is predominantly expressed in renal TECs and is equipped with a variety of biological functions. The antiaging role of Klotho is related to its downregulation of cytokines and growth factor signaling, such as interferon-γ (IFN-γ), insulin-like growth factor-1 (IGF-1), and TGF-β, and works by inducing antioxidative stress mediated by insulin, via IGF-1 signaling [86] (Figure 3(a)). Furthermore, it is related to iron imbalance [85]. The level of Klotho expression in diabetic patients and mice was significantly reduced, which was related to increased urinary calcium excretion [87]. Klotho levels were decreased with increasing albumin excretion in patients [88] and were significantly associated with a decline in eGFR [89]. These findings are similar to those of other studies [90]. Thus, Klotho may be a predictive biomarker for the progression of DN.
Klotho was observed to attenuate renal fibrosis in DN. The expression of Klotho in renal tubules declined in streptozotocin- (STZ-) diabetic rats, and Klotho alleviated HG-induced profibrotic genes, TGF-β signaling, and cell hypertrophy in rat renal fibroblast cell line NRK-49F cells. Moreover, Klotho attenuated HG-induced FN expression and cell hypertrophy via ERK1/2 and p38 kinase-dependent pathways [91] (Figure 3(a)). In addition, in spontaneously diabetic mice, it was also observed that the upregulation of Klotho attenuated renal hypertrophy, albuminuria, glomerular mesangial expansion, as well as attenuated glomerular macrophage infiltration and suppressed proinflammatory cytokines [92]. Additionally, Klotho downregulated early growth response factor 1 by inhibiting TGF-β1/Smad3 signaling in HG-induced human mesangial cells to combat renal fibrosis [93] (Figure 3(a)).
Klotho has also been shown to be involved in anti-inflammation in DN. In db/db mice, renal Klotho gene and protein expression were significantly downregulated, and overexpression of Klotho repressed NF-κB activation and subsequent production of inflammatory cytokines in response to TNF-α stimulation. These findings suggest that Klotho serves as an anti-inflammatory modulator that negatively regulates the production of NF-κB linked inflammatory proteins [94] (Figure 3(a)).
Klotho deficiency may be associated with increased OS, and anti-OS is a potential treatment target for DN. OS is more serious in patients with DN compared to that of the healthy controls [95]. AGE-triggered cellular senescence was at least partially due to the activation of OS, and Klotho overexpression protected TECs from injury induced by AGEs and H2O2; thus, Klotho can attenuate the OS [96].
Klotho inhibits the progression of DN by attenuating vascular endothelial dysfunction, calpain activation, and chronic inflammation. In addition, Klotho has been shown to inhibit diabetic renal tubular hypertrophy by inhibiting IGF-1 signaling [92, 97] (Figure 3(a)). Taken together, Klotho participates in multiple antiaging pathways to protect renal function in DN.
5.3. Autophagy and Kidney Aging
Autophagy is a degradation and recycling system in the process of growth, development, and aging. Autophagy has two main roles in the cell: (1) self-digestion in nutrient-deficient conditions to achieve the reuse of energy; (2) degradation of damaged or excess organelles and macromolecules to maintain cellular metabolism under stress [98]. A large number of studies have demonstrated that autophagy inhibition increases with aging and progresses in aging-related diseases [99, 100]. As a convergent mechanism of multiple longevity models, the activity of basal autophagy is elevated in many longevity paradigms of lifespan extension or delayed aging [101]. Autophagy appears to be a causal effector of existing antiaging manipulations such as the longevity drugs, such as resveratrol, rapamycin, and spermidine [102]. Rapidly accumulating evidence has revealed that autophagy is involved in renal physiology, kidney aging, and several kidney diseases, and plays a renoprotective role in various animal models.
Notably, serum levels of Beclin-1, a regulator of autophagy, are reduced in patients with DM and DN. Beclin-1 is also related to the stage of DN and correlates with the degree of albuminuria, which indicates autophagy inhibition in patients with diabetic renal lesions [103]. In addition, the basal activity of autophagy is inhibited in intrinsic cells in diabetic kidneys [104]. The potential mechanisms might be due to the appearance of oxidative stress and inflammation secondary to stimuli such as AGEs and urinary proteins [105–107]. Autophagy regulates TGF-β expression and suppresses kidney fibrosis through the autophagic degradation of mature TGF-β [108], which contributes to the occurrence of diabetic diffuse glomerulosclerosis and the excessive deposition of fibrotic materials in DN [109]. The LC3-II/LC3I ratio, Atg5 level, and Atg7 expression in the diabetic kidney with Sirt1 knockdown sharply declined [110], whereas Sirt1-induced autophagy was enhanced in an experimental model of DN [111]. Additionally, other studies have indicated that AMPK downregulation [112] and mTORC1 upregulation [113] are two key players in orchestrating events in autophagy and aging, which are crucial for the onset or progression of DN [114, 115].
Impaired autophagy has been demonstrated in vivo in DN patients that LC3 and P62 accumulation in the kidney of DN patients [116] and DN animal models [117], as well as in vitro [106]. Actually, the antisenescence of autophagy remains controversial. Oxidative stress-induced senescence is linked to autophagy impairment [118]. Mitochondrial dysfunction induced by high glucose is the main cause of oxidative stress and triggers senescence of TECs which can be modulated by mitophagy [119]. It was reported that atg5-deficient podocytes developed a series of aging-related alterations, such as lipofuscin accumulation and damaged mitochondria increase, the load of oxidized proteins, and the occurrence of ubiquitin and p62/SQSTM1-positive protein aggregates [120]. However, another study illustrated that silencing atg-5 reduced the hallmarks of stresses-induced TEC senescence [121]. It was clarified that general autophagy played an antisenescence role, but under stresses, once cells over a certain time point in senescence, autophagy showed prosenescence because of removing stresses that senescent cells must treat with [122]. More interestingly, it was reported that increased lysine63 ubiquitination not the whole level of ubiquitin was related to impaired autophagy and apoptosis of TEC induced by hyperglycemia [116]. So how autophagy in DN influences cell fates remains complicated. For atherosclerosis, defective autophagy promoted senescence and apoptosis in endothelial cells. Defects in autophagic machinery seemed to initiate apoptosis, while the expression of p53 was likely to onset senescence. Anyhow, senescence and apoptosis were two complementary cell fates controlled by autophagy [123]. Nevertheless, in DN, more studies are needed to conduct to elucidate how autophagy promotes cell fates.
5.4. Lysosome and Kidney Aging
Lysosomes are the main catabolic organelles essential for cell homeostasis that are found in all animal cell types except for erythrocytes and play a pivotal role in regulating a variety of processes, such as calcium signaling and nutrient responses to autophagic degradation of intracellular components. Lysosomes have been reported to have important significance in the control of lifespan [124]. Specifically, lysosomal dysfunction induces failure of cellular homeostasis during aging, which reduces the overall degradative capacity of cells and influences cellular and organismal life and death [124, 125]. Cellular senescence is partly determined by the lysosomal function related to mitochondria. Lysosomal dysfunction induces the imbalance of mitochondrial turnover, resulting in the generation of more ROS, which in turn targets lysosomes [126]. In ROS-senescence, mitochondrial dysfunction plays an initiating role, while lysosomal dysfunction is more directly responsible for senescence [118].
It is well established that lysosomal-cathepsins translocation, caused by lysosomal membrane permeabilization (LMP) (Figure 3), induces lysosomal-dependent cell death (LDCD), which has been observed in some age-related diseases, such as Parkinson's disease [127, 128]. In the study of DN, renal TECs with AGEs stimulation triggered the lysosomal membrane permeabilization, resulting in a decrease of activity of cathepsin B and cathepsin L, lysosomal acidification, and defective degradation of DQ-ovalbumin. However, these effects of AGEs can be blocked by antibodies against AGE-specific receptors or by antioxidants, which indicates that in the condition of DN, oxidative stress may play an important role in lysosomal dysfunction and further lead to tubular cell senescence and apoptosis [106]. Furthermore, urinary LAMP-2 levels are significantly decreased in patients with DN, which correlated with the urinary albumin to creatinine ratio (ACR) and GFR and might lead to the accumulation of autophagic vacuoles [129]. Lysosomal cathepsins are responsible for initiating and executing cell death during aging [130] (specifically in the kidney), and dysregulation of cathepsins B, D, L, and S is shown to be responsible for the onset or progress of kidney diseases [131]. Altered cathepsin D was captured in the tubulointerstitium of renal tissue from patients with DN, and, more precisely, cathepsin D upregulation suppressed the LMP and loss of mitochondrial membrane potential triggered by AGEs, which suggests a protective role in DN [132].
Recent data unveil mTOR activation, nuclear translocation of transcription factor EB (TFEB) inhibition, and the interaction between mTOR and TFEB in glomeruli from db/db mice and podocytes treated with AGEs, which is an irreplaceable factor that involved in the pathogenesis of DN [133]. Previous work has confirmed that TFEB is the master gene in coordinating lysosomal expression and its regulated network [134] and regulates lysosomal biogenesis and cellular clearance [135, 136]. Multiple lines of evidence indicate that mTORC1 resides at the lysosomal surface [135], in response to nutrient-sensing pathways [137], and accelerates cellular and organismal senescence [138, 139]. Hence, in the context of aging and longevity, it will be captivating to eavesdrop on the “cross-talk” between kidney disease and aging through lysosome-based signaling pathways.
6. Main Consequences of Accelerated Kidney Aging in DN
There are two main consequences from the accelerated kidney aging in DN. First, more rapid GFR declines. Kidney aging is featured with the reducing GFR. Due to chronic hyperglycemia, hypertension, and proteinuria, 34.8% of patients have a progressive disease with an annual GFR decline of 3.57 ± 1.45 mL/min/1.73 m2/year due to chronic hyperglycemia, hypertension, and proteinuria [140]. Significantly, DN patients with heavy proteinuria experience a rapid renal deterioration with the rate of decline of 46–60 mL/min/1.73 m2/year [141]. Those represent a more accelerated decline compared to normal biological aging at an average rate of 1 mL per year after 30 years old [142]. Second, kidney aging triggers system aging, contributing to the mortality of DN. Patients with DM and CKD have a sharply higher risk for cardiovascular diseases (CVD) compared to diabetic patients with no CKD and CVD, not ESRD seems to be the main cause of death [143]. In Japan, DN has been the main cause of chronic hemodialysis and in populations with chronic hemodialysis, the morbidity of frailty in the DN group was significantly higher than that in the non-DN group [144]. Alarmingly, 32.7% of individuals with DN have frailty significantly increasing the risk of developing ESRD and mortality [145]. A variety of other phenotypes of premature aging still have been observed in patients with CKD, such as the following: vascular calcification, cardiac insufficiency, osteoporosis caused by calcium and phosphorus metabolism disorders, muscle atrophy, and cognitive dysfunction [43, 146, 147]. As a specific CKD, aging-related DN is closely related to systemic aging. A range of factors play a part role in the aging-related decline in renal function, including increased levels of oxidative stress and inflammatory reactions, activation of the RASS, and the stress resistance responses, excessive secretion of angiotensin II, mTOR overactivation, deficiency of Klotho and vitamin D [21, 44]. These factors mentioned above could have a role in impairing the antiaging pathway and may underlie premature aging in DN [96, 148–150]. In 852 healthy adults aged 30–98 years, Han et al. reported that declines of naturally aging-related renal function and cardiac diastolic function are not independent processes [151]. In addition, sarcopenia in elderly patients with kidney diseases had a higher prevalence compared with that of younger ones [152], which may provide a better understanding that kidney aging accelerates systemic multiple organ dysfunction.
7. Potential Therapeutic Strategy Targeting Accelerated Kidney Aging in DM
Animal models have shown that diet interventions retard systemic and kidney aging, especially diets with low-AGE contents and enrichment of antioxidants [153]. A 40% adult-onset calorie restriction tended to suspend the age-related structural alterations of kidney-like glomerulosclerosis, interstitial-fibrosis formation, and vascular-wall thickening, which was associated with the decrease of the accumulation of mitochondrial enzyme abnormalities [154]. Short-term caloric restriction was demonstrated to play a protective role against renal senescence via increasing autophagic activity and reducing oxidative stress [155], which may be related to the modulation of AMPK/mTOR signaling [156], attenuating inflammatory process via downregulation of NF-κB [157], as well as the suppression of apoptosis [158]. For humans, some clinical studies have reported the effects of diet restriction for diabetic kidneys. Calorie restriction exhibits renoprotection via amelioration of glomerular hyperfiltration of patients with T2DM with abdominal obesity [159]. With a period of four months of low protein-diet intervention, renal function with a restricted glucose control improved among diabetic patients with macroalbuminuria [160]. However, there is a lack of evidence to show whether there is a beneficial effect of long-term diet restriction for diabetic patients with kidney lesions [161].
Considered a caloric restriction mimetic, resveratrol, a natural polyphenol extracted from grapes and several plants, is characterized as a powerful free-radical scavenger and antioxidant and has been recognized to have an effect on antiaging and life extension. A clinical trial showed that resveratrol might be an effective complementary selection with ARBs to reduce albuminuria in patients with DN [162]. Animal experiments have illustrated that resveratrol restrains the oxidative stress markers in diabetic rats, decreased the expression of renal TGF-β1 and FN [163], which might be associated with the activation of Sirt1 and PGC-1α [164], and increased the expression of Foxo1 [165] and PPARδ [166]. Multiple signaling pathways, including PI3K/Akt, c-Jun N-terminal kinase (JNK)/NF-κB, Akt/NF-κB, and p38 MAPK/TGF-β, have been demonstrated to underlie the renoprotective mechanisms of resveratrol [167] and its antiaging effect in diabetic kidneys [168]. Interestingly, it has been reported that resveratrol has the potential capacity to increase the expression of antiaging proteins such as Klotho and Sirt1 to alleviate the vascular calcification in CKD.
Metformin application remains controversial for DN due to its risk on hyperlactatemia and renal impairment in moderate-to-severe CKD, especially in the elderly [169, 170]. Actually, dosage adjustment of metformin appears to be safe and efficacious for moderate-to-severe CKD, and evidence has shown its potential benefits in lowering the risk of death and cardiovascular event in stage 3 CKD and sustaining calcium-phosphorus homeostasis to prevent vascular calcification [171, 172]. Metformin has shown antiaging benefits in diseases including DM and CKD [173–175] and in reducing the all-cause mortality and diseases of aging, including tumors and cardiovascular diseases of diabetics, even compared to nondiabetic [176]. The activation of AMPK plays a vital role in the mechanisms underlying the beneficial effects of metformin for DN. Hyperglycemia gave rise to suppression of phosphorylation and activity of AMPK, leading to multiple pathophysiological changes [177]. Metformin has elucidated the protective effect on podocytes, glomerular mesangial cells, and proximal tubular epithelial cells. In vitro, metformin showed antiapoptosis of podocytes induced by high glucose due to activation of AMPK and inhibition of mTOR signaling [178], and the activation of AMPK seemed to be associated with the activation of P2 receptors via upregulation extracellular ATP concentration [179]. Additionally, metformin adjusted nephrin protein expression [180] and repressed oxidative injury to restore podocytes [181] and also relieved insulin resistance of podocytes through activating Sirt1 and AMPK in diabetic rats [182]. Metformin alleviated inflammation of mesangial cells [183], which was related to upregulated glucagon-like peptide-1 (GLP-1) receptor expression [184]. The Sirt1/Foxo1 signal pathway was focused on demonstrating the antioxidative stress effect of metformin subsequently with the activation of autophagy in diabetic rats and high-glucose-induced mesangial cells [185, 186]. Increasing PGC-1α expression in high-glucose-induced TECs and suppression of AKT and mTOR activation in proteinuria induced TECs, subsequently followed by augmented autophagy and mitochondrial dynamics or ER stress that contributed to the renoprotective effects of metformin in DN [187, 188]. Meanwhile, metformin and rapamycin reversed high-glucose-induced premature senescence of renal cells, as well as induced downregulation of Connexin43 via activation of AMPK and the inhibition of mTOR [189], while the P21 expression was suppressed via modulation of AMPK by metformin-independent repression of mTOR [190].
It is important to note that not only metformin but other antidiabetic agents show emerging renoprotection targeting renal cellular senescence. It has been reported that in patients with type 2 diabetes and kidney disease, compared to a placebo, the inhibitor of (sodium-glucose cotransporter-2) SGLT2, canagliflozin, indeed decreased the relative risk of ESRD, a doubling of the creatinine level or death from renal caused by 34% [191]. This result indicates that SGLT2 inhibitors might be the new hope of the patients with DN after the use of renin-angiotensin system blockers over the past 18 years. The underlying mechanism may be related to its antisenescence of renal cells resulting from the fact that SGLT2 increased the expression of senescent markers in proximal tubules [76] and endothelial cells [192] in DM, indicating that SGLT2 inhibitors might retard renal accelerated aging in DM to preserve kidney function. Other antidiabetic agents reported to protect against accelerated aging are dipeptidyl peptidase 4 (DPP4) inhibition and GLP-1 receptor agonists, which act on the modulation of incretin that protect against age-related diseases including DN [193]. Actually, DPP4 inhibition demonstrated clear antiaging effects.
The lifespan of klotho−/− mice was prolonged, and their body weight was significantly related to greater kidney weight in the intervention of linagliptin [194]. Although no conclusive evidence has demonstrated that DPP4 inhibition improves diabetic kidney lesions, it may control blood glucose and albuminuria as well as be tolerated in patients with DM and CKD, indicating potential renal benefits [195]. It has been reported that rectification of the imbalance between DPP4 and GLP-1 is helpful to vascular aging [196]. Thus, besides DPP4 inhibition, GLP-1 receptor agonists have also shown antiaging potential. GLP-1 receptor agonists showed renoprotection independent of glycemic control, such as inhibition of cellular apoptosis, inflammation, and oxidative injury via the upregulation of Sirt1 [197]. Nevertheless, the renoprotective effect had seemingly acted indirectly on the kidney but was associated with a systemic immunomodulatory effect [198].
PPARγ expresses a low level in kidneys, and it has been shown that its expression and activity reduce during aging and results in the loss of aging-associated function [199]. Pioglitazone presented antiaging via the upregulation of Sirt1 and Klotho, decreased the p53 protein level in aged ApoE−/− mice [200], and alleviated aging-related renal injury via modulation on mitochondrial function [201]. Clinical studies suggested that low-dose pioglitazone was an effective renoprotective method in DN [202, 203]. As a result, pioglitazone showed its potential protection against accelerated senescence in DN.
Considering that oxidative stress plays a major role in the progress of DN and accelerates the kidney aging, chronic antioxidant supplements are eagerly sought after for their long-term benefits. Antioxidants (including vitamin C, vitamin E, and zinc) may protect against early renal damage [204]. High-dose vitamin E supplements in the treatment of DN resulted in a notable decrease in urine protein, which may be partly associated with the alleviation of autophagic stress in TECs [117, 205]. Nicotinamide adenine dinucleotide (NAD) functions as a coenzyme in redox reactions and mediates many biological processes, including metabolism and aging, as well as metabolic diseases like DM [206, 207]. As a hallmark of aging, NADs are related to the inducing of autophagy, repairing DNA and activation of Sirt1 and NADs, which are regulated by AMPK [208]. The age-dependent decrease of NADs happens in many tissues, including kidneys [206]. NAD precursors can delay aging and counteract a broad spectrum of age-related diseases. It has been reported that NAD replenishment contributed to retarding the renal lesion in diabetic rats. However, the precision of NAD+ supplementation is needed due to the regulation of SASP leading to tumor-promoting effects [209]. Actually, a number of studies are focused on antioxidant supplementation in the treatment of DN, but conclusive evidence is still lacking to demonstrate their long-term clinical benefits.
Owing to the fact that fewer senescence cells can even lead to reduced survival in older individuals, senotherapies, such as selective elimination of senescent cells (senolytics) or the disruption of the senescent cells' secretome (senostatics), are gaining significant attention from researchers to retard the progression of aging-related diseases [210, 211]. So far, dozens of senolytics have been reported, which have been considered adjunctive therapies for aging-related diseases such as tumors [212], idiopathic pulmonary fibrosis [213], Alzheimer's disease [214], and renal disease [215]. These reports suggest that senolytics may be a beneficial supplementary therapy for DM patients with chronic kidney injuries. A recent clinical study reported that 3 d of oral Dasatinib and Quercetin alleviated adipose tissue senescent cell burden and decreased skin epidermal p16INK4A+ and p21CIP1+ cells and circulating SASP factors in patients with DN, indicating that senolytics relieved senescent cell burden [216]. Hence, it is hypothesized that a similar intervention could be complementary to clinic therapy in the treatment of DN in the future. Actually, the application of senotherapies in aging-related disease remains uncertain [217], including their specific-kidney protection, so more clinical and further studies are needed.
Simultaneously, traditional Chinese medicine (TCM) is another popular option for DN because of its definite curative effect. The renal protection offered by Shenkang injection, a classic compound prescription, has also been demonstrated to retard high glucose-induced senescence of renal tubular cells [218]. Some extracts of herbs have exhibited antiaging properties, such as curcumin [219, 220], a glycoprotein isolated from Fupenzi [221], tea polyphenols [222], the flavonoid 4,4′-dimethoxychalcone (DMC) [102], and berberine [223]. These results imply that TCMs appear to have potential advantages to protect renal function against kidney aging in DM in the future. The potential mechanisms of drugs with antiaging properties applied to DN are summarized in Table 2.
Table 2.
Examples of potential mechanisms underlying renal benefits of drugs with antiaging effects.
| Drugs | Beneficial effects | Mechanisms | In vivo | In vitro | Ref. |
|---|---|---|---|---|---|
| Resveratrol | Attenuation of renal fibrosis | Regulation of AMPK/NOX4/ROS signaling | db/db mice | [255] | |
| Resveratrol | Renoprotection | Enhancement of hypoxia-induced autophagy via Sirt1 | STZ-induced diabetic rats, | Hypoxic-condition-induced rat proximal tubular epithelial cells NRK-52e | [110] |
| Resveratrol | Suppression of renal inflammation and mesangial cell proliferation | Modulation on Akt/NF-𝜅B pathway | STZ-induced diabetic rats; | HG-induced rat mesangial cells | [256] |
| Resveratrol | Regulating oxidative stress and mitochondrial function | Modulation of the Sirt1/FoxO1 signal pathway | STZ-induced diabetic rats | HG-induced rat mesangial cells | [165, 257] |
| Resveratrol | Renoprotection | Extenuating the oxidative stress and downregulation of RAGE expression | STZ-induced diabetic rats | [258] | |
| Resveratrol | Ameliorating lipotoxicity, oxidative stress, apoptosis, endothelial dysfunction; glomerular matrix expansion and inflammation | Activating the AMPK-Sirt1-PGC-1α axis and PPARα through increases in AdipoR1 and AdipoR2 expression | db/db mice | HG-induced human glomerular endothelial cells and NMS2 mesangial cells | [259, 260] |
| Resveratrol | Protection on podocytes | Activation of autophagy involved with miR-383-5p | db/db mice | HG-induced human podocytes | [261] |
| Resveratrol | Protection on mesangial cells | Negative regulation of the p38 MAPK/TGF-β1 pathway | STZ-induced diabetic rats | HG-induced rat mesangial cells | [167] |
| Resveratrol | Protection on podocytes | Against apoptosis by increasing autophagy via miRNA-18a-5p expression | db/db mice | HG-induced human podocytes | [262] |
| Resveratrol | Effect on endoplasmic reticulum stress | Reducing expressions of 78 kDa glucose-regulated protein (GRP78), protein kinase RNA-like endoplasmic reticulum kinase (PERK), and activating transcription factor 4 (ATF4) and C/EBP-homologous protein (CHOP) | STZ induced diabetic rats | [263] | |
| Resveratrol | Protection on podocytes | Reducing oxidative damage and apoptosis of podocytes via Sirt1/PGC-1α mitochondrial protection | HG-induced immortalized mouse podocytes | [164] | |
| Resveratrol | Renoprotection and reducing albuminuria | Suppression of the angiotensin II (Ang II)/angiotensin II type 1 receptor (AT1R) axis and enhancing the angiotensin 1-7 (Ang 1-7)/Mas receptor (MasR) axis; anti-inflammation and oxidative stress | Aged C57BL/6 mice | [30] | |
| Resveratrol | Regulation on endothelial dysfunction | Modulation of Sirt1 and PPARγ | db/db mice | [166] | |
| Metformin | Restore the insulin responsiveness of podocytes | Regulating Sirt1 and AMPK activities | HG-induced rat podocytes | [182] | |
| Metformin | Exhibiting an anti-apoptotic impact on podocytes | Activation of AMPK and inhibition of mTOR signaling | HG-induced immortalized human podocytes | [178] | |
| Metformin | Renoprotective effect | Increasing SOD activity and decreasing malondialdehyde level; decreasing the expression levels of TGF-β1 | STZ-induced diabetic rats | [264] | |
| Metformin | Improving diabetic tubulopathy | Increasing in PGC1α activity by modulating mitochondrial dynamics and autophagy | STZ-induced diabetic mice, | HG-induced human renal proximal tubular epithelial cell line HKC8 | [188] |
| Metformin | Against proteinuria cytotoxicity | Suppression of Akt and mTOR activation, inhibition of EMT and apoptosis and augmentation of autophagy and ER defense response through AMPK-independent and AMPK-dependent mechanisms | Albumin-induced rat renal proximal tubular cells | [187] | |
| Metformin | Protection on podocytes | Upregulating the renal tissue nephron expression | STZ-induced rats | [180] | |
| Metformin | Protection on podocytes | Increases extracellular ATP concentration, leading to activation of P2 receptors and consequent modulation of the podocytes' metabolism through AMPK and NAD(P)H oxidase | HG-induced mouse podocytes | [179] | |
| Metformin | Alleviation of cell senescence | Downregulation of Connexin43 via activation of AMPK and the inhibition of mTOR | HG-induced primary rat glomerular mesangial cells | [189] | |
| Metformin | Alleviation of high-glucose-induced oxidative stress | Regulating p-p38MAPK protein expression | HG-induced rat glomerular mesangial cells | [265] | |
| Metformin | Alleviation of inflammation | Inhibits nuclear factor-κB activation and inflammatory cytokines expression including monocyte chemoattractant protein-1 (MCP-1), intercellular adhesion molecular depend on AMPK | HG-induced rat glomerular mesangial cells | [183] | |
| Metformin | Inhibition of apoptosis and inflammatory and fibrotic reactions in tubular cells | Reducing ROS generation via suppression of RAGE expression through AMP-activated protein kinase activation | AGEs induced human proximal tubular epithelial cells | [266] | |
| Metformin | Relieving oxidative stress, slowed down abnormal cell proliferation | Enhancing autophagy and through Sirt1/FoxO1 pathway via AMPK | STZ-induced diabetic rats; | HG-induced rat mesangial cells, | [185, 186] |
| Metformin | Exerting anti-inflammatory | Upregulating GLP-1R expression via AMPK | db/db mice | HG-induced rat mesangial cell line (HBZY-1) | [184] |
| Metformin | Attenuating hypoxia | Reducing uncoupling protein-2- (UCP2-) mediated mitochondrial proton LEAK | STZ-induced diabetic rats | [267] | |
| Metformin | Alleviating cell senescence | Reducing p21 expression by activating AMPK. | Human embryonic kidney (HEK293) cell line | [190] | |
| Metformin | Improving epithelial-to-mesenchymal transition | Inhibiting early growth response- (Egr-) 1; inhibiting MCP-1 expression via BMP and activin membrane-bound inhibitor- (BAMBI-) mediated inhibition of ERK1/2 | TGF-β1-induced rat renal tubular epithelial cell line (NRK-52E) | [268, 269] | |
| GLP-1 | Protection of podocytes | Against apoptosis, inhibition reactive oxygen species production and proinflammatory cytokine secretion, through Sirt1 activation | HG-induced mouse podocytes | [197] | |
| GLP-1R agonist (Exendin-4) | Against renal fibrosis | Inhibiting the transfer of extracellular vesicle miR-192 | HG-induced renal tubular epithelial cells | [270] | |
| GLP-1R agonist (Exendin-4) | Inhibiting cell proliferation and fibronectin secretion | Reversing ERK phosphorylation and enhancing expression of mTOR via AMPK | HG-induced rat mesangial cells | [271] | |
| GLP-1 analog (liraglutide) | Against renal inflammatory and protection on endothelial cells | Inhibiting STAT3/JAK2 expression via SIRT1 | db/db mice | AGEs- or HG-induced endothelial cells | [272] |
| GLP-1R analog (liraglutide) | Ameliorating early renal injury | Increasing the expression of FoxO1 mRNA and reducing renal phosphorylation levels of Akt and FoxO1 protein | STZ-induced diabetic rats | [273] | |
| GLP-1R analog (liraglutide) | Renoprotective effect | Inhibiting autophagy and apoptosis dependent on GLP-1R | HG-induced human renal tubular epithelial cell line (HK-2) | [274] | |
| GLP-1 analog (liraglutide) | Against oxidative stress and albuminuria | Via a PKA-mediated inhibition of renal NAD(P)H oxidase | STZ-induced diabetic rats | HG-induced human mesangial cells | [275] |
| DPP 4 inhibitor (sitagliptin) | Attenuation of glomerular lesions | Alleviation of oxidative injury | STZ-induced diabetic rats | [276] | |
| SGLT2i (dapagliflozin) | Attenuation of renal fibrosis | Elevating O-GlcNAcylation and tubular hypoxia | STZ-induced diabetic rats | HG-induced human proximal tubular epithelial cell line (HK-2) | [277] |
| SGLT2i (dapagliflozin) | Against inflammation and postponing the progression of renal injury | Inhibition of HMGB1-RAGE-NF-κB signaling pathway | HG-induced human proximal tubular epithelial cell line (HK-2) | [278] | |
| SGLT2i (canagliflozin) | Against renal inflammation, extracellular matrix turnover and fibrosis | Reduction in TNFR1, IL-6, MMP7 and FN1 | HG-induced human proximal tubular epithelial cell line (HK-2) | [279] | |
| SGLT2i (Ipragliflozin) | Improvements in glomerular damage | Normalizing the levels of accumulated tricarboxylic acid cycle intermediates and increased oxidative stress | db/db mice | [280] | |
| SGLT2i (Empagliflozin) | Anti-inflammatory and antifibrotic effects | Suppressing AGE-RAGE axis | STZ-induced diabetic rats | [281] | |
| Pioglitazone | Reprotection in DM | Decreasing expression of hypoxia-inducible factor-1a (HIF-1a) and vascular endothelial growth factor (VEGF) | STZ-induced diabetic rats | [282] | |
| Pioglitazone | Ameliorating aging-related renal injury | Increasing klotho, decreasing oxidative stress, and mitochondrial injury; regulating p66Shc phosphorylation, which integrates many signaling pathways that affect mitochondrial function and longevity, by reducing protein kinase C | Aging male Sprague-Dawley rats | [201] | |
| Dasatinib and quercetin | Decreasing human senescent cell burden | Alleviating adipose tissue senescent cell burden, decreasing skin epidermal p16INK4A+ and p21CIP1+ cells and circulating SASP factors in patients with DN | Human tissues | [216] |
Abbreviations: STZ: streptozotocin; DN: diabetic nephropathy; HG: high glucose; GLP-1: glucagon-like peptide-1; DPP4: dipeptidyl peptidase 4; SGLT2i: sodium-glucose cotransporter-2 inhibitor; AMPK/NOX4/ROS: AMP-activated protein kinase/NADPH oxidase-4/reactive oxygen species; Akt/NF-κB: protein kinase B/nuclear factor kappa-B; FoxO1: forkhead box O 1; PGC-1: peroxisome proliferator-activated receptor- (PPAR-) α coactivador-1; AdipoR: adiponectin receptor protein; MAPK: mitogen-activated protein kinase; TGF-β: transforming growth factor-β: mTOR: mammalian target of rapamycin; SOD: superoxide dismutase; EMT: epithelial-mesenchymal transdifferentiation; RAGE: receptor for advanced glycation end products (AGEs); HMGB1: high mobility group box 1-receptor; ERK: extracellular signal-regulated kinase; STAT3: signal transducer and activator of transcription; JAK2: janus kinase 2; TNFR1: TNF receptor 1; MMP7: matrix metalloproteinase 7; FN1: fibronectin 1.
8. Conclusion
Accelerated senescence of kidneys plays a vital role in DN progression. To delay renal lesions in DM or DN patients, accelerated or premature kidney aging must be delayed. The underlying mechanisms of accelerated kidney aging in DM or DN remain complex and multiple. Hyperglycemia, inflammation, oxidative stress, and hypertension induce renal inherited cellular senescence and the downregulation of antiaging proteins, such as Sirt1 and Klotho, and the inactivation of the lysosome-dependent autophagy pathway.
Antihyperglycemia is the most important factor in the treatment of DM and the prevention of DN. Metformin, pioglitazone, and the inhibitor of SGLT2 have been reported to exhibit antisenescence effects independent of antihyperglycemia. Owing to its certain renoprotection for DN patients, inhibitors of SGLT2 are given priority to consideration besides ACEI/ARBs. Additionally, metformin and pioglitazone should be more considered in the treatment of DM and in the early stage of DN. In addition, for DM or DN patients, antioxidant-rich food and antisenescence-compound-rich food are recommended. However, it should be noted their intake should be considered according to their sugar content. Concurrently, moderate calorie restriction and a level of exercise are encouraged for patients with DM and early DN.
Autophagy is the common downstream of aging-related pathways and is central to DN progression. Unfortunately, the regulation and activity mechanisms of autophagy remain incompletely understood with respect to DN. It has been proved that Sirt1 has been regulating autophagy through the deacetylation of Atg5, Atg7, and Atg8 [224]. Autophagy extended the lifespan of mice knock-out Klotho [225], and Klotho attenuated renal lesions by regulating the autophagy clearance [226]. Based on these studies, we identified a key link between antiaging proteins like Sirt1, Klotho in DN, but further studies are still necessary to illustrate how the antiaging proteins regulate the autophagy and the exact sites for autophagy modulation.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (Grant No. 81774298 and 81774278) and the Fundamental Research Funds for the Central Universities (Grant No. 2019-JYB-XS-043), but this research did not receive any public funding from commercial, or not-for-profit, sectors.
Contributor Information
Yaoxian Wang, Email: a2249@bucm.edu.cn.
Wei Jing Liu, Email: liuweijing-1977@hotmail.com.
Conflicts of Interest
The authors declare no conflicts of interest.
Authors' Contributions
YXW, WJL (Wei Jing Liu), JG, and HJZ contributed to the conception, design, and drafting of the manuscript. JG, WTZ, and WJL (Weijiao Lou) contributed to the optimization of the design. CHX and WJH helped optimize the drafting of the manuscript. FZ and XTH helped screen the literature. JG and WJL (Wei Jing Liu) contributed to the revised version. The final version was approved for submission by all authors. Jing Guo and Hui Juan Zheng contributed equally to this work.
References
- 1.Federation ID. IDF Diabetes Atlas. 9th. Brussels: Belgium: International Diabetes Federation; 2019. [Google Scholar]
- 2.Hsu R. K., Powe N. R. Recent trends in the prevalence of chronic kidney disease: not the same old song. Current Opinion in Nephrology and Hypertension. 2017;26(3):187–196. doi: 10.1097/MNH.0000000000000315. [DOI] [PubMed] [Google Scholar]
- 3.Zhang L., Long J., Jiang W., et al. Trends in chronic kidney disease in China. The New England Journal of Medicine. 2016;375(9):905–906. doi: 10.1056/NEJMc1602469. [DOI] [PubMed] [Google Scholar]
- 4.Romagnani P., Remuzzi G., Glassock R., et al. Chronic kidney disease. Nature Reviews Disease Primers. 2017;3(1, article 17088) doi: 10.1038/nrdp.2017.88. [DOI] [PubMed] [Google Scholar]
- 5.Burton D. G. A., Faragher R. G. A. Obesity and type-2 diabetes as inducers of premature cellular senescence and ageing. Biogerontology. 2018;19(6):447–459. doi: 10.1007/s10522-018-9763-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinez C. J., Sangros G. J., Garcia S. F., et al. Chronic renal disease in Spain: prevalence and related factors in persons with diabetes mellitus older than 64 years. Nefrología. 2018;38:401–413. doi: 10.1016/j.nefro.2017.11.025. [DOI] [PubMed] [Google Scholar]
- 7.Thomas M. C., Cooper M. E., Zimmet P. Changing epidemiology of type 2 diabetes mellitus and associated chronic kidney disease. Nature Reviews. Nephrology. 2016;12(2):73–81. doi: 10.1038/nrneph.2015.173. [DOI] [PubMed] [Google Scholar]
- 8.Anders H. J., Huber T. B., Isermann B., Schiffer M. CKD in diabetes: diabetic kidney disease versus nondiabetic kidney disease. Nature Reviews Nephrology. 2018;14(6):361–377. doi: 10.1038/s41581-018-0001-y. [DOI] [PubMed] [Google Scholar]
- 9.Schmitt R., Melk A. Molecular mechanisms of renal aging. Kidney International. 2017;92(3):569–579. doi: 10.1016/j.kint.2017.02.036. [DOI] [PubMed] [Google Scholar]
- 10.Musso C. G., Oreopoulos D. G. Aging and physiological changes of the kidneys including changes in glomerular filtration rate. Nephron. Physiology. 2011;119(Supplement 1):1–5. doi: 10.1159/000328010. [DOI] [PubMed] [Google Scholar]
- 11.Denic A., Lieske J. C., Chakkera H. A., et al. The substantial loss of nephrons in healthy human kidneys with aging. Journal of the American Society of Nephrology. 2016;28(1):313–320. doi: 10.1681/asn.2016020154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Glassock R. J., Rule A. D. Aging and the kidneys: anatomy, physiology and consequences for defining chronic kidney disease. Nephron. 2016;134(1):25–29. doi: 10.1159/000445450. [DOI] [PubMed] [Google Scholar]
- 13.Duan Z.-Y., Cai G.-Y., Chen Y.-Z., et al. Aging promotes progression of IgA nephropathy: a systematic review and meta-analysis. American Journal of Nephrology. 2013;38(3):241–252. doi: 10.1159/000354646. [DOI] [PubMed] [Google Scholar]
- 14.Liu J., Yang J.-R., Chen X.-M., Cai G.-Y., Lin L.-R., He Y.-N. Impact of ER stress-regulated ATF4/p 16 signaling on the premature senescence of renal tubular epithelial cells in diabetic nephropathy. American Journal of Physiology-Cell Physiology. 2015;308(8):C621–C630. doi: 10.1152/ajpcell.00096.2014. [DOI] [PubMed] [Google Scholar]
- 15.Thomas M. C., Brownlee M., Susztak K., et al. Diabetic kidney disease. Nature Reviews. Disease Primers. 2015;1(1):p. 15018. doi: 10.1038/nrdp.2015.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baldea A. J. Effect of aging on renal function plus monitoring and support. Surgical Clinics of North America. 2015;95(1):71–83. doi: 10.1016/j.suc.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 17.Verzola D., Gandolfo M. T., Gaetani G., et al. Accelerated senescence in the kidneys of patients with type 2 diabetic nephropathy. American Journal of Physiology-Renal Physiology. 2008;295(5):F1563–F1573. doi: 10.1152/ajprenal.90302.2008. [DOI] [PubMed] [Google Scholar]
- 18.Vashistha H., Marrero L., Reiss K., et al. Aging phenotype (s) in kidneys of diabetic mice are p66ShcA dependent. American Journal of Physiology. Renal Physiology. 2018;315(6):F1833–F1842. doi: 10.1152/ajprenal.00608.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanajou D., Ghorbani Haghjo A., Argani H., Aslani S. AGE-RAGE axis blockade in diabetic nephropathy: current status and future directions. European Journal of Pharmacology. 2018;833:158–164. doi: 10.1016/j.ejphar.2018.06.001. [DOI] [PubMed] [Google Scholar]
- 20.Matoba K., Takeda Y., Nagai Y., Kawanami D., Utsunomiya K., Nishimura R. Unraveling the role of inflammation in the pathogenesis of diabetic kidney disease. International Journal of Molecular Sciences. 2019;20(14):p. 3393. doi: 10.3390/ijms20143393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choudhury D., Levi M. Kidney aging--inevitable or preventable? Nature reviews Nephrology. 2011;7(12):706–717. doi: 10.1038/nrneph.2011.104. [DOI] [PubMed] [Google Scholar]
- 22.Nass N., Bartling B., Navarrete Santos A., et al. Advanced glycation end products, diabetes and ageing. Zeitschrift für Gerontologie und Geriatrie. 2007;40(5):349–356. doi: 10.1007/s00391-007-0484-9. [DOI] [PubMed] [Google Scholar]
- 23.Fournet M., Bonté F., Desmoulière A. Glycation damage: a possible hub for major pathophysiological disorders and aging. Aging and Disease. 2018;9(5):880–900. doi: 10.14336/AD.2017.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kazi R. S., Banarjee R. M., Deshmukh A. B., Patil G. V., Jagadeeshaprasad M. G., Kulkarni M. J. Glycation inhibitors extend yeast chronological lifespan by reducing advanced glycation end products and by back regulation of proteins involved in mitochondrial respiration. Journal of Proteomics. 2017;156:104–112. doi: 10.1016/j.jprot.2017.01.015. [DOI] [PubMed] [Google Scholar]
- 25.Meissner C., Ritz-Timme S. Molecular pathology and age estimation. Forensic Science International. 2010;203(1-3):34–43. doi: 10.1016/j.forsciint.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 26.Ohashi S., Abe H., Takahashi T., et al. Advanced glycation end products increase collagen-specific chaperone protein in mouse diabetic nephropathy. The Journal of Biological Chemistry. 2004;279(19):19816–19823. doi: 10.1074/jbc.M310428200. [DOI] [PubMed] [Google Scholar]
- 27.Yamagishi S., Nakamura N., Suematsu M., Kaseda K., Matsui T. Advanced glycation end products: a molecular target for vascular complications in diabetes. Molecular Medicine. 2015;21(Supplement 1):S32–S40. doi: 10.2119/molmed.2015.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Inagi R. RAGE and glyoxalase in kidney disease. Glycoconjugate Journal. 2016;33(4):619–626. doi: 10.1007/s10719-016-9689-8. [DOI] [PubMed] [Google Scholar]
- 29.Zain M., Awan F. R. Renin angiotensin aldosterone system (RAAS): its biology and drug targets for treating diabetic nephropathy. Pakistan Journal of Pharmaceutical Sciences. 2014;27(5):1379–1391. [PubMed] [Google Scholar]
- 30.Jang I.-A., Kim E., Lim J., et al. Effects of resveratrol on the renin-angiotensin system in the aging kidney. Nutrients. 2018;10(11):p. 1741. doi: 10.3390/nu10111741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoon H. E., Choi B. S. The renin-angiotensin system and aging in the kidney. The Korean Journal of Internal Medicine. 2014;29(3):291–295. doi: 10.3904/kjim.2014.29.3.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garagliano J. M., Katsurada A., Miyata K., et al. Advanced glycation end products stimulate angiotensinogen production in renal proximal tubular cells. The American Journal of the Medical Sciences. 2019;357(1):57–66. doi: 10.1016/j.amjms.2018.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sagoo M. K., Gnudi L. Diabetic nephropathy: is there a role for oxidative stress? Free Radical Biology and Medicine. 2018;116:50–63. doi: 10.1016/j.freeradbiomed.2017.12.040. [DOI] [PubMed] [Google Scholar]
- 34.Liochev S. I. Reactive oxygen species and the free radical theory of aging. Free Radical Biology and Medicine. 2013;60:1–4. doi: 10.1016/j.freeradbiomed.2013.02.011. [DOI] [PubMed] [Google Scholar]
- 35.Hu F., Xue M., Li Y., et al. Early growth response 1 (Egr1) is a transcriptional activator of NOX4 in oxidative stress of diabetic kidney disease. Journal of Diabetes Research. 2018;2018:10. doi: 10.1155/2018/3405695.3405695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen X., Fang M. Oxidative stress mediated mitochondrial damage plays roles in pathogenesis of diabetic nephropathy rat. European Review for Medical and Pharmacological Sciences. 2018;22:5248–5254. doi: 10.26355/eurrev_201808_15723. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Y., Feng J., Wang Q., et al. Hyperglycaemia stress-induced renal injury is caused by extensive mitochondrial fragmentation, attenuated MKP1 signalling, and activated JNK-CaMKII-Fis1 biological axis. Cellular Physiology and Biochemistry. 2018;51(4):1778–1798. doi: 10.1159/000495681. [DOI] [PubMed] [Google Scholar]
- 38.Takahashi T., Harris R. C. Role of endothelial nitric oxide synthase in diabetic nephropathy: lessons from diabetic eNOS knockout mice. Journal of Diabetes Research. 2014;2014:17. doi: 10.1155/2014/590541.590541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee H. B. Reactive oxygen species-regulated signaling pathways in diabetic nephropathy. Journal of the American Society of Nephrology. 2003;14(90003):241S–2245. doi: 10.1097/01.ASN.0000077410.66390.0F. [DOI] [PubMed] [Google Scholar]
- 40.Lindblom R., Higgins G., Coughlan M., de Haan J. B. Targeting mitochondria and reactive oxygen species-driven pathogenesis in diabetic nephropathy. The Review of Diabetic Studies. 2015;12(1-2):134–156. doi: 10.1900/RDS.2015.12.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu Q., Wang W. W., Zhang M. Z., et al. ROS induces epithelial-mesenchymal transition via the TGF-β1/PI3K/Akt/mTOR pathway in diabetic nephropathy. Experimental and Therapeutic Medicine. 2019;17(1):835–846. doi: 10.3892/etm.2018.7014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng Y. S., Chao J., Chen C., Lv L. L., Han Y. C., Liu B. C. The PKCβ-p66shc-NADPH oxidase pathway plays a crucial role in diabetic nephropathy. The Journal of Pharmacy and Pharmacology. 2019;71(3):338–347. doi: 10.1111/jphp.13043. [DOI] [PubMed] [Google Scholar]
- 43.Kooman J. P., Kotanko P., Schols A. M. W. J., Shiels P. G., Stenvinkel P. Chronic kidney disease and premature ageing. Nature Reviews Nephrology. 2014;10(12):732–742. doi: 10.1038/nrneph.2014.185. [DOI] [PubMed] [Google Scholar]
- 44.Stenvinkel P., Larsson T. E. Chronic kidney disease: a clinical model of premature aging. American Journal of Kidney Diseases. 2013;62(2):339–351. doi: 10.1053/j.ajkd.2012.11.051. [DOI] [PubMed] [Google Scholar]
- 45.Moreno J. A., Gomez-Guerrero C., Mas S., et al. Targeting inflammation in diabetic nephropathy: a tale of hope. Expert Opinion on Investigational Drugs. 2018;27(11):917–930. doi: 10.1080/13543784.2018.1538352. [DOI] [PubMed] [Google Scholar]
- 46.Rahimi Z. The role of renin angiotensin aldosterone system genes in diabetic nephropathy. Canadian Journal of Diabetes. 2016;40(2):178–183. doi: 10.1016/j.jcjd.2015.08.016. [DOI] [PubMed] [Google Scholar]
- 47.Lee E. A., Seo J. Y., Jiang Z., et al. Reactive oxygen species mediate high glucose-induced plasminogen activator inhibitor-1 up-regulation in mesangial cells and in diabetic kidney. Kidney International. 2005;67(5):1762–1771. doi: 10.1111/j.1523-1755.2005.00274.x. [DOI] [PubMed] [Google Scholar]
- 48.Campbell R. C., Ruggenenti P., Remuzzi G. Proteinuria in diabetic nephropathy: treatment and evolution. Current Diabetes Reports. 2003;3(6):497–504. doi: 10.1007/s11892-003-0014-0. [DOI] [PubMed] [Google Scholar]
- 49.O’Sullivan E. D., Hughes J., Ferenbach D. A. Renal aging: causes and consequences. Journal of the American Society of Nephrology. 2017;28(2):407–420. doi: 10.1681/ASN.2015121308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shiels P. G., McGuinness D., Eriksson M., Kooman J. P., Stenvinkel P. The role of epigenetics in renal ageing. Nature Reviews Nephrology. 2017;13(8):471–482. doi: 10.1038/nrneph.2017.78. [DOI] [PubMed] [Google Scholar]
- 51.Valentijn F. A., Falke L. L., Nguyen T. Q., Goldschmeding R. Cellular senescence in the aging and diseased kidney. Journal of Cell Communication and Signaling. 2018;12(1):69–82. doi: 10.1007/s12079-017-0434-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schmitt R., Susnik N., Melk A. Molecular aspects of renal senescence. Current Opinion in Organ Transplantation. 2015;20(4):412–416. doi: 10.1097/MOT.0000000000000214. [DOI] [PubMed] [Google Scholar]
- 53.Yang H., Fogo A. B. Cell senescence in the aging kidney. Journal of the American Society of Nephrology. 2010;21(9):1436–1439. doi: 10.1681/ASN.2010020205. [DOI] [PubMed] [Google Scholar]
- 54.Docherty M.-H., O’Sullivan E. D., Bonventre J. V., Ferenbach D. A. Cellular senescence in the kidney. Journal of the American Society of Nephrology. 2019;30(5):726–736. doi: 10.1681/ASN.2018121251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Satriano J., Mansoury H., Deng A., et al. Transition of kidney tubule cells to a senescent phenotype in early experimental diabetes. American Journal of Physiology-Cell Physiology. 2010;299(2):C374–C380. doi: 10.1152/ajpcell.00096.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baker D. J., Childs B. G., Durik M., et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature. 2016;530(7589):184–189. doi: 10.1038/nature16932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baker D. J., Wijshake T., Tchkonia T., et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479(7372):232–236. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hudgins A. D., Tazearslan C., Tare A., Zhu Y., Huffman D., Suh Y. Age- and tissue-specific expression of senescence biomarkers in mice. Frontiers in Genetics. 2018;9 doi: 10.3389/fgene.2018.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kitada K., Nakano D., Ohsaki H., et al. Hyperglycemia causes cellular senescence via a SGLT2- and p21-dependent pathway in proximal tubules in the early stage of diabetic nephropathy. Journal of Diabetes and its Complications. 2014;28(5):604–611. doi: 10.1016/j.jdiacomp.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guo Q., Li X., Han H., et al. Histone lysine methylation in TGF-β1 mediated p21 gene expression in rat mesangial cells. BioMed Research International. 2016;2016:9. doi: 10.1155/2016/6927234.6927234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yamada T., Ishihara H., Tamura A., et al. WFS1-deficiency increases endoplasmic reticulum stress, impairs cell cycle progression and triggers the apoptotic pathway specifically in pancreatic beta-cells. Human Molecular Genetics. 2006;15(10):1600–1609. doi: 10.1093/hmg/ddl081. [DOI] [PubMed] [Google Scholar]
- 62.Liu J., Huang K., Cai G.-Y., et al. Receptor for advanced glycation end-products promotes premature senescence of proximal tubular epithelial cells via activation of endoplasmic reticulum stress-dependent p21 signaling. Cellular Signalling. 2014;26(1):110–121. doi: 10.1016/j.cellsig.2013.10.002. [DOI] [PubMed] [Google Scholar]
- 63.Ruster C., Bondeva T., Franke S., Forster M., Wolf G. Advanced glycation end-products induce cell cycle arrest and hypertrophy in podocytes. Nephrology Dialysis Transplantation. 2008;23(7):2179–2191. doi: 10.1093/ndt/gfn085. [DOI] [PubMed] [Google Scholar]
- 64.Wolf G., Schanze A., Stahl R. A. K., Shankland S. J., Amann K. p27Kip1 knockout mice are protected from diabetic nephropathy: evidence for p27Kip1 haplotype insufficiency1. Kidney International. 2005;68(4):1583–1589. doi: 10.1111/j.1523-1755.2005.00570.x. [DOI] [PubMed] [Google Scholar]
- 65.Awazu M. The lack of cyclin kinase inhibitor p27Kip1 ameliorates progression of diabetic nephropathy. Journal of the American Society of Nephrology. 2003;14(3):699–708. doi: 10.1097/01.ASN.0000051726.41601.C0. [DOI] [PubMed] [Google Scholar]
- 66.Lopes-Paciencia S., Saint-Germain E., Rowell M. C., Ruiz A. F., Kalegari P., Ferbeyre G. The senescence-associated secretory phenotype and its regulation. Cytokine. 2019;117:15–22. doi: 10.1016/j.cyto.2019.01.013. [DOI] [PubMed] [Google Scholar]
- 67.Wang W.-J., Cai G.-Y., Chen X.-M. Cellular senescence, senescence-associated secretory phenotype, and chronic kidney disease. Oncotarget. 2017;8, article 64520 doi: 10.18632/oncotarget.17327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Melk A., Ramassar V., Helms L. M., et al. Telomere shortening in kidneys with age. Journal of the American Society of Nephrology. 2000;11:444–453. doi: 10.1681/ASN.V113444. [DOI] [PubMed] [Google Scholar]
- 69.Ameh O. I., Okpechi I. G., Dandara C., Kengne A. P. Association between telomere length, chronic kidney disease, and renal traits: a systematic review. OMICS. 2017;21(3):143–155. doi: 10.1089/omi.2016.0180. [DOI] [PubMed] [Google Scholar]
- 70.D-w C., C-m J., Wan C., et al. Upregulation of MiR-126 delays the senescence of human glomerular mesangial cells induced by high glucose via telomere-p 53-p 21-Rb signaling pathway. Current Medical Science. 2018;38:758–764. doi: 10.1007/s11596-018-1942-x. [DOI] [PubMed] [Google Scholar]
- 71.Chuang P. Y., Cai W., Li X., et al. Reduction in podocyte SIRT1 accelerates kidney injury in aging mice. American Journal of Physiology-Renal Physiology. 2017;313(3):F621–F628. doi: 10.1152/ajprenal.00255.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhou L., Xu D.-y., Sha W.-g., et al. High glucose induces renal tubular epithelial injury via Sirt1/NF-kappaB/microR-29/Keap1 signal pathway. Journal of Translational Medicine. 2015;13(1):p. 352. doi: 10.1186/s12967-015-0710-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kume S., Kitada M., Kanasaki K., Maegawa H., Koya D. Anti-aging molecule, Sirt 1: a novel therapeutic target for diabetic nephropathy. Archives of Pharmacal Research. 2013;36(2):230–236. doi: 10.1007/s12272-013-0019-4. [DOI] [PubMed] [Google Scholar]
- 74.Kume S., Uzu T., Horiike K., et al. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. Journal of Clinical Investigation. 2010;120(4):1043–1055. doi: 10.1172/JCI41376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Waldman M., Cohen K., Yadin D., et al. Regulation of diabetic cardiomyopathy by caloric restriction is mediated by intracellular signaling pathways involving ‘SIRT1 and PGC-1α’. Cardiovascular Diabetology. 2018;17(1):p. 111. doi: 10.1186/s12933-018-0754-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kitada M., Takeda A., Nagai T., Ito H., Kanasaki K., Koya D. Dietary restriction ameliorates diabetic nephropathy through anti-inflammatory effects and regulation of the autophagy via restoration of Sirt1 in diabetic Wistar fatty (fa/fa) rats: a model of type 2 diabetes. Experimental Diabetes Research. 2011;2011:11. doi: 10.1155/2011/908185.908185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huang K., Huang J., Xie X., et al. Sirt1 resists advanced glycation end products-induced expressions of fibronectin and TGF- β 1 by activating the Nrf2/ARE pathway in glomerular mesangial cells. Free Radical Biology and Medicine. 2013;65:528–540. doi: 10.1016/j.freeradbiomed.2013.07.029. [DOI] [PubMed] [Google Scholar]
- 78.Xue M., Li Y., Hu F., et al. High glucose up-regulates microRNA-34a-5p to aggravate fibrosis by targeting SIRT1 in HK-2 cells. Biochemical and Biophysical Research Communications. 2018;498(1):38–44. doi: 10.1016/j.bbrc.2017.12.048. [DOI] [PubMed] [Google Scholar]
- 79.Tang B. L. Sirt 1 and the mitochondria. Molecules and Cells. 2016;39:87–95. doi: 10.14348/molcells.2016.2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Huang K., Gao X., Wei W. The crosstalk between Sirt1 and Keap1/Nrf2/ARE anti-oxidative pathway forms a positive feedback loop to inhibit FN and TGF-β1 expressions in rat glomerular mesangial cells. Experimental Cell Research. 2017;361(1):63–72. doi: 10.1016/j.yexcr.2017.09.042. [DOI] [PubMed] [Google Scholar]
- 81.Zhang L., Chen Z., Gong W., et al. Paeonol ameliorates diabetic renal fibrosis through promoting the activation of the Nrf 2/ARE pathway via up-regulating Sirt 1. Frontiers in Pharmacology. 2018;9:p. 521. doi: 10.3389/fphar.2018.00512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang Q., Deng Q., Zhang J., et al. Activation of the Nrf2-ARE pathway ameliorates hyperglycemia-mediated mitochondrial dysfunction in podocytes partly through Sirt1. Cellular Physiology and Biochemistry. 2018;48(1):1–15. doi: 10.1159/000491658. [DOI] [PubMed] [Google Scholar]
- 83.Sopjani M., Rinnerthaler M., Kruja J., Dermaku-Sopjani M. Intracellular signaling of the aging suppressor protein klotho. Current Molecular Medicine. 2015;15(1):27–37. doi: 10.2174/1566524015666150114111258. [DOI] [PubMed] [Google Scholar]
- 84.Dërmaku-Sopjani M., Kolgeci S., Abazi S., Sopjani M. Significance of the anti-aging protein klotho. Molecular Membrane Biology. 2013;30(8):369–385. doi: 10.3109/09687688.2013.837518. [DOI] [PubMed] [Google Scholar]
- 85.Kim J.-H., Hwang K.-H., Park K.-S., Kong I. D., Cha S.-K. Biological role of anti-aging protein klotho. Journal of Lifestyle Medicine. 2015;5(1):1–6. doi: 10.15280/jlm.2015.5.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xu Y., Sun Z. Molecular basis of klotho: from gene to function in aging. Endocrine Reviews. 2015;36(2):174–193. doi: 10.1210/er.2013-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Osamu Asai K. N. T. T. Decreased renal a-klotho expression in early diabetic nephropathy in humans and mice and its possible role in urinary calcium excretion. International Society of Nephrology. 2012;81:539–547. doi: 10.1038/ki.2011.423. [DOI] [PubMed] [Google Scholar]
- 88.Inci A., Sari F., Coban M., et al. Soluble klotho and fibroblast growth factor 23 levels in diabetic nephropathy with different stages of albuminuria. Journal of Investigative Medicine. 2016;64(6):1128–1133. doi: 10.1136/jim-2016-000142. [DOI] [PubMed] [Google Scholar]
- 89.Kim S. S., Song S. H., Kim I. J., et al. Decreased plasma α-klotho predict progression of nephropathy with type 2 diabetic patients. Journal of Diabetes and its Complications. 2016;30(5):887–892. doi: 10.1016/j.jdiacomp.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 90.Wu C., Wang Q., Lv C., et al. The changes of serum sKlotho and NGAL levels and their correlation in type 2 diabetes mellitus patients with different stages of urinary albumin. Diabetes Research and Clinical Practice. 2014;106(2):343–350. doi: 10.1016/j.diabres.2014.08.026. [DOI] [PubMed] [Google Scholar]
- 91.Huang J.-S., Chuang C.-T., Liu M.-H., Lin S.-H., Guh J.-Y., Chuang L.-Y. Klotho attenuates high glucose-induced fibronectin and cell hypertrophy via the ERK1/2-p38 kinase signaling pathway in renal interstitial fibroblasts. Molecular and Cellular Endocrinology. 2014;390(1-2):45–53. doi: 10.1016/j.mce.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 92.Kadoya H., Satoh M., Haruna Y., Sasaki T., Kashihara N. Klotho attenuates renal hypertrophy and glomerular injury in Ins2Akita diabetic mice. Clinical and Experimental Nephrology. 2016;20(5):671–678. doi: 10.1007/s10157-015-1202-3. [DOI] [PubMed] [Google Scholar]
- 93.Li Y., Hu F., Xue M., et al. Klotho down-regulates Egr-1 by inhibiting TGF-β1/Smad3 signaling in high glucose treated human mesangial cells. Biochemical and Biophysical Research Communications. 2017;487(2):216–222. doi: 10.1016/j.bbrc.2017.04.036. [DOI] [PubMed] [Google Scholar]
- 94.Zhao Y., Banerjee S., Dey N., et al. Klotho depletion contributes to increased inflammation in kidney of the db/db mouse model of diabetes via RelA (serine)536 phosphorylation. Diabetes. 2011;60:1908–1916. doi: 10.2337/db10-1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Inci A., Olmaz R., Sar F., Coban M., Ellidag H. Y., Sar K. M. Increased oxidative stress in diabetic nephropathy and its relationship with soluble klotho levels. Hippokratia. 2016;20:p. 198. [PMC free article] [PubMed] [Google Scholar]
- 96.Liu Y. N., Zhou J., Li T., et al. Sulodexide protects renal tubular epithelial cells from oxidative stress-induced injury via upregulating klotho expression at an early stage of diabetic kidney disease. Journal of Diabetes Research. 2017;2017:10. doi: 10.1155/2017/4989847.4989847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nagasu H., Satoh M., Kuwabara A., et al. Overexpression of klotho protein modulates uninephrectomy-induced compensatory renal hypertrophy by suppressing IGF-I signals. Biochemical and Biophysical Research Communications. 2011;407(1):39–43. doi: 10.1016/j.bbrc.2011.02.089. [DOI] [PubMed] [Google Scholar]
- 98.Mizushima N. Autophagy: process and function. Genes & Development. 2007;21(22):2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 99.Lionaki E., Markaki M., Tavernarakis N. Autophagy and ageing: insights from invertebrate model organisms. Ageing Research Reviews. 2013;12(1):413–428. doi: 10.1016/j.arr.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 100.Rubinsztein D. C., Marino G., Kroemer G. Autophagy and aging. Cell. 2011;146(5):682–695. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 101.Nakamura S., Yoshimori T. Autophagy and longevity. Molecules and Cells. 2018;41(1):65–72. doi: 10.14348/molcells.2018.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Carmona-Gutierrez D., Zimmermann A., Kainz K., et al. The flavonoid 4,4′-dimethoxychalcone promotes autophagy-dependent longevity across species. Nature Communications. 2019;10(1):p. 651. doi: 10.1038/s41467-019-08555-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Naguib M., Rashed L. A. Serum level of the autophagy biomarker Beclin-1 in patients with diabetic kidney disease. Diabetes Research and Clinical Practice. 2018;143:56–61. doi: 10.1016/j.diabres.2018.06.022. [DOI] [PubMed] [Google Scholar]
- 104.Liu W. J., Huang W. F., Ye L., et al. The activity and role of autophagy in the pathogenesis of diabetic nephropathy. European Review for Medical and Pharmacological Sciences. 2018;22(10):3182–3189. doi: 10.26355/eurrev_201805_15079. [DOI] [PubMed] [Google Scholar]
- 105.Liu W. J., Luo M.-N., Tan J., et al. Autophagy activation reduces renal tubular injury induced by urinary proteins. Autophagy. 2013;10:243–256. doi: 10.4161/auto.27004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Liu W. J., Shen T. T., Chen R. H., et al. Autophagy-lysosome pathway in renal tubular epithelial cells is disrupted by advanced glycation end products in diabetic nephropathy. Journal of Biological Chemistry. 2015;290(33):20499–20510. doi: 10.1074/jbc.M115.666354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhang J., Xiang X., Shu S., et al. Advanced oxidation protein products inhibit the autophagy of renal tubular epithelial cells. Experimental and Therapeutic Medicine. 2018;15:3908–3916. doi: 10.3892/etm.2018.5875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ding Y., Kim S., Lee S. Y., Koo J. K., Wang Z., Choi M. E. Autophagy regulates TGF-β expression and suppresses kidney fibrosis induced by unilateral ureteral obstruction. Journal of the American Society of Nephrology. 2014;25(12):2835–2846. doi: 10.1681/ASN.2013101068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liu F., Chen H. Y., Huang X. R., et al. C-reactive protein promotes diabetic kidney disease in a mouse model of type 1 diabetes. Diabetologia. 2011;54(10):2713–2723. doi: 10.1007/s00125-011-2237-y. [DOI] [PubMed] [Google Scholar]
- 110.Ma L., Fu R., Duan Z., et al. Sirt1 is essential for resveratrol enhancement of hypoxia-induced autophagy in the type 2 diabetic nephropathy rat. Pathology, Research and Practice. 2016;212(4):310–318. doi: 10.1016/j.prp.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 111.Wang X., Gao Y., Tian N., et al. Astragaloside IV represses high glucose-induced mesangial cells activation by enhancing autophagy via SIRT1 deacetylation of NF-kappaB p 65 subunit. Drug Design, Development and Therapy. 2018;12:2971–2980. doi: 10.2147/DDDT.S174058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yao F., Zhang M., Chen L. 5'-monophosphate-activated protein kinase (AMPK) improves autophagic activity in diabetes and diabetic complications. Acta Pharmaceutica Sinica B. 2016;6(1):20–25. doi: 10.1016/j.apsb.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Liu Y., Zhang J., Wang Y., Zeng X. Apelin involved in progression of diabetic nephropathy by inhibiting autophagy in podocytes. Cell Death and Disease. 2017;8(8, article e3006) doi: 10.1038/cddis.2017.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Salminen A., Kaarniranta K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Research Reviews. 2012;11(2):230–241. doi: 10.1016/j.arr.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 115.Walters H., Cox L. mTORC inhibitors as broad-spectrum therapeutics for age-related diseases. International Journal of Molecular Sciences. 2018;19, article E2325 doi: 10.3390/ijms19082325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pontrelli P., Oranger A., Barozzino M., et al. Deregulation of autophagy under hyperglycemic conditions is dependent on increased lysine 63 ubiquitination: a candidate mechanism in the progression of diabetic nephropathy. Journal of Molecular Medicine (Berlin, Germany) 2018;96(7):645–659. doi: 10.1007/s00109-018-1656-3. [DOI] [PubMed] [Google Scholar]
- 117.Zhao Y., Zhang W., Jia Q., et al. High dose vitamin E attenuates diabetic nephropathy via alleviation of autophagic stress. Frontiers in Physiology. 2019;9 doi: 10.3389/fphys.2018.01939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tai H., Wang Z., Gong H., et al. Autophagy impairment with lysosomal and mitochondrial dysfunction is an important characteristic of oxidative stress-induced senescence. Autophagy. 2016;13:99–113. doi: 10.1080/15548627.2016.1247143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chen K., Dai H., Yuan J., et al. Optineurin-mediated mitophagy protects renal tubular epithelial cells against accelerated senescence in diabetic nephropathy. Cell Death & Disease. 2018;9(2):p. 105. doi: 10.1038/s41419-017-0127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jiang M., Liu K., Luo J., Dong Z. Autophagy is a renoprotective mechanism during in vitro hypoxia and in vivo ischemia-reperfusion injury. The American Journal of Pathology. 2010;176(3):1181–1192. doi: 10.2353/ajpath.2010.090594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Baisantry A., Bhayana S., Wrede C., et al. The impact of autophagy on the development of senescence in primary tubular epithelial cells. Cell Cycle. 2016;15(21):2973–2979. doi: 10.1080/15384101.2016.1234547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kwon Y., Kim J. W., Jeoung J. A., Kim M. S., Kang C. Autophagy is pro-senescence when seen in close-up, but anti-senescence in long-shot. Molecules and Cells. 2017;40(9):607–612. doi: 10.14348/molcells.2017.0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Grootaert M. O. J., Roth L., Schrijvers D. M., De Meyer G. R. Y., Martinet W. Defective autophagy in atherosclerosis: to die or to senesce? Oxidative Medicine and Cellular Longevity. 2018;2018:12. doi: 10.1155/2018/7687083.7687083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Carmona-Gutierrez D., Hughes A. L., Madeo F., Ruckenstuhl C. The crucial impact of lysosomes in aging and longevity. Ageing Research Reviews. 2016;32:2–12. doi: 10.1016/j.arr.2016.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Leeman D. S., Hebestreit K., Ruetz T., et al. Lysosome activation clears aggregates and enhances quiescent neural stem cell activation during aging. Science. 2018;359(6381):1277–1283. doi: 10.1126/science.aag3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Park J. T., Lee Y.-S., Cho K. A., Park S. C. Adjustment of the lysosomal-mitochondrial axis for control of cellular senescence. Ageing Research Reviews. 2018;47:176–182. doi: 10.1016/j.arr.2018.08.003. [DOI] [PubMed] [Google Scholar]
- 127.Serrano-Puebla A., Boya P. Lysosomal membrane permeabilization in cell death: new evidence and implications for health and disease. Annals of the New York Academy of Sciences. 2016;1371(1):30–44. doi: 10.1111/nyas.12966. [DOI] [PubMed] [Google Scholar]
- 128.Wang F., Gomez-Sintes R., Boya P. Lysosomal membrane permeabilization and cell death. Traffic. 2018;19(12):918–931. doi: 10.1111/tra.12613. [DOI] [PubMed] [Google Scholar]
- 129.Kaburagi Y., Takahashi E., Kajio H., et al. Urinary afamin levels are associated with the progression of diabetic nephropathy. Diabetes Research and Clinical Practice. 2019;147:37–46. doi: 10.1016/j.diabres.2018.02.034. [DOI] [PubMed] [Google Scholar]
- 130.Stoka V., Turk V., Turk B. Lysosomal cathepsins and their regulation in aging and neurodegeneration. Ageing Research Reviews. 2016;32:22–37. doi: 10.1016/j.arr.2016.04.010. [DOI] [PubMed] [Google Scholar]
- 131.Cocchiaro P., De Pasquale V., Della Morte R., et al. The multifaceted role of the lysosomal protease cathepsins in kidney disease. Frontiers in Cell and Developmental Biology. 2017;5:p. 114. doi: 10.3389/fcell.2017.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Du F., Wang T., Li S., et al. Cathepsin D protects renal tubular cells from damage induced by high glucose independent of its enzymatic activity. American Journal of Translational Research. 2017;9(12):5528–5537. [PMC free article] [PubMed] [Google Scholar]
- 133.Zhao X., Chen Y., Tan X., et al. Advanced glycation end-products suppress autophagic flux in podocytes by activating mammalian target of rapamycin and inhibiting nuclear translocation of transcription factor EB. The Journal of Pathology. 2018;245(2):235–248. doi: 10.1002/path.5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Palmieri M., Impey S., Kang H., et al. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Human Molecular Genetics. 2011;20(19):3852–3866. doi: 10.1093/hmg/ddr306. [DOI] [PubMed] [Google Scholar]
- 135.Sancak Y., Bar-Peled L., Zoncu R., Markhard A. L., Nada S., Sabatini D. M. Ragulator-rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141(2):290–303. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sardiello M. Transcription factor EB: from master coordinator of lysosomal pathways to candidate therapeutic target in degenerative storage diseases. Annals of the New York Academy of Sciences. 2016;1371(1):3–14. doi: 10.1111/nyas.13131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Demetriades C., Doumpas N., Teleman A. A. Regulation of TORC1 in response to amino acid starvation via lysosomal recruitment of TSC2. Cell. 2014;156(4):786–799. doi: 10.1016/j.cell.2014.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Antikainen H., Driscoll M., Haspel G., Dobrowolski R. TOR-mediated regulation of metabolism in aging. Aging Cell. 2017;16(6):1219–1233. doi: 10.1111/acel.12689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wei Y., Zhang Y.-J., Cai Y. Growth or longevity: the TOR’s decision on lifespan regulation. Biogerontology. 2013;14(4):353–363. doi: 10.1007/s10522-013-9435-6. [DOI] [PubMed] [Google Scholar]
- 140.Altemtam N., Russell J., El Nahas M. A study of the natural history of diabetic kidney disease (DKD) Nephrology Dialysis Transplantation. 2012;27(5):1847–1854. doi: 10.1093/ndt/gfr561. [DOI] [PubMed] [Google Scholar]
- 141.Lim C. T. S., Nordin N. Z., Fadhlina N. Z., et al. Rapid decline of renal function in patients with type 2 diabetes with heavy proteinuria: a report of three cases. BMC Nephrology. 2019;20(1):p. 22. doi: 10.1186/s12882-019-1203-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhou X. J., Rakheja D., Yu X., Saxena R., Vaziri N. D., Silva F. G. The aging kidney. Kidney International. 2008;74(6):710–720. doi: 10.1038/ki.2008.319. [DOI] [PubMed] [Google Scholar]
- 143.Maqbool M., Cooper M. E., Jandeleit-Dahm K. A. M. Cardiovascular disease and diabetic kidney disease. Seminars in Nephrology. 2018;38(3):217–232. doi: 10.1016/j.semnephrol.2018.02.003. [DOI] [PubMed] [Google Scholar]
- 144.Kakio Y., Uchida H. A., Takeuchi H., et al. Diabetic nephropathy is associated with frailty in patients with chronic hemodialysis. Geriatrics & Gerontology International. 2018;18(12):1597–1602. doi: 10.1111/ggi.13534. [DOI] [PubMed] [Google Scholar]
- 145.Chien K.-L., Chan D.-C., Huang J.-W., Wang J., Chao C.-T. Frailty predicts an increased risk of end-stage renal disease with risk competition by mortality among 165, 461 diabetic kidney disease patients. Aging and Disease. 2019;10 doi: 10.14336/AD.2019.0216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Covic A., Vervloet M., Massy Z. A., et al. Bone and mineral disorders in chronic kidney disease: implications for cardiovascular health and ageing in the general population. The Lancet Diabetes & Endocrinology. 2018;6(4):319–331. doi: 10.1016/S2213-8587(17)30310-8. [DOI] [PubMed] [Google Scholar]
- 147.Kooman J. P., van der Sande F. M., Leunissen K. M. L. Kidney disease and aging: a reciprocal relation. Experimental Gerontology. 2017;87(Part B):156–159. doi: 10.1016/j.exger.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 148.Burns K. D. Angiotensin II and its receptors in the diabetic kidney. American Journal of Kidney Diseases. 2000;36(3):449–467. doi: 10.1053/ajkd.2000.16192. [DOI] [PubMed] [Google Scholar]
- 149.Lin Y.-C., Chang Y.-H., Yang S.-Y., Wu K.-D., Chu T.-S. Update of pathophysiology and management of diabetic kidney disease. Journal of the Formosan Medical Association. 2018;117(8):662–675. doi: 10.1016/j.jfma.2018.02.007. [DOI] [PubMed] [Google Scholar]
- 150.Wang Y., Yang S., Zhou Q., Zhang H., Yi B. Effects of vitamin D supplementation on renal function, inflammation and glycemic control in patients with diabetic nephropathy: a systematic review and meta-analysis. Kidney & Blood Pressure Research. 2019;44(1):72–87. doi: 10.1159/000498838. [DOI] [PubMed] [Google Scholar]
- 151.Han L.-L., Bai X.-J., Lin H.-L., Sun X.-F., Chen X.-M. Association between kidney and cardiac diastolic function in Chinese subjects without overt disease: correlation with ageing and inflammatory markers. European Journal of Clinical Investigation. 2011;41(10):1077–1086. doi: 10.1111/j.1365-2362.2011.02503.x. [DOI] [PubMed] [Google Scholar]
- 152.D’Alessandro C., Piccoli G., Barsotti M., et al. Prevalence and correlates of sarcopenia among elderly CKD outpatients on tertiary care. Nutrients. 2018;10(12, article 1951) doi: 10.3390/nu10121951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bolignano D., Mattace-Raso F., Sijbrands E. J. G., Zoccali C. The aging kidney revisited: a systematic review. Ageing Research Reviews. 2014;14:65–80. doi: 10.1016/j.arr.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 154.McKiernan S. H., Tuen V. C., Baldwin K., Wanagat J., Djamali A., Aiken J. M. Adult-onset calorie restriction delays the accumulation of mitochondrial enzyme abnormalities in aging rat kidney tubular epithelial cells. American Journal of Physiology-Renal Physiology. 2007;292(6):F1751–F1760. doi: 10.1152/ajprenal.00307.2006. [DOI] [PubMed] [Google Scholar]
- 155.Ning Y.-C., Cai G.-Y., Zhuo L., et al. Short-term calorie restriction protects against renal senescence of aged rats by increasing autophagic activity and reducing oxidative damage. Mechanisms of Ageing and Development. 2013;134(11-12):570–579. doi: 10.1016/j.mad.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 156.Dong D., Cai G. Y., Ning Y. C., et al. Alleviation of senescence and epithelial-mesenchymal transition in aging kidney by short-term caloric restriction and caloric restriction mimetics via modulation of AMPK/mTOR signaling. Oncotarget. 2017;8(10):16109–16121. doi: 10.18632/oncotarget.14884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Jung K. J., Lee E. K., Kim J. Y., et al. Effect of short term calorie restriction on pro-inflammatory NF-kB and AP-1 in aged rat kidney. Inflammation Research. 2009;58(3):143–150. doi: 10.1007/s00011-008-7227-2. [DOI] [PubMed] [Google Scholar]
- 158.Lee J. H., Jung K. J., Kim J. W., Kim H. J., Yu B. P., Chung H. Y. Suppression of apoptosis by calorie restriction in aged kidney. Experimental Gerontology. 2004;39(9):1361–1368. doi: 10.1016/j.exger.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 159.Ruggenenti P., Abbate M., Ruggiero B., et al. Renal and systemic effects of calorie restriction in patients with type 2 diabetes with abdominal obesity: a randomized controlled trial. Diabetes. 2016;66(1):75–86. doi: 10.2337/db16-0607. [DOI] [PubMed] [Google Scholar]
- 160.Velazquez L. L., Sil A. M., Goycochea R. M., Torres T. M., Castaneda L. R. Effect of protein restriction diet on renal function and metabolic control in patients with type 2 diabetes: a randomized clinical trial. Nutrición Hospitalaria. 2008;23:141–147. [PubMed] [Google Scholar]
- 161.Dussol B., Iovanna C., Raccah D., et al. A randomized trial of low-protein diet in type 1 and in type 2 diabetes mellitus patients with incipient and overt nephropathy. Journal of Renal Nutrition. 2005;15(4):398–406. doi: 10.1053/j.jrn.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 162.Sattarinezhad A., Roozbeh J., Shirazi Yeganeh B., Omrani G. R., Shams M. Resveratrol reduces albuminuria in diabetic nephropathy: a randomized double- blind placebo-controlled clinical trial. Diabetes & Metabolism. 2019;45(1):53–59. doi: 10.1016/j.diabet.2018.05.010. [DOI] [PubMed] [Google Scholar]
- 163.Hussein M. M. A., Mahfouz M. K. Effect of resveratrol and rosuvastatin on experimental diabetic nephropathy in rats. Biomedicine & Pharmacotherapy. 2016;82:685–692. doi: 10.1016/j.biopha.2016.06.004. [DOI] [PubMed] [Google Scholar]
- 164.Zhang T., Chi Y., Ren Y., Du C., Shi Y., Li Y. Resveratrol reduces oxidative stress and apoptosis in podocytes via Sir2-related enzymes, sirtuins1 (SIRT1)/peroxisome proliferator-activated receptor γ co-activator 1α (PGC-1α) Axis. Medical Science Monitor. 2019;25:1220–1231. doi: 10.12659/MSM.911714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wu L., Zhang Y., Ma X., Zhang N., Qin G. The effect of resveratrol on FoxO1 expression in kidneys of diabetic nephropathy rats. Molecular Biology Reports. 2012;39(9):9085–9093. doi: 10.1007/s11033-012-1780-z. [DOI] [PubMed] [Google Scholar]
- 166.Cheang W. S., Wong W. T., Wang L., et al. Resveratrol ameliorates endothelial dysfunction in diabetic and obese mice through sirtuin 1 and peroxisome proliferator-activated receptor δ. Pharmacological Research. 2019;139:384–394. doi: 10.1016/j.phrs.2018.11.041. [DOI] [PubMed] [Google Scholar]
- 167.Qiao Y., Gao K., Wang Y., Wang X., Cui B. Resveratrol ameliorates diabetic nephropathy in rats through negative regulation of the p38 MAPK/TGF-β1 pathway. Experimental and Therapeutic Medicine. 2017;13(6):3223–3230. doi: 10.3892/etm.2017.4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Abharzanjani F., Afshar M., Hemmati M., Moossavi M. Short-term high dose of quercetin and resveratrol alters aging markers in human kidney cells. International Journal of Preventive Medicine. 2017;8:p. 64. doi: 10.4103/ijpvm.IJPVM_139_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Hsu W. H., Hsiao P.-J., Lin P.-C., Chen S.-C., Lee M.-Y., Shin S.-J. Effect of metformin on kidney function in patients with type 2 diabetes mellitus and moderate chronic kidney disease. Oncotarget. 2018;9(4):5416–5423. doi: 10.18632/oncotarget.23387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Gosmanova E. O., Shahzad S. R., Sumida K., Kovesdy C. P., Gosmanov A. R. Metformin is associated with increase in lactate level in elderly patients with type 2 diabetes and CKD stage 3: a case-control study. Journal of Diabetes and its Complications. 2020;34(1, article 107474) doi: 10.1016/j.jdiacomp.2019.107474. [DOI] [PubMed] [Google Scholar]
- 171.Charytan D. M., Solomon S. D., Ivanovich P., et al. Metformin use and cardiovascular events in patients with type 2 diabetes and chronic kidney disease. Diabetes, Obesity & Metabolism. 2019;21(5):1199–1208. doi: 10.1111/dom.13642. [DOI] [PubMed] [Google Scholar]
- 172.Neven E., Vervaet B., Brand K., et al. Metformin prevents the development of severe chronic kidney disease and its associated mineral and bone disorder. Kidney International. 2018;94(1):102–113. doi: 10.1016/j.kint.2018.01.027. [DOI] [PubMed] [Google Scholar]
- 173.Valencia W. M., Palacio A., Tamariz L., Florez H. Metformin and ageing: improving ageing outcomes beyond glycaemic control. Diabetologia. 2017;60(9):1630–1638. doi: 10.1007/s00125-017-4349-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Barzilai N., Crandall J. P., Kritchevsky S. B., Espeland M. A. Metformin as a tool to target aging. Cell Metabolism. 2016;23(6):1060–1065. doi: 10.1016/j.cmet.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Miles J. M., Rule A. D., Borlaug B. A. Use of metformin in diseases of aging. Current Diabetes Reports. 2014;14(6):p. 490. doi: 10.1007/s11892-014-0490-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Campbell J. M., Bellman S. M., Stephenson M. D., Lisy K. Metformin reduces all-cause mortality and diseases of ageing independent of its effect on diabetes control: a systematic review and meta-analysis. Ageing Research Reviews. 2017;40:31–44. doi: 10.1016/j.arr.2017.08.003. [DOI] [PubMed] [Google Scholar]
- 177.Eisenreich A., Leppert U. Update on the protective renal effects of metformin in diabetic nephropathy. Current Medicinal Chemistry. 2017;24(31):3397–3412. doi: 10.2174/0929867324666170404143102. [DOI] [PubMed] [Google Scholar]
- 178.Langer S., Kreutz R., Eisenreich A. Metformin modulates apoptosis and cell signaling of human podocytes under high glucose conditions. Journal of Nephrology. 2016;29(6):765–773. doi: 10.1007/s40620-015-0258-1. [DOI] [PubMed] [Google Scholar]
- 179.Piwkowska A., Rogacka D., Jankowski M., Angielski S. Metformin reduces NAD(P)H oxidase activity in mouse cultured podocytes through purinergic dependent mechanism by increasing extracellular ATP concentration. Acta Biochimica Polonica. 2013;60(4):607–612. [PubMed] [Google Scholar]
- 180.Zhai L., Gu J., Yang D., Hu W., Wang W., Ye S. Metformin ameliorates podocyte damage by restoring renal tissue nephrin expression in type 2 diabetic rats. Journal of Diabetes. 2017;9(5):510–517. doi: 10.1111/1753-0407.12437. [DOI] [PubMed] [Google Scholar]
- 181.Kim J., Shon E., Kim C. S., Kim J. S. Renal podocyte injury in a rat model of type 2 diabetes is prevented by metformin. Experimental Diabetes Research. 2012;2012:9. doi: 10.1155/2012/210821.210821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Rogacka D., Audzeyenka I., Rychlowski M., et al. Metformin overcomes high glucose-induced insulin resistance of podocytes by pleiotropic effects on SIRT1 and AMPK. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2018;1864(1):115–125. doi: 10.1016/j.bbadis.2017.10.014. [DOI] [PubMed] [Google Scholar]
- 183.Gu J., Ye S., Shan W., Sun W., Hu Y. Metformin inhibits nuclear factor-κB activation and inflammatory cytokines expression induced by high glucose via adenosine monophosphate-activated protein kinase activation in rat glomerular mesangial cells in vitro. National Medical Journal of China. 2014;127:1755–1760. [PubMed] [Google Scholar]
- 184.Kang Z., Zeng J., Zhang T., et al. Hyperglycemia induces NF‐κB activation and MCP-1 expression via downregulating GLP-1R expression in rat mesangial cells: inhibition by metformin. Cell Biology International. 2019;43(8):940–953. doi: 10.1002/cbin.11184. [DOI] [PubMed] [Google Scholar]
- 185.Ren H., Shao Y., Wu C., Ma X., Lv C., Wang Q. Metformin alleviates oxidative stress and enhances autophagy in diabetic kidney disease via AMPK/SIRT1-FoxO1 pathway. Molecular and Cellular Endocrinology. 2020;500, article 110628 doi: 10.1016/j.mce.2019.110628. [DOI] [PubMed] [Google Scholar]
- 186.Xu J., Liu L. Q., Xu L. L., Xing Y., Ye S. Metformin alleviates renal injury in diabetic rats by inducing Sirt1/FoxO1 autophagic signal axis. Clinical and Experimental Pharmacology & Physiology. 2020;47(4):599–608. doi: 10.1111/1440-1681.13226. [DOI] [PubMed] [Google Scholar]
- 187.Allouch S., Munusamy S. Metformin attenuates albumin-induced alterations in renal tubular cells in vitro. Journal of Cellular Physiology. 2017;232(12):3652–3663. doi: 10.1002/jcp.25838. [DOI] [PubMed] [Google Scholar]
- 188.Lee S. Y., Kang J. M., Kim D. J., et al. PGC1α activators mitigate diabetic Tubulopathy by improving mitochondrial dynamics and quality control. Journal Diabetes Research. 2017;2017, article 6483572:15. doi: 10.1155/2017/6483572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Guo Y.-N., Wang J.-C., Cai G.-Y., et al. AMPK-mediated downregulation of connexin43 and premature senescence of mesangial cells under high-glucose conditions. Experimental Gerontology. 2014;51:71–81. doi: 10.1016/j.exger.2013.12.016. [DOI] [PubMed] [Google Scholar]
- 190.Molnar Z., Millward A. B., Tse W., Demaine A. G. p21WAF1/CIP1 expression is differentially regulated by metformin and rapamycin. International Journal of Chronic Diseases. 2014;2014:17. doi: 10.1155/2014/327640.327640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Perkovic V., Jardine M. J., Neal B., et al. Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. The New England Journal of Medicine. 2019;380(24):2295–2306. doi: 10.1056/NEJMoa1811744. [DOI] [PubMed] [Google Scholar]
- 192.Khemais‐Benkhiat S., Belcastro E., Idris‐Khodja N., et al. Angiotensin II-induced redox-sensitive SGLT1 and 2 expression promotes high glucose-induced endothelial cell senescence. Journal of Cellular and Molecular Medicine. 2020;24(3):2109–2122. doi: 10.1111/jcmm.14233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Zhang D., Ma M., Liu Y. Protective effects of incretin against age-related diseases. Current Drug Delivery. 2019;16(9):793–806. doi: 10.2174/1567201816666191010145029. [DOI] [PubMed] [Google Scholar]
- 194.Hasegawa Y., Hayashi K., Takemoto Y., et al. DPP-4 inhibition with linagliptin ameliorates the progression of premature aging in klotho-/- mice. Cardiovascular Diabetology. 2017;16(1):p. 154. doi: 10.1186/s12933-017-0639-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Coppolino G., Leporini C., Rivoli L., et al. Exploring the effects of DPP-4 inhibitors on the kidney from the bench to clinical trials. Pharmacological Research. 2018;129:274–294. doi: 10.1016/j.phrs.2017.12.001. [DOI] [PubMed] [Google Scholar]
- 196.Cheng X. W., Narisawa M., Jin E., Yu C., Xu W., Piao L. Dose rectification of an imbalance between DPP4 and GLP-1 ameliorates chronic stress-related vascular aging and atherosclerosis? Clinical and Experimental Pharmacology & Physiology. 2018;45(5):467–470. doi: 10.1111/1440-1681.12903. [DOI] [PubMed] [Google Scholar]
- 197.Shi J. X., Huang Q. Glucagon‑like peptide‑1 protects mouse podocytes against high glucose‑induced apoptosis, and suppresses reactive oxygen species production and proinflammatory cytokine secretion, through sirtuin 1 activation in vitro. Molecular Medicine Reports. 2018;18(2):1789–1797. doi: 10.3892/mmr.2018.9085. [DOI] [PubMed] [Google Scholar]
- 198.Moellmann J., Klinkhammer B. M., Onstein J., et al. Glucagon-like peptide 1 and its cleavage products are renoprotective in murine diabetic nephropathy. Diabetes. 2018;67(11):2410–2419. doi: 10.2337/db17-1212. [DOI] [PubMed] [Google Scholar]
- 199.Yang J., Zhou Y., Guan Y. PPARγ as a therapeutic target in diabetic nephropathy and other renal diseases. Current Opinion in Nephrology and Hypertension. 2012;21(1):97–105. doi: 10.1097/MNH.0b013e32834de526. [DOI] [PubMed] [Google Scholar]
- 200.Shen D., Li H., Zhou R., Liu M.-j., Yu H., Wu D.-F. Pioglitazone attenuates aging-related disorders in aged apolipoprotein E deficient mice. Experimental Gerontology. 2018;102:101–108. doi: 10.1016/j.exger.2017.12.002. [DOI] [PubMed] [Google Scholar]
- 201.Yang H.-C., Deleuze S., Zuo Y., Potthoff S. A., Ma L.-J., Fogo A. B. The PPARγ agonist pioglitazone ameliorates aging-related progressive renal injury. Journal of the American Society of Nephrology. 2009;20(11):2380–2388. doi: 10.1681/ASN.2008111138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Pourshabanan P., Momeni A., Mahmoudnia L., Kheiri S. Effect of pioglitazone on decreasing of proteinuria in type 2 diabetic patients with nephropathy. Diabetes and Metabolic Syndrome: Clinical Research and Reviews. 2019;13:132–136. doi: 10.1016/j.dsx.2018.04.013. [DOI] [PubMed] [Google Scholar]
- 203.Satirapoj B., Watanakijthavonkul K., Supasyndh O. Safety and efficacy of low dose pioglitazone compared with standard dose pioglitazone in type 2 diabetes with chronic kidney disease: a randomized controlled trial. PLoS One. 2018;13(10, article e0206722) doi: 10.1371/journal.pone.0206722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Bolignano D., Cernaro V., Gembillo G., Baggetta R., Buemi M., D’Arrigo G. Antioxidant agents for delaying diabetic kidney disease progression: a systematic review and meta-analysis. PLoS One. 2017;12(6, article e0178699) doi: 10.1371/journal.pone.0178699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Aghadavod E., Soleimani A., Hamidi G., Keneshlou F., Heidari A., Asemi Z. Effects of high-dose vitamin E supplementation on markers of cardiometabolic risk and oxidative stress in patients with diabetic nephropathy: a randomized double-blinded controlled trial. Iranian Journal of Kidney Diseases. 2018;12:p. 156. [PubMed] [Google Scholar]
- 206.Fang E. F., Lautrup S., Hou Y., et al. NAD+ in aging: molecular mechanisms and translational implications. Trends in Molecular Medicine. 2017;23(10):899–916. doi: 10.1016/j.molmed.2017.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Okabe K., Yaku K., Tobe K., Nakagawa T. Implications of altered NAD metabolism in metabolic disorders. Journal of Biomedical Science. 2019;26(1):p. 34. doi: 10.1186/s12929-019-0527-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Fang E. F., Kassahun H., Croteau D. L., et al. NAD+ replenishment improves lifespan and healthspan in ataxia telangiectasia models via mitophagy and DNA repair. Cell Metabolism. 2016;24(4):566–581. doi: 10.1016/j.cmet.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Nacarelli T., Lau L., Fukumoto T., et al. NAD+ metabolism governs the proinflammatory senescence-associated secretome. Nature Cell Biology. 2019;21(3):397–407. doi: 10.1038/s41556-019-0287-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Childs B. G., Gluscevic M., Baker D. J., et al. Senescent cells: an emerging target for diseases of ageing. Nature reviews Drug discovery. 2017;16(10):718–735. doi: 10.1038/nrd.2017.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.He S., Sharpless N. E. Senescence in health and disease. Cell. 2017;169(6):1000–1011. doi: 10.1016/j.cell.2017.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Short S., Fielder E., Miwa S., von Zglinicki T. Senolytics and senostatics as adjuvant tumour therapy. eBioMedicine. 2019;41:683–692. doi: 10.1016/j.ebiom.2019.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Justice J. N., Nambiar A. M., Tchkonia T., et al. Senolytics in idiopathic pulmonary fibrosis: results from a first-in-human, open-label, pilot study. eBioMedicine. 2019;40:554–563. doi: 10.1016/j.ebiom.2018.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Zhang P., Kishimoto Y., Grammatikakis I., et al. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nature Neuroscience. 2019;22:719–728. doi: 10.1038/s41593-019-0372-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Knoppert S. N., Valentijn F. A., Nguyen T. Q., Goldschmeding R., Falke L. L. Cellular senescence and the kidney: potential therapeutic targets and tools. Frontiers in Pharmacology. 2019;10:p. 770. doi: 10.3389/fphar.2019.00770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Hickson L. T. J., Langhi Prata L. G. P., Bobart S. A., et al. Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of dasatinib plus quercetin in individuals with diabetic kidney disease. eBioMedicine. 2019;47:446–456. doi: 10.1016/j.ebiom.2019.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Blagosklonny M. V. Paradoxes of senolytics. Aging. 2018;10(12):4289–4293. doi: 10.18632/aging.101750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Fu B., Yang J., Chen J., et al. Preventive effect of Shenkang injection against high glucose-induced senescence of renal tubular cells. Frontiers of Medicine. 2018;13:267–272. doi: 10.1007/s11684-017-0586-8. [DOI] [PubMed] [Google Scholar]
- 219.Bielak-Zmijewska A., Grabowska W., Ciolko A., et al. The role of curcumin in the modulation of ageing. International Journal of Molecular Sciences. 2019;20, article E1239 doi: 10.3390/ijms20051239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Li W., He Y., Zhang R., Zheng G., Zhou D. The curcumin analog EF24 is a novel senolytic agent. Aging. 2019;11:771–782. doi: 10.18632/aging.101787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Ulusu N. N., Gök M., Şakul A. A. S., Ari N., Stefek M., Karasu Ç. Antioxidant SMe1EC2 modulates pentose phosphate pathway and glutathione-dependent enzyme activities in tissues of aged diabetic rats. Interdisciplinary Toxicology. 2017;10(4):148–154. doi: 10.1515/intox-2017-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Cao D., Zhao M., Wan C., et al. Role of tea polyphenols in delaying hyperglycemia-induced senescence in human glomerular mesangial cells via miR-126/Akt–p53–p21 pathways. International Urology and Nephrology. 2019;51(6):1071–1078. doi: 10.1007/s11255-019-02165-7. [DOI] [PubMed] [Google Scholar]
- 223.Xu Z., Feng W., Shen Q., et al. Rhizoma coptidis and berberine as a natural drug to combat aging and aging-related diseases via anti-oxidation and AMPK activation. Aging and Disease. 2017;8(6):760–777. doi: 10.14336/AD.2016.0620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.In Hye L., Liu C., Raul M., et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proceedings of the National Academy of Sciences. 2008;105:3374–3379. doi: 10.1073/pnas.0712145105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Fernández Á. F., Sebti S., Wei Y., et al. Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature. 2018;558(7708):136–140. doi: 10.1038/s41586-018-0162-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Lim S. W., Shin Y. J., Luo K., et al. Effect of klotho on autophagy clearance in tacrolimus-induced renal injury. The FASEB Journal. 2018;33:2694–2706. doi: 10.1096/fj.201800751R. [DOI] [PubMed] [Google Scholar]
- 227.Coppé J.-P., Desprez P.-Y., Krtolica A., Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annual Review of Pathology. 2010;5(1):99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Chang M. S., Hsu Y. H. The role of IL-20 in chronic kidney disease and diabetic nephropathy: pathogenic and therapeutic implications. Journal of Leukocyte Biology. 2018;104(5):919–923. doi: 10.1002/JLB.MR1217-489R. [DOI] [PubMed] [Google Scholar]
- 229.Fathy S. A., Mohamed M. R., Ali M. A. M., El-Helaly A. E., Alattar A. T. Influence of IL-6, IL-10, IFN-γ and TNF-α genetic variants on susceptibility to diabetic kidney disease in type 2 diabetes mellitus patients. Biomarkers. 2018;24:43–45. doi: 10.1080/1354750X.2018.1501761. [DOI] [PubMed] [Google Scholar]
- 230.Kolseth I. B. M., Reine T. M., Parker K., et al. Increased levels of inflammatory mediators and proinflammatory monocytes in patients with type I diabetes mellitus and nephropathy. Journal of Diabetes and its Complications. 2017;31(1):245–252. doi: 10.1016/j.jdiacomp.2016.06.029. [DOI] [PubMed] [Google Scholar]
- 231.Ma J., Li Y. J., Chen X., Kwan T., Chadban S. J., Wu H. Interleukin 17A promotes diabetic kidney injury. Scientific Reports. 2019;9(1):p. 2264. doi: 10.1038/s41598-019-38811-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Nawaz S. S., Joy S. S., Al Farsi Y., George T. P., Siddiqui K. Potential role of serum fetuin-a in relation with pro-inflammatory, chemokine and adhesion molecules in diabetic kidney disease: a case–control study. Molecular Biology Reports. 2019;46(1):1239–1246. doi: 10.1007/s11033-019-04592-2. [DOI] [PubMed] [Google Scholar]
- 233.Sierra-Mondragon E., Molina-Jijon E., Namorado-Tonix C., Rodríguez-Muñoz R., Pedraza-Chaverri J., Reyes J. L. All-trans retinoic acid ameliorates inflammatory response mediated by TLR4/NF- κB during initiation of diabetic nephropathy. The Journal of Nutritional Biochemistry. 2018;60:47–60. doi: 10.1016/j.jnutbio.2018.06.002. [DOI] [PubMed] [Google Scholar]
- 234.So B.-I., Song Y.-S., Fang C.-H., et al. G-CSF prevents progression of diabetic nephropathy in rat. PLoS One. 2013;8(10, article e77048) doi: 10.1371/journal.pone.0077048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Sun M., Bu W., Li Y., et al. Danzhi Jiangtang capsule ameliorates kidney injury via inhibition of the JAK-STAT signaling pathway and increased antioxidant capacity in STZ-induced diabetic nephropathy rats. BioScience Trends. 2018;12(6):595–604. doi: 10.5582/bst.2018.01255. [DOI] [PubMed] [Google Scholar]
- 236.Tsai Y.-C., Kuo P.-L., Kuo M.-C., et al. The interaction of miR-378i-Skp2 regulates cell senescence in diabetic nephropathy. Journal of Clinical Medicine. 2018;7(12):p. 468. doi: 10.3390/jcm7120468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Wang Z., Wei M., Wang M., et al. Inhibition of macrophage migration inhibitory factor reduces diabetic nephropathy in type II diabetes mice. Inflammation. 2014;37(6):2020–2029. doi: 10.1007/s10753-014-9934-x. [DOI] [PubMed] [Google Scholar]
- 238.Xu H.-l., Wang X.-t., Cheng Y., et al. Ursolic acid improves diabetic nephropathy _via_ suppression of oxidative stress and inflammation in streptozotocin-induced rats. Biomedicine & Pharmacotherapy. 2018;105:915–921. doi: 10.1016/j.biopha.2018.06.055. [DOI] [PubMed] [Google Scholar]
- 239.Xu K., Guo L., Bu H., Wang H. Daphnetin inhibits high glucose-induced extracellular matrix accumulation, oxidative stress and inflammation in human glomerular mesangial cells. Journal of Pharmacological Sciences. 2019;139(2):91–97. doi: 10.1016/j.jphs.2018.11.013. [DOI] [PubMed] [Google Scholar]
- 240.Valiño-Rivas L., Cuarental L., Grana O., et al. TWEAK increases CD74 expression and sensitizes to DDT proinflammatory actions in tubular cells. PLoS One. 2018;13(6, article e0199391) doi: 10.1371/journal.pone.0199391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Roy M. S., Janal M. N., Crosby J., Donnelly R. Markers of endothelial dysfunction and inflammation predict progression of diabetic nephropathy in African Americans with type 1 diabetes. Kidney International. 2015;87(2):427–433. doi: 10.1038/ki.2014.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Takamiya Y., Fukami K., Yamagishi S.-i., et al. Experimental diabetic nephropathy is accelerated in matrix metalloproteinase-2 knockout mice. Nephrology Dialysis Transplantation. 2013;28(1):55–62. doi: 10.1093/ndt/gfs387. [DOI] [PubMed] [Google Scholar]
- 243.Wu J., Mei C., Vlassara H., Striker G. E., Zheng F. Oxidative stress-induced JNK activation contributes to proinflammatory phenotype of aging diabetic mesangial cells. American Journal of Physiology-Renal Physiology. 2009;297(6):F1622–F1631. doi: 10.1152/ajprenal.00078.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Zha D., Yao T., Bao L., Gao P., Wu X. Telmisartan attenuates diabetic nephropathy progression by inhibiting the dimerization of angiotensin type-1 receptor and adiponectin receptor-1. Life Sciences. 2019;221:109–120. doi: 10.1016/j.lfs.2019.01.044. [DOI] [PubMed] [Google Scholar]
- 245.Yi B., Hu X., Zhang H., et al. Nuclear NF-κB p65 in peripheral blood mononuclear cells correlates with urinary MCP-1, RANTES and the severity of type 2 diabetic nephropathy. PLoS One. 2014;9(6, article e99633) doi: 10.1371/journal.pone.0099633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Lv J., Wang Z., Wang Y., et al. Renoprotective effect of the Shen-Yan-Fang-Shuai formula by inhibiting TNF-α/NF-κB signaling pathway in diabetic rats. Journal of Diabetes Research. 2017;2017:11. doi: 10.1155/2017/4319057.4319057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Watanabe J., Takiyama Y., Honjyo J., et al. Role of IGFBP7 in diabetic nephropathy: TGF-β1 induces IGFBP7 via Smad2/4 in human renal proximal tubular epithelial cells. PLoS One. 2016;11(3, article e0150897) doi: 10.1371/journal.pone.0150897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Zhang Z., Li L., Li Z. K., et al. Effect and mechanism of mulberry leaf polysaccharide on type 1 diabetic nephropathy in rats. Zhonghua Yi Xue Za Zhi. 2018;98(22):1792–1796. doi: 10.3760/cma.j.issn.0376-2491.2018.22.013. [DOI] [PubMed] [Google Scholar]
- 249.El-Saeed A. M., El-Mohasseb G. F. Circulating fibroblast growth factors 21 and 23 as biomarkers of progression in diabetic nephropathy in type 2 diabetes with normoalbuminuria. The Egyptian Journal of Immunology. 2017;24(2):93–99. [PubMed] [Google Scholar]
- 250.Farías-Basulto A., Martínez-Ramírez H. R., Gómez-García E. F., et al. Circulating levels of soluble klotho and fibroblast growth factor 23 in diabetic patients and its association with early nephropathy. Archives of Medical Research. 2018;49(7):451–455. doi: 10.1016/j.arcmed.2019.01.008. [DOI] [PubMed] [Google Scholar]
- 251.Cakirca G., Turgut F. H. Serum matrix metalloproteinase-9, tissue inhibitor of metalloproteinase-1 and matrix metalloproteinase-9/neutrophil gelatinase-associated lipocalin complex levels in patients with early-stage diabetic nephropathy. Iranian Journal of Kidney Diseases. 2018;12(5):299–304. [PubMed] [Google Scholar]
- 252.Garcia-Tejeda A. U., Sampieri C. L., Suarez-Torres I., et al. Association of urinary activity of MMP-9 with renal impairment in Mexican patients with type 2 diabetes mellitus. PeerJ. 2018;6, article e6067 doi: 10.7717/peerj.6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Yu F.-N., Hu M.-L., Wang X.-F., et al. Effects of microRNA-370 on mesangial cell proliferation and extracellular matrix accumulation by binding to canopy 1 in a rat model of diabetic nephropathy. Journal of Cellular Physiology. 2019;234(5):6898–6907. doi: 10.1002/jcp.27448. [DOI] [PubMed] [Google Scholar]
- 254.Yaribeygi H., Mohammadi M. T., Rezaee R., Sahebkar A. Fenofibrate improves renal function by amelioration of NOX-4, IL-18, and p53 expression in an experimental model of diabetic nephropathy. Journal of Cellular Biochemistry. 2018;119:7458–7469. doi: 10.1002/jcb.27055. [DOI] [PubMed] [Google Scholar]
- 255.He T., Xiong J., Nie L., et al. Resveratrol inhibits renal interstitial fibrosis in diabetic nephropathy by regulating AMPK/NOX4/ROS pathway. Journal of Molecular Medicine. 2016;94(12):1359–1371. doi: 10.1007/s00109-016-1451-y. [DOI] [PubMed] [Google Scholar]
- 256.Xu F., Wang Y., Cui W., et al. Resveratrol prevention of diabetic nephropathy is associated with the suppression of renal inflammation and mesangial cell proliferation: possible roles of Akt/NF-kappaB pathway. International Journal of Endocrinology. 2014;2014:9. doi: 10.1155/2014/289327.289327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Xu Y., Nie L., Yin Y.-G., et al. Resveratrol protects against hyperglycemia-induced oxidative damage to mitochondria by activating SIRT1 in rat mesangial cells. Toxicology and Applied Pharmacology. 2012;259(3):395–401. doi: 10.1016/j.taap.2011.09.028. [DOI] [PubMed] [Google Scholar]
- 258.Moridi H., Karimi J., Sheikh N., et al. Resveratrol-dependent down-regulation of receptor for advanced glycation end-products and oxidative stress in kidney of rats with diabetes. International Journal of Endocrinology and Metabolism. 2015;13, article e23542 doi: 10.5812/ijem.23542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Park H. S., Lim J. H., Kim M. Y., et al. Resveratrol increases AdipoR1 and AdipoR2 expression in type 2 diabetic nephropathy. Journal of Translational Medicine. 2016;14(1):p. 176. doi: 10.1186/s12967-016-0922-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Kim M. Y., Lim J. H., Youn H. H., et al. Resveratrol prevents renal lipotoxicity and inhibits mesangial cell glucotoxicity in a manner dependent on the AMPK–SIRT1–PGC1α axis in db/db mice. Diabetologia. 2013;56(1):204–217. doi: 10.1007/s00125-012-2747-2. [DOI] [PubMed] [Google Scholar]
- 261.Xu X. H., Ding D. F., Yong H. J., et al. Resveratrol transcriptionally regulates miRNA-18a-5p expression ameliorating diabetic nephropathy via increasing autophagy. European Review for Medical and Pharmacological Sciences. 2017;21:4952–4965. [PubMed] [Google Scholar]
- 262.Huang S. S., Ding D. F., Chen S., et al. Resveratrol protects podocytes against apoptosis via stimulation of autophagy in a mouse model of diabetic nephropathy. Scientific Reports. 2017;7(1):p. 45692. doi: 10.1038/srep45692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Yuan D., Liu X. M., Fang Z., Du L. L., Chang J., Lin S. H. Protective effect of resveratrol on kidney in rats with diabetic nephropathy and its effect on endoplasmic reticulum stress. European Review for Medical and Pharmacological Sciences. 2018;22(5):1485–1493. doi: 10.26355/eurrev_201803_14497. [DOI] [PubMed] [Google Scholar]
- 264.Zhang S., Xu H., Yu X., Wu Y., Sui D. Metformin ameliorates diabetic nephropathy in a rat model of low-dose streptozotocin-induced diabetes. Experimental and Therapeutic Medicine. 2017;14(1):383–390. doi: 10.3892/etm.2017.4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Yao X. M., Ye S. D., Xiao C. C., Gu J. F., Yang D., Wang S. Metformin alleviates high glucose-mediated oxidative stress in rat glomerular mesangial cells by modulation of p38 mitogen-activated protein kinase expression in vitro. Molecular Medicine Reports. 2015;12:520–526. doi: 10.3892/mmr.2015.3446. [DOI] [PubMed] [Google Scholar]
- 266.Ishibashi Y., Matsui T., Takeuchi M., Yamagishi S. Metformin inhibits advanced glycation end products (AGEs)-induced renal tubular cell injury by suppressing reactive oxygen species generation via reducing receptor for AGEs (RAGE) expression. Hormone and Metabolic Research. 2012;44(12):891–895. doi: 10.1055/s-0032-1321878. [DOI] [PubMed] [Google Scholar]
- 267.Christensen M., Schiffer T. A., Gustafsson H., Krag S. P., Nørregaard R., Palm F. Metformin attenuates renal medullary hypoxia in diabetic nephropathy through inhibition uncoupling protein-2. Diabetes/Metabolism Research and Reviews. 2019;35(2, article e3091) doi: 10.1002/dmrr.3091. [DOI] [PubMed] [Google Scholar]
- 268.Guan M., Li W., Xu L., et al. Metformin improves epithelial-to-mesenchymal transition induced by TGF-β1 in renal tubular epithelial NRK-52E cells via inhibiting Egr-1. Journal Diabetes Research. 2018;2018, article 1031367:8. doi: 10.1155/2018/1031367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Liang D., Song Z., Liang W., Li Y., Liu S. Metformin inhibits TGF-beta 1-induced MCP-1 expression through BAMBI-mediated suppression of MEK/ERK1/2 signalling. Nephrology (Carlton) 2019;24(4):481–488. doi: 10.1111/nep.13430. [DOI] [PubMed] [Google Scholar]
- 270.Jia Y., Zheng Z., Guan M., et al. Exendin-4 ameliorates high glucose-induced fibrosis by inhibiting the secretion of miR-192 from injured renal tubular epithelial cells. Experimental & Molecular Medicine. 2018;50(5):1–13. doi: 10.1038/s12276-018-0084-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Xu W.-W., Guan M.-P., Zheng Z.-J., et al. Exendin-4 alleviates high glucose-induced rat mesangial cell dysfunction through the AMPK pathway. Cellular Physiology and Biochemistry. 2014;33(2):423–432. doi: 10.1159/000358623. [DOI] [PubMed] [Google Scholar]
- 272.Zitman-Gal T., Einbinder Y., Ohana M., Katzav A., Kartawy A., Benchetrit S. Effect of liraglutide on the Janus kinase/signal transducer and transcription activator (JAK/STAT) pathway in diabetic kidney disease in db/db mice and in cultured endothelial cells. Journal of Diabetes. 2019;11(8):656–664. doi: 10.1111/1753-0407.12891. [DOI] [PubMed] [Google Scholar]
- 273.Chen P., Shi X., Xu X., et al. Liraglutide ameliorates early renal injury by the activation of renal FoxO1 in a type 2 diabetic kidney disease rat model. Diabetes Research and Clinical Practice. 2018;137:173–182. doi: 10.1016/j.diabres.2017.09.006. [DOI] [PubMed] [Google Scholar]
- 274.Zhao X., Liu G., Shen H., et al. Liraglutide inhibits autophagy and apoptosis induced by high glucose through GLP-1R in renal tubular epithelial cells. International Journal of Molecular Medicine. 2015;35(3):684–692. doi: 10.3892/ijmm.2014.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Hendarto H., Inoguchi T., Maeda Y., et al. GLP-1 analog liraglutide protects against oxidative stress and albuminuria in streptozotocin-induced diabetic rats via protein kinase A-mediated inhibition of renal NAD(P)H oxidases. Metabolism. 2012;61(10):1422–1434. doi: 10.1016/j.metabol.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 276.Marques C., Goncalves A., Pereira P. M. R., et al. The dipeptidyl peptidase 4 inhibitor sitagliptin improves oxidative stress and ameliorates glomerular lesions in a rat model of type 1 diabetes. Life Sciences. 2019;234:p. 116738. doi: 10.1016/j.lfs.2019.116738. [DOI] [PubMed] [Google Scholar]
- 277.Hodrea J., Balogh D. B., Hosszu A., et al. ReducedO-GlcNAcylation and tubular hypoxia contribute to the antifibrotic effect of SGLT2 inhibitor dapagliflozin in the diabetic kidney. American Journal of Physiology. Renal Physiology. 2020;318(4):F1017–F1029. doi: 10.1152/ajprenal.00021.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Yao D., Wang S., Wang M., Lu W. Renoprotection of dapagliflozin in human renal proximal tubular cells via the inhibition of the high mobility group box 1receptor for advanced glycation end products nuclear factor kappaB signaling pathway. Molecular Medicine Reports. 2018;18:3625–3630. doi: 10.3892/mmr.2018.9393. [DOI] [PubMed] [Google Scholar]
- 279.Heerspink H. J. L., Perco P., Mulder S., et al. Canagliflozin reduces inflammation and fibrosis biomarkers: a potential mechanism of action for beneficial effects of SGLT2 inhibitors in diabetic kidney disease. Diabetologia. 2019;62(7):1154–1166. doi: 10.1007/s00125-019-4859-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Tanaka S., Sugiura Y., Saito H., et al. Sodium-glucose cotransporter 2 inhibition normalizes glucose metabolism and suppresses oxidative stress in the kidneys of diabetic mice. Kidney International. 2018;94(5):912–925. doi: 10.1016/j.kint.2018.04.025. [DOI] [PubMed] [Google Scholar]
- 281.Ojima A., Matsui T., Nishino Y., Nakamura N., Yamagishi S. Empagliflozin, an inhibitor of sodium-glucose cotransporter 2 exerts anti-inflammatory and antifibrotic effects on experimental diabetic nephropathy partly by suppressing AGEs-receptor axis. Hormone and Metabolic Research. 2015;47:686–692. doi: 10.1055/s-0034-1395609. [DOI] [PubMed] [Google Scholar]
- 282.Xu X., Chen P., Zheng Q., Wang Y., Chen W. Effect of pioglitazone on diabetic nephropathy and expression of HIF-1α and VEGF in the renal tissues of type 2 diabetic rats. Diabetes Research and Clinical Practice. 2011;93(1):63–69. doi: 10.1016/j.diabres.2011.03.019. [DOI] [PubMed] [Google Scholar]