

Candida albicans is an important fungal pathogen causing common superficial infections as well as invasive diseases with an extremely high morbidity and mortality. Antifungal therapies are limited in efficiency and availability. In this research, we describe the regulation of riboflavin production in C. albicans. Since riboflavin biosynthesis is essential to this organism, we can appreciate that targeting it would be a promising new strategy to combat these fungal infections. We provide evidence that one particular enzyme in the production process, CaRib1, would be most promising as an antifungal drug target, as it plays a central role in regulation and proves to be essential in a mouse model of systemic infection.
KEYWORDS: Candida albicans, antifungal agents, drug targets, riboflavin, vitamin B2
ABSTRACT
Candida albicans is a major cause of fungal infections, both superficial and invasive. The economic costs as well as consequences for patient welfare are substantial. Only a few treatment options are available due to the high resemblance between fungal targets and host molecules, as both are eukaryotes. Riboflavin is a yellow pigment, also termed vitamin B2. Unlike animals, fungi can synthesize this essential component themselves, thereby leading us to appreciate that targeting riboflavin production is a promising novel strategy against fungal infections. Here, we report that the GTP cyclohydrolase encoded by C. albicans RIB1 (CaRIB1) is essential and rate-limiting for production of riboflavin in the fungal pathogen. We confirm the high potential of CaRib1 as an antifungal drug target, as its deletion completely impairs in vivo infectibility by C. albicans in model systems. Furthermore, the stimulating effect of iron deprivation and PKA activation on riboflavin production seems to involve CaRib1 and the upstream transcription factor CaSef1. Gathering insights in the synthesis mechanism of riboflavin in pathogenic fungi, like C. albicans, will allow us to design a novel strategy and specifically target this process to combat fungal infections.
IMPORTANCE Candida albicans is an important fungal pathogen causing common superficial infections as well as invasive diseases with an extremely high morbidity and mortality. Antifungal therapies are limited in efficiency and availability. In this research, we describe the regulation of riboflavin production in C. albicans. Since riboflavin biosynthesis is essential to this organism, we can appreciate that targeting it would be a promising new strategy to combat these fungal infections. We provide evidence that one particular enzyme in the production process, CaRib1, would be most promising as an antifungal drug target, as it plays a central role in regulation and proves to be essential in a mouse model of systemic infection.
INTRODUCTION
Candida albicans is an opportunistic commensal, becoming pathogenic when the host immune system is compromised. About 70% of women worldwide suffer from vaginal candidiasis at least once in their life. Five to 10% suffer from recurrent vaginal infections, greatly diminishing patient welfare (1, 2). In cases of severe immune deprivation, such as after organ transplantation, the fungus can penetrate host tissue to reach the bloodstream and, from there, reach vital organs. Systemic infections cause death in 40 to 60% of affected patients (3). The limited availability of antifungal therapies and the increased onset of resistance against used drugs hamper efficient eradication of infections and treatment of patients. Despite many attempts made by the scientific community, very few antifungal drugs with a novel mode of action have reached the clinical phase of drug testing over recent years, indicating a need for alternative drug targets (4). A main hurdle when exploring metabolic pathways as potential drug targets is the high level of conservation between the fungal actors and the human ones, as both are eukaryotes. Drugs targeting conserved processes generally exhibit significant side effects (5).
A process that is gaining interest as a potential target for novel antifungal drugs is riboflavin synthesis (6, 7). Riboflavin is considered a vitamin, as it is essential for human and animal growth, yet it has to be supplemented in the food and feed as no de novo synthesis occurs in these organisms. Riboflavin, or vitamin B2, serves as a precursor for flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD). Both coenzymes are necessary to form flavoproteins, which are involved in redox metabolism (8). The connection between FMN/FAD and iron-sulfur clusters, occurring in the electron transport chain, where reduction of Fe-S is accompanied by oxidation of FMN or FAD, is noteworthy (9). Commercial production of riboflavin is performed by microbial species, such as Bacillus subtilis and Candida famata (10, 11). Vitamin metabolism, and riboflavin production more specifically, has been investigated in terms of antimicrobial drug development. For organisms that cannot synthesize riboflavin themselves, such as Lactobacilli species, Enterococcus faecalis, and Listeria monocytogenes, riboflavin transport is indispensable (12). Other bacteria, such as Escherichia coli, Salmonella, and Mycobacterium, strictly depend on the endogenous production of riboflavin, as efficient transporter proteins are not available (13, 14). In these organisms, riboflavin synthase and lumazine synthase have been investigated thoroughly as attractive targets for antibacterial drug development. By slightly adapting the enzyme’s natural substrates, one can yield analogues, termed antimetabolites, which have an inhibitory effect on the enzyme’s activity (15). In this way, trifluoromethylated pyrazoles were found to inhibit the riboflavin synthase of Mycobacterium tuberculosis (16). Similarly, chemicals were found to inhibit the riboflavin synthase or lumazine synthase of Bacillus subtilis, E. coli, Brucella abortus, and others (17–19). Using a similar strategy, the laboratory of Markus Fisher has solved the crystal structure of two enzymes involved in riboflavin synthesis in C. albicans, more specifically Rib3 and Rib4 (20, 21). A number of inhibitors were modeled against the crystal structure of the enzymes, yet no in vivo evidence of inhibition was presented. Several enzymes encoded by RIB genes are involved in converting GTP and ribulose-5-phosphate into riboflavin and further into FMN and FAD. Many C. albicans genes are still uncharacterized and were assigned their name based on sequence homology with genes from the model organism Saccharomyces cerevisiae (22). The biosynthesis pathway of riboflavin, as established for S. cerevisiae, is depicted in Fig. S1 in the supplemental material (23, 24). C. albicans RIB1 (CaRIB1), CaRIB2, CaRIB4, and CaRIB7 are involved in conversion of GTP to 6,7-dimethyl-8-ribityllumazine. CaRIB3 is involved in conversion of ribulose-5-phosphate to 3,4-dihydroxy-2-butanone 4-phosphate. CaRIB5 is the last enzyme involved in the production of riboflavin. CaFMN1 converts riboflavin into FMN.
Schematic representation of the riboflavin biosynthesis pathway in S. cerevisiae. Based on homology with S. cerevisiae, C. albicans enzymes are depicted alongside the reactions, the enzymes for which the open reading frames are uncharacterized are shown in orange, and the others are in blue. More details are given in the text. This figure uses data from references 23 and 24. Download FIG S1, EPS file, 2.7 MB (2.8MB, eps) .
Copyright © 2020 Demuyser et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
In this report, we identify CaRib1 as a GTP cyclohydrolase by heterologous expression in S. cerevisiae and show that it catalyzes an essential and rate-limiting step in riboflavin synthesis. Deleting the gene completely impairs cytotoxicity against mammalian cells as well as infectibility in a systemic mouse model. We confirm that a low cellular iron content as well as high activity of protein kinase A (PKA) positively influence production of riboflavin (25) and show that expression of CaRIB1 is elevated under both conditions. CaSef1, a transcription factor, regulates riboflavin synthesis in an iron- and PKA-dependent manner and is essential in a systemic mouse model of infection.
RESULTS
Riboflavin production can be measured through fluorescence emission of spent medium.
Riboflavin secreted to the culture medium was measured spectrophotometrically by analyzing fluorescence emission at 530 nm of the cell-free supernatant, with excitation at 450 nm. Figure S2 in the supplemental material shows the excitation and emission spectra of SC5314 culture supernatant (Fig. S2b) compared to pure riboflavin dissolved in the same medium (Fig. S2a). The standard curve depicted in Fig. S2c was used to quantify the riboflavin content as measured spectrophotometrically. All experiments in the manuscript were performed in LoFloglu medium mimicking complete synthetic medium with glucose but lacking riboflavin, unless stated otherwise.
Fluorescence characteristics of riboflavin and culture supernatant. (a) Excitation and emission spectra of riboflavin, dissolved in LoFloglu medium, were determined. (b) Likewise, excitation and emission spectra of sterilized 24-h-old SC5314 culture supernatants were evaluated. (c) A standard curve of the fluorescence emission of riboflavin was set up. RFU, relative fluorescence units. Download FIG S2, EPS file, 1.5 MB (1.5MB, eps) .
Copyright © 2020 Demuyser et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
CaRib1 is a central player in riboflavin synthesis.
To determine the rate-limiting enzymes in riboflavin biosynthesis, we generated strains overexpressing all eight genes putatively involved in the pathway. The laboratory reference SC5314 was used as the background strain. To objectively compare the effect of the increased dosage of the different genes, equal copy numbers of plasmid integration were verified. We used a setup based on quantitative PCR (qPCR) analysis, similar to that published by our laboratory earlier (26). Riboflavin production was monitored by fluorescence emission of the cell-free supernatant of cultures grown for 24 h. As shown in Fig. 1a, only overexpression of CaRIB1 leads to an increase in the produced and secreted levels of riboflavin, indicating that Rib1 catalyzes the rate-limiting step of the production process. CaRIB1 is an ScRIB1 ortholog that is uncharacterized in C. albicans. It encodes a putative GTP cyclohydrolase II enzyme that converts GTP into 2,5-diamino-6-ribosylamino-4(H)-pyrimidinedione 5′-phosphate as one of the first steps in the riboflavin synthesis pathway (27). Overexpression of CaFMN1 decreases riboflavin production, since this gene encodes a putative riboflavin kinase, converting riboflavin to the nonfluorescent flavin mononucleotide.
FIG 1.

CaRib1 is a central regulator of riboflavin biosynthesis in C. albicans. (a) Strains overexpressing the genes encoding riboflavin synthesis enzymes were grown for 24 h, and fluorescence at 530 nm of the sterile supernatant was measured upon excitation at 450 nm. The relative ratio of fluorescence to OD600 is shown as average and SEM. Upon overexpression of CaRIB1, production of riboflavin increases. (b) Growth of the Carib1Δ/Δ strain as well as the reintegrant and the wild-type SC5314 strain was monitored over time. Deletion of CaRIB1 inhibits growth in the absence of external riboflavin. Reintegration of the gene reverts this phenotype. (c) Growth of the Carib1Δ/Δ strain was assessed after 24 h in the absence and presence of external riboflavin. Data are shown as averages and SEM. Adding riboflavin partially restores the growth of this deletion strain. (d) Growth of the S. cerevisiae rib1Δ/Δ strain as well as the CaRIB1 reintegrant and the wild-type S288c strain was monitored over time. CaRIB1 is able to fully complement the S. cerevisiae orthologue in terms of growth in the absence of external riboflavin. Statistical analyses shown in panels a and c were performed by one-way analysis of variance (ANOVA), with Bonferroni correction. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Deletion of both CaRIB1 alleles in the SC5314 background strain completely inhibits growth in the absence of externally added riboflavin, indicating that it is an essential gene under conditions of low available riboflavin. Reintegration of the gene placed behind the constitutive CaACT1 promoter reverts this growth deficit, as shown in Fig. 1b. Upon addition of increasing concentrations of riboflavin, growth of the Carib1Δ/Δ deletion strain is restored partially (Fig. 1c), indicating that a mechanism for riboflavin transport through the cell membrane must exist. Finally, we show that heterologous expression of CaRIB1 can functionally complement the S. cerevisiae deletion strain, Scrib1Δ/Δ, as can be seen in Fig. 1d. This reinforces the role of CaRib1 as a GTP cyclohydrolase II involved in riboflavin synthesis.
CaRib1 is essential for virulence in relevant model systems.
CaRIB1 is essential under conditions where low levels of external riboflavin are present. The concentration of this vitamin in human serum ranges between 0.04 and 0.24 mg/liter, which is significantly lower than the concentration needed for in vitro growth restoration of the deletion strain (28). To determine whether Rib1 is also essential in a context resembling the in vivo situation, we evaluated the effect of its deletion in the laboratory strain on toxicity toward mammalian HeLa cells. Toxicity is measured by spectrophotometric analysis of lactate dehydrogenase (LDH) release in the cell supernatant. Figure 2a shows that the Carib1Δ/Δ deletion strain imposes no toxicity toward the HeLa cells, while reintegration of the gene partially restores this phenotype. We confirm that the reduced toxicity is not caused by a reduction of microbial cell adhesion to the mammalian cells (Fig. 2b).
FIG 2.
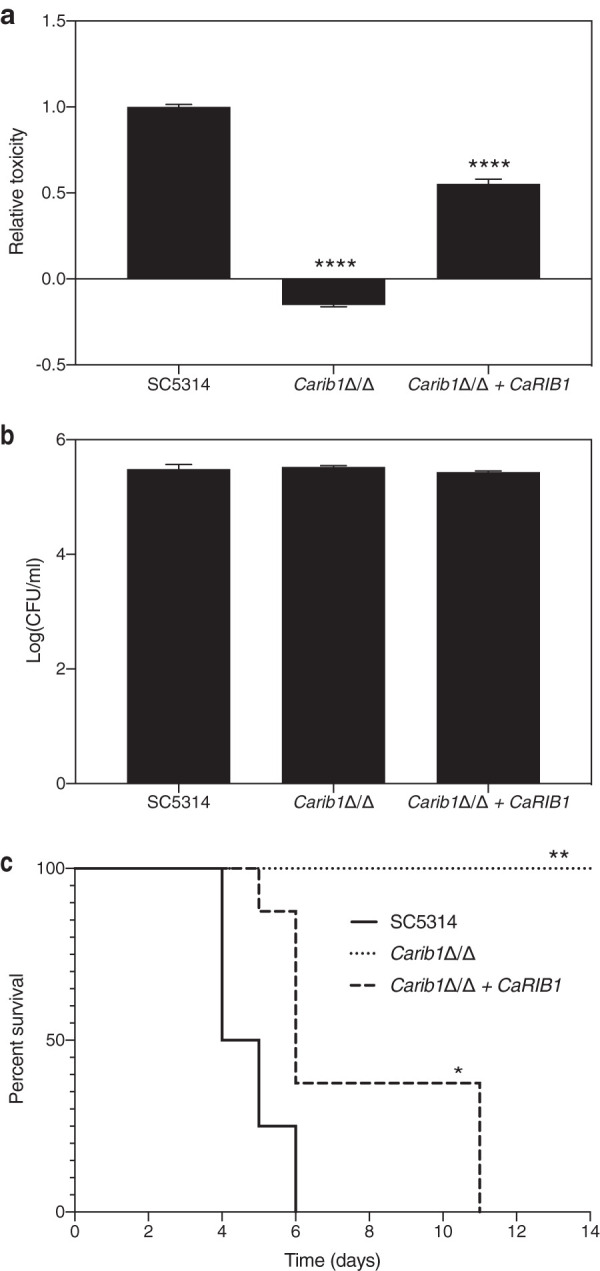
CaRib1 is essential for full virulence in relevant model systems. (a) The Carib1Δ/Δ deletion strain as well as the wild-type and the reintegrant strains were added to mammalian HeLa cells. After 24 h of incubation at 37°C and 5% CO2, cytotoxicity was determined based on LDH release in the culture supernatant. Deletion of CaRIB1 causes the complete absence of cytotoxicity toward mammalian HeLa cells (determined by LDH activity and shown as average and SEM). (b) The Carib1Δ/Δ deletion, wild-type, and reintegrant strains were added to mammalian HeLa cells. After 90 min of incubation at 37°C and 5% CO2, adhesion was determined based on number of CFU. Average values and SEM are shown. There is no significant difference between the deletion strain and the wild type. (c) Mice were infected with the same strains through tail vein injection. The C. albicans strain without CaRib1 is completely avirulent in a mouse model of systemic infection. Statistical analyses for panels a and b were performed by one-way ANOVA, with Bonferroni correction. ****, P < 0.0001. Statistical analysis for panel c was performed by a log rank Mantel-Cox test. *, P < 0.05; **, P < 0.01.
The inability of the Carib1Δ/Δ deletion strain to grow in the absence of adequate amounts of riboflavin is confirmed in a mouse model of systemic infection. Mice were infected with 8.5 × 105 cells per mouse by intravenous injection and were inspected daily for morbidity and mortality. Upon signs of severe morbidity, such as over 15% weight loss, the mice were sacrificed. As can be seen in Fig. 2c, the strain lacking both CaRIB1 alleles is completely avirulent in this mouse model, while the wild-type and reintegrant strains start causing mortality after 4 or 5 days, respectively. These results indicate that CaRib1 is a promising novel antifungal drug target.
The iron content of the growth medium modulates riboflavin production.
A connection between flavin metabolism and iron was established earlier, as FAD and FMN are involved in the redox state of Fe-S clusters (9). To determine the effect of iron on the production or secretion of riboflavin to the medium, we cultured the SC5314 laboratory strain in the presence of different concentrations of FeCl3 or Fe2(SO4)3 and iron chelator bathophenanthrolinedisulfonate (BPS) or ferrozine. As can be seen in Fig. 3a and Fig. S3a, the addition of iron diminishes the riboflavin content in the culture supernatant. Addition of increasing concentrations of ferrozine or BPS has the opposite effect, as shown visually (Fig. S4) and quantitatively (Fig. 3b and Fig. S3b). Thus, we can conclude that iron levels negatively correlate with production of riboflavin in C. albicans.
FIG 3.

Riboflavin production alters upon iron addition or sequestration and PKA activation. SC5314 cultures were grown for 24 h in the presence of FeCl3 (a), an iron chelator, ferrozine (b), or dbcAMP (c). (d) CaTPK1 and CaTPK2 overexpression strains were grown for 24 h, as well as the empty vector (EV) control strain. The fluorescence was measured at 530 nm when excited upon 450 nm. The average and the SEM of relative fluorescence to OD600 ratios are shown. Statistical analysis was performed using a one-way analysis of variance (ANOVA) statistical test with Bonferroni correction (comparison to culture without added compound). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Riboflavin production alters upon iron addition or sequestration. SC5314 cultures were grown for 24 h in the presence of Fe2(SO4)3 (a) or an iron chelator, BPS (b). The fluorescence was measured at 530 nm when excited upon 450 nm. The average and the SEM of relative fluorescence to OD600 ratios are shown. Statistical analysis was performed using a one-way analysis of variance (ANOVA) statistical test with Bonferroni correction (comparison to culture without added compound). *, P < 0.05; **, P < 0.01. Download FIG S3, EPS file, 1.3 MB (1.6MB, eps) .
Copyright © 2020 Demuyser et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Empirical observation of riboflavin production in the presence of iron chelators. SC5314 cultures were grown for 24 h in the presence of 0, 0.2, 1, and 5 mM ferrozine (a) or 0, 0.02, 0.05, and 0.1 mg/ml BPS (c). Blank medium with chelator was incubated to compare ferrozine (b) and BPS (d). Images were taken of the cultures (upper), and cultures were spun down and imaged using blue light at 460 nm (lower). Download FIG S4, TIF file, 2.6 MB (2.6MB, tif) .
Copyright © 2020 Demuyser et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Riboflavin production and autofluorescence increase upon PKA stimulation.
Activity of the cyclic AMP (cAMP)-PKA pathway has been linked to riboflavin production in certain fungi (29, 30). We confirm that increasing signaling through this pathway augments the amount of riboflavin found in the culture supernatant of the C. albicans wild-type strain. Figure 3c shows the effect of adding increasing concentrations of dibutyryl cAMP (dbcAMP) to the growth medium of the SC5314 wild-type strain. This compound is an analogue of cAMP and activates PKA. We also generated mutant strains overexpressing the genes encoding either of the catalytic PKA subunits of C. albicans, CaTpk1 and CaTpk2. We verified equal amounts of copy number insertions of the overexpression plasmids. Both CaTPK1 and CaTPK2 overexpression strains showed increased amounts of riboflavin in the culture supernatant after 24 h of growth (Fig. 3d). Both results indicate that PKA activity positively affects riboflavin production.
Riboflavin is a naturally fluorescent product, excitable at 450 nm and emitting light at a wavelength of 530 nm. As it is of importance in fluorescence experimentation, we verified whether increased production of riboflavin, through overactivation of the cAMP-PKA signaling pathway, would have an effect on the autofluorescence level of the cells. As shown in Fig. S5a, addition of increasing amounts of dbcAMP significantly increases the cellular fluorescence at a wavelength of 531/40 nm, as measured by flow cytometry (excitation at 488 nm). We also showed increasing autofluorescence levels in the CaTPK1 and CaTPK2 overexpression strains (Fig. S5b).
Autofluorescence levels alter upon activation of the cAMP-PKA pathway. (a) SC5314 cultures were grown for 24 h in the presence of dbcAMP. (b) CaTPK1 and CaTPK2 overexpression strains were grown for 24 h, as was the empty vector (EV) control strain. The fluorescence was measured at 531/40-nm emission when excited with a 488-nm light. The average and the SEM of relative fluorescence to OD600 ratios are shown. Statistical analysis was performed using a one-way ANOVA statistical test with Bonferroni correction (comparison to culture without added compound or EV strain). RFU, relative fluorescence units. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Download FIG S5, EPS file, 1.3 MB (1.3MB, eps) .
Copyright © 2020 Demuyser et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Iron deprivation and PKA overactivation alter transcription of CaRIB synthesis genes.
We determined whether activation of cAMP-PKA signaling and iron deprivation could cause increased production of riboflavin via transcriptional regulation. To do so, we assessed gene expression of several riboflavin synthesis-associated genes in the presence of an iron chelator in the SC5314 wild-type strain as well as in CaTPK1 and CaTPK2 overexpression strains.
Figure 4a shows the effect of an iron chelator, ferrozine, on the expression of the main riboflavin biosynthesis enzyme-encoding genes. We show that expression of CaRIB1 and CaRIB4 is upregulated upon iron deprivation. This was confirmed by using the alternative iron chelator BPS (Fig. S6). As mentioned earlier, the open reading frame (ORF) of CaRIB1 was uncharacterized, yet the orthologue in S. cerevisiae is known to encode the first enzyme in riboflavin biosynthesis, a GTP cyclohydrolase II (27). CaRIB4 encodes the lumazine synthase (31). CaRIB7 is uncharacterized as well, and the S. cerevisiae orthologue encodes the 5-amino-6-(5-phosphoribosylamino)uracil reductase (27). In the presence of ferrozine and BPS, the expression of this gene is downregulated. In the presence of ferrozine, expression of CaRIB2 also is downregulated (Fig. 4a); however, this is not the case in the presence of BPS (Fig. S6). The S. cerevisiae orthologue of CaRIB2 encodes a 2,5-diamino-6-ribitylamino-4(3H)-pyrimidinone 5′-phosphate deaminase enzyme (27). Figure 4b shows the effect of PKA activation via CaTPK overexpression on the expression of the main riboflavin biosynthesis enzyme-encoding genes. Only expression of CaRIB1 was significantly upregulated in the CaTPK2 overexpression strain. Remarkably, expression of CaRIB5 was significantly downregulated in both overexpression strains. CaRIB1 expression is increased upon both iron limitation and PKA activation. This indicates that CaRib1 is involved in riboflavin synthesis as a central point of regulation.
FIG 4.

Expression of genes involved in riboflavin biosynthesis alters upon iron deprivation and PKA activation. (a) SC5314 cultures were grown for 8 h in the presence of ferrozine. (b) The empty vector (EV) control strain and CaTPK1 and CaTPK2 overexpression cultures were grown for 8 h. Gene expression was analyzed using qRT-PCR. Results are displayed as the average and SEM relative to the wild-type control. Statistical analysis was conducted on log2(Y)-transformed data using one-way ANOVA with Bonferroni correction. *, P < 0.05; **, P < 0.01; ****, P < 0.0001.
Expression of genes involved in riboflavin biosynthesis alters upon iron deprivation. SC5314 cultures were grown for 8 h in the presence of BPS. Gene expression was analyzed using qRT-PCR. Results are displayed as the average relative to the wild-type control and together with the SEM. Statistical analysis was conducted on log2(Y)-transformed data using one-way ANOVA with Bonferroni correction. *, P < 0.05; **, P < 0.01. Download FIG S6, EPS file, 1.8 MB (1.8MB, eps) .
Copyright © 2020 Demuyser et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
CaSef1 regulates riboflavin synthesis in an iron- and PKA-dependent manner.
CaSEF1 encodes a Zn2-Cys6 transcription factor that is reported to be involved in the regulation of iron uptake (32). In other organisms, Sef1 is also involved in regulation of riboflavin synthesis (8). This transcription factor was shown to play an essential role in regulation of RIB genes, although the exact mechanisms are not known yet (32, 33). We show that the expression of the gene encoding the transcriptional regulator CaSef1 is strongly induced under iron deprivation conditions, as presented in Fig. 5a. Expression of CaSEF1 was also significantly upregulated in both CaTPK1 and CaTPK2 overexpression strains, as can be seen in Fig. 5b.
FIG 5.

CaSef1 and its phosphorylation are necessary for riboflavin production in the absence of iron and presence of PKA activation. (a) SC5314 cultures were grown for 8 h in the presence of ferrozine. (b) The empty vector (EV) control strain and CaTPK1 and CaTPK2 overexpression cultures were grown for 8 h. Gene expression was analyzed using qRT-PCR. Results are displayed as the average and SEM relative to the wild-type control. Statistical analysis was conducted on log2(Y)-transformed data using one-way ANOVA with Bonferroni correction. SC5314, Casef1Δ/Δ, the CaSef1 phosphorylation mutant, and the reintegrant strain were grown for 24 h in the presence of ferrozine (c) or dbcAMP (d). The fluorescence was measured at 530 nm when excited upon 450 nm. The average and SEM of relative fluorescence to OD600 ratios are shown. Statistical analysis was performed using a two-way ANOVA statistical test with Bonferroni correction (comparison to culture without added compound). **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
To confirm the involvement of Sef1 in riboflavin synthesis in C. albicans and to verify whether this is modulated by iron levels and PKA activity, we compared riboflavin production in a Casef1Δ/Δ deletion strain to that of the SC5314 wild type in the presence of dbcAMP or ferrozine. As can be seen in Fig. 5c and d, respectively, deletion of CaSEF1 has no effect on riboflavin production in the absence of iron chelation or PKA stimulation. However, in the presence of low iron levels or upon PKA activation, the deletion strain is unable to synthesize as much riboflavin as the wild-type strain. This indicates that Sef1 is an essential link between the upstream input signals, iron limitation, and PKA activation and the downstream response, riboflavin synthesis.
We discovered, using the ScanSite tool (34), that CaSef1 contains two putative PKA phosphorylation sites, S132 and S676, and generated a mutant strain in which both serines are mutated to alanines, thereby preventing phosphorylation. Figure 5c and d show that this strain does produce significantly less riboflavin than the wild-type strain, indicating that the hypothesized phosphorylation of CaSef1 by PKA is essential under both PKA-activating as well as iron-limiting conditions.
CaSef1 but not its phosphorylation by PKA is essential for virulence in a mouse model of systemic infection.
It was shown earlier that CaSef1 is essential under iron-limiting conditions (32). To determine whether this transcription factor is also essential for full virulence in an in vivo context, we assessed toxicity against mammalian cells, imposed by the mutant strain compared to the wild-type SC5314 strain, as described earlier. Figure 6a reveals that neither the deletion strain nor the phosphorylation mutant show a decrease in toxicity imposed on HeLa cells. This indicates that neither CaSef1 nor its putative phosphorylation by PKA is essential for virulence in this model. Figure 6b confirms that no difference in adhesion to the mammalian cells can be detected.
FIG 6.
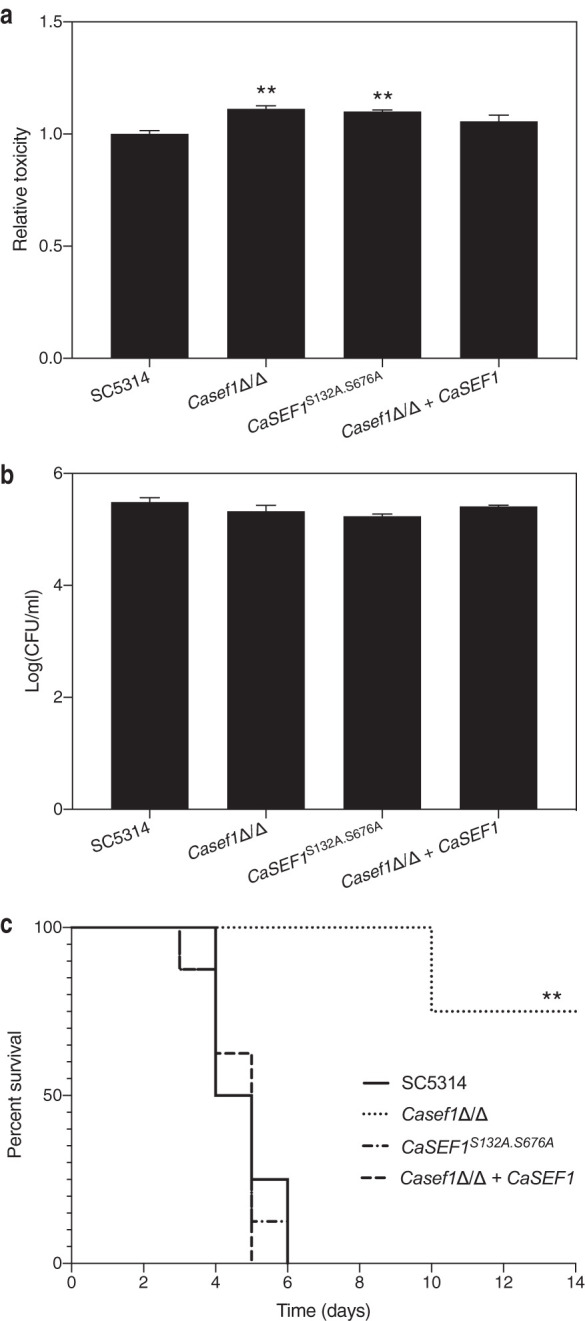
CaSef1 is essential for full virulence in a systemic mouse model of infection. (a) The Casef1Δ/Δ deletion strain as well as the wild-type, the phosphorylation mutant, and the reintegrant strains were added to mammalian HeLa cells. After 24 h of incubation at 37°C and 5% CO2, cytotoxicity was determined based on LDH release in the culture supernatant. Data are shown as average and SEM. Neither deletion nor mutation of CaSEF1 causes a decrease of cytotoxicity toward mammalian HeLa cells (determined by LDH activity). (b) The Casef1Δ/Δ deletion, phosphorylation mutant, wild-type, and reintegrant strains were added to mammalian HeLa cells. After 90 min of incubation at 37°C and 5% CO2, adhesion was determined based on number of CFU. Average values and SEM are shown. There is no significant difference between the mutant strains and the wild type. (c) Mice were infected with the same strains through tail vein injection. The C. albicans strain without CaSef1 is less virulent in a mouse model of systemic infection. Statistical analyses of panels a and b were performed by one-way ANOVA, with Bonferroni correction. **, P < 0.01. Statistical analysis of panel c was performed by a log-rank Mantel-Cox test. **, P < 0.01.
Since researchers reported earlier that the Casef1Δ/Δ deletion strain is required for full virulence in a mouse model of systemic infection, we tested this strain as well as the phosphorylation mutant in a similar model system (32). Figure 6c indeed shows that deleting both alleles of the CaSEF1 gene impairs full virulence in mice and that reintegration of the gene behind the constitutive CaACT1 promoter restores this phenotype. However, preventing putative phosphorylation by PKA at sites S132 and S676 does not alter virulence compared to that of the wild-type SC5314 strain. This implies that phosphorylation of CaSef1 at these specific sites does not play a major role in the in vivo context that we tested.
DISCUSSION
In the manuscript, we show that riboflavin production is a promising target for development of novel drugs against C. albicans. CaRib1 plays a central role in this process. By heterologous expression in S. cerevisiae, we confirmed the function of this enzyme in the riboflavin synthesis process, being a GTP cyclohydrolase. Deletion of this gene renders the strain nonviable in the absence of externally added riboflavin. Upon addition of riboflavin to the culture medium, the strain can be rescued, indicating that transport is possible. Thus, the potential of using CaRib1 as a drug target depends on the concentration of riboflavin that is present in the host niche where C. albicans is expected to be infectious. Based on the literature, it can be stated that the concentration of riboflavin in human blood are significantly lower than the minimal concentration needed to sustain growth in vitro. This is confirmed by our in vivo data, where we show that the Carib1Δ/Δ deletion strain is not able to cause any morbidity or mortality in a systemic mouse model of infection. Whether the concentration of riboflavin is adequate in other host niches and whether targeting CaRib1 would also be promising in other types of infections remain to be investigated. Apart from the essential character of CaRib1, this enzyme is interesting as an antifungal drug target in another respect as well. Upon overexpression of all RIB genes separately and measurement of the effect on riboflavin production, only overexpression of CaRIB1 leads to an increase in production, indicating that CaRib1 catalyzes a rate-limiting step in the process. Thus, targeting CaRib1 is thought to be more effective than targeting any of the other enzymes.
Upstream regulation of riboflavin production is not fully investigated in C. albicans. However, given the established importance of CaRib1 in the production process, it would be reasonable that upstream regulation targets this essential and rate-limiting factor in the process. We show that this is indeed the case. It was established earlier that iron deficiency stimulates production of riboflavin (25). We confirm this observation and show that under these conditions, expression of CaRIB1 is upregulated. It has been hypothesized by others that riboflavin acts as a type of siderophore important in the uptake of iron (8). This would explain why it is secreted in the culture medium, as it would have to scavenge iron externally. Whether this is indeed the case, however, will have to be investigated further. We also demonstrate that activating PKA increases riboflavin synthesis and, thereby, cellular autofluorescence. These findings have practical implications for cell biologists who seek to visualize green fluorescent protein-labeled proteins, as the wavelengths we used for excitation and emission detection of autofluorescence are the approximate wavelengths of this often-used fluorophore. Caution should be taken when performing quantitative fluorescence microscopy with C. albicans under PKA-altering conditions. Since CaRIB1 expression is upregulated upon activation of PKA, it is likely that regulation of riboflavin production happens mainly through CaRib1. A possible factor linking upstream inducing factors and CaRIB1 gene expression is the transcription factor CaSef1. This transcriptional regulator has been implicated in regulation of riboflavin synthesis in other organisms (8). We show that deletion of both alleles of the CaSEF1 gene impairs overproduction of riboflavin in the presence of an iron chelator or PKA activator. This indicates that the transcription factor is essential in signaling upstream cues, such as iron limitation or PKA activation, down to the riboflavin biosynthesis enzymes. We also noticed that under these specific conditions, expression of CaSEF1 itself is upregulated. It was reported earlier that CaSef1 can indeed regulate its own expression in a feedback loop through CaHap43 and CaSfu1, two other transcription factors involved in regulation of iron uptake (32). It has been shown as well that CaSef1 is activated under iron-limiting conditions. The mechanism by which PKA activity could be linked to CaSef1 remained largely elusive. We can discern two hypothetical models for PKA to affect riboflavin synthesis. A first way in which PKA can influence riboflavin production is indirect by affecting iron uptake in the cell. It was shown for S. cerevisiae that ScTpk2 negatively regulates iron uptake (35). Therefore, the overexpression of this gene could mimic iron deficiency. Second, it is possible that PKA influences the synthesis of this metabolite. Using the online protein motif search tool ScanSite to search for putative phosphorylation sequences in CaSef1, we found two potential PKA-specific phosphorylation sequences (34). Direct phosphorylation of CaSef1 would present a possible regulatory mechanism of riboflavin synthesis by PKA. We generated a mutant strain in which CaSef1 has two phosphorylatable serines replaced by alanines. In vitro data show that PKA phosphorylation could indeed be essential for its function, as the CaSEF1S132A.S676A mutant strain resembles the deletion mutant to a large extent. However, our in vivo data do not support this conclusion. The CaSEF1S132A.S676A mutant strain resembles the wild type and imposes high levels of virulence on the mice. It is possible, though, that other in vivo model systems show alternative results or that other phosphorylation sites exist. Another potential link between PKA and CaSef1 includes the protein kinase CaSsn3. PKA-regulated activation of CaSsn3 as well as CaSsn3-regulated activation of CaSef1 have been reported for C. albicans (36, 37). Figure 7 schematically depicts the hypothetical model of how riboflavin production can be regulated by iron availability as well as PKA activity. Further experimentation is necessary to unravel the exact regulation of riboflavin synthesis.
FIG 7.

Schematic representation of hypothetical iron- and PKA-dependent regulation of riboflavin synthesis. For four situations, high/low iron and high/low PKA activity, the hypothetical regulation of riboflavin synthesis is illustrated based on data from the manuscript as well as Chen et al. (32). Details can be found in the text. The higher the intensity/darkness of the depicted symbol or arrow, the higher the activity of the enzyme or reaction. Full arrows depict known interactions, striped arrows depict hypothetical, partially verified interactions, and dotted arrows depict purely hypothetical interactions. P, phosphate; PKA, protein kinase A.
The role of riboflavin production in microbial pathogenesis has been recognized relatively recently. It was shown for several pathogens, including C. albicans, that the production of this vitamin in nutrient-poor host niches is essential for survival and, thus, is important for virulence (6, 38–40). As riboflavin production is an essential process in C. albicans and no orthologues of the enzymes involved are present in human cells, riboflavin production is an attractive target for new antifungal drugs (6, 7).
MATERIALS AND METHODS
Strains and plasmids.
The C. albicans strains used in this study are listed in Table S1 in the supplemental material. Plasmids are summarized in Table S2 and primers in Table S3. For constructing overexpression and reintegrant C. albicans strains, CaTPK1, CaTPK2, CaSEF1, and CaRIB genes and CaFMN1 were cloned in the integrative plasmid CIp10-NAT1 (26, 41). CaTPK1 was integrated in the NheI-ClaI-cut Clp10-NAT1 vector, while CaTPK2, CaSEF1, CaFMN1, and all CaRIB genes were integrated in the NheI-AatII-cut Clp10-NAT1 vector. For integration, plasmids and the empty plasmid CIp10-NAT1 were linearized by StuI and transformed using a lithium acetate method (42). All overexpression strains, CaTPK1ox, CaTPK2ox, CaRIBox, and CaFMN1ox, and the empty vector control strain (EV) were generated in the SC5314 background. Integration was verified by diagnostic PCR. For each transformation, we obtained 4 independent transformants with equal copy number insertions. C. albicans rib1Δ/Δ and sef1Δ/Δ deletion strains as well as the CaSEF1S132A.S676A phosphorylation mutant were constructed in the background strain SC5314 using the CRISPR system described by Nguyen et al. (43). With their HIS-FLP split marker system, gRNA expression cassettes were amplified from plasmids pADH110 and pADH147 and cotransformed with plasmid pADH99 (CAS9 harboring) after digestion by MssI. Carib1Δ/Δ and Casef1Δ/Δ mutants were constructed with the guide RNAs 5′-TACATTGGTAGATTGACACC and 5′-TTACAATTAGGTCTACATCG, respectively. Duplexed oligonucleotide donor DNA (89 bp), comprising upstream and downstream sequences of the targeted gene, were provided as repair templates. For constructing the CaSEF1S132A.S676A phosphorylation mutant, serine (TCA) to alanine (GCC) mutations were introduced simultaneously using the guide RNAs 5′-TGGATTTGTGACCCTTTGCG and 5′-AAGACGATCAGTACTTGATA, targeting S132 and S676, respectively. Two 89-bp duplexed oligonucleotides containing the desired GCC mutation and 43-bp upstream and downstream homologous flanking sites were used simultaneously as donor DNA, generating strain CaSEF1S132A.S676A. After verifying the modification with diagnostic PCR, the NAT1 marker and CRISPR components were removed by induction of the FLP recombinase in YP maltose (2%). All deletions and mutations were confirmed by Sanger sequencing (Eurofins Genomics). By integrating plasmid CIp10-CaRIB1-NAT1 or CIp10-CaSEF1-NAT1 in their respective deletion strain, reintegrant strains Carib1Δ/Δ + CaRIB1 and Casef1Δ/Δ + CaSEF1 were constructed. Integration was verified by diagnostic PCR. For each transformation, we obtained at least 3 independent transformants with equal copy number insertions. For heterologous expression of CaRIB1 in the diploid S. cerevisiae strain S288c, a stepwise CRISPR/Cas9-based replacement methodology was applied. The CAS9- and gRNA-expressing plasmids were created by DiCarlo et al. and modified by Cen and coworkers (44, 45). First, both ScRIB1 alleles were replaced by deletion cassettes constitutively expressing dominant hygromycin and nourseothricin resistance markers and flanked by G1 gRNA recognition sites. The selection markers were amplified from plasmids pTOPO-G1-HPH-G1 and pTOPO-G1-NatMX-G1, with primers containing tails homologous to the ScRIB1 flanking regions. Correct integration of the cassettes was confirmed by PCR. Next, CAS9-expressing plasmid pTEF-CAS9-BLE was transformed. A G1-specific protospacer was assembled into pgRNA-uni-KanMX with the NEBuilder HiFi DNA assembly cloning kit (New England Biolabs) by following the manufacturer’s instructions. The design of this gRNA was based on the finding of Farboud and Meyer that Cas9-mediated DNA cleavage was enhanced at this G1 site (5′-GGCTGATTTTCGCAGTTCGGGGG) (46). This gRNA was cotransformed together with the appropriate donor DNA. To construct Scrib1Δ/Δ, donor DNA contained 89-bp duplexed oligonucleotide donor DNA, comprising upstream and downstream sequences. For heterologous expression of CaRIB1 in S288c, donor DNA was PCR amplified from SC5314 genomic DNA, with primers containing tails homologous to sequences upstream and downstream of ScRIB1. gRNA- and CAS9-expressing plasmids were lost by propagation under nonselective conditions. The genotype of transformants was confirmed by Sanger sequencing (Eurofins Genomics). C. albicans and S. cerevisiae strains were propagated in YPD (1% yeast extract [Merck], 2% Bacto peptone [Oxoid], 2% dextrose). For solid media, 2% Difco granulated agar was added. Where required, 200 mg/liter riboflavin (Sigma) was added. For selecting C. albicans transformants, 200 mg/liter nourseothricin (Jena Bioscience) was added. For transforming S. cerevisiae, 100 mg/liter nourseothricin (Jena Bioscience), 300 mg/liter hygromycin B (InvivoGen), 10 mg/liter phleomycin (InvivoGen), and/or 200 mg/liter Geneticin G418 sulfate (ThermoFisher) were added.
Strains used in this study. Download Table S1, DOCX file, 0.02 MB (19.9KB, docx) .
Copyright © 2020 Demuyser et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Plasmids used in this study. Download Table S2, DOCX file, 0.02 MB (21.8KB, docx) .
Copyright © 2020 Demuyser et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Primers used in this study. Download Table S3, DOCX file, 0.02 MB (18.2KB, docx) .
Copyright © 2020 Demuyser et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Growth conditions: media and chemicals.
C. albicans strains were grown in LoFloglu medium. This medium contains 0.079% complete supplement mixture (CSM; MP Biomedicals), 0.19% yeast nitrogen base without amino acids and lacking folic acid and riboflavin (Formedium), 0.5% ammonium sulfate (Fluka), and 2% glucose. The pH was adapted to 5.5 in liquid medium. For some experiments, the growth medium of the wild-type strain SC5314 was supplemented with different concentrations of FeCl3 (Sigma), Fe2(SO4)3 (Sigma), BPS (Sigma), ferrozine (Sigma), or dibutyryl-cAMP (Sigma).
Riboflavin measurement in cell supernatant.
C. albicans strains were grown overnight at 30°C in 3 ml LoFloglu medium. The overnight cultures were adapted to an optical density at 600 nm (OD600) of 0.1 in 3 ml LoFloglu medium and grown at 30°C. In selected experiments, FeCl3 (0.1, 0.2, and 0.5 mM), Fe2(SO4)3 (0.1, 0.2, and 0.5 mM), BPS (0.02, 0.05, and 0.1 mg/ml), ferrozine (0.2, 1, and 5 mM), or dbcAMP (0.5, 2, and 5 mg/ml) was added to the new medium. After 24 h of growth, 1 ml of the cell cultures was transferred to Eppendorf tubes and the OD600 was measured. After centrifugation, 0.8 ml of the cell supernatant was transferred to a new tube. The centrifugation step was repeated, and 0.6 ml cell supernatant was retrieved and used for further analysis in the Synergy H1 hybrid multimode microplate reader (BioTek). Wells of a black-walled 96-well microtiter plate (Greiner) were filled with 0.2 ml cell supernatant, and absorbance at 445 nm was measured. Fluorescence measurements were performed using emission at 530 nm upon excitation at 450 nm. For all quantitative experiments, the ratio of this fluorescence to the OD600 is shown. In some of the experiments, the presence of riboflavin in the culture supernatant was imaged using blue light of 460 nm.
Autofluorescence measurement by FACS.
C. albicans strains were grown overnight at 30°C in 3 ml LoFloglu medium. The overnight cultures were adapted to an OD600 of 0.1 in 3 ml LoFloglu medium and grown at 30°C. For the wild-type SC5314 strain, dbcAMP (2 and 5 mg/ml) was added to the new medium. After 24 h the cells were washed 3 times and adjusted to an OD600 of 1 in LoFloglu medium. Flow cytometry was performed using the BD influx flow cytometer. The samples were analyzed using a blue laser (488 nm) in combination with an emission filter of 531/40 nm.
Copy number determination by qPCR.
After transformation of SC5314 with the overexpression plasmids, genomic DNA was extracted from the transformants. Quantitative PCR (qPCR) was performed using GoTaq polymerase (Promega) and the StepOnePlus real-time PCR device (ThermoFisher). qPCRs were conducted on 2.5 ng genomic DNA per reaction, with primers for the CaACT1 promoter region of the CIp10 plasmid and Ca18S, CaTEF1, and CaACT1 as reference genes. Primers used for copy number determination are listed in Table S3. qBasePlus software (Biogazelle) was used for copy number analysis.
RNA extraction and gene expression analysis by qRT-PCR.
C. albicans strains were grown overnight at 30°C in 5 ml LoFloglu medium. The overnight cultures were adapted to an OD600 of 0.1 in 50 ml LoFloglu medium and grown at 30°C for 8 h. For the wild-type SC5314 strain, BPS (0.05 mg/ml) and ferrozine (5 mM) were also added to the medium. Cells were harvested and washed with ice-cold water. After resuspension in TRIzol (ThermoFisher), cells were broken with glass beads using a FastPrep machine (MP Biomedicals). The addition of chloroform, isopropanol, and three washes with 70% ethanol resulted in the isolation of pure RNA. The RNA concentration was quantified using a NanoDrop spectrophotometer (ND-1000; Isogen Life Science). Equal amounts of RNA were treated with DNase (New England Biolabs) and then converted to cDNA using the iScript cDNA synthesis kit (Bio-Rad). Real-time quantitative PCRs (qRT-PCR) were performed using GoTaq polymerase (Promega) and the StepOnePlus real-time PCR device (ThermoFisher). Primers used for qPCR are listed in Table S3. qBasePlus software was used for gene expression analysis. Statistical analysis of the results was performed using GraphPad Prism. Expression data were log2(Y) transformed to enable the use of standard statistical methods. Graphs show the untransformed data with the standard errors of the means (SEM). The statistical method used is depicted under each graph.
Mammalian cell culture.
HeLa cells, derived from a human cervical carcinoma, were kindly donated by Frank Claessens (KU Leuven). These cells were cultured in Dulbecco’s modified Eagle medium (DMEM) from Gibco, complemented with GlutaMAX (Gibco), 10% fetal bovine serum, and gentamicin (Gibco). Cells were seeded in cell culture flasks and incubated at 37°C and 5% CO2 in air.
Mammalian cell toxicity determination.
Cell toxicity imposed by C. albicans cells was measured through release of LDH in the culture supernatant by using the CyQUANT LDH cytotoxicity assay from ThermoFisher. HeLa cells were seeded in special 96-well microtiter plates (Nunclon Delta Surface; ThermoFisher) at 105 cells per well in 100 μl and allowed to adhere to the bottom for 24 h at 37°C and under 5% CO2. Candida cells were added to a final concentration of 105 cells per well and allowed to infect the cells for another 24 h under the same conditions. According to the manufacturer’s guidelines, lysis buffer was added to a few wells as a positive control and phosphate-buffered saline (PBS) as a negative control. After incubation for 45 min, 50 μl of the cell supernatant was transferred to a normal 96-well microtiter plate, and 50 μl of reaction mixture was added. After 30 min of incubation at room temperature and protected from light, 50 μl stop buffer was added and the OD was determined spectrophotometrically at 490 and 680 nm. The toxicity in wells where lysis buffer was added was set to 100%, and the toxicity imposed by the Candida cells was determined relative to this control.
Adhesion to mammalian cells.
Adhesion of Candida cells to mammalian HeLa cells was assessed using the following procedure. HeLa cells were seeded at a density of 105 cells per well in 100 μl and allowed to adhere and grow for 48 h at 37°C and under 5% CO2. Candida cells were added to a final concentration of 105 cells per well and allowed to infect the cells for 90 min under the same conditions. The cells were washed twice using PBS, and 25 μl trypsin was added per well and incubated for 10 min. After addition of 175 μl DMEM, cells were resuspended, diluted, and plated onto YPD agar plates. Numbers of CFU were determined.
Mouse model of systemic infection.
All experiments involving mice were approved by the Ethical Committee for Animal Experimentation of KU Leuven. Each treatment group contained eight female BALB/c mice of 6 to 8 weeks of age. The sample size was determined based on analysis by G*Power software (47). The Candida strains were grown overnight in YPD, washed twice using PBS, and injected at 8.5 × 105 cells per mouse via the tail vein. After infection, the mice were inspected daily for morbidity and mortality. Upon signs of severe morbidity, such as over 15% weight loss, severely hunched back, or ruffled fur, the mice were euthanized using isoflurane gas and cervical dislocation.
Statistical analysis.
The sample size of the in vitro experiments varied from 3 to 4 samples within each group. For the SC5314 wild-type strain, individual colonies were taken as replicates. For mutant strains, independent transformants were isolated and used. Displayed parameters, statistical methods used, and corrections applied are clarified in the figure legends.
ACKNOWLEDGMENTS
L.D. was supported by a personal research grant from the FWO (Fund for Scientific Research Flanders- 11P9814N). This work was further supported by a grant from the KU Leuven Research Fund (C14/17/063). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
We thank J. Thevelein for kindly providing us with the pTOPO-G1-HPH-G1 and pTOPO-G1-NatMX-G1 plasmids.
L.D., I.P., and P.V. conceived the experiments. L.D. and I.P. performed the experiments. All authors contributed to discussion of the results.
We have no conflicts of interest to declare.
REFERENCES
- 1.Sobel JD. 2007. Vulvovaginal candidosis. Lancet 369:1961–1971. doi: 10.1016/S0140-6736(07)60917-9. [DOI] [PubMed] [Google Scholar]
- 2.Goncalves B, Ferreira C, Alves CT, Henriques M, Azeredo J, Silva S. 2016. Vulvovaginal candidiasis: epidemiology, microbiology and risk factors. Crit Rev Microbiol 42:905–927. doi: 10.3109/1040841X.2015.1091805. [DOI] [PubMed] [Google Scholar]
- 3.Brown GD, Denning DW, Gow NA, Levitz SM, Netea MG, White TC. 2012. Hidden killers: human fungal infections. Sci Transl Med 4:165rv13. doi: 10.1126/scitranslmed.3004404. [DOI] [PubMed] [Google Scholar]
- 4.Gonzalez-Lara MF, Sifuentes-Osornio J, Ostrosky-Zeichner L. 2017. Drugs in clinical development for fungal infections. Drugs 77:1505–1518. doi: 10.1007/s40265-017-0805-2. [DOI] [PubMed] [Google Scholar]
- 5.Deray G. 2002. Amphotericin B nephrotoxicity. J Antimicrob Chemother 49(Suppl 1):37–41. doi: 10.1093/jac/49.suppl_1.37. [DOI] [PubMed] [Google Scholar]
- 6.Becker JM, Kauffman SJ, Hauser M, Huang L, Lin M, Sillaots S, Jiang B, Xu D, Roemer T. 2010. Pathway analysis of Candida albicans survival and virulence determinants in a murine infection model. Proc Natl Acad Sci U S A 107:22044–22049. doi: 10.1073/pnas.1009845107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dietl AM, Meir Z, Shadkchan Y, Osherov N, Haas H. 2018. Riboflavin and pantothenic acid biosynthesis are crucial for iron homeostasis and virulence in the pathogenic mold Aspergillus fumigatus. Virulence 9:1036–1049. doi: 10.1080/21505594.2018.1482181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abbas CA, Sibirny AA. 2011. Genetic control of biosynthesis and transport of riboflavin and flavin nucleotides and construction of robust biotechnological producers. Microbiol Mol Biol Rev 75:321–360. doi: 10.1128/MMBR.00030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gnandt E, Dorner K, Strampraad MFJ, de Vries S, Friedrich T. 2016. The multitude of iron-sulfur clusters in respiratory complex I. Biochim Biophys Acta 1857:1068–1072. doi: 10.1016/j.bbabio.2016.02.018. [DOI] [PubMed] [Google Scholar]
- 10.Wang Z, Chen T, Ma X, Shen Z, Zhao X. 2011. Enhancement of riboflavin production with Bacillus subtilis by expression and site-directed mutagenesis of zwf and gnd gene from Corynebacterium glutamicum. Bioresour Technol 102:3934–3940. doi: 10.1016/j.biortech.2010.11.120. [DOI] [PubMed] [Google Scholar]
- 11.Dmytruk K, Lyzak O, Yatsyshyn V, Kluz M, Sibirny V, Puchalski C, Sibirny A. 2014. Construction and fed-batch cultivation of Candida famata with enhanced riboflavin production. J Biotechnol 172:11–17. doi: 10.1016/j.jbiotec.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 12.Vogl C, Grill S, Schilling O, Stulke J, Mack M, Stolz J. 2007. Characterization of riboflavin (vitamin B2) transport proteins from Bacillus subtilis and Corynebacterium glutamicum. J Bacteriol 189:7367–7375. doi: 10.1128/JB.00590-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dahl SG, Sylte I, Ravna AW. 2004. Structures and models of transporter proteins. J Pharmacol Exp Ther 309:853–860. doi: 10.1124/jpet.103.059972. [DOI] [PubMed] [Google Scholar]
- 14.Mack M, Grill S. 2006. Riboflavin analogs and inhibitors of riboflavin biosynthesis. Appl Microbiol Biotechnol 71:265–275. doi: 10.1007/s00253-006-0421-7. [DOI] [PubMed] [Google Scholar]
- 15.Long Q, Ji L, Wang H, Xie J. 2010. Riboflavin biosynthetic and regulatory factors as potential novel anti-infective drug targets. Chem Biol Drug Des 75:339–347. doi: 10.1111/j.1747-0285.2010.00946.x. [DOI] [PubMed] [Google Scholar]
- 16.Zhao Y, Bacher A, Illarionov B, Fischer M, Georg G, Ye QZ, Fanwick PE, Franzblau SG, Wan B, Cushman M. 2009. Discovery and development of the covalent hydrates of trifluoromethylated pyrazoles as riboflavin synthase inhibitors with antibiotic activity against Mycobacterium tuberculosis. J Org Chem 74:5297–5303. doi: 10.1021/jo900768c. [DOI] [PubMed] [Google Scholar]
- 17.Cushman M, Jin G, Sambaiah T, Illarionov B, Fischer M, Ladenstein R, Bacher A. 2005. Design, synthesis, and biochemical evaluation of 1,5,6,7-tetrahydro-6,7-dioxo-9-D-ribitylaminolumazines bearing alkyl phosphate substituents as inhibitors of lumazine synthase and riboflavin synthase. J Org Chem 70:8162–8170. doi: 10.1021/jo051332v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morgunova E, Illarionov B, Saller S, Popov A, Sambaiah T, Bacher A, Cushman M, Fischer M, Ladenstein R. 2010. Structural study and thermodynamic characterization of inhibitor binding to lumazine synthase from Bacillus anthracis. Acta Crystallogr D Biol Crystallogr 66:1001–1011. doi: 10.1107/S0907444910029690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Serer MI, Carrica MDC, Trappe J, Lopez Romero S, Bonomi HR, Klinke S, Cerutti ML, Goldbaum FA. 2019. A high-throughput screening for inhibitors of riboflavin synthase identifies novel antimicrobial compounds to treat brucellosis. FEBS J 286:2522–2535. doi: 10.1111/febs.14829. [DOI] [PubMed] [Google Scholar]
- 20.Echt S, Bauer S, Steinbacher S, Huber R, Bacher A, Fischer M. 2004. Potential anti-infective targets in pathogenic yeasts: structure and properties of 3,4-dihydroxy-2-butanone 4-phosphate synthase of Candida albicans. J Mol Biol 341:1085–1096. doi: 10.1016/j.jmb.2004.06.053. [DOI] [PubMed] [Google Scholar]
- 21.Morgunova E, Saller S, Haase I, Cushman M, Bacher A, Fischer M, Ladenstein R. 2007. Lumazine synthase from Candida albicans as an anti-fungal target enzyme: structural and biochemical basis for drug design. J Biol Chem 282:17231–17241. doi: 10.1074/jbc.M701724200. [DOI] [PubMed] [Google Scholar]
- 22.Skrzypek MS, Binkley J, Binkley G, Miyasato SR, Simison M, Sherlock G. 2017. The Candida Genome Database (CGD): incorporation of Assembly 22, systematic identifiers and visualization of high throughput sequencing data. Nucleic Acids Res 45:D592–D596. doi: 10.1093/nar/gkw924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gudipati V, Koch K, Lienhart WD, Macheroux P. 2014. The flavoproteome of the yeast Saccharomyces cerevisiae. Biochim Biophys Acta 1844:535–544. doi: 10.1016/j.bbapap.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fischer M, Bacher A. 2005. Biosynthesis of flavocoenzymes. Nat Prod Rep 22:324–350. doi: 10.1039/b210142b. [DOI] [PubMed] [Google Scholar]
- 25.Knight SA, Lesuisse E, Stearman R, Klausner RD, Dancis A. 2002. Reductive iron uptake by Candida albicans: role of copper, iron and the TUP1 regulator. Microbiology 148:29–40. doi: 10.1099/00221287-148-1-29. [DOI] [PubMed] [Google Scholar]
- 26.Demuyser L, Swinnen E, Fiori A, Herrera-Malaver B, Verstrepen K, Van Dijck P. 2017. Mitochondrial cochaperone Mge1 is involved in regulating susceptibility to fluconazole in Saccharomyces cerevisiae and Candida species. mBio 8:e00201-17. doi: 10.1128/mBio.00201-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oltmanns O, Bacher A. 1972. Biosynthesis of riboflavine in Saccharomyces cerevisiae: the role of genes rib 1 and rib 7. J Bacteriol 110:818–822. doi: 10.1128/JB.110.3.818-822.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Priemer DS. 2018. Vitamin B2 (riboflavin). https://emedicine.medscape.com/article/2088605-overview. Accessed 16 March 2020.
- 29.Bai C, Xu XL, Wang HS, Wang YM, Chan FY, Wang Y. 2011. Characterization of a hyperactive Cyr1 mutant reveals new regulatory mechanisms for cellular cAMP levels in Candida albicans. Mol Microbiol 82:879–893. doi: 10.1111/j.1365-2958.2011.07859.x. [DOI] [PubMed] [Google Scholar]
- 30.Stahmann KP, Arst HN, Jr, Althofer H, Revuelta JL, Monschau N, Schlupen C, Gatgens C, Wiesenburg A, Schlosser T. 2001. Riboflavin, overproduced during sporulation of Ashbya gossypii, protects its hyaline spores against ultraviolet light. Environ Microbiol 3:545–550. doi: 10.1046/j.1462-2920.2001.00225.x. [DOI] [PubMed] [Google Scholar]
- 31.Nett JE, Lepak AJ, Marchillo K, Andes DR. 2009. Time course global gene expression analysis of an in vivo Candida biofilm. J Infect Dis 200:307–313. doi: 10.1086/599838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen C, Pande K, French SD, Tuch BB, Noble SM. 2011. An iron homeostasis regulatory circuit with reciprocal roles in Candida albicans commensalism and pathogenesis. Cell Host Microbe 10:118–135. doi: 10.1016/j.chom.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dmytruk KV, Voronovsky AY, Sibirny AA. 2006. Insertion mutagenesis of the yeast Candida famata (Debaryomyces hansenii) by random integration of linear DNA fragments. Curr Genet 50:183–191. doi: 10.1007/s00294-006-0083-0. [DOI] [PubMed] [Google Scholar]
- 34.Obenauer JC, Cantley LC, Yaffe MB. 2003. Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res 31:3635–3641. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robertson LS, Causton HC, Young RA, Fink GR. 2000. The yeast A kinases differentially regulate iron uptake and respiratory function. Proc Natl Acad Sci U S A 97:5984–5988. doi: 10.1073/pnas.100113397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wartenberg A, Linde J, Martin R, Schreiner M, Horn F, Jacobsen ID, Jenull S, Wolf T, Kuchler K, Guthke R, Kurzai O, Forche A, d'Enfert C, Brunke S, Hube B. 2014. Microevolution of Candida albicans in macrophages restores filamentation in a nonfilamentous mutant. PLoS Genet 10:e1004824. doi: 10.1371/journal.pgen.1004824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen C, Noble SM. 2012. Post-transcriptional regulation of the Sef1 transcription factor controls the virulence of Candida albicans in its mammalian host. PLoS Pathog 8:e1002956. doi: 10.1371/journal.ppat.1002956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bonomi HR, Marchesini MI, Klinke S, Ugalde JE, Zylberman V, Ugalde RA, Comerci DJ, Goldbaum FA. 2010. An atypical riboflavin pathway is essential for Brucella abortus virulence. PLoS One 5:e9435. doi: 10.1371/journal.pone.0009435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Flieger M, Bandouchova H, Cerny J, Chudíčková M, Kolarik M, Kovacova V, Martínková N, Novák P, Šebesta O, Stodůlková E, Pikula J. 2016. Vitamin B2 as a virulence factor in Pseudogymnoascus destructans skin infection. Sci Rep 6:33200. doi: 10.1038/srep33200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garfoot AL, Zemska O, Rappleye CA. 2014. Histoplasma capsulatum depends on de novo vitamin biosynthesis for intraphagosomal proliferation. Infect Immun 82:393–404. doi: 10.1128/IAI.00824-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fonzi WA, Irwin MY. 1993. Isogenic strain construction and gene mapping in Candida albicans. Genetics 134:717–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gietz RD, Schiestl RH, Willems AR, Woods RA. 1995. Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast 11:355–360. doi: 10.1002/yea.320110408. [DOI] [PubMed] [Google Scholar]
- 43.Nguyen N, Quail MMF, Hernday AD. 2017. An efficient, rapid, and recyclable system for CRISPR-mediated genome editing in Candida albicans. mSphere 2:e00149-17. doi: 10.1128/mSphereDirect.00149-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.DiCarlo JE, Norville JE, Mali P, Rios X, Aach J, Church GM. 2013. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res 41:4336–4343. doi: 10.1093/nar/gkt135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cen Y, Timmermans B, Souffriau B, Thevelein JM, Van Dijck P. 2017. Comparison of genome engineering using the CRISPR-Cas9 system in C. glabrata wild-type and lig4 strains. Fungal Genet Biol 107:44–50. doi: 10.1016/j.fgb.2017.08.004. [DOI] [PubMed] [Google Scholar]
- 46.Farboud B, Meyer BJ. 2015. Dramatic enhancement of genome editing by CRISPR/Cas9 through improved guide RNA design. Genetics 199:959–971. doi: 10.1534/genetics.115.175166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Faul F, Erdfelder E, Lang AG, Buchner A. 2007. G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods 39:175–191. doi: 10.3758/bf03193146. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Schematic representation of the riboflavin biosynthesis pathway in S. cerevisiae. Based on homology with S. cerevisiae, C. albicans enzymes are depicted alongside the reactions, the enzymes for which the open reading frames are uncharacterized are shown in orange, and the others are in blue. More details are given in the text. This figure uses data from references 23 and 24. Download FIG S1, EPS file, 2.7 MB (2.8MB, eps) .
Copyright © 2020 Demuyser et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Fluorescence characteristics of riboflavin and culture supernatant. (a) Excitation and emission spectra of riboflavin, dissolved in LoFloglu medium, were determined. (b) Likewise, excitation and emission spectra of sterilized 24-h-old SC5314 culture supernatants were evaluated. (c) A standard curve of the fluorescence emission of riboflavin was set up. RFU, relative fluorescence units. Download FIG S2, EPS file, 1.5 MB (1.5MB, eps) .
Copyright © 2020 Demuyser et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Riboflavin production alters upon iron addition or sequestration. SC5314 cultures were grown for 24 h in the presence of Fe2(SO4)3 (a) or an iron chelator, BPS (b). The fluorescence was measured at 530 nm when excited upon 450 nm. The average and the SEM of relative fluorescence to OD600 ratios are shown. Statistical analysis was performed using a one-way analysis of variance (ANOVA) statistical test with Bonferroni correction (comparison to culture without added compound). *, P < 0.05; **, P < 0.01. Download FIG S3, EPS file, 1.3 MB (1.6MB, eps) .
Copyright © 2020 Demuyser et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Empirical observation of riboflavin production in the presence of iron chelators. SC5314 cultures were grown for 24 h in the presence of 0, 0.2, 1, and 5 mM ferrozine (a) or 0, 0.02, 0.05, and 0.1 mg/ml BPS (c). Blank medium with chelator was incubated to compare ferrozine (b) and BPS (d). Images were taken of the cultures (upper), and cultures were spun down and imaged using blue light at 460 nm (lower). Download FIG S4, TIF file, 2.6 MB (2.6MB, tif) .
Copyright © 2020 Demuyser et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Autofluorescence levels alter upon activation of the cAMP-PKA pathway. (a) SC5314 cultures were grown for 24 h in the presence of dbcAMP. (b) CaTPK1 and CaTPK2 overexpression strains were grown for 24 h, as was the empty vector (EV) control strain. The fluorescence was measured at 531/40-nm emission when excited with a 488-nm light. The average and the SEM of relative fluorescence to OD600 ratios are shown. Statistical analysis was performed using a one-way ANOVA statistical test with Bonferroni correction (comparison to culture without added compound or EV strain). RFU, relative fluorescence units. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Download FIG S5, EPS file, 1.3 MB (1.3MB, eps) .
Copyright © 2020 Demuyser et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Expression of genes involved in riboflavin biosynthesis alters upon iron deprivation. SC5314 cultures were grown for 8 h in the presence of BPS. Gene expression was analyzed using qRT-PCR. Results are displayed as the average relative to the wild-type control and together with the SEM. Statistical analysis was conducted on log2(Y)-transformed data using one-way ANOVA with Bonferroni correction. *, P < 0.05; **, P < 0.01. Download FIG S6, EPS file, 1.8 MB (1.8MB, eps) .
Copyright © 2020 Demuyser et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Strains used in this study. Download Table S1, DOCX file, 0.02 MB (19.9KB, docx) .
Copyright © 2020 Demuyser et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Plasmids used in this study. Download Table S2, DOCX file, 0.02 MB (21.8KB, docx) .
Copyright © 2020 Demuyser et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Primers used in this study. Download Table S3, DOCX file, 0.02 MB (18.2KB, docx) .
Copyright © 2020 Demuyser et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.